Nitric Oxide-Mediated Enhancement and Reversal of Resistance of Anticancer Therapies


Abstract
1. Introduction
1.1. Cancer and Conventional Therapies
1.2. Resistance to Various Therapies and Mechanisms
1.3. Reversal of Resistance
2. Nitric Oxide and Cancer
2.1. Introduction
2.2. Dual Role of NO in Cancer Biology
2.3. Role in Apoptosis
2.3.1. As a Pro-Apoptotic Regulator
2.3.2. As an Anti-Apoptotic Regulator
2.4. Role as an Immune Mediator
3. Nitric Oxide in Overcoming Immune and Chemo Resistance
4. NO Induction and NO Donors in Cancer Therapy
5. Future Perspectives and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, B.; Carlsson, G.; Machover, D.; Petrelli, N.; Roth, A.; Schmoll, H.J.; Tveit, K.M.; Gibson, F. A review of the evolution of systemic chemotherapy in the management of colorectal cancer. Clin. Colorectal Cancer 2015, 14, 1–10. [Google Scholar] [CrossRef]
- Pilkington, G.; Boland, A.; Brown, T.; Oyee, J.; Bagust, A.; Dickson, R. A systematic review of the clinical effectiveness of first-line chemotherapy for adult patients with locally advanced or metastatic non-small cell lung cancer. Thorax 2015, 70, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Hajatdoost, L.; Sedaghat, K.; Walker, E.; Thomas, J.; Kosari, S. Chemotherapy in Pancreatic Cancer: A Systematic Review. Medicina 2018, 54, 48. [Google Scholar] [CrossRef] [PubMed]
- Lilenbaum, R.C.; Herndon, J.E.; List, M.A.; Desch, C.; Watson, D.M.; Miller, A.A.; Perry, M.C.; Saville, W.; Chahinian, P.; Weeks, J.C.; et al. Single-Agent Versus Combination Chemotherapy in Advanced Non–Small-Cell Lung Cancer: The Cancer and Leukemia Group B (study 9730). J. Clin. Oncol. 2005, 23, 190–196. [Google Scholar] [CrossRef]
- Carrick, S.; Parker, S.; Thornton, C.E.; Ghersi, D.; Simes, J.; Wilcken, N. Single agent versus combination chemotherapy for metastatic breast cancer. Cochrane Database Syst. Rev. 2009. [Google Scholar] [CrossRef]
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Front. Oncol. 2013, 3, 113. [Google Scholar] [CrossRef]
- Kapiteijn, E.; Marijnen, C.A.; Nagtegaal, I.D.; Putter, H.; Steup, W.H.; Wiggers, T.; Rutten, H.J.; Pahlman, L.; Glimelius, B.; van Krieken, J.H.J.; et al. Preoperative radiotherapy combined with total mesorectal excision for resectable rectal cancer. N. Engl. J. Med. 2001, 345, 638–646. [Google Scholar] [CrossRef]
- Bosset, J.F.; Collette, L.; Calais, G.; Mineur, L.; Maingon, P.; Radosevic-Jelic, L.; Daban, A.; Bardet, E.; Beny, A.; Ollier, J.C. Chemotherapy with preoperative radiotherapy in rectal cancer. N. Engl. J. Med. 2006, 355, 1114–1123. [Google Scholar] [CrossRef]
- Bernstein, M.B.; Krishnan, S.; Hodge, J.W.; Chang, J.Y. Immunotherapy and stereotactic ablative radiotherapy (ISABR): A curative approach? Nat. Rev. Clin. Oncol. 2016, 13, 516–524. [Google Scholar] [CrossRef]
- Kang, J.; Demaria, S.; Formenti, S. Current clinical trials testing the combination of immunotherapy with radiotherapy. J. Immunother. Cancer 2016, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Weichselbaum, R.R.; Liang, H.; Deng, L.; Fu, Y.X. Radiotherapy and immunotherapy: A beneficial liaison? Nat. Rev. Clin. Oncol. 2017, 14, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 2016, 14, 73. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; Matson, V.; Flood, B.; Spranger, S.; Gajewski, T.F. Innate immune signaling and regulation in cancer immunotherapy. Cell Res. 2017, 27, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Cording, S.; Medvedovic, J.; Aychek, T.; Eberl, G. Innate lymphoid cells in defense, immunopathology and immunotherapy. Nat. Immunol. 2016, 17, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Crinier, A.; Vivier, E.; Bléry, M. Helper-like innate lymphoid cells and cancer immunotherapy. In Seminars in Immunology; Academic Press: San Diego, CA, USA, 2019. [Google Scholar]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther.-Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Taldone, T.; Gozman, A.; Maharaj, R.; Chiosis, G. Targeting Hsp90: Small-molecule inhibitors and their clinical development. Curr. Opin. Pharmacol. 2008, 8, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Vogler, M.; Dinsdale, D.; Dyer, M.J.; Cohen, G.M. Bcl-2 inhibitors: Small molecules with a big impact on cancer therapy. Cell Death Differ. 2009, 16, 360–367. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Dong, G.; Wu, S.; Fang, K.; Miao, Z.; Wang, W.; Sheng, C. Small molecules simultaneously inhibiting p53-murine double minute 2 (MDM2) interaction and histone deacetylases (HDACs): Discovery of novel multitargeting antitumor agents. J. Med. Chem. 2018, 61, 7245–7260. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhou, M.; Li, Z.; Li, H.; Polaczek, P.; Dai, H.; Wu, Q.; Liu, C.; Karanja, K.K.; Popuri, V.; et al. A selective small molecule DNA2 inhibitor for sensitization of human cancer cells to chemotherapy. EBioMedicine 2016, 6, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Bruens, S.T.; Milanese, C.; Verkaik, N.; Mastroberardino, P.; Gyenis, A.; Chang, J.; Derks, K.; Wiemer, E.; Jenster, G. Mapping mechanisms of radiotherapy resistance in prostate cancer. Cancer Res. 2016, 76, 1657. [Google Scholar]
- Chabner, B.A.; Roberts, T.G., Jr. Chemotherapy and the war on cancer. Nat. Rev. Cancer 2005, 5, 65–72. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Long, G.V.; Scolyer, R.A.; Teng, M.W.; Smyth, M.J. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat. Rev. 2017, 52, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.; Paret, C.; El Malki, K.; Alt, F.; Wingerter, A.; Neu, M.A.; Kron, B.; Russo, A.; Lehmann, N.; Roth, L.; et al. CD19 isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J. Immunother. 2017, 40, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Chang, C.M.; Yuan, M.; McKenna, W.G.; Shu, H.K.G. Resistance to small molecule inhibitors of epidermal growth factor receptor in malignant gliomas. Cancer Res. 2003, 63, 7443–7450. [Google Scholar] [CrossRef]
- Jia, Y.; Yun, C.H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR (T790M) and EGFR (C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129–132. [Google Scholar] [CrossRef]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef]
- Löscher, W.; Potschka, H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat. Rev. Neurosci. 2005, 6, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Hays, E.; Bonavida, B. YY1 regulates cancer cell immune resistance by modulating PD-L1 expression. Drug Resist. Updates 2019, 43, 10–28. [Google Scholar] [CrossRef] [PubMed]
