Cysteine Glutathionylation Acts as a Redox Switch in Endothelial Cells


Abstract
:1. Introduction
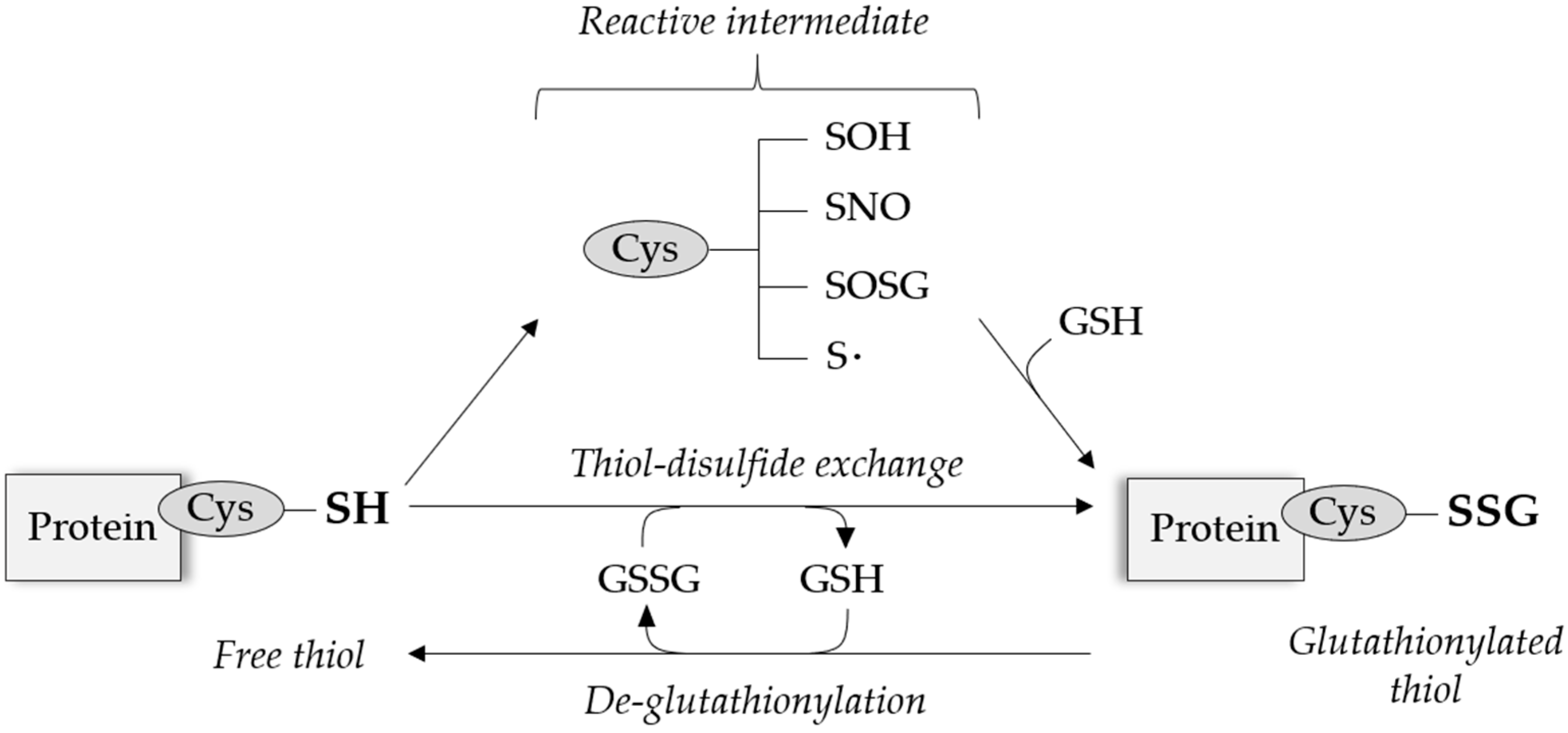
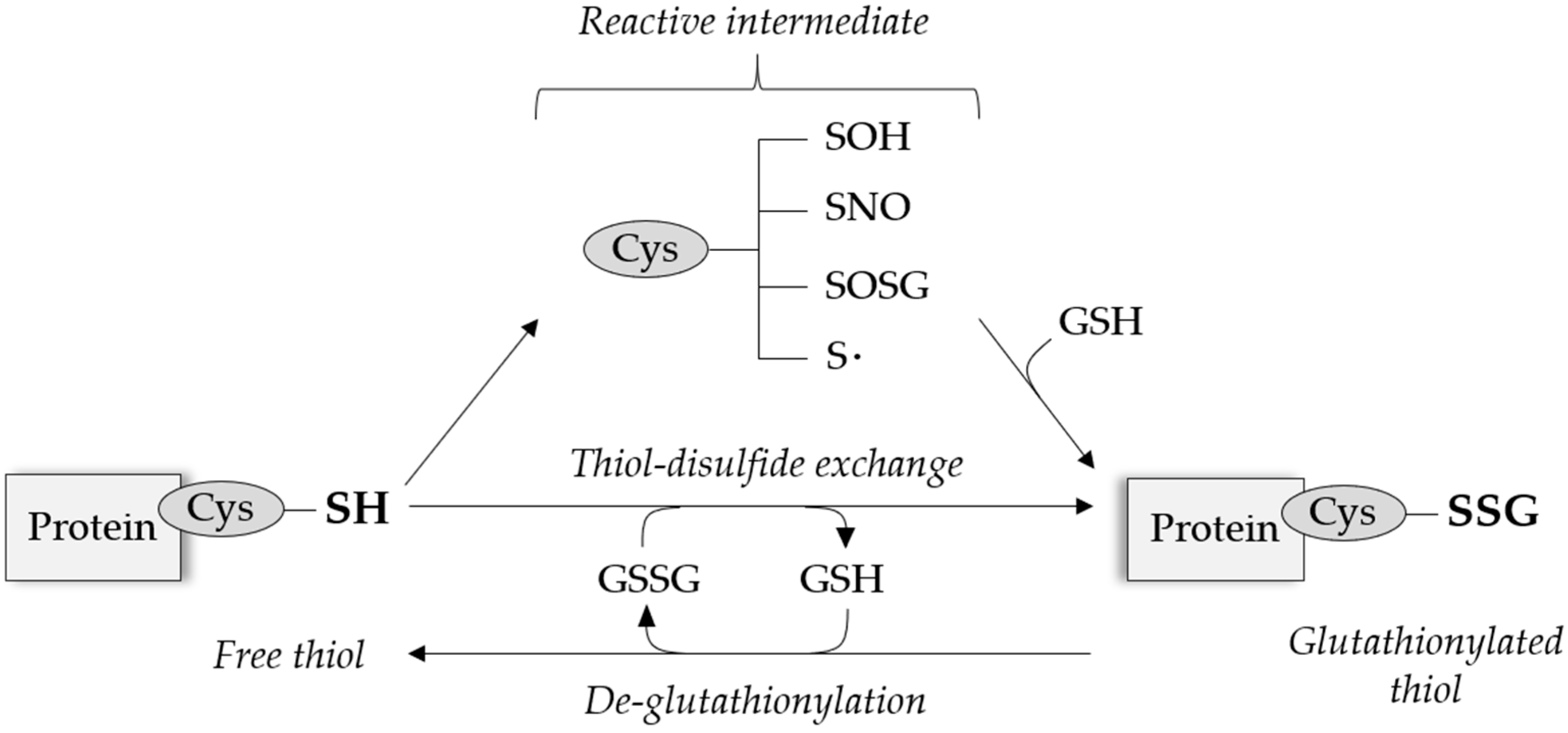
2. Protein S-Glutathionylation
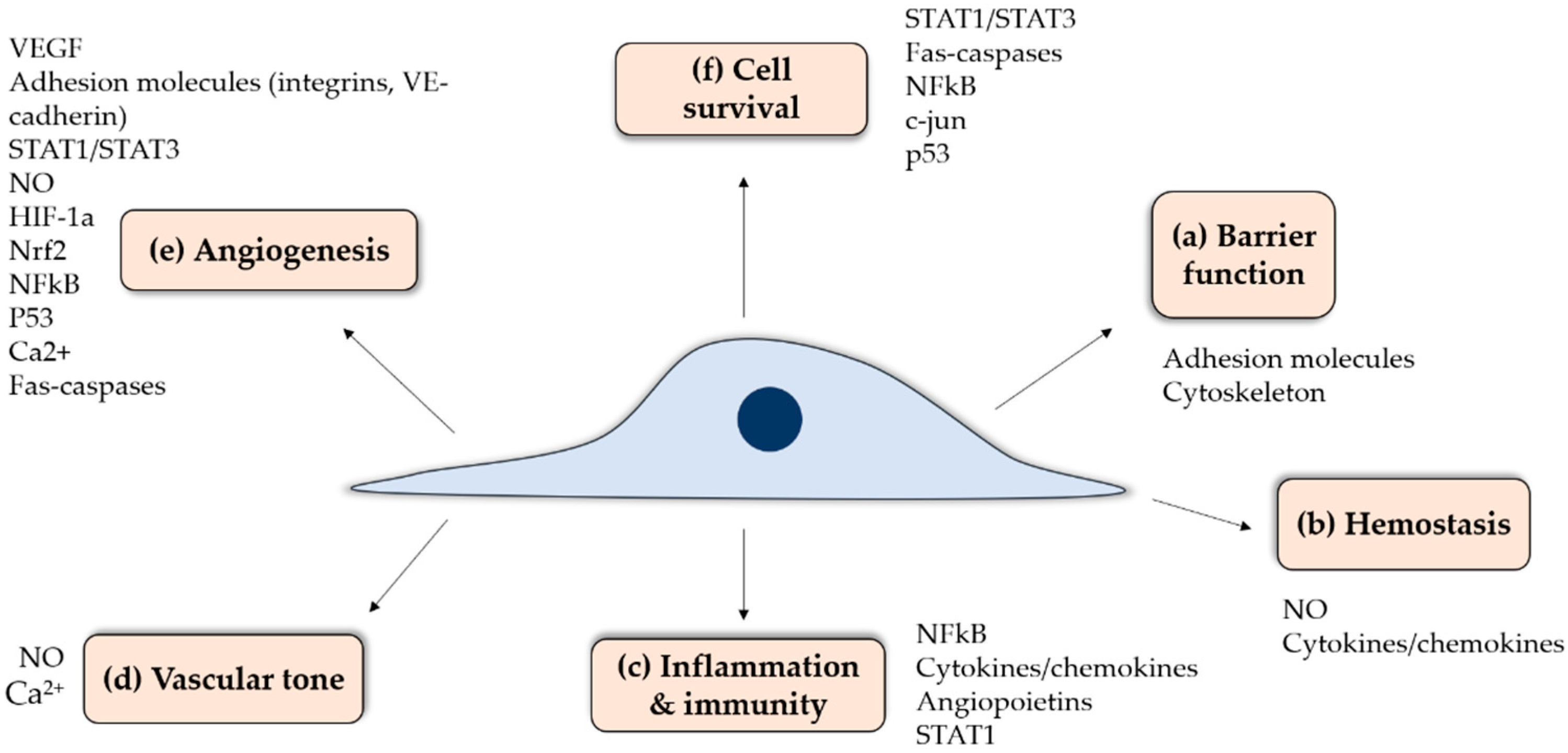
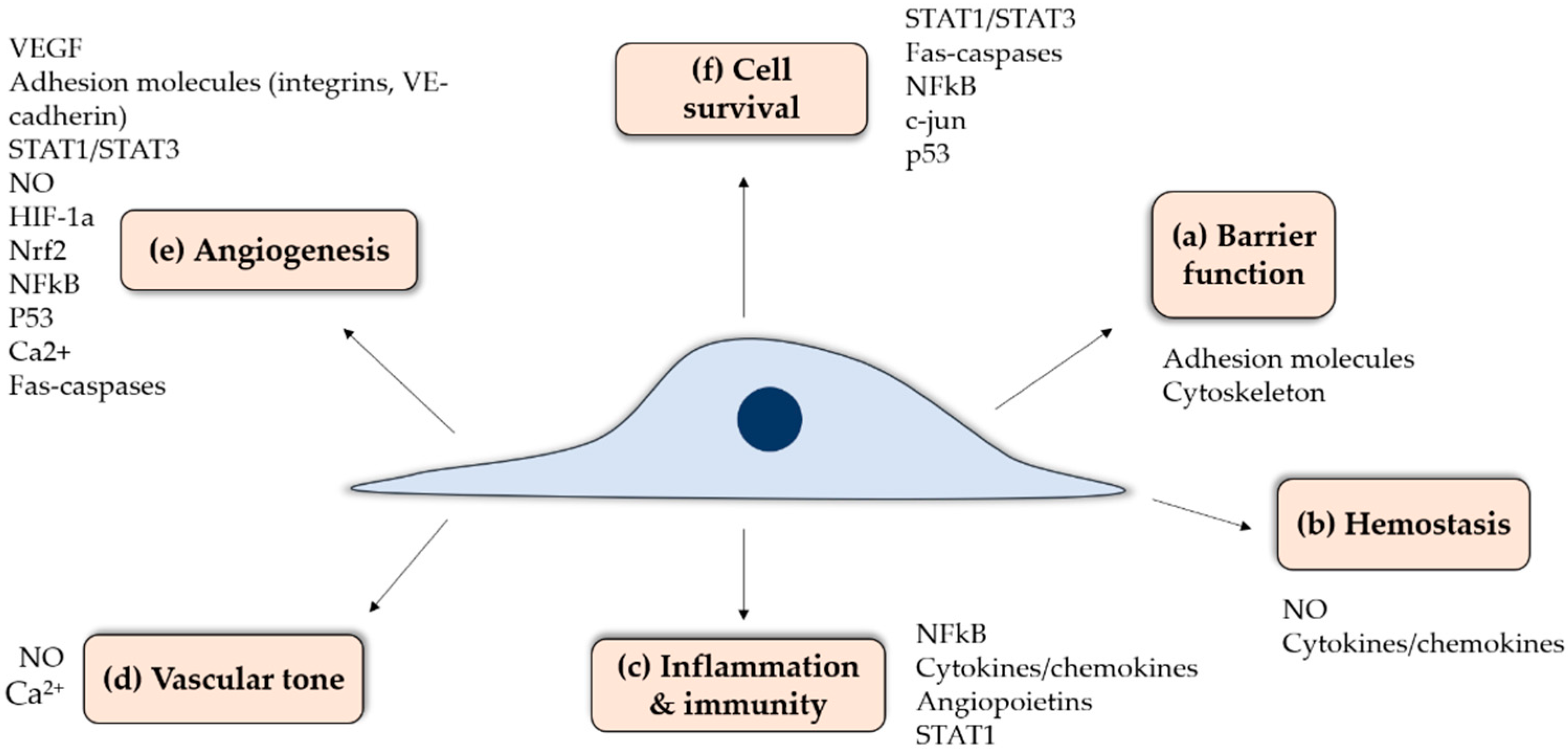
3. Role of Endothelium in Vascular Physiology
4. Transcription Regulation by S-Glutathionylation: Epigenetics Regulators and Transcription Factors
4.1. Epigenetic Regulators
4.1.1. Histone Proteins
4.1.2. Histone-Modifying Enzymes
4.2. Transcription Factors
4.2.1. S-Glutathionylation-Mediated Inhibition of Transcription Factors
4.2.2. S-Glutathionylation-Mediated Activation of Transcription Factors
4.2.3. Opposite Effects of S-Glutathionylation on Various Signal Transducer and Activator of Transcription (STAT) Proteins
5. Redox Control of Phosphorylation by S-Glutathionylation: Phosphatases, GTPases and Kinases
5.1. Phosphatases
5.2. GTPases
5.3. Kinases
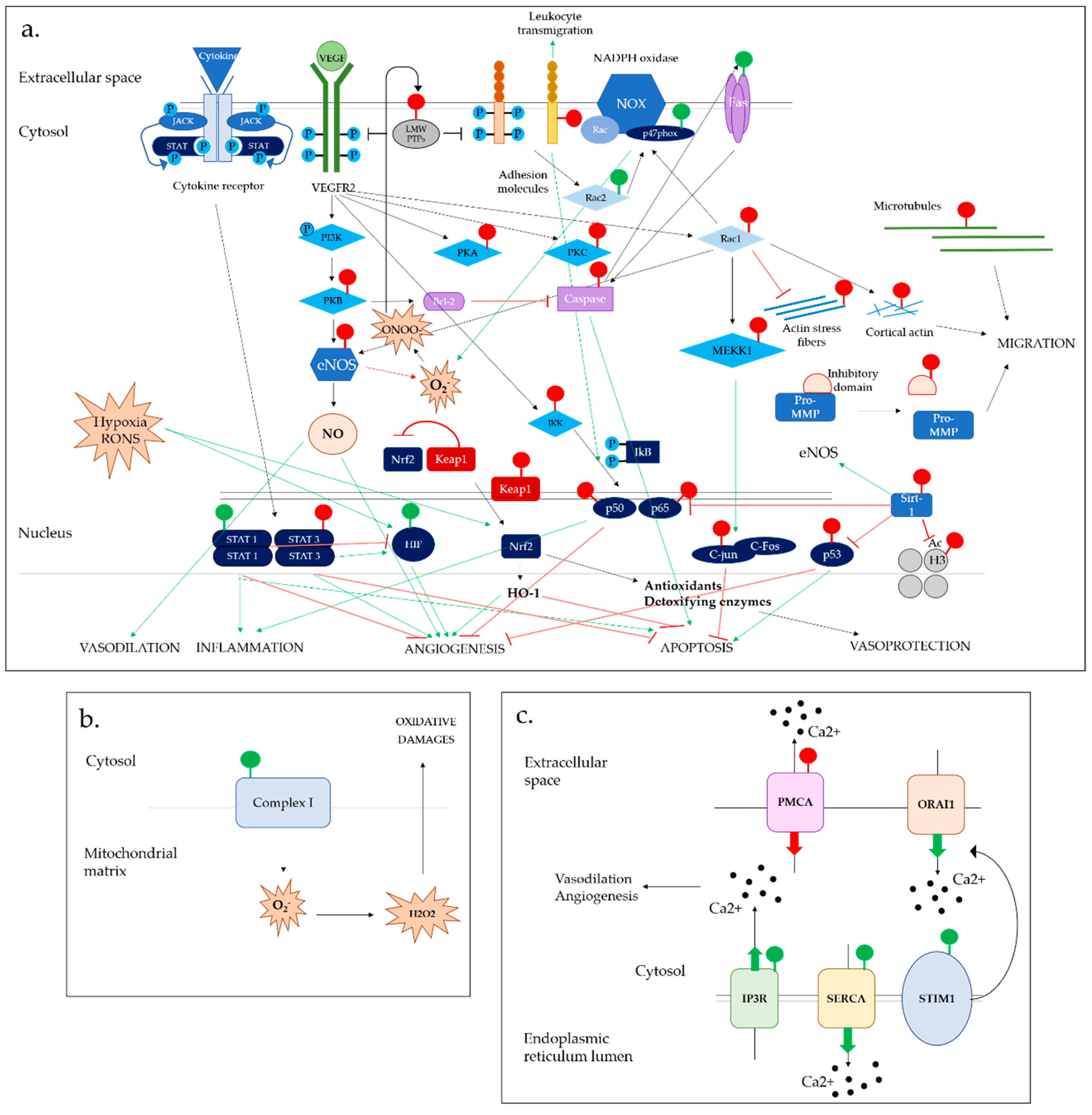
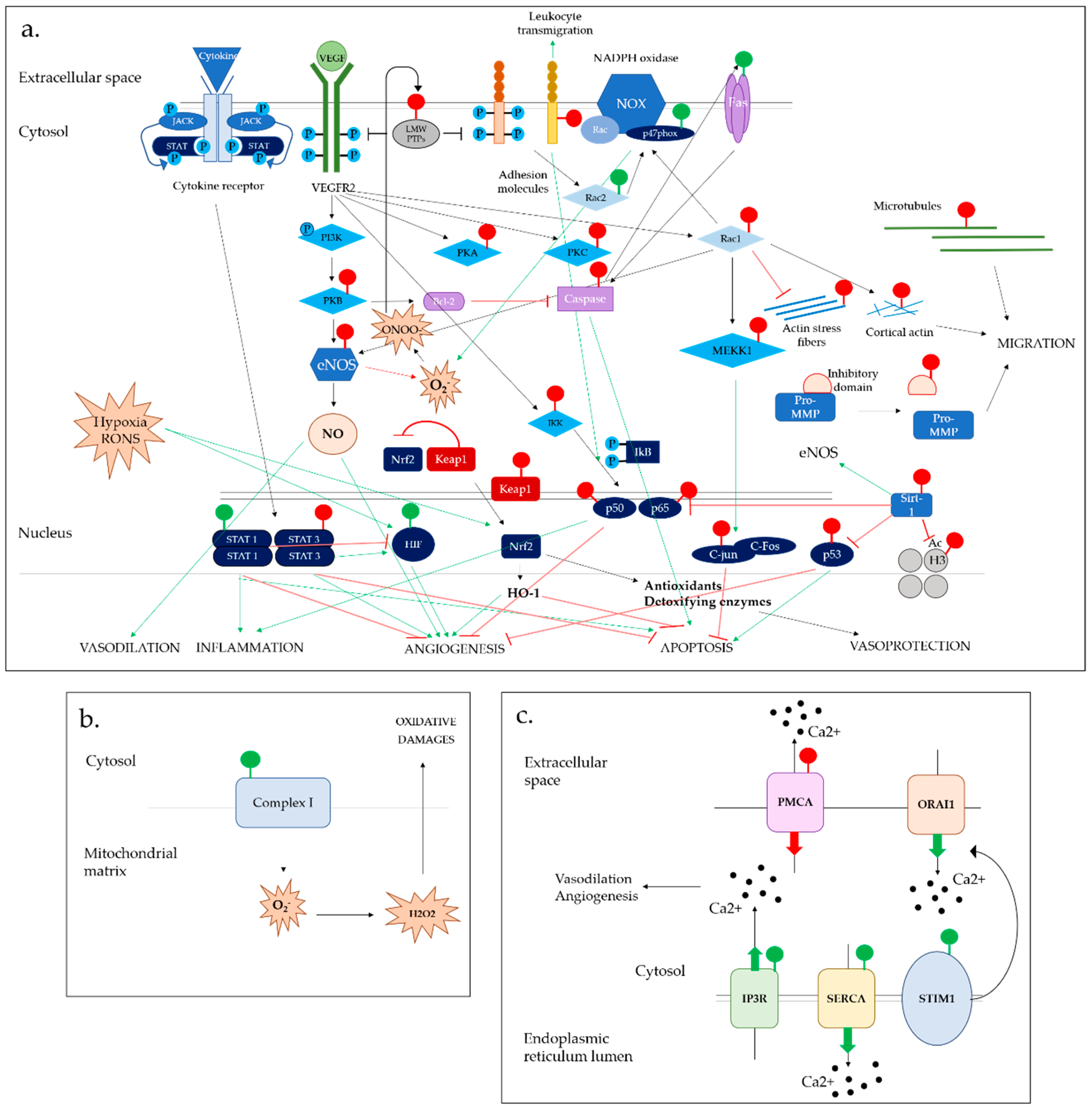
6. S-Glutathionylation Effects on RONS Production
6.1. NADPH Oxidase Complex
6.2. Endothelial Nitric Oxide Synthase System
7. S-Glutathionylation Effects on Ca2+ Homeostasis
7.1. Calcium-Dependent IP3R & PMCA Channels
7.2. SERCA2b Calcium Pump
7.3. STIM1 Molecule and ORAI1 Channel
8. S-Glutathionylation Effects on Cell Death and Autophagy
8.1. Apoptotic Signalling
8.2. Autophagy
9. Redox Regulation of Cell Structure and Dynamics by S-Glutathionylation
9.1. Metalloproteases
9.2. Adhesion Proteins
9.3. Cytoskeletal Proteins
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| [Ca2+]i | cytosolic calcium ion concentrations |
| AngII | angiotensin II |
| Ca2+ | calcium ion |
| CO | carbon oxide |
| CVD | cardiovascular diseases |
| Cys | cysteine |
| EC | endothelial cell |
| ECM | extracellular matrix |
| eNOS | endothelial nitric oxide synthase |
| ER | endoplasmic reticulum |
| Grx | glutaredoxin |
| GSH | glutathione |
| HDAC | histone deacetylase |
| HIF-1α | hypoxia-inducible factor 1α |
| ICAM-1 | intercellular adhesion molecule 1 |
| LMW-PTP | Low-molecular-weight protein tyrosin phosphatase |
| MAPK | mitogen-activated protein kinase |
| MEK | mitogen-activated protein kinase kinase |
| MMP | matrix metalloproteinases |
| NAD+ | nicotinamide adenine dinucleotide |
| NFκB | nuclear factor-kappa B |
| NO | nitric oxide |
| NOX | NADPH oxidase |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| O2 | superoxide |
| oxPTM | oxidative post-translational modifications |
| PKB | protein kinase B |
| PTM | post-translational modifications |
| PTP | protein tyrosine phosphatase |
| Rac1 | ras-related C3 botulinum toxin substrate 1 |
| RONS | reactive oxygen and nitrogen species |
| Sirt-1 | sirtuin 1 |
| STAT | signal transducer and activator of transcription |
| TGF-β | transforming growth factor beta |
| TIMPs | tissue inhibitors of metalloproteinases |
| TNF-α | tumor necrosis factor alpha |
| VCAM-1 | vascular cell adhesion protein 1 |
| VEGF | vascular endothelial growth factor |
| VEGFR1 | vascular endothelial growth factor receptor 1 |
References
- Cervantes Gracia, K.; Llanas-Cornejo, D.; Husi, H. CVD and Oxidative Stress. J. Clin. Med. 2017, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Panth, N.; Paudel, K.R.; Parajuli, K. Reactive Oxygen Species: A Key Hallmark of Cardiovascular Disease. Adv. Med. 2016, 2016, 9152732. [Google Scholar] [CrossRef] [PubMed]
- Sorriento, D.; De Luca, N.; Trimarco, B.; Iaccarino, G. The Antioxidant Therapy: New Insights in the Treatment of Hypertension. Front. Physiol. 2018, 9, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murdoch, C.E.; Shuler, M.; Haeussler, D.J.F.; Kikuchi, R.; Bearelly, P.; Han, J.; Watanabe, Y.; Fuster, J.J.; Walsh, K.; Ho, Y.-S.; et al. Glutaredoxin-1 Up-regulation Induces Soluble Vascular Endothelial Growth Factor Receptor 1, Attenuating Post-ischemia Limb Revascularization. J. Boil. Chem. 2014, 289, 8633–8644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolin, J.D. Redox Control of Allergic Airway Disease: Impact of Glutaredoxin-1 on Epithelial Driven Inflammation and Allergen-Induced Airway Remodeling. Bachelor’s Thesis, The University of Vermon, Burlington, VT, USA, 2015. [Google Scholar]
- Kuipers, I.; Louis, R.; Manise, M.; Dentener, M.A.; Irvin, C.G.; Janssen-Heininger, Y.M.; Brightling, C.E.; Wouters, E.F.; Reynaert, N.L. Increased glutaredoxin-1 and decreased protein S-glutathionylation in sputum of asthmatics. Eur. Respir. J. 2013, 41, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Poston, L.; Briley, A.; Seed, P.; Kelly, F.; Shennan, A.; Seed, P. Vitamin C and vitamin E in pregnant women at risk for pre-eclampsia (VIP trial): Randomised placebo-controlled trial. Lancet 2006, 367, 1145–1154. [Google Scholar] [CrossRef]
- Mieyal, J.J.; Gallogly, M.M.; Qanungo, S.; Sabens, E.A.; Shelton, M.D. Molecular Mechanisms and Clinical Implications of Reversible Protein S-Glutathionylation. Antioxid. Redox Signal. 2008, 10, 1941–1988. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Uys, J.D.; Tew, K.D.; Townsend, D.M. S-Glutathionylation: From Molecular Mechanisms to Health Outcomes. Antioxid. Redox Signal. 2011, 15, 233–270. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.J.L.; Pinto, J.T.; Callery, P.S. Reversible and irreversible protein glutathionylation: Biological and clinical aspects. Expert Opin. Drug Metab. Toxicol. 2011, 7, 891–910. [Google Scholar] [CrossRef]
- Dominko, K.; Đikić, D. Glutathionylation: A regulatory role of glutathione in physiological processes. Arch. Ind. Hyg. Toxicol. 2018, 69, 1–24. [Google Scholar] [CrossRef]
- Grek, C.L.; Zhang, J.; Manevich, Y.; Townsend, D.M.; Tew, K.D. Causes and Consequences of Cysteine S-Glutathionylation. J. Boil. Chem. 2013, 288, 26497–26504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Luca, A.; Moroni, N.; Serafino, A.; Primavera, A.; Pastore, A.; Pedersen, J.Z.; Petruzzelli, R.; Farrace, M.G.; Pierimarchi, P.; Pierimarchi, G.; et al. Treatment of doxorubicin-resistant MCF7/Dx cells with nitric oxide causes histone glutathionylation and reversal of drug resistance. Biochem. J. 2011, 440, 175–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Giménez, J.L.; Olaso, G.; Hake, S.B.; Bönisch, C.; Wiedemann, S.M.; Markovic, J.; Dasi, F.; Gimeno, A.; Perez-Quilis, C.; Palacios, Ò.; et al. Histone H3 Glutathionylation in Proliferating Mammalian Cells Destabilizes Nucleosomal Structure. Antioxid. Redox Signal. 2013, 19, 1305–1320. [Google Scholar] [CrossRef] [Green Version]
- Autiero, I.; Costantini, S.; Colonna, G. Human Sirt-1: Molecular Modeling and Structure-Function Relationships of an Unordered Protein. PLoS ONE 2009, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zee, R.S.; Yoo, C.B.; Pimentel, D.R.; Perlman, D.H.; Burgoyne, J.R.; Hou, X.; McComb, M.E.; Costello, C.E.; Cohen, R.A.; Bachschmid, M.M. Redox Regulation of Sirtuin-1 by S-Glutathiolation. Antioxid. Redox Signal. 2010, 13, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Fry, J.L.; Han, J.; Hou, X.; Pimentel, D.R.; Matsui, R.; Cohen, R.A.; Bachschmid, M.M. A Redox-resistant Sirtuin-1 Mutant Protects against Hepatic Metabolic and Oxidant Stress. J. Boil. Chem. 2014, 289, 7293–7306. [Google Scholar] [CrossRef] [Green Version]
- Liao, B.-C.; Hsieh, C.-W.; Lin, Y.-C.; Wung, B.-S. The Glutaredoxin/Glutathione System Modulates NF-κB Activity by Glutathionylation of p65 in Cinnamaldehyde-Treated Endothelial Cells. Toxicol. Sci. 2010, 116, 151–163. [Google Scholar] [CrossRef]
- Pineda-Molina, E.; Klatt, P.; Vázquez, J.; Marina, A.; De Lacoba, M.G.; Pérez-Sala, D.; Lamas, S. Glutathionylation of the p50 Subunit of NF-κB: A Mechanism for Redox-Induced Inhibition of DNA Binding. Biochemistry 2001, 40, 14134–14142. [Google Scholar] [CrossRef]
- Klatt, P.; Molina, E.P.; De Lacoba, M.G.; Padilla, C.A.; Martínez-Galisteo, E.; Bárcena, J.A.; Lamas, S. Redox regulation of c-Jun DNA binding by reversible S-glutathiolation. FASEB J. 1999, 13, 1481–1490. [Google Scholar] [CrossRef]
- Klatt, P.; Molina, E.P.; Lamas, S. Nitric Oxide Inhibits c-Jun DNA Binding by Specifically TargetedS-Glutathionylation. J. Boil. Chem. 1999, 274, 15857–15864. [Google Scholar] [CrossRef] [Green Version]
- Velu, C.S.; Niture, S.K.; Doneanu, C.E.; Pattabiraman, N.; Srivenugopal, K.S. Human p53 is Inhibited by Glutathionylation of Cysteines Present in the Proximal DNA-Binding Domain During Oxidative Stress. Biochemistry 2007, 46, 7765–7780. [Google Scholar] [CrossRef] [PubMed]
- Jeon, D.; Park, H.J.; Kim, H.S. Protein S-glutathionylation induced by hypoxia increases hypoxia-inducible factor-1α in human colon cancer cells. Biochem. Biophys. Res. Commun. 2018, 495, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Murdoch, C.E.; Sano, S.; Ido, Y.; Bachschmid, M.M.; Cohen, R.A.; Matsui, R. Glutathione adducts induced by ischemia and deletion of glutaredoxin-1 stabilize HIF-1α and improve limb revascularization. Proc. Natl. Acad. Sci. USA 2016, 113, 6011–6016. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Kole, S.; Precht, P.; Pazin, M.J.; Bernier, M. S-Glutathionylation impairs signal transducer and activator of transcription 3 activation and signaling. Endocrinology 2009, 150, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Butturini, E.; Darra, E.; Chiavegato, G.; Cellini, B.; Cozzolino, F.; Pucci, P.; Dell’Orco, D.; Monti, M.; Mariotto, S. S-Glutathionylation at Cys328 and Cys542 Impairs STAT3 Phosphorylation. ACS Chem. Boil. 2014, 9, 1885–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.-C.; Huang, Y.-T.; Hsieh, C.-W.; Yang, P.-M.; Wung, B.-S. Carbon Monoxide Induces Heme Oxygenase-1 to Modulate STAT3 Activation in Endothelial Cells via S-Glutathionylation. PLoS ONE 2014, 9, e100677. [Google Scholar] [CrossRef] [PubMed]
- Butturini, E.; Cozzolino, F.; Boriero, D.; Carcereri de Prati, A.; Monti, M.; Rossin, M.; Canetti, D.; Cellini, B.; Pucci, P.; Mariotto, S. S-glutathionylation exerts opposing roles in the regulation of STAT1 and STAT3 signaling in reactive microglia. Free Radic Biol. Med. 2018, 590, 191–201. [Google Scholar]