- Baritaki, S.; Huerta-Yepez, S.; Sakai, T.; Spandidos, D.A.; Bonavida, B. Chemotherapeutic drugs sensitize cancer cells to TRAIL-mediated apoptosis: Up-regulation of DR5 and inhibition of Yin Yang 1. Mol. Cancer Ther. 2007, 6, 1387–1399. [Google Scholar] [CrossRef] [PubMed]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Vormehr, M.; Van de Roemer, N.; Diken, M.; Löwer, M.; Diekmann, J.; Schrörs, B.; Vascotto, F.; Castle, J.C.; Tadmor, A.D.; et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015, 520, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Lyford-Pike, S.; Peng, S.; Young, G.D.; Taube, J.M.; Westra, W.H.; Akpeng, B.; Bruno, T.C.; Richmon, J.D.; Wang, H.; Bishop, J.A.; et al. Evidence for a role of the PD-1: PD-L1 pathway in immune resistance of HPV-associated head and neck squamous cell carcinoma. Cancer Res. 2013, 73, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN promotes resistance to T cell–mediated immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yuan, Y.; Chen, W.; Putra, J.; Suriawinata, A.A.; Schenk, A.D.; Miller, H.E.; Guleria, I.; Barth, R.J.; Huang, Y.H.; et al. Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc. Natl. Acad. Sci. USA 2015, 112, 6682–6687. [Google Scholar] [CrossRef] [PubMed]
- Almand, B.; Clark, J.I.; Nikitina, E.; van Beynen, J.; English, N.R.; Knight, S.C.; Carbone, D.P.; Gabrilovich, D.I. Increased production of immature myeloid cells in cancer patients: A mechanism of immunosuppression in cancer. J. Immunol. 2001, 166, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kortylewski, M.; Pardoll, D. Tumour immunology: Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Kudo-Saito, C.; Shirako, H.; Takeuchi, T.; Kawakami, Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell 2009, 15, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Pinzon-Charry, A.; Maxwell, T.; López, J.A. Dendritic cell dysfunction in cancer: A mechanism for immunosuppression. Immunol. Cell Biol. 2005, 83, 451–461. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Chakraborty, M.; Abrams, S.I.; Camphausen, K.; Liu, K.; Scott, T.; Coleman, C.N.; Hodge, J.W. Irradiation of tumor cells up-regulates Fas and enhances CTL lytic activity and CTL adoptive immunotherapy. J. Immunol. 2003, 170, 6338–6347. [Google Scholar] [CrossRef]
- Schiavone, M.B.; Broach, V.; Shoushtari, A.N.; Carvajal, R.D.; Alektiar, K.; Kollmeier, M.A.; Abu-Rustum, N.R.; Leitao, M.M., Jr. Combined immunotherapy and radiation for treatment of mucosal melanomas of the lower genital tract. Gynecol. Oncol. Rep. 2016, 16, 42–46. [Google Scholar] [CrossRef]
- Hiniker, S.M.; Reddy, S.A.; Maecker, H.T.; Subrahmanyam, P.B.; Rosenberg-Hasson, Y.; Swetter, S.M.; Saha, S.; Shura, L.; Knox, S.J. A prospective clinical trial combining radiation therapy with systemic immunotherapy in metastatic melanoma. Int. J. Radiat. Oncol. Biol. Phys. 2016, 96, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, D.J.; Lawson, D.H.; Richards, J.M.; Conry, R.M.; Miller, D.M.; Treisman, J.; Gailani, F.; Riley, L.; Conlon, K.; Pockaj, B.; et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N. Engl. J. Med. 2011, 364, 2119–2127. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bondarenko, I.; Luft, A.; Serwatowski, P.; Barlesi, F.; Chacko, R.; Sebastian, M.; Neal, J.; Lu, H.; Cuillerot, J.M.; et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non–small-cell lung cancer: Results from a randomized, double-blind, multicenter phase II study. J. Clin. Oncol. 2012, 30, 2046–2054. [Google Scholar] [CrossRef] [PubMed]
- Pfirschke, C.; Engblom, C.; Rickelt, S.; Cortez-Retamozo, V.; Garris, C.; Pucci, F.; Yamazaki, T.; Poirier-Colame, V.; Newton, A.; Redouane, Y.; et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity 2016, 44, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Bonavida, B.; Garban, H. Nitric oxide-mediated sensitization of resistant tumor cells to apoptosis by chemo-immunotherapeutics. Redox Biol. 2015, 6, 486–494. [Google Scholar] [CrossRef]
- Heckler, M.; Osterberg, N.; Guenzle, J.; Thiede-Stan, N.K.; Reichardt, W.; Weidensteiner, C.; Saavedra, J.E.; Weyerbrock, A. The nitric oxide donor JS-K sensitizes U87 glioma cells to repetitive irradiation. Tumor Biol. 2017, 39, 1010428317703922. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Yepez, S.; Vega, M.; Escoto-Chavez, S.E.; Murdock, B.; Sakai, T.; Baritaki, S.; Bonavida, B. Nitric oxide sensitizes tumor cells to TRAIL-induced apoptosis via inhibition of the DR5 transcription repressor Yin Yang 1. Nitric Oxide 2009, 20, 39–52. [Google Scholar] [CrossRef]
- Qiu, M.; Chen, L.; Tan, G.; Ke, L.; Zhang, S.; Chen, H.; Liu, J. A reactive oxygen species activation mechanism contributes to JS-K-induced apoptosis in human bladder cancer cells. Sci. Rep. 2015, 5, 15104. [Google Scholar] [CrossRef]
- Pariente, R.; Pariente, J.A.; Rodríguez, A.B.; Espino, J. Melatonin sensitizes human cervical cancer H e L a cells to cisplatin-induced cytotoxicity and apoptosis: Effects on oxidative stress and DNA fragmentation. J. Pineal Res. 2016, 60, 55–64. [Google Scholar] [CrossRef]
- Lu, H.; Li, X.; Lu, Y.; Qiu, S.; Fan, Z. ASCT2 (SLC1A5) is an EGFR-associated protein that can be co-targeted by cetuximab to sensitize cancer cells to ROS-induced apoptosis. Cancer Lett. 2016, 381, 23–30. [Google Scholar] [CrossRef]
- Velez, J.; Pan, R.; Lee, J.T.; Enciso, L.; Suarez, M.; Duque, J.E.; Jaramillo, D.; Lopez, C.; Morales, L.; Bornmann, W.; et al. Biguanides sensitize leukemia cells to ABT-737-induced apoptosis by inhibiting mitochondrial electron transport. Oncotarget 2016, 7, 51435–51449. [Google Scholar] [CrossRef] [PubMed]
- Doshi, K.A.; Trotta, R.; Natarajan, K.; Rassool, F.V.; Tron, A.E.; Huszar, D.; Perrotti, D.; Baer, M.R. Pim kinase inhibition sensitizes FLT3-ITD acute myeloid leukemia cells to topoisomerase 2 inhibitors through increased DNA damage and oxidative stress. Oncotarget 2016, 7, 48280–48295. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Aguirre, V.; Velez-Pardo, C.; Jimenez-Del-Rio, M. Fructose sensitizes Jurkat cells oxidative stress-induced apoptosis via caspase-dependent and caspase-independent mechanisms. Cell Biol. Int. 2016, 40, 1162–1173. [Google Scholar] [CrossRef] [PubMed]
- Weiming, X.U.; Liu, L.Z.; Loizidou, M.; Ahmed, M.; Charles, I.G. The role of nitric oxide in cancer. Cell Res. 2002, 12, 311–320. [Google Scholar]
- Bonavida, B. (Ed.) Nitric Oxide and Cancer: Pathogenesis and Therapy; Springer: Los Angeles, CA, USA, 2015. [Google Scholar]
- Nathan, C.; Xie, Q.W. Regulation of biosynthesis of nitric oxide. J. Biol. Chem. 1994, 269, 13725–13728. [Google Scholar]
- Choudhari, S.K.; Chaudhary, M.; Bagde, S.; Gadbail, A.R.; Joshi, V. Nitric oxide and cancer: A review. World J. Surg. Oncol. 2013, 11, 118. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Fu, J.; Zhang, Y. Nitric oxide donor-based cancer therapy: Advances and prospects. J. Med. Chem. 2017, 60, 7617–7635. [Google Scholar] [CrossRef]
- Thomsen, L.L.; Miles, D.W.; Happerfield, L.; Bobrow, L.G.; Knowles, R.G.; Moncada, S. Nitric oxide synthase activity in human breast cancer. Br. J. Cancer 1995, 72, 41–44. [Google Scholar] [CrossRef]
- Ambs, S.; Glynn, S.A. Candidate pathways linking inducible nitric oxide synthase to a basal-like transcription pattern and tumor progression in human breast cancer. Cell Cycle 2001, 10, 619–624. [Google Scholar] [CrossRef]
- Basudhar, D.; Somasundaram, V.; de Oliveira, G.A.; Kesarwala, A.; Heinecke, J.L.; Cheng, R.Y.; Glynn, S.A.; Ambs, S.; Wink, D.A.; Ridnour, L.A. Nitric oxide synthase-2-derived nitric oxide drives multiple pathways of breast cancer progression. Antioxid. Redox Signal. 2017, 26, 1044–1058. [Google Scholar] [CrossRef]
- Thomsen, L.L.; Lawton, F.G.; Knowles, R.G.; Beesley, J.E.; Riveros-Moreno, V.; Moncada, S. Nitric oxide synthase activity in human gynecological cancer. Cancer Res. 1994, 54, 1352–1354. [Google Scholar]
- Gallo, O.; Fini-Storchi, I.; Vergari, W.A.; Masini, E.; Morbidelli, L.; Ziche, M.; Franchi, A. Role of nitric oxide in angiogenesis and tumor progression in head and neck cancer. J. Natl. Cancer Inst. 1998, 90, 587–596. [Google Scholar] [CrossRef]
- Ambs, S.; Merriam, W.G.; Bennett, W.P.; Felley-Bosco, E.; Ogunfusika, M.O.; Oser, S.M.; Klein, S.; Shields, P.G.; Billiar, T.R.; Harris, C.C. Frequent nitric oxide synthase-2 expression in human colon adenomas: Implication for tumor angiogenesis and colon cancer progression. Cancer Res. 1998, 58, 334–341. [Google Scholar]
- Klotz, T.; Bloch, W.; Volberg, C.; Engelmann, U.; Addicks, K. Selective expression of inducible nitric oxide synthase in human prostate carcinoma. Cancer Interdiscip. Int. J. Am. Cancer Soc. 1998, 82, 1897–1903. [Google Scholar] [CrossRef]
- Swana, H.S.; Smith, S.D.; Perrotta, P.L.; Saito, N.; Wheeler, M.A.; Weiss, R.M. Inducible nitric oxide synthase with transitional cell carcinoma of the bladder. J. Urol. 1999, 161, 630–634. [Google Scholar] [CrossRef]
- Jaiswal, M.; LaRusso, N.F.; Gores, G.J. Nitric oxide in gastrointestinal epithelial cell carcinogenesis: Linking inflammation to oncogenesis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2001, 281, G626–G634. [Google Scholar] [CrossRef]
- Tu, Y.T.; Tao, J.; Liu, Y.Q.; Li, Y.; Huang, C.Z.; Zhang, X.B.; Lin, Y. Expression of endothelial nitric oxide synthase and vascular endothelial growth factor in human malignant melanoma and their relation to angiogenesis. Clin. Exp. Dermatol. Exp. Dermatol. 2006, 31, 413–418. [Google Scholar] [CrossRef]
- Wang, J.; He, P.; Gaida, M.; Yang, S.; Schetter, A.J.; Gaedcke, J.; Ghadimi, B.M.; Ried, T.; Yfantis, H.; Lee, D.; et al. Inducible nitric oxide synthase enhances disease aggressiveness in pancreatic cancer. Oncotarget 2016, 7, 52993–53004. [Google Scholar] [CrossRef]
- Kawasaki, K.; Smith, R.S.; Hsieh, C.M.; Sun, J.; Chao, J.; Liao, J.K. Activation of the phosphatidylinositol 3-kinase/protein kinase Akt pathway mediates nitric oxide-induced endothelial cell migration and angiogenesis. Mol. Cell. Biol. 2003, 23, 5726–5737. [Google Scholar] [CrossRef]
- Fukumura, D.; Gohongi, T.; Kadambi, A.; Izumi, Y.; Ang, J.; Yun, C.O.; Buerk, D.G.; Huang, P.L.; Jain, R.K. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc. Natl. Acad. Sci. USA 2001, 98, 2604–2609. [Google Scholar] [CrossRef]
- Ziche, M.; Morbidelli, L. Nitric oxide and angiogenesis. J. Neuro-Oncol. 2000, 50, 139–148. [Google Scholar] [CrossRef]
- Fukumura, D.; Kashiwagi, S.; Jain, R.K. The role of nitric oxide in tumour progression. Nat. Rev. Cancer 2006, 6, 521–534. [Google Scholar] [CrossRef]
- Li, J.; Billiar, T.R.; Talanian, R.V.; Kim, Y.M. Nitric oxide reversibly inhibits seven members of the caspase family via S-nitrosylation. Biochem. Biophys. Res. Commun. 1997, 240, 419–424. [Google Scholar] [CrossRef]
- Vasudevan, D.; Hickok, J.R.; Bovee, R.C.; Pham, V.; Mantell, L.L.; Bahroos, N.; Kanabar, P.; Cao, X.J.; Maienschein-Cline, M.; Garcia, B.A.; et al. Nitric oxide regulates gene expression in cancers by controlling histone posttranslational modifications. Cancer Res. 2015, 75, 5299–5308. [Google Scholar] [CrossRef]
- Matsuzaki, H.; Tamatani, M.; Mitsuda, N.; Namikawa, K.; Kiyama, H.; Miyake, S.I.; Tohyama, M. Activation of Akt kinase inhibits apoptosis and changes in Bcl-2 and Bax expression induced by nitric oxide in primary hippocampal neurons. J. Neurochem. 1999, 73, 2037–2046. [Google Scholar]
- Thomas, D.D.; Heinecke, J.L.; Ridnour, L.A.; Cheng, R.Y.; Kesarwala, A.H.; Switzer, C.H.; McVicar, D.W.; Roberts, D.D.; Glynn, S.; Fukuto, J.M.; et al. Signaling and stress: The redox landscape in NOS2 biology. Free Radic. Biol. Med. 2015, 87, 204–225. [Google Scholar] [CrossRef]
- Lopez-Rivera, E.; Jayaraman, P.; Parikh, F.; Davies, M.A.; Ekmekcioglu, S.; Izadmehr, S.; Milton, D.R.; Chipuk, J.E.; Grimm, E.A.; Estrada, Y.; et al. Inducible nitric oxide synthase drives mTOR pathway activation and proliferation of human melanoma by reversible nitrosylation of TSC2. Cancer Res. 2014, 74, 1067–1078. [Google Scholar] [CrossRef]
- Yang, G.Y.; Taboada, S.; Liao, J. Induced nitric oxide synthase as a major player in the oncogenic transformation of inflamed tissue. Methods Mol. Biol. 2009, 512, 119–156. [Google Scholar]
- Hibbs, J.B.; Taintor, R.R.; Vavrin, Z. Macrophage cytotoxicity: Role for L-arginine deiminase and imino nitrogen oxidation to nitrite. Science 1987, 235, 473–476. [Google Scholar] [CrossRef]
- Albina, J.E.; Reichner, J.S. Role of nitric oxide in mediation of macrophage cytotoxicity and apoptosis. Cancer Metastasis Rev. 1998, 17, 39–53. [Google Scholar] [CrossRef]
- Chang, C.F.; Diers, A.R.; Hogg, N. Cancer cell metabolism and the modulating effects of nitric oxide. Free Radic. Biol. Med. 2015, 79, 324–336. [Google Scholar] [CrossRef]
- Kwon, N.S.; Stuehr, D.J.; Nathan, C.F. Inhibition of tumor cell ribonucleotide reductase by macrophage-derived nitric oxide. J. Exp. Med. 1991, 174, 761–767. [Google Scholar] [CrossRef]
- MESSMER, U.K.; BRÜNE, B. Nitric oxide-induced apoptosis: p53-dependent and p53-independent signalling pathways. Biochem. J. 1996, 319, 299–305. [Google Scholar] [CrossRef]
- Chung, H.T.; Pae, H.O.; Choi, B.M.; Billiar, T.R.; Kim, Y.M. Nitric oxide as a bioregulator of apoptosis. Biochem. Biophys. Res. Commun. 2001, 282, 1075–1079. [Google Scholar] [CrossRef]
- Xie, K.; Huang, S.; Dong, Z.; Juang, S.H.; Gutman, M.; Xie, Q.W.; Nathan, C.; Fidler, I.J. Transfection with the inducible nitric oxide synthase gene suppresses tumorigenicity and abrogates metastasis by K-1735 murine melanoma cells. J. Exp. Med. 1995, 181, 1333–1343. [Google Scholar] [CrossRef]