- Carvalho, A.N.; Marques, C.; Guedes, R.C.; Castro-Caldas, M.; Rodrigues, E.; Van Horssen, J.; Gama, M.J.; Castro-Caldas, M.; Horssen, J. S-Glutathionylation of Keap1: A new role for glutathioneS-transferase pi in neuronal protection. FEBS Lett. 2016, 590, 1455–1466. [Google Scholar] [CrossRef]
- Wang, L.; Qu, G.; Gao, Y.; Su, L.; Ye, Q.; Jiang, F.; Zhao, B.; Miao, J. A small molecule targeting glutathione activates Nrf2 and inhibits cancer cell growth through promoting Keap-1 S-glutathionylation and inducing apoptosis. RSC Adv. 2018, 8, 792–804. [Google Scholar] [CrossRef]
- Reynaert, N.L.; van der Vliet, A.; Guala, A.S.; McGovern, T.; Hristova, M.; Pantano, C.; Heintz, N.H.; Heim, J.; Ho, Y.-S.; Matthews, D.E.; et al. Dynamic redox control of NF- kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory B kinase beta. Proc. Natl. Acad. Sci. USA 2006, 103, 13086–13091. [Google Scholar] [CrossRef]
- Abdelsaid, M.A.; El-Remessy, A.B. S-glutathionylation of LMW-PTP regulates VEGF-mediated FAK activation and endothelial cell migration. J. Cell Sci. 2012, 125, 4751–4760. [Google Scholar] [CrossRef] [PubMed]
- Barrett, W.C.; DeGnore, J.P.; Keng, Y.-F.; Zhang, Z.-Y.; Yim, M.B.; Chock, P.B. Roles of Superoxide Radical Anion in Signal Transduction Mediated by Reversible Regulation of Protein-tyrosine Phosphatase 1B. J. Boil. Chem. 1999, 274, 34543–34546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, W.C.; DeGnore, J.P.; König, S.; Fales, H.M.; Keng, Y.-F.; Zhang, Z.-Y.; Yim, M.B.; Chock, P.B. Regulation of PTP1B via Glutathionylation of the Active Site Cysteine 215. Biochemistry 1999, 38, 6699–6705. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Weisbrod, R.M.; Shao, D.; Watanabe, Y.; Yin, X.; Bachschmid, M.M.; Seta, F.; Janssen-Heininger, Y.M.; Matsui, R.; Zang, M.; et al. The redox mechanism for vascular barrier dysfunction associated with metabolic disorders: Glutathionylation of Rac1 in endothelial cells. Redox Boil. 2016, 9, 306–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, G.A.; Zhou, B.; Cox, A.D.; Campbell, S.L. Rho GTPases, oxidation, and cell redox control. Small GTPases 2014, 5, e28579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kil, I.S.; Shin, S.W.; Park, J.-W. S-glutathionylation regulates GTP-binding of Rac2. Biochem. Biophys. Res. Commun. 2012, 425, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, L.; Hobbs, G.A.; Aghajanian, A.; Campbell, S.L. Redox Regulation of Ras and Rho GTPases: Mechanism and Function. Antioxid. Redox Signal. 2013, 18, 250–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavreul, N.; Pimental, D.R.; Adachi, T.; Ido, Y.; Schöneich, C.; Cohen, R.A. S-glutathiolation by peroxynitrite of p21ras at cysteine-118 mediates its direct activation and downstream signaling in endothelial cells. FASEB J. 2006, 20, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, A.N.; Cobb, M.H. Protein kinase function and glutathionylation. Biochem. J. 2004, 381, e1. [Google Scholar] [CrossRef] [PubMed]
- Humphries, K.M.; Juliano, C.; Taylor, S.S. Regulation of cAMP-dependent Protein Kinase Activity by Glutathionylation. J. Boil. Chem. 2002, 277, 43505–43511. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Pan, S.; Berk, B.C. Glutaredoxin Mediates Akt and eNOS Activation by Flow in a Glutathione Reductase-Dependent Manner. Arter. Thromb. Vasc. Boil. 2007, 27, 1283–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Jann, J.; Xavier, C.; Wu, H. Glutaredoxin 1 (Grx1) Protects Human Retinal Pigment Epithelial Cells from Oxidative Damage by Preventing AKT Glutathionylation. Investig. Opthalmol. Vis. Sci. 2015, 56, 2821. [Google Scholar] [CrossRef] [PubMed]
- Cross, J.V.; Templeton, D.J. Oxidative stress inhibits MEKK1 by site-specific glutathionylation in the ATP-binding domain. Biochem. J. 2004, 381, 675–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, N.E.; Stewart, J.R.; Ioannides, C.G.; O’Brian, C.A. Oxidant-InducedS-Glutathiolation Inactivates Protein Kinase C-α (PKC-α): A Potential Mechanism of PKC Isozyme Regulation. Biochemistry 2000, 39, 10319–10329. [Google Scholar] [CrossRef] [PubMed]
- Nagarkoti, S.; Dubey, M.; Awasthi, D.; Kumar, V.; Chandra, T.; Kumar, S.; Dikshit, M. S-Glutathionylation of p47phox sustains superoxide generation in activated neutrophils. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2018, 1865, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.R.; Hurrell, F.; Murphy, M.P.; Shannon, R.J.; Lin, T.-K.; Hirst, J. Reversible Glutathionylation of Complex I Increases Mitochondrial Superoxide Formation. J. Boil. Chem. 2003, 278, 19603–19610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.A.; Wang, T.Y.; Varadharaj, S.; Reyes, L.A.; Hemann, C.; Hassan Talukder, M.A.; Chen, Y.R.; Druhan, L.J.; Zweier, J.L. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 2010, 468, 1115–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Pascali, F.; Hemann, C.; Samons, K.; Chen, C.-A.; Zweier, J.L. Hypoxia and Reoxygenation Induce Endothelial Nitric Oxide Synthase Uncoupling in Endothelial Cells through Tetrahydrobiopterin Depletion and S-Glutathionylation. Biochemistry 2014, 53, 3679–3688. [Google Scholar] [CrossRef]