- Xu, L.; Xie, K.; Fidler, I.J. Therapy of human ovarian cancer by transfection with the murine interferon β gene: Role of macrophage-inducible nitric oxide synthase. Hum. Gene Ther. 1998, 9, 2699–2708. [Google Scholar] [CrossRef]
- Scott, D.J.; Hull, M.A.; Cartwright, E.J.; Lam, W.K.; Tisbury, A.; Poulsom, R.; Markham, A.F.; Bonifer, C.; Coletta, P.L. Lack of inducible nitric oxide synthase promotes intestinal tumorigenesis in the ApcMin/+ mouse. Gastroenterology 2001, 121, 889–899. [Google Scholar] [CrossRef]
- Hussain, S.P.; Trivers, G.E.; Hofseth, L.J.; He, P.; Shaikh, I.; Mechanic, L.E.; Doja, S.; Jiang, W.; Subleski, J.; Shorts, L.; et al. Nitric oxide, a mediator of inflammation, suppresses tumorigenesis. Cancer Res. 2004, 64, 6849–6853. [Google Scholar] [CrossRef]
- Williams, J.L.; Kashfi, K.; Ouyang, N.; del Soldato, P.; Kopelovich, L.; Rigas, B. NO-donating aspirin inhibits intestinal carcinogenesis in Min (APCMin/+) mice. Biochem. Biophys. Res. Commun. 2004, 313, 784–788. [Google Scholar] [CrossRef]
- Xie, K.; Dong, Z.; Fidler, I.J. Activation of nitric oxide synthase gene for inhibition of cancer metastasis. J. Leukoc. Biol. 1996, 59, 797–803. [Google Scholar] [CrossRef]
- Xie, K.; Huang, S.; Dong, Z.; Juang, S.H.; Wang, Y.; Fidler, I.J. Destruction of bystander cells by tumor cells transfected with inducible nitric oxide (NO) synthase gene. J. Natl. Cancer Inst. 1997, 89, 421–427. [Google Scholar] [CrossRef]
- Choi, B.M.; Pae, H.O.; Jang, S.I.; Kim, Y.M.; Chung, H.T. Nitric oxide as a pro-apoptotic as well as anti-apoptotic modulator. BMB Rep. 2002, 35, 116–126. [Google Scholar] [CrossRef]
- Messmer, U.K.; Lapetina, E.G.; Brüne, B. Nitric oxide-induced apoptosis in RAW 264.7 macrophages is antagonized by protein kinase C-and protein kinase A-activating compounds. Mol. Pharmacol. 1995, 47, 757–765. [Google Scholar]
- Bustamante, J.; Bersier, G.; Romero, M.; Badin, R.A.; Boveris, A. Nitric oxide production and mitochondrial dysfunction during rat thymocyte apoptosis. Arch. Biochem. Biophys. 2000, 376, 239–247. [Google Scholar] [CrossRef]
- Ghatan, S.; Larner, S.; Kinoshita, Y.; Hetman, M.; Patel, L.; Xia, Z.; Youle, R.J.; Morrison, R.S. P38 MAP kinase mediates BAX translocation in nitric oxide–induced apoptosis in neurons. J. Cell Biol. 2000, 150, 335–348. [Google Scholar] [CrossRef]
- Cui, S.; Reichner, J.S.; Mateo, R.B.; Albina, J.E. Activated murine macrophages induce apoptosis in tumor cells through nitric oxide-dependent or-independent mechanisms. Cancer Res. 1994, 54, 2462–2467. [Google Scholar]
- Garbán, H.J.; Bonavida, B. Nitric oxide sensitizes ovarian tumor cells to Fas-induced apoptosis. Gynecol. Oncol. 1999, 73, 257–264. [Google Scholar] [CrossRef]
- Huerta-Yepez, S.; Vega, M.; Jazirehi, A.; Garban, H.; Hongo, F.; Cheng, G.; Bonavida, B. Nitric oxide sensitizes prostate carcinoma cell lines to TRAIL-mediated apoptosis via inactivation of NF-κB and inhibition of Bcl-xL expression. Oncogene 2004, 23, 4993–5003. [Google Scholar] [CrossRef]
- Wang, X.; Zalcenstein, A.; Oren, M. Nitric oxide promotes p53 nuclear retention and sensitizes neuroblastoma cells to apoptosis by ionizing radiation. Cell Death Differ. 2003, 10, 468–476. [Google Scholar] [CrossRef]
- Yoo, Y.M.; Jung, E.M.; Ahn, C.; Jeung, E.B. Nitric oxide prevents H2O2-induced apoptosis in SK-N-MC human neuroblastoma cells. Int. J. Biol. Sci. 2018, 14, 1974–1984. [Google Scholar] [CrossRef]
- Aktan, F. iNOS-mediated nitric oxide production and its regulation. Life Sci. 2004, 75, 639–653. [Google Scholar] [CrossRef]
- Coleman, J.W. Nitric oxide in immunity and inflammation. Int. Immunopharmacol. 2001, 1, 1397–1406. [Google Scholar] [CrossRef]
- Brunet, L.R. Nitric oxide in parasitic infections. Int. Immunopharmacol. 2001, 1, 1457–1467. [Google Scholar] [CrossRef]
- MacMicking, J.; Xie, Q.W.; Nathan, C. Nitric oxide and macrophage function. Annu. Rev. Immunol. 1997, 15, 323–350. [Google Scholar] [CrossRef]
- Karupiah, G.; Xie, Q.W.; Buller, R.M.; Nathan, C.; Duarte, C.; MacMicking, J.D. Inhibition of viral replication by interferon-gamma-induced nitric oxide synthase. Science 1993, 261, 1445–1448. [Google Scholar] [CrossRef]
- Ren, H.; Wu, J.; Colletta, A.; Meyerhoff, M.E.; Xi, C. Efficient eradication of mature Pseudomonas aeruginosa biofilm via controlled delivery of nitric oxide combined with antimicrobial peptide and antibiotics. Front. Microbiol. 2016, 7, 1260. [Google Scholar] [CrossRef]
- Wo, Y.; Brisbois, E.J.; Bartlett, R.H.; Meyerhoff, M.E. Recent advances in thromboresistant and antimicrobial polymers for biomedical applications: Just say yes to nitric oxide (NO). Biomater. Sci. 2016, 4, 1161–1183. [Google Scholar] [CrossRef]
- Namivandi-Zangeneh, R.; Sadrearhami, Z.; Bagheri, A.; Sauvage-Nguyen, M.; Ho, K.K.K.; Kumar, N.; Wong, E.H.; Boyer, C. Nitric Oxide-Loaded Antimicrobial Polymer for the Synergistic Eradication of Bacterial Biofilm. ACS Macro Lett. 2018, 7, 592–597. [Google Scholar] [CrossRef]
- Wei, X.Q.; Charles, I.G.; Smith, A.; Ure, J.; Feng, G.J.; Huang, F.P.; Xu, D.; Muller, W.; Moncada, S.; Liew, F.Y. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature 1995, 375, 408–411. [Google Scholar] [CrossRef]
- Sahrbacher, U.C.; Lechner, F.; Eugster, H.P.; Frei, K.; Lassmann, H.; Fontana, A. Mice with an inactivation of the inducible nitric oxide synthase gene are susceptible to experimental autoimmune encephalomyelitis. Eur. J. Immunol. 1998, 28, 1332–1338. [Google Scholar] [CrossRef]
- Al-Ramadi, B.K.; Meissler, J.J., Jr.; Huang, D.; Eisenstein, T.K. Immunosuppression induced by nitric oxide and its inhibition by interleukin-4. Eur. J. Immunol. 1992, 22, 2249–2254. [Google Scholar] [CrossRef]
- Dai, W.J.; Gottstein, B. Nitric oxide-mediated immunosuppression following murine Echinococcus multilocularis infection. Immunology 1999, 97, 107–116. [Google Scholar] [CrossRef]
- Abrahamsohn, I.A.; Coffman, R.L. Cytokine and nitric oxide regulation of the immunosuppression in Trypanosoma cruzi infection. J. Immunol. 1995, 155, 3955–3963. [Google Scholar]
- Rockett, K.A.; Awburn, M.M.; Rockett, E.J.; Cowden, W.B.; Clark, I.A. Possible role of nitric oxide in malarial immunosuppression. Parasite Immunol. 1994, 16, 243–249. [Google Scholar] [CrossRef]
- Lejeune, P.; Lagadec, P.; Onier, N.; Pinard, D.; Ohshima, H.; Jeannin, J.F. Nitric oxide involvement in tumor-induced immunosuppression. J. Immunol. 1994, 152, 5077–5083. [Google Scholar]
- Sato, K.; Ozaki, K.; Oh, I.; Meguro, A.; Hatanaka, K.; Nagai, T.; Muroi, K.; Ozawa, K. Nitric oxide plays a critical role in suppression of T-cell proliferation by mesenchymal stem cells. Blood 2007, 109, 228–234. [Google Scholar] [CrossRef]