- Shang, Q.; Bao, L.; Guo, H.; Hao, F.; Luo, Q.; Chen, J.; Guo, C. Contribution of glutaredoxin-1 to S-glutathionylation of endothelial nitric oxide synthase for mesenteric nitric oxide generation in experimental necrotizing enterocolitis. Transl. Res. 2017, 188, 92–105. [Google Scholar] [CrossRef]
- Galougahi, K.K.; Liu, C.; Gentile, C.; Kok, C.; Nunez, A.; Garcia, A.; Fry, N.A.S.; Davies, M.J.; Hawkins, C.L.; Rasmussen, H.H.; et al. Glutathionylation Mediates Angiotensin II–Induced eNOS Uncoupling, Amplifying NADPH Oxidase—Dependent Endothelial Dysfunction. J. Am. Heart Assoc. 2014, 3, 1–11. [Google Scholar] [CrossRef]
- Wu, F.; Szczepaniak, W.S.; Shiva, S.; Liu, H.; Wang, Y.; Wang, L.; Wang, Y.; Kelley, E.E.; Chen, A.F.; Gladwin, M.T.; et al. Nox2-dependent glutathionylation of endothelial NOS leads to uncoupled superoxide production and endothelial barrier dysfunction in acute lung injury. Am. J. Physiol. Cell. Mol. Physiol. 2014, 307, L987–L997. [Google Scholar] [CrossRef] [PubMed]
- Henschke, P.N.; Elliott, S.J. Oxidized glutathione decreases luminal Ca2+ content of the endothelial cell ins(1,4,5) P3 -sensitive Ca2+ store. Biochem. J. 2015, 312, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Lock, J.T.; Sinkins, W.G.; Schilling, W.P. Protein S-glutathionylation enhances Ca2+-induced Ca2+ release via the IP3 receptor in cultured aortic endothelial cells. J. Physiol. 2012, 590, 3431–3447. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Kang, J.; Kwon, H.; Frueh, D.; Yoo, S.H.; Wagner, G.; Park, S. Effects of Redox Potential and Ca2+ on the Inositol 1,4,5-Trisphosphate Receptor L3-1 Loop Region. J. Biol. Chem. 2008, 283, 25567–25575. [Google Scholar] [CrossRef] [PubMed]
- Lock, J.T.; Sinkins, W.G.; Schilling, W.P. Effect of protein S-glutathionylation on Ca2+ homeostasis in cultured aortic endothelial cells. AJP Heart Circ. Physiol. 2011, 300, H493–H506. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, A.M.; Thompson, M.D.; Bolotina, V.M.; Tong, X.; Cohen, R.A. Nox4- and Nox2-dependent oxidant production is required for VEGF-induced SERCA cysteine-674 S-glutathiolation and endothelial cell migration. Free Radic. Boil. Med. 2012, 53, 2327–2334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, M.D.; Mei, Y.; Weisbrod, R.M.; Silver, M.; Shukla, P.C.; Bolotina, V.M.; Cohen, R.A.; Tong, X. Glutathione adducts on sarcoplasmic/endoplasmic reticulum Ca2+ ATPase Cys-674 regulate endothelial cell calcium stores and angiogenic function as well as promote ischemic blood flow recovery. J. Biol. Chem. 2014, 289, 19907–19916. [Google Scholar] [CrossRef]
- Mei, Y.; Thompson, M.D.; Shiraishi, Y.; Cohen, R.A.; Tong, X. Sarcoplasmic/endoplasmic reticulum Ca2+ ATPase C674 promotes ischemia- and hypoxia-induced angiogenesis via coordinated endothelial cell and macrophage function. J. Mol. Cell. Cardiol. 2014, 76, 275–282. [Google Scholar] [CrossRef]
- Sharov, V.S.; Dremina, E.S.; Galeva, N.A.; Williams, T.D.; Schöneich, C. Quantitative mapping of oxidation-sensitive cysteine residues in SERCA in vivo and in vitro by HPLC–electrospray-tandem MS: Selective protein oxidation during biological aging. Biochem. J. 2006, 394, 605–615. [Google Scholar] [CrossRef]
- Tong, X.Y.; Hou, X.; Jourd’Heuil, D.; Weisbrod, R.M.; Cohen, R.A. Upregulation of Nox4 by TGFβ1 Oxidizes SERCA and Inhibits NO in Arterial Smooth Muscle of the Prediabetic Zucker Rat. Circ. Res. 2010, 107, 975–983. [Google Scholar] [CrossRef]
- Qin, F.; Siwik, D.A.; Pimentel, D.R.; Morgan, R.J.; Biolo, A.; Tu, V.H.; Kang, Y.J.; Cohen, R.A.; Colucci, W.S. Cytosolic H2O2 mediates hypertrophy, apoptosis, and decreased SERCA activity in mice with chronic hemodynamic overload. Am. J. Physiol. Circ. Physiol. 2014, 306, H1453–H1463. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, B.J.; Irrinki, K.M.; Mallilankaraman, K.; Lien, Y.-C.; Wang, Y.; Bhanumathy, C.D.; Subbiah, R.; Ritchie, M.F.; Soboloff, J.; Baba, Y.; et al. S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J. Cell Boil. 2010, 190, 391–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anathy, V.; Aesif, S.W.; Guala, A.S.; Havermans, M.; Reynaert, N.L.; Ho, Y.-S.; Budd, R.C.; Janssen-Heininger, Y.M. Redox amplification of apoptosis by caspase-dependent cleavage of glutaredoxin 1 and S-glutathionylation of Fas. J. Cell Boil. 2009, 184, 241–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, S.; Berk, B.C. Glutathiolation Regulates Tumor Necrosis Factor-α–Induced Caspase-3 Cleavage and Apoptosis. Circ. Res. 2006, 100, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Pinto, J.T.; Deng, H.; Richie, J.P. Inhibition of Caspase-3 Activity and Activation by Protein Glutathionylation. Biochem. Pharmacol. 2008, 75, 2234–2244. [Google Scholar] [CrossRef] [PubMed]
- McGarry, D.J.; Chakravarty, P.; Wolf, C.R.; Henderson, C.J. Altered Protein S-Glutathionylation Identifies a Potential Mechanism of Resistance to Acetaminophen-Induced Hepatotoxicity. J. Pharmacol. Exp. Ther. 2015, 355, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Yepes, J.; Burns, M.; Anandhan, A.; Khalimonchuk, O.; Del Razo, L.M.; Quintanilla-Vega, B.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Oxidative Stress, Redox Signaling, and Autophagy: Cell Death Versus Survival. Antioxid. Redox Signal. 2014, 21, 66–85. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Akaike, T.; Sawa, T.; Miyamoto, Y.; Van Der Vliet, A.; Maeda, H. Activation of Matrix Metalloproteinases by Peroxynitrite-induced ProteinS-Glutathiolation via DisulfideS-Oxide Formation. J. Boil. Chem. 2001, 276, 29596–29602. [Google Scholar] [CrossRef]
- Mukherjee, T.K.; Mishra, A.K.; Mukhopadhyay, S.; Hoidal, J.R. High Concentration of Antioxidants N-Acetylcysteine and Mitoquinone-Q Induces Intercellular Adhesion Molecule 1 and Oxidative Stress by Increasing Intracellular Glutathione. J. Immunol. 2014, 178, 1835–1844. [Google Scholar] [CrossRef]
- Stojkov, D.; Amini, P.; Oberson, K.; Sokollik, C.; Duppenthaler, A.; Simon, H.-U.; Yousefi, S. ROS and glutathionylation balance cytoskeletal dynamics in neutrophil extracellular trap formation. J. Cell Boil. 2017, 216, 4073–4090. [Google Scholar] [CrossRef]
- Fiaschi, T.; Cozzi, G.; Raugei, G.; Formigli, L.; Ramponi, G.; Chiarugi, P. Redox regulation of β-actin during integrin-mediated cell adhesion. J. Biol. Chem. 2006, 281, 22983–22991. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Boja, E.S.; Tan, W.; Tekle, E.; English, S.; Mieyal, J.J.; Chock, P.B.; Fales, H.M. Reversible Glutathionylation Regulates Actin Polymerization in A431 Cells. J. Boil. Chem. 2001, 276, 47763–47766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Seefeldt, T.; Young, A.; Zhang, X.; Zhao, Y.; Ruffolo, J.; Kaushik, R.S.; Guan, X. Microtubule S-glutathionylation as a potential approach for antimitotic agents. BMC Cancer 2012, 12, 245. [Google Scholar] [CrossRef] [PubMed]
- Landino, L.M.; Moynihan, K.L.; Todd, J.V.; Kennett, K.L. Modulation of the redox state of tubulin by the glutathione/glutaredoxin reductase system. Biochem. Biophys. Res. Commun. 2004, 314, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.S.; Marsden, P.A. Epigenetics in the Vascular Endothelium: Looking from a different perspective in the epigenomics era. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2297–2306. [Google Scholar] [CrossRef] [PubMed]
- Berezin, A.E. Epigenetic Mechanisms of Endothelial Progenitor Cell Dysfunction. J. Clin. Epigenet. 2016, 2. [Google Scholar] [CrossRef] [Green Version]
- Fraineau, S.; Brand, M.; Palii, C.G.; Allan, D.S. Epigenetic regulation of endothelial-cell-mediated vascular repair. FEBS J. 2015, 282, 1605–1629. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Ray, S.; Home, P.; Saha, B.; Wang, S.; Sheibani, N.; Tawfik, O.; Cheng, N.; Paul, S. Regulation of Angiogenesis by Histone Chaperone HIRA-mediated Incorporation of Lysine 56-acetylated Histone H3.3 at Chromatin Domains of Endothelial Genes. J. Boil. Chem. 2010, 285, 41567–41577. [Google Scholar] [CrossRef] [Green Version]
- Fish, J.E.; Matouk, C.C.; Rachlis, A.; Lin, S.; Tai, S.C.; D’Abreo, C.; Marsden, P.A. The Expression of Endothelial Nitric-oxide Synthase Is Controlled by a Cell-specific Histone Code. J. Boil. Chem. 2005, 280, 24824–24838. [Google Scholar] [CrossRef] [Green Version]
- Urbich, C.; Dimmeler, S.; Zeiher, A.M.; Rossig, L.; Diehl, F. The histone methyltransferase MLL is an upstream regulator of endothelial-cell sprout formation. Blood 2006, 109, 1472–1478. [Google Scholar]
- Kreuz, S.; Fischle, W. Oxidative stress signaling to chromatin in health and disease. Epigenomics 2016, 8, 843–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downs, J.A.; Nussenzweig, M.C.; Nussenzweig, A. Chromatin dynamics and the preservation of genetic information. Nature 2007, 447, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Panyim, S.; Sommer, K.R.; Chalkley, R. Oxidation of the cysteine-containing histone F3. Detection of an evolutionary mutation in a conservative histone. Biochemistry 1971, 10, 3911–3917. [Google Scholar] [CrossRef] [PubMed]
- Blackshire, B. The effects of histone glutathionylation on chromatin structure. FASEB J. 2014, 28, 942–945. [Google Scholar]