- van der Veen, R.C. Nitric oxide and T helper cell immunity. Int. Immunopharmacol. 2001, 1, 1491–1500. [Google Scholar] [CrossRef]
- Hirano, K.; Hosoi, A.; Matsushita, H.; Iino, T.; Ueha, S.; Matsushima, K.; Seto, Y.; Kakimi, K. The nitric oxide radical scavenger carboxy-PTIO reduces the immunosuppressive activity of myeloid-derived suppressor cells and potentiates the antitumor activity of adoptive cytotoxic T lymphocyte immunotherapy. Oncoimmunology 2015, 4, e1019195. [Google Scholar] [CrossRef][Green Version]
- Rattan, R.; Sakr, S.; Ali-Fehmi, R.; Abdulfatah, E.; Hanna, R.K.; Giri, S.; Munkarah, A.R. S-nitrosoglutathione, a physiologic nitric oxide carrier, reduces immunosupression in ovarian cancer. Gynecol. Oncol. 2018, 149, 47. [Google Scholar] [CrossRef]
- Fang, F.C. Perspectives series: Host/pathogen interactions. Mechanisms of nitric oxide-related antimicrobial activity. J. Clin. Investig. 1997, 99, 2818–2825. [Google Scholar] [CrossRef]
- Mishra, B.B.; Lovewell, R.R.; Olive, A.J.; Zhang, G.; Wang, W.; Eugenin, E.; Smith, C.M.; Phuah, J.Y.; Long, J.E.; Dubuke, M.L.; et al. Nitric oxide prevents a pathogen-permissive granulocytic inflammation during tuberculosis. Nat. Microbiol. 2017, 2, 17072. [Google Scholar] [CrossRef]
- Billiar, T.R.; Kim’ff, Y.M. Cytoprotective actions of nitric oxide in hepatic inflammation. In Nitric Oxide and the Cell: Proliferation, Differentiation, and Death; Princeton University Press: Princeton, NJ, USA, 2017; Volume 4893, pp. 101–108. [Google Scholar]
- Lee, W.J.; Tateya, S.; Cheng, A.M.; Rizzo-DeLeon, N.; Wang, N.F.; Handa, P.; Wilson, C.L.; Clowes, A.W.; Sweet, I.R.; Bomsztyk, K.; et al. M2 macrophage polarization mediates anti-inflammatory effects of endothelial nitric oxide signaling. Diabetes 2015, 64, 2836–2846. [Google Scholar] [CrossRef]
- Ross, R.; Reske-Kunz, A.B. The role of NO in contact hypersensitivity. Int. Immunopharmacol. 2001, 1, 1469–1478. [Google Scholar] [CrossRef]
- Ohshima, H.; Bartsch, H. Chronic infections and inflammatory processes as cancer risk factors: Possible role of nitric oxide in carcinogenesis. Mutat. Res./Fundam. Mol. Mech. Mutagenes. 1994, 305, 253–264. [Google Scholar] [CrossRef]
- Hussain, S.P.; He, P.; Subleski, J.; Hofseth, L.J.; Trivers, G.E.; Mechanic, L.; Hofseth, A.B.; Bernard, M.; Schwank, J.; Nguyen, G.; et al. Nitric oxide is a key component in inflammation-accelerated tumorigenesis. Cancer Res. 2008, 68, 7130–7136. [Google Scholar] [CrossRef]
- Beckman, J.S. The double-edged role of nitric oxide in brain function and superoxide-mediated injury. J. Dev. Physiol. 1991, 15, 53–59. [Google Scholar]
- Bittrich, H.; Mátzig, A.K.; Kráker, I.; Appel, K.E. NO2-induced DNA single strand breaks are inhibited by antioxidative vitamins in V79 cells. Chem. Biol. Interact. 1993, 86, 199–211. [Google Scholar] [CrossRef]
- Nguyen, T.; Brunson, D.; Crespi, C.L.; Penman, B.W.; Wishnok, J.S.; Tannenbaum, S.R. DNA damage and mutation in human cells exposed to nitric oxide in vitro. Proc. Natl. Acad. Sci. USA 1992, 89, 3030–3034. [Google Scholar] [CrossRef]
- Wink, D.A.; Kasprzak, K.S.; Maragos, C.M.; Elespuru, R.K.; Misra, M.; Dunams, T.M.; Cebula, T.A.; Koch, W.H.; Andrews, A.W.; Allen, J.S. DNA deaminating ability and genotoxicity of nitric oxide and its progenitors. Science 1991, 254, 1001–1003. [Google Scholar] [CrossRef]
- Baritaki, S. Reversal of Multiple Cancer Oncogenic Pleiotropic Properties by NO-Modulating Therapies. In Therapeutic Application of Nitric Oxide in Cancer and Inflammatory Disorders; Academic Press: San Diego, CA, USA, 2019; pp. 29–58. [Google Scholar]
- Adams, C.; McCarthy, H.O.; Coulter, J.A.; Worthington, J.; Murphy, C.; Robson, T.; Hirst, D.G. Nitric oxide synthase gene therapy enhances the toxicity of cisplatin in cancer cells. J. Gene Med. 2009, 11, 160–168. [Google Scholar] [CrossRef]
- Bonavida, B.; Baritaki, S.; Huerta-Yepez, S.; Vega, M.I.; Chatterjee, D.; Yeung, K. Novel therapeutic applications of nitric oxide donors in cancer: Roles in chemo-and immunosensitization to apoptosis and inhibition of metastases. Nitric Oxide 2008, 19, 152–157. [Google Scholar] [CrossRef]
- Hongo, F.; Garban, H.; Huerta-Yepez, S.; Vega, M.; Jazirehi, A.R.; Mizutani, Y.; Miki, T.; Bonavida, B. Inhibition of the transcription factor Yin Yang 1 activity by S-nitrosation. Biochem. Biophys. Res. Commun. 2005, 336, 692–701. [Google Scholar] [CrossRef]
- Knox, S.J.; Ning, S.; Peehl, D.; Oronsky, B.; Scicinski, J. RRx-001 combined with anti-PD-L1 antibody increases the complete response rate in a preclinical myeloma model. Mol. Cancer Ther. 2015, 14, C181. [Google Scholar]
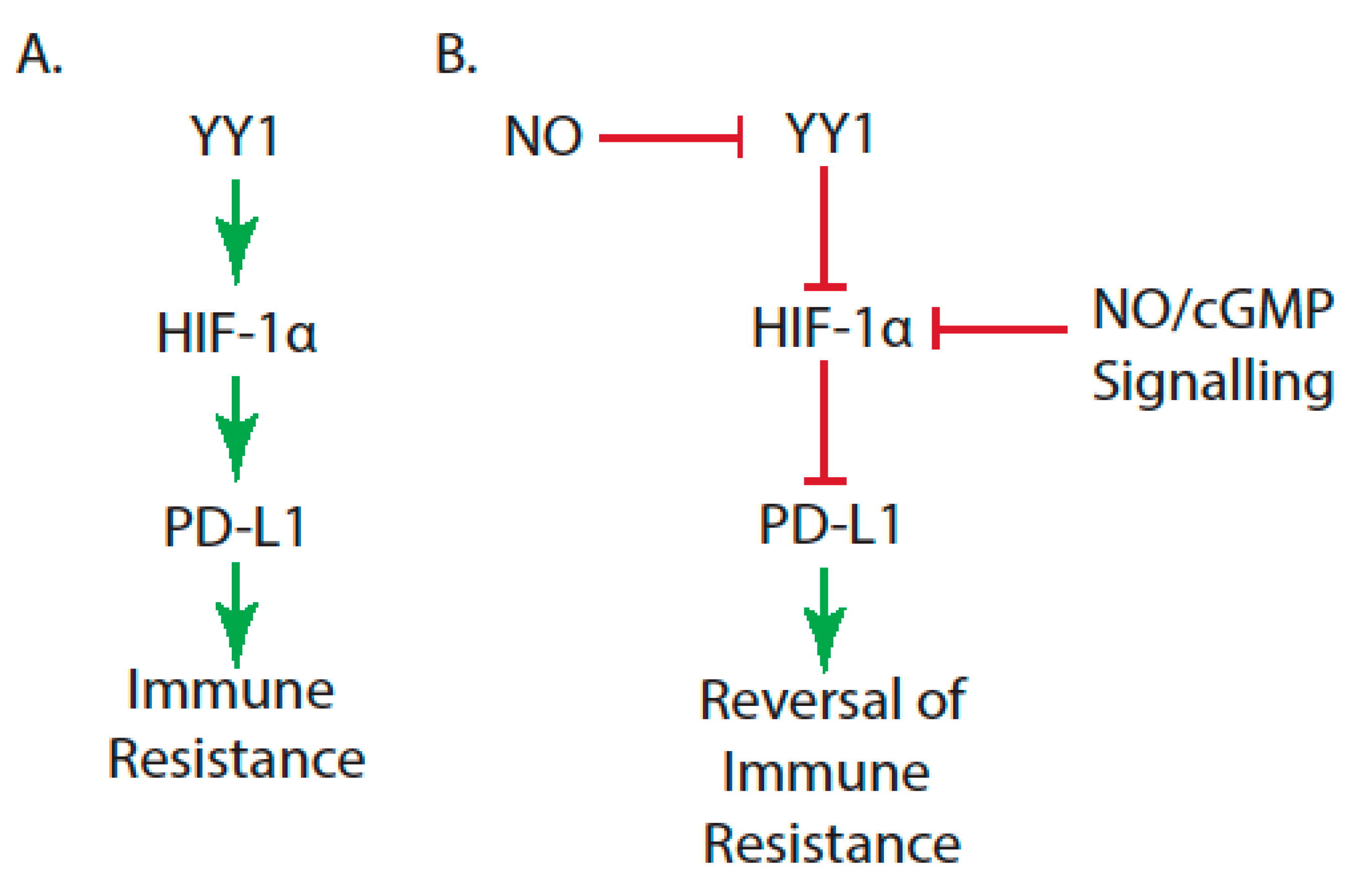
- Graham, C.; Barsoum, I.; Kim, J.; Black, M.; Siemens, R.D. Mechanisms of hypoxia-induced immune escape in cancer and their regulation by nitric oxide. Redox Biol. 2015, 5, 417. [Google Scholar] [CrossRef]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef]
- Wu, S.; Kasim, V.; Kano, M.R.; Tanaka, S.; Ohba, S.; Miura, Y.; Miyata, K.; Liu, X.; Matsuhashi, A.; Chung, U.I.; et al. Transcription factor YY1 contributes to tumor growth by stabilizing hypoxia factor HIF-1α in a p53-independent manner. Cancer Res. 2013, 73, 1787–1799. [Google Scholar] [CrossRef]
- Ren, Z.; Gu, X.; Lu, B.; Chen, Y.; Chen, G.; Feng, J.; Lin, J.; Zhang, Y.; Peng, H. Anticancer efficacy of a nitric oxide-modified derivative of bifendate against multidrug-resistant cancer cells. J. Cell. Mol. Med. 2016, 20, 1095–1105. [Google Scholar] [CrossRef]
- Stewart, G.D.; Nanda, J.; Katz, E.; Bowman, K.J.; Christie, J.G.; Brown, D.G.; McLaren, D.B.; Riddick, A.C.; Ross, J.A.; Jones, G.D.; et al. DNA strand breaks and hypoxia response inhibition mediate the radiosensitisation effect of nitric oxide donors on prostate cancer under varying oxygen conditions. Biochem. Pharmacol. 2011, 81, 203–210. [Google Scholar] [CrossRef]
- Bonavida, B. Nitric oxide donors sensitize resistant cancer cells to apoptosis induced by chemotherapy: Molecular mechanisms of sensitization. In Nitric Oxide (Donor/Induced) in Chemosensitizing; Academic Press: San Diego, CA, USA, 2017; pp. 15–34. [Google Scholar]
- Kielbik, M.; Szulc-Kielbik, I.; Nowak, M.; Sulowska, Z.; Klink, M. Evaluation of nitric oxide donors impact on cisplatin resistance in various ovarian cancer cell lines. Toxicol. In Vitro 2016, 36, 26–37. [Google Scholar] [CrossRef]
- Kielbik, M.; Klink, M.; Brzezinska, M.; Szulc, I.; Sulowska, Z. Nitric oxide donors: Spermine/NO and diethylenetriamine/NO induce ovarian cancer cell death and affect STAT3 and AKT signaling proteins. Nitric Oxide 2013, 35, 93–109. [Google Scholar] [CrossRef]
- Li, L.; Zhu, L.; Hao, B.; Gao, W.; Wang, Q.; Li, K.; Wang, M.; Huang, M.; Liu, Z.; Yang, Q.; et al. iNOS-derived nitric oxide promotes glycolysis by inducing pyruvate kinase M2 nuclear translocation in ovarian cancer. Oncotarget 2017, 8, 33047–33063. [Google Scholar] [CrossRef]
- Scicinski, J.; Oronsky, B.; Ning, S.; Knox, S.; Peehl, D.; Kim, M.M.; Langecker, P.; Fanger, G. NO to cancer: The complex and multifaceted role of nitric oxide and the epigenetic nitric oxide donor, RRx-001. Redox Biol. 2015, 6, 1–8. [Google Scholar] [CrossRef]
- Oronsky, B.; Scicinski, J.; Ning, S.; Peehl, D.; Oronsky, A.; Cabrales, P.; Bednarski, M.; Knox, S. RRx-001, a novel dinitroazetidine radiosensitizer. Investig. New Drugs 2016, 34, 371–377. [Google Scholar] [CrossRef]
- Oronsky, B.; Reid, T.R.; Larson, C.; Carter, C.A.; Brzezniak, C.E.; Oronsky, A.; Cabrales, P. RRx-001 protects against cisplatin-induced toxicities. J. Cancer Res. Clin. Oncol. 2017, 143, 1671–1677. [Google Scholar] [CrossRef]
- Song, J.M.; Upadhyaya, P.; Kassie, F. Nitric oxide-donating aspirin (NO-Aspirin) suppresses lung tumorigenesis in vitro and in vivo and these effects are associated with modulation of the EGFR signaling pathway. Carcinogenesis 2018, 39, 911–920. [Google Scholar] [CrossRef]
- Stevens, E.V.; Carpenter, A.W.; Shin, J.H.; Liu, J.; Der, C.J.; Schoenfisch, M.H. Nitric oxide-releasing silica nanoparticle inhibition of ovarian cancer cell growth. Mol. Pharm. 2010, 7, 775–785. [Google Scholar] [CrossRef][Green Version]
- Duong, H.T.; Kamarudin, Z.M.; Erlich, R.B.; Li, Y.; Jones, M.W.; Kavallaris, M.; Boyer, C.; Davis, T.P. Intracellular nitric oxide delivery from stable NO-polymeric nanoparticle carriers. Chem. Commun. 2013, 49, 4190–4192. [Google Scholar] [CrossRef]
- Munaweera, I.; Shi, Y.; Koneru, B.; Patel, A.; Dang, M.H.; Di Pasqua, A.J.; Balkus, K.J., Jr. Nitric oxide-and cisplatin-releasing silica nanoparticles for use against non-small cell lung cancer. J. Inorg. Biochem. 2015, 153, 23–31. [Google Scholar] [CrossRef]
- Guo, R.; Tian, Y.; Wang, Y.; Yang, W. Near-infrared laser-triggered nitric oxide nanogenerators for the reversal of multidrug resistance in cancer. Adv. Funct. Mater. 2017, 27, 1606398. [Google Scholar] [CrossRef]
- Liu, T.; Qiao, Z.; Wang, J.; Zhang, P.; Zhang, Z.; Guo, D.S.; Yang, X. Molecular imprinted S-nitrosothiols nanoparticles for nitric oxide control release as cancer target chemotherapy. Colloids Surf. B Biointerfaces 2019, 173, 356–365. [Google Scholar] [CrossRef]
- Ali, A.A.; McCrudden, C.M.; McCarthy, H.O. Evaluation of the Impact of Nitric Oxide on Resistance to Platinum-Based Chemotherapeutics. In Nitric Oxide (Donor/Induced) in Chemosensitizing; Academic Press: San Diego, CA, USA, 2017; pp. 71–90. [Google Scholar]
- Liu, B.; Huang, X.; Li, Y.; Liao, W.; Li, M.; Liu, Y.; He, R.; Feng, D.; Zhu, R.; Kurihara, H. JS-K, a nitric oxide donor, induces autophagy as a complementary mechanism inhibiting ovarian cancer. BMC Cancer 2019, 19, 645. [Google Scholar] [CrossRef]
- Smeda, M.; Kieronska, A.; Adamski, M.G.; Proniewski, B.; Sternak, M.; Mohaissen, T.; Przyborowski, K.; Derszniak, K.; Kaczor, D.; Stojak, M.; et al. Nitric oxide deficiency and endothelial–mesenchymal transition of pulmonary endothelium in the progression of 4T1 metastatic breast cancer in mice. Breast Cancer Res. 2018, 20, 86. [Google Scholar] [CrossRef]
- Tan, J.; Zeng, Q.; Jiang, X.Z.; He, L.Y.; Wang, J.R.; Yao, K.; Wang, C.H. Apoptosis of bladder transitional cell carcinoma T24 cells induced by adenovirus-mediated inducible nitric oxide synthase gene transfection. Chin. J. Cancer Res. 2013, 25, 593–599. [Google Scholar]
- Ye, S.; Yang, W.; Wang, Y.; Ou, W.; Ma, Q.; Yu, C.; Ren, J.; Zhong, G.; Shi, H.; Yuan, Z.; et al. Cationic liposome-mediated nitric oxide synthase gene therapy enhances the antitumor effects of cisplatin in lung cancer. Int. J. Mol. Med. 2013, 31, 33–42. [Google Scholar] [CrossRef]
- Garrido, P.; Shalaby, A.; Walsh, E.M.; Keane, N.; Webber, M.; Keane, M.M.; Sullivan, F.J.; Kerin, M.J.; Callagy, G.; Ryan, A.E.; et al. Impact of inducible nitric oxide synthase (iNOS) expression on triple negative breast cancer outcome and activation of EGFR and ERK signaling pathways. Oncotarget 2017, 8, 80568–80588. [Google Scholar] [CrossRef]
- Ekmekcioglu, S.; Grimm, E.A.; Roszik, J. Targeting iNOS to increase efficacy of immunotherapies. Hum. Vaccines Immunother. 2017, 13, 1105–1108. [Google Scholar] [CrossRef]
- Melo, F.; Gonçalves, D.; Sousa, R.; Gonçalves, J.; Silva, D.; Icimoto, M.; Gimenez, M.; De Angelis, K.; Llesuy, S.; Fernandes, D.; et al. Imbalance between nitric oxide and superoxide anion induced by uncoupled nitric oxide synthase contributes to human metastatic melanoma development. Free Radic. Biol. Med. 2018, 128, S70. [Google Scholar] [CrossRef]
- Hsieh, M.J.; Lin, C.W.; Chiou, H.L.; Yang, S.F.; Chen, M.K. Dehydroandrographolide, an iNOS inhibitor, extracted from from Andrographis paniculata (Burm. f.) Nees, induces autophagy in human oral cancer cells. Oncotarget 2015, 6, 30831–30849. [Google Scholar] [CrossRef]
- Marigo, I.; Zilio, S.; Desantis, G.; Mlecnik, B.; Agnellini, A.H.; Ugel, S.; Sasso, M.S.; Qualls, J.E.; Kratochvill, F.; Zanovello, P.; et al. T cell cancer therapy requires CD40-CD40L activation of tumor necrosis factor and inducible nitric-oxide-synthase-producing dendritic cells. Cancer Cell 2016, 30, 377–390. [Google Scholar] [CrossRef]
- Das, D.S.; Ray, A.; Das, A.; Song, Y.; Tian, Z.; Oronsky, B.; Richardson, P.; Scicinski, J.; Chauhan, D.; Anderson, K.C. A novel hypoxia-selective epigenetic agent RRx-001 triggers apoptosis and overcomes drug resistance in multiple myeloma cells. Leukemia 2016, 30, 2187–2197. [Google Scholar] [CrossRef]
- Chen, J.; Wang, T.; Xu, S.; Zhang, P.; Lin, A.; Wu, L.; Yao, H.; Xie, W.; Zhu, Z.; Xu, J. Discovery of novel antitumor nitric oxide-donating β-elemene hybrids through inhibiting the PI3K/Akt pathway. Eur. J. Med. Chem. 2017, 135, 414–423. [Google Scholar] [CrossRef]
- Müller, E.; Speth, M.; Christopoulos, P.F.; Lunde, A.; Avdagic, A.; Øynebråten, I.; Corthay, A. Both type I and type II interferons can activate antitumor M1 macrophages when combined with TLR stimulation. Front. Immunol. 2018, 9, 2520. [Google Scholar] [CrossRef]
- Fraix, A.; Sortino, S. Combination of PDT photosensitizers with NO photodononors. Photochem. Photobiol. Sci. 2018, 17, 1709–1727. [Google Scholar] [CrossRef]
- Barcińska, E.; Wierzbicka, J.; Zauszkiewicz-Pawlak, A.; Jacewicz, D.; Dabrowska, A.; Inkielewicz-Stepniak, I. Role of oxidative and nitro-oxidative damage in silver nanoparticles cytotoxic effect against human pancreatic ductal adenocarcinoma cells. Oxidative Med. Cell. Longev. 2018, 2018, 8251961. [Google Scholar] [CrossRef]