- Colussi, C.; Mozzetta, C.; Gurtner, A.; Illi, B.; Rosati, J.D.; Straino, S.; Ragone, G.; Pescatori, M.; Zaccagnini, G.; Antonini, A.; et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc. Natl. Acad. Sci. USA 2008, 105, 19183–19187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Wu, Y.; Yang, P. High glucose-induced oxidative stress represses sirtuin deacetylase expression and increases histone acetylation leading to neural tube defects. J. Neurochem. 2016, 137, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.; Escande, C.; Denicola, A. Potential modulation of sirtuins by oxidative stress. Oxid. Med. Cell. Longev. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto-Komatsu, A.; Hirase, T.; Asaka, M.; Node, K. Angiotensin II induces microtubule reorganization mediated by a deacetylase SIRT2 in endothelial cells. Hypertens. Res. 2011, 34, 949–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maizel, J.; Xavier, S.; Chen, J.; Lin, C.H.S.; Vasko, R.; Goligorsky, M.S. Sirtuin 1 ablation in endothelial cells is associated with impaired angiogenesis and diastolic dysfunction. Am. J. Physiol. Circ. Physiol. 2014, 307, H1691–H1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghisays, F.; Brace, C.S.; Yackly, S.M.; Kwon, H.J.; Mills, K.F.; Kashentseva, E.; Dimitriev, I.P.; Curiel, D.T.; Imai, S.-I.; Ellenberger, T. The N-terminal Domain of SIRT1 Is a Positive Regulator of Endogenous SIRT1-dependent Deacetylation and Transcriptional Outputs. Cell Rep. 2015, 10, 1665–1673. [Google Scholar] [CrossRef]
- Yeung, F.; E Hoberg, J.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; A Frye, R.; Mayo, M.W. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Strand, S.; Schlufter, F.; Siuda, D.; Reifenberg, G.; Kleinert, H.; Förstermann, U.; Li, H. Role of SIRT1 and FOXO factors in eNOS transcriptional activation by resveratrol. Nitric Oxide 2013, 32, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Mattagajasingh, I.; Kim, C.-S.; Naqvi, A.; Yamamori, T.; Hoffman, T.A.; Jung, S.-B.; DeRicco, J.; Kasuno, K.; Irani, K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 14855–14860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempe, S.; Kestler, H.; Lasar, A.; Wirth, T. NF-κB controls the global pro-inflammatory response in endothelial cells: Evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 2005, 33, 5308–5319. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.-N.; Zhu, N.; Liu, C.; Wu, H.-T.; Gui, Y.; Liao, D.-F.; Qin, L. Wnt5a and its signaling pathway in angiogenesis. Clin. Chim. Acta 2017, 471, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Qanungo, S.; Starke, D.W.; Pai, H.V.; Mieyal, J.J.; Nieminen, A.-L. Glutathione Supplementation Potentiates Hypoxic Apoptosis by S-Glutathionylation of p65-NFκB. J. Boil. Chem. 2007, 282, 18427–18436. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis and c-Jun. J. Natl. Cancer Inst. 2004, 96, 644. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, L.; Han, W.; Shen, T.; Ma, C.; Liu, Y.; Nie, X.; Liu, M.; Ran, Y.; Zhu, D. Activation of JNK/c-Jun is required for the proliferation, survival, and angiogenesis induced by EET in pulmonary artery endothelial cells. J. Lipid Res. 2012, 53, 1093–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vleugel, M.M.; Greijer, A.E.; Bos, R.; Van Der Wall, E.; Van Diest, P.J. c-Jun activation is associated with proliferation and angiogenesis in invasive breast cancer. Hum. Pathol. 2006, 37, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Yamakuchi, M.; Panta, S.; Hashiguchi, T. p53 and Vascular Dysfunction: MicroRNA in Endothelial Cells, Vasculitis in Practice—An Update on Special Situations—Clinical and Therapeutic Considerations. IntechOpen 2018. [Google Scholar] [CrossRef]
- Chen, B.; Lu, Y.; Chen, Y.; Cheng, J. The role of Nrf2 in oxidative stress-induced endothelial injuries. J. Endocrinol. 2015, 225, R83–R99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florczyk, U.; Jazwa, A.; Maleszewska, M.; Mendel, M.; Szade, K.; Kozakowska, M.; Grochot-Przeczek, A.; Viscardi, M.; Czauderna, S.; Bukowska-Strakova, K.; et al. Nrf2 Regulates Angiogenesis: Effect on Endothelial Cells, Bone Marrow-Derived Proangiogenic Cells and Hind Limb Ischemia. Antioxid. Redox Signal. 2013, 20, 1693–1708. [Google Scholar] [CrossRef] [PubMed]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Shui, Q.Y.; Garcia, J.G.N.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 564–565. [Google Scholar] [CrossRef] [PubMed]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genome Res. 1998, 12, 149–162. [Google Scholar]
- Rawlings, J.S. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartoli, M.; Gu, X.; Tsai, N.T.; Venema, R.C.; Brooks, S.E.; Marrero, M.B.; Caldwell, R.B. Vascular Endothelial Growth Factor Activates STAT Proteins in Aortic Endothelial Cells. J. Boil. Chem. 2000, 275, 33189–33192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.-H.; Murphy, D.A.; Lassoued, W.; Thurston, G.; Feldman, M.D.; Lee, W.M. Activated STAT3 is a mediator and biomarker of VEGF endothelial activation. Cancer Boil. Ther. 2008, 7, 1994–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Laso, V.; Sastre, C.; Méndez-Barbero, N.; Egido, J.; Martín-Ventura, J.L.; Gómez-Guerrero, C.; Blanco-Colio, L.M. TWEAK blockade decreases atherosclerotic lesion size and progression through suppression of STAT1 signaling in diabetic mice. Sci. Rep. 2017, 7, 46679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Zhang, Y.; Xu, L.; Lin, Y.; Yang, X.; Bai, L.; Chen, Y.; Zhao, S.; Fan, J.; Cheng, X.; et al. Protein Inhibitor of Activated STAT3 Suppresses Oxidized LDL-induced Cell Responses during Atherosclerosis in Apolipoprotein E-deficient Mice. Sci. Rep. 2016, 6, 36790. [Google Scholar] [CrossRef]