- Islam, W.; Fang, J.; Imamura, T.; Etrych, T.; Subr, V.; Ulbrich, K.; Maeda, H. Augmentation of the Enhanced Permeability and Retention Effect with Nitric Oxide–Generating Agents Improves the Therapeutic Effects of Nanomedicines. Mol. Cancer Ther. 2018, 17, 2643–2653. [Google Scholar] [CrossRef]
- Porshneva, K.; Papiernik, D.; Psurski, M.; Nowak, M.; Matkowski, R.; Ekiert, M.; Milczarek, M.; Banach, J.; Jarosz, J.; Wietrzyk, J. Combination therapy with DETA/NO and clopidogrel inhibits metastasis in murine mammary gland cancer models via improved vasoprotection. Mol. Pharm. 2018, 15, 5277–5290. [Google Scholar] [CrossRef]
- Hou, L.; Zhang, Y.; Yang, X.; Tian, C.; Yan, Y.; Zhang, H.; Shi, J.; Zhang, H.; Zhang, Z. Intracellular NO-Generator Based on Enzyme Trigger for Localized Tumor-Cytoplasm Rapid Drug Release and Synergetic Cancer Therapy. ACS Appl. Mater. Interfaces 2018, 11, 255–268. [Google Scholar] [CrossRef]
- Salaroglio, I.C.; Gazzano, E.; Abdullrahman, A.; Mungo, E.; Castella, B.; Abd, G.E.F.A.E.; Massaia, M.; Donadelli, M.; Rubinstein, M.; Riganti, C.; et al. Increasing intratumor C/EBP-β LIP and nitric oxide levels overcome resistance to doxorubicin in triple negative breast cancer. J. Exp. Clin. Cancer Res. 2018, 37, 286. [Google Scholar] [CrossRef]
- Fauskanger, M.H.F.; Haabeth, O.A.W.; Skjeldal, F.M.; Bogen, B.; Tveita, A.A. Tumor killing by CD4+ T cells is mediated via induction of iNOS-dependent macrophage cytotoxicity. Front. Immunol. 2018, 9, 1684. [Google Scholar] [CrossRef]
- Kang, Y.; Kim, J.; Park, J.; Lee, Y.M.; Saravanakumar, G.; Park, K.M.; Choi, W.; Kim, K.; Lee, E.; Kim, C.; et al. Tumor vasodilation by N-Heterocyclic carbene-based nitric oxide delivery triggered by high-intensity focused ultrasound and enhanced drug homing to tumor sites for anti-cancer therapy. Biomaterials 2019, 217, 119297. [Google Scholar] [CrossRef]
- Feng, T.; Wan, J.; Li, P.; Ran, H.; Chen, H.; Wang, Z.; Zhang, L. A novel NIR-controlled NO release of sodium nitroprusside-doped Prussian blue nanoparticle for synergistic tumor treatment. Biomaterials 2019, 214, 119213. [Google Scholar] [CrossRef]
- Bechmann, N.; Kniess, T.; Pietzsch, J. Nitric Oxide-Releasing Selective Estrogen Receptor Modulators: A Bifunctional Approach to Improve the Therapeutic Index. J. Med. Chem. 2019. [Google Scholar] [CrossRef]
- Gurunathan, S.; Jeyaraj, M.; Kang, M.H.; Kim, J.H. Graphene Oxide–Platinum Nanoparticle Nanocomposites: A Suitable Biocompatible Therapeutic Agent for Prostate Cancer. Polymers 2019, 11, 733. [Google Scholar] [CrossRef]
- Reis, A.K.C.A.; Stern, A.; Monteiro, H.P. S-nitrosothiols and H2S donors: Potential chemo-therapeutic agents in cancer. Redox Biol. 2019, 101190. [Google Scholar] [CrossRef]
- Lin, A.; Gorbanev, Y.; De Backer, J.; Van Loenhout, J.; Van Boxem, W.; Lemière, F.; Cos, P.; Dewilde, S.; Smits, E.; Bogaerts, A. Non-Thermal Plasma as a Unique Delivery System of Short-Lived Reactive Oxygen and Nitrogen Species for Immunogenic Cell Death in Melanoma Cells. Adv. Sci. 2019, 6, 1802062. [Google Scholar] [CrossRef]
- Sun, F.; Wang, Y.; Luo, X.; Ma, Z.; Xu, Y.; Zhang, X.; Lv, T.; Zhang, Y.; Wang, M.; Huang, Z.; et al. Anti-CD24 antibody-nitric oxide conjugate (ANC) selectively and potently suppresses hepatic carcinoma. Cancer Res. 2019, 2839. [Google Scholar] [CrossRef]
- Wilson, A.; Menon, V.; Khan, Z.; Alam, A.; Litovchick, L.; Yakovlev, V. Nitric oxide-donor/PARP-inhibitor combination: A new approach for sensitization to ionizing radiation. Redox Biol. 2019, 24, 101169. [Google Scholar] [CrossRef]
- Moniruzzaman, R.; Rehman, M.U.; Zhao, Q.L.; Jawaid, P.; Mitsuhashi, Y.; Sakurai, K.; Heshiki, W.; Ogawa, R.; Tomihara, K.; Saitoh, J.I.; et al. Combination of 5-aminosalicylic acid and hyperthermia synergistically enhances apoptotic cell death in HSC-3 cells due to intracellular nitric oxide/peroxynitrite generation. Cancer Lett. 2019, 451, 58–67. [Google Scholar] [CrossRef]
- Xu, Y.; Ren, H.; Liu, J.; Wang, Y.; Meng, Z.; He, Z.; Miao, W.; Chen, G.; Li, X. A switchable NO-releasing nanomedicine for enhanced cancer therapy and inhibition of metastasis. Nanoscale 2019, 11, 5474–5488. [Google Scholar] [CrossRef]
- Dong, X.; Liu, H.J.; Feng, H.Y.; Yang, S.C.; Liu, X.L.; Lai, X.; Lu, Q.; Lovell, J.F.; Chen, H.Z.; Fang, C. Enhanced Drug Delivery by Nanoscale Integration of a Nitric Oxide Donor to Induce Tumor Collagen Depletion. Nano Lett. 2019, 19, 997–1008. [Google Scholar] [CrossRef]
- Chen, M.; Song, F.; Liu, Y.; Tian, J.; Liu, C.; Li, R.; Zhang, Q. A dual pH-sensitive liposomal system with charge-reversal and NO generation for overcoming multidrug resistance in cancer. Nanoscale 2019, 11, 3814–3826. [Google Scholar] [CrossRef]
- Yang, D.; Li, T.; Li, Y.; Zhang, S.; Li, W.; Liang, H.; Xing, Z.; Du, L.; He, J.; Kuang, C.; et al. H2S suppresses indoleamine 2, 3-dioxygenase 1 and exhibits immunotherapeutic efficacy in murine hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 88. [Google Scholar] [CrossRef]
- Rapozzi, V.; Della Pietra, E.; Bonavida, B. Dual roles of nitric oxide in the regulation of tumor cell response and resistance to photodynamic therapy. Redox Biol. 2015, 6, 311–317. [Google Scholar] [CrossRef]
- Bradley, S.A.; Steinert, J.R. Characterisation and comparison of temporal release profiles of nitric oxide generating donors. J. Neurosci. Methods 2015, 245, 116–124. [Google Scholar] [CrossRef]
- García-Ortiz, A.; Serrador, J.M. Nitric oxide signaling in T cell-mediated immunity. Trends Mol. Med. 2018, 24, 412–427. [Google Scholar] [CrossRef]
- Sukhatme, V.; Bouche, G.; Meheus, L.; Sukhatme, V.P.; Pantziarka, P. Repurposing Drugs in Oncology (ReDO)—Nitroglycerin as an anti-cancer agent. Ecancermedicalscience 2015, 9, 568. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Immune Resistance Mechanism | Reference |
|---|---|
| Absence of good bacteria in the gut including Akkermansia muciniphila | [32] |
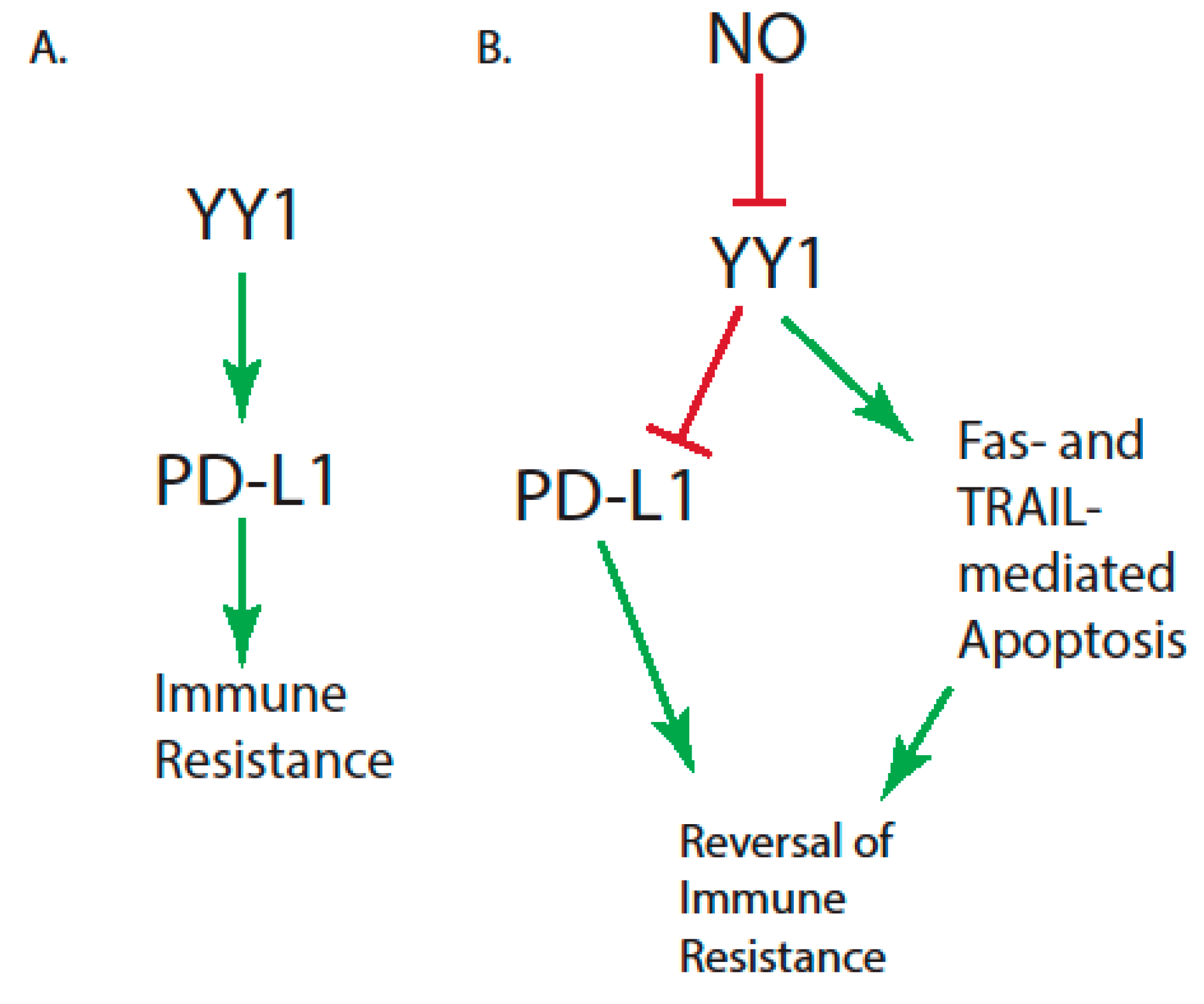
| High levels of Yin-Yang 1 (YY1), which modulate programmed death ligand 1 (PD-L1) expression | [35,36] |
| Absence of tumor antigens | [37] |
| Downregulation or mutation of MHCs and decreased antigen presentation | [38] |
| T-cell exhaustion mediated by up-regulation of PD-L1 and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) expression | [39,40] |
| Loss of Phosphatase and Tensin Homolog (PTEN) expression and activation of the PI3K-AKT pathway | [41] |
| High levels of Interferon gamma (IFN-γ), which drives expression of PD-L1 | [42] |
| Lack of T-cells with tumor antigen-specific receptors | [43] |
| Presence of inhibitory receptors on immune cells (V-domain Immunoglobulin Suppressor of T-cell Activation (VISTA,) Lymphocyte Activating Gene 3 (LAG-3,) and T-cell Immunoglobulin and Mucin Protein 3 (TIM-3)) | [44] |
| Immunosuppression caused by: The increased production of immature myeloid cells in cancer patients Signal Transducer and Activator of Transcription 3 (STAT3) activity Snail during cancer metastasis Dendritic cell dysfunction | [45,46,47,48], respectively |
| Sensitizing Factor | Type of Sensitization | Type of Tumor Cell | Reference |
|---|---|---|---|
| Nitric Oxide Donors (Inhibit YY1 and NF-kB and upregulate DR5) | Tumor Necrosis Factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis | Prostate carcinoma cells | [56,58] |
| Reactive oxygen species | JS-K-induced cell apoptosis | Bladder cancer cells | [59] |
| Melatonin | Reactive oxygen species-induced apoptosis | HeLa cervical cancer cells | [60] |
| Cetuximab (EGFR antibody) | Reactive oxygen species-induced apoptosis | Head and neck squamous cell carcinoma | [61] |
| Biguanides and Rotenone (superoxide inducers) | ABT-737-induced apoptosis | Leukemia cells | [62] |
| AZD1208 (Pan-Pim kinase inhibitor) and Topoisomerase 2 inhibitor (chemotherapy drug) | Reactive oxygen species-induced apoptosis | Acute Myeloid leukemia | [63] |
| Mitochondria targeting molecules that shift cells from Glucose to Fructose metabolism | Rotenone and reactive oxygen species-induced apoptosis | Jurkat leukemia cells | [64] |
| NO-Dependent Therapies | Antitumor Effect | Reference |
|---|---|---|
| NO production by tumor-infiltrating myeloid cells | Important for adoptively transferred CD8+ cytotoxic T cells to destroy tumors | [175] |
| RRx-001 (NO donor) | Cancer cell cytotoxicity and protection of cisplatin-induced toxicities Induction of apoptosis and reversal of drug resistance in multiple myeloma cells | [157,159,176] |
| NO-donating β-elemene hybrids | Inhibited tumor growth in liver tumors | [177] |
| Type I IFNs, IFN-a and IFN-b | Synergized with Toll-like Receptor (TLR) agonists for transcription of iNOS mRNA and secretion of NO and inhibited cancer cell growth of lewis lung carcinoma | [178] |
| NO-donating aspirin | Suppressed tumorigenesis in vitro and in vivo through modulation of the Epidermal Growth Factor Receptor (EGFR) signaling pathway in lung tumors | [160] |
| Coupling of photodynamic therapy with photocontrolled release of NO | Synergistic therapeutic effects via various mechanisms | [179] |
| Increase in NOS expression and nitric oxide levels triggered by silver nanoparticles | Induced apoptosis of pancreatic ductal adenocarcinoma | [180] |
| NO generators nitroglycerin, hydroxyurea, and l-arginine | Improved the therapeutic effects of the polymer-conjugated pirarubicin and increased delivery of nanomedicine to solid tumor models in end-stage breast cancer | [181] |
| NO-donor DETA/NO combined with clopidogrel | Improved vasoprotective and antiplatelet activity and reduced lung metastatic foci formation in metastatic mammary gland cancer | [182] |
| Intracellular enzyme-triggered NO-generator | Tumor cytoplasm-specific disruption and localized doxorubicin rapid drug release, increased apoptosis by NO | [183] |
| Endogenous production of NO by chloroquine and bortezomib | Enhanced doxorubicin’s cytotoxicity by inducing C/EBP-β LIP induction and inhibiting P-glycoprotein activity in triple-negative breast cancer | [184] |
| NO release into tumor cells by iNOS within tumor-infiltrating macrophages | Intracellular accumulation of toxic secondary oxidants, such as peroxynitrate, increased apoptosis through activation of the mitochondrial pathway | [185] |
| JS-K (NO donor) | Induced autophagy and inhibited tumor growth of ovarian cancer | [165] |
| N-heterocyclic carbene-based NO donors delivered by high-intensity ultrasound | High heat and tumor growth inhibition | [186] |
| Near-infrared laser-controlled NO release of sodium nitroprusside-doped Prussian blue nanoparticle | Photothermal effect in vivo and in vitro of breast cancer cells | [187] |
| Near-infrared laser-triggered NO nanogenerators | Reversal of multidrug resistance (MDR) via inhibition of the expression of P-glycol in an in vivo humanized MDR cancer model | [164] |
| NO-releasing selective estrogen receptor modulators | Anti-proliferative effect in breast cancer and melanoma cells | [188] |
| Graphene oxide platinum nanoparticle nanocomposites | Increased pro-apoptotic genes and decreased anti-apoptotic genes in prostate cancer | [189] |
| S-nitrosothiols and H2S donors | Effective in killing cancer cells but not normal cells | [190] |
| Nonthermal plasma delivery of NO | Immunogenic cell death of melanoma cells | [191] |
| Anti-CD24 Antibody-NO conjugate | Induced apoptosis of tumor cells and suppressed tumor growth in vitro and in vivo in hepatic carcinoma | [192] |
| NO-donor and Parp inhibitor combination | Sensitized cells to ionizing radiation treatment in BRCA1/2-proficient tumors | [193] |
| NO production from a combination of 5-aminosalicylic acid and hyperthermia | Induced apoptotic cell death of oral squamous cell carcinoma | [194] |
| Switchable NO-releasing nanoparticle activated by near-infrared radiation | Induced tumor vascular permeability, improved drug accumulation, blocks metastasis, and directly kills cancer cells | [195] |
| Nanoparticles loaded with doxorubicin and the NO-donor, S-nitrosothiol | Activated endogenous matrix metalloproteinases, which degrade collagen in the tumor extracellular matrix | [196] |
| pH-sensitive liposomal polymer that delivers the NO- donor DEANONOate and paclitaxel into cancer cells | Reversed a negative charge to a positive charge in the tumor microenvironment leading to the improvement of cell uptake of paclitaxel and the release of DETANONOate in the lysosome of multi-drug-resistant cancer cells | [197] |
| H2S donors | Increases iNOS and NO and restricts tumor development of hepatocellular carcinoma | [198] |
| Combination of a NO donor and photodynamic therapy | Increased cytotoxic effect in vitro and in vivo | [199] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hays, E.; Bonavida, B. Nitric Oxide-Mediated Enhancement and Reversal of Resistance of Anticancer Therapies. Antioxidants 2019, 8, 407. https://doi.org/10.3390/antiox8090407
Hays E, Bonavida B. Nitric Oxide-Mediated Enhancement and Reversal of Resistance of Anticancer Therapies. Antioxidants. 2019; 8(9):407. https://doi.org/10.3390/antiox8090407
Chicago/Turabian StyleHays, Emily, and Benjamin Bonavida. 2019. "Nitric Oxide-Mediated Enhancement and Reversal of Resistance of Anticancer Therapies" Antioxidants 8, no. 9: 407. https://doi.org/10.3390/antiox8090407
APA StyleHays, E., & Bonavida, B. (2019). Nitric Oxide-Mediated Enhancement and Reversal of Resistance of Anticancer Therapies. Antioxidants, 8(9), 407. https://doi.org/10.3390/antiox8090407