- Xue, C.; Xie, J.; Zhao, D.; Lin, S.; Zhou, T.; Shi, S.; Shao, X.; Lin, Y.; Zhu, B.; Cai, X. The JAK/STAT3 signalling pathway regulated angiogenesis in an endothelial cell/adipose-derived stromal cell co-culture, 3D gel model. Cell Prolif. 2017, 50, 1–10. [Google Scholar] [CrossRef]
- Battle, T.E.; Lynch, R.A.; Frank, D.A. Signal Transducer and Activator of Transcription 1 Activation in Endothelial Cells Is a Negative Regulator of Angiogenesis. Cancer Res. 2006, 66, 3649–3657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, S.G.; Bae, Y.S.; Lee, S.-R.; Kwon, J. Hydrogen Peroxide: A Key Messenger That Modulates Protein Phosphorylation Through Cysteine Oxidation. Sci. Signal. 2000, 2000, pe1. [Google Scholar] [CrossRef] [PubMed]
- Klomsiri, C.; Karplus, P.A.; Poole, L.B. Cysteine-Based Redox Switches in Enzymes. Antioxid. Redox Signal. 2010, 14, 1065–1077. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, P. The Redox Regulation of LMW-PTP During Cell Proliferation or Growth Inhibition. IUBMB Life 2001, 52, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, E.; Raugei, G.; Chiarugi, P.; Ramponi, G. A novel redox-based switch: LMW-PTP oxidation enhances Grb2 binding and leads to ERK activation. Biochem. Biophys. Res. Commun. 2006, 348, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Xing, K.; Raza, A.; Löfgren, S.; Fernando, M.R.; Ho, Y.S.; Lou, M.F. LMW-PTP and Its Possible Physiological Functions of redox signaling in the eye lens. Biochim. Biophys. Acta. 2007, 1774, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Patrushev, N.; Inomata, H.; Mehta, D.; Urao, N.; Kim, H.W.; Razvi, M.; Kini, V.; Mahadev, K.; Goldstein, B.J.; et al. Role of Protein Tyrosine Phosphatase 1B in VEGF Signaling and Cell-Cell Adhesions in Endothelial Cells. Circ. Res. 2008, 102, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Besnier, M.; Galaup, A.; Nicol, L.; Henry, J.-P.; Coquerel, D.; Gueret, A.; Mulder, P.; Brakenhielm, E.; Thuillez, C.; Germain, S.; et al. Enhanced angiogenesis and increased cardiac perfusion after myocardial infarction in protein tyrosine phosphatase 1B-deficient mice. FASEB J. 2014, 28, 3351–3361. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Sankar, S.; Lin, C.; Kontos, C.D.; Schroff, A.D.; Cha, E.H.; Feng, S.-M.; Li, S.-F.; Yu, Z.; Van Etten, R.L.; et al. HCPTPA, a Protein Tyrosine Phosphatase That Regulates Vascular Endothelial Growth Factor Receptor-mediated Signal Transduction and Biological Activity. J. Boil. Chem. 1999, 274, 38183–38188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilzer, P.; Chang, K.; Marvel, J.; Rowold, E.; Jaudes, P.; Ullensvang, S.; Kilo, C.; Williamson, J.R. Albumin Permeation of New Vessels Is Increased in Diabetic Rats. Diabetes 1985, 34, 333–336. [Google Scholar] [CrossRef]
- Williamson, J.R.; Chang, K.; Tilton, R.G.; Prater, C.; Jeffrey, J.R.; Weigel, C.; Sherman, W.R.; Eades, D.M.; Kilo, C. Increased Vascular Permeability in Spontaneously Diabetic BB/W Rats and in Rats with Mild Versus Severe Streptozocin-Induced Diabetes - Prevention by Aldose Reductase Inhibitors and Castration. Diabetes 1987, 36, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Usatyuk, P.V.; Vepa, S.; Watkins, T.; He, D.; Parinandi, N.L.; Natarajan, V. Redox Regulation of Reactive Oxygen Species-Induced p38 MAP Kinase Activation and Barrier Dysfunction in Lung Microvascular Endothelial Cells. Antioxid. Redox Signal. 2003, 5, 723–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandenbroucke, E.; Mehta, D.; Minshall, R.; Malik, A.B. Regulation of Endothelial Junctional Permeability. Ann. N. Y. Acad. Sci. 2008, 1123, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J. Rho GTPase signalling in cell migration. Curr. Opin. Cell Boil. 2015, 36, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvakumar, B.; Hess, D.T.; Goldschmidt-clermont, P.J.; Stamler, J.S. Co-regulation of constitutive nitric oxide synthase and NADPH oxidase by the small GTPase Rac. Measurement 2009, 582, 2195–2202. [Google Scholar] [CrossRef] [PubMed]
- Haeussler, D.J.; Pimentel, D.R.; Hou, X.; Burgoyne, J.R.; Cohen, R.A.; Bachschmid, M.M. Endomembrane H-Ras Controls Vascular Endothelial Growth Cell Migration. J. Biol. Chem. 2013, 288, 15380–15389. [Google Scholar] [CrossRef] [PubMed]
- Küppers, V.; Vockel, M.; Nottebaum, A.F.; Vestweber, D. Phosphatases and kinases as regulators of the endothelial barrier function. Cell Tissue Res. 2014, 355, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Bogatcheva, N.V.; Dudek, S.M.; Garcia, J.G.N.; Verin, A.D. Mitogen-Activated Protein Kinases in Endothelial Pathophysiology. J. Investig. Med. 2003, 51, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.; Shah, A.M. NADPH oxidase and endothelial cell function. Clin. Sci. 2005, 109, 217–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haddad, P.; Dussault, S.; Groleau, J.; Turgeon, J.; Michaud, S.-E.; Ménard, C.; Perez, G.; Maingrette, F.; Rivard, A.; Ménard, C. Nox2-Containing NADPH Oxidase Deficiency Confers Protection from Hindlimb Ischemia in Conditions of Increased Oxidative Stress. Arter. Thromb. Vasc. Boil. 2009, 29, 1522–1528. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.E.; Cave, A.C.; Zhang, M.; Shah, A.M. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc. Res. 2006, 71, 208–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirker, A.; Murdoch, C.E.; Protti, A.; Sawyer, G.J.; Santos, C.X.; Martin, D.; Zhang, X.; Brewer, A.C.; Zhang, M.; Shah, A.M. Cell-specific effects of Nox2 on the acute and chronic response to myocardial infarction. J. Mol. Cell. Cardiol. 2016, 98, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murdoch, C.E.; Chaubey, S.; Zeng, L.; Yu, B.; Ivetic, A.; Walker, S.J.; Vanhoutte, D.; Heymans, S.; Grieve, D.J.; Cave, A.C.; et al. Endothelial NADPH Oxidase-2 Promotes Interstitial Cardiac Fibrosis and Diastolic Dysfunction Through Proinflammatory Effects and Endothelial-Mesenchymal Transition. J. Am. Coll. Cardiol. 2014, 63, 2734–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzik, T.J.; Sadowski, J.; Guzik, B.; Jopek, A.; Kapelak, B.; Przybyłowski, P.; Wierzbicki, K.; Korbut, R.; Harrison, D.G.; Channon, K.M. Coronary Artery Superoxide Production and Nox Isoform Expression in Human Coronary Artery Disease. Arter. Thromb. Vasc. Boil. 2006, 26, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mythreye, K.; Goldschmidt-Clermont, P.J.; Satterwhite, L.L.; Moldovan, L. Reactive oxygen species in vascular endothelial cell motility. Roles of NAD(P)H oxidase and Rac1. Cardiovasc. Res. 2006, 71, 236–246. [Google Scholar]
- Ushio-Fukai, M. Redox signaling in angiogenesis: Role of NADPH oxidase. Cardiovasc. Res. 2006, 71, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Li, J.-M.; Mullen, A.M.; Yun, S.; Wientjes, F.; Brouns, G.Y.; Thrasher, A.J.; Shah, A.M. Essential Role of the NADPH Oxidase Subunit p47 phox in Endothelial Cell Superoxide Production in Response to Phorbol Ester and Tumor Necrosis Factor-α. Circ. Res. 2002, 90, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Bernard, L.; Clempus, R. Vascular NAD (P) H oxidases: Specific features, expression, and regulation. Am. J. Physiol. 2003, 285, R277–R297. [Google Scholar]
- Tang, X.; Luo, Y.-X.; Chen, H.-Z.; Liu, D.-P. Mitochondria, endothelial cell function, and vascular diseases. Front. Physiol. 2014, 5, 1–17. [Google Scholar] [CrossRef]
- Tousoulis, D.; Kampoli, A.-M.; Tentolouris Nikolaos Papageorgiou, C.; Stefanadis, C. The Role of Nitric Oxide on Endothelial Function. Curr. Vasc. Pharmacol. 2011, 10, 4–18. [Google Scholar] [CrossRef]
- Crabtree, M.J.; Brixey, R.; Batchelor, H.; Hale, A.B.; Channon, K.M. Integrated Redox Sensor and Effector Functions for Endothelial Nitric-oxide Synthase (eNOS) Uncoupling. J. Biol. Chem. 2013, 288, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Tran, Q.-K.; Ohashi, K.; Watanabe, H. Calcium signalling in endothelial cells. Cardiovasc. Res. 2000, 48, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Raturi, A.; Ortiz-Sandoval, C.; Simmen, T. Redox dependence of endoplasmic reticulum (ER) Ca2+ signaling. Histol. Histopathol. 2014, 29, 543–552. [Google Scholar] [PubMed]
- Tong, X.Y.; Evangelista, A.; Cohen, R.A. Targeting the Redox Regulation of SERCA in Vascular Physiology and Disease. Curr. Opin. Pharmacol. 2010, 10, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Weisbrod, R.M.; Pimentel, D.R.; Ying, J.; Sharov, V.S.; Schöneich, C.; Cohen, R.A. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 2004, 10, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, A.M.; Thompson, M.D.; Weisbrod, R.M.; Pimental, D.R.; Tong, X.Y.; Bolotina, V.M.; Cohen, R.A. Redox Regulation of SERCA2 Is Required for Vascular Endothelial Growth Factor-Induced Signaling and Endothelial Cell Migration. Antioxid. Redox Signal. 2012, 17, 1099–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazbeck, P.; Tauseef, M.; Kruse, K.; Amin, M.R.; Sheikh, R.; Feske, S.; Komarova, K.; Mehta, D. STIM1 Phosphorylation at Y361 Recruits Orai1 to STIM1 Puncta and Induces Ca2+ Entry. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Watson, E.C.; Grant, Z.L.; Coultas, L. Endothelial cell apoptosis in angiogenesis and vessel regression. Cell. Mol. Life Sci. 2017, 74, 4387–4403. [Google Scholar] [CrossRef]
- Paone, S.; Baxter, A.A.; Hulett, M.D.; Poon, I.K.H. Endothelial cell apoptosis and the role of endothelial cell-derived extracellular vesicles in the progression of atherosclerosis. Cell. Mol. Life Sci. 2018, 76, 1093–1106. [Google Scholar] [CrossRef]
- Warren, M.C.; A Bump, E.; Medeiros, D.; Braunhut, S.J. Oxidative stress–induced apoptosis of endothelial cells. Free Radic. Boil. Med. 2000, 29, 537–547. [Google Scholar] [CrossRef]
- Kobayashi, N.; Delano, F.A.; Schmid-Schoönbein, G.W. Oxidative Stress Promotes Endothelial Cell Apoptosis and Loss of Microvessels in the Spontaneously Hypertensive Rats. Arter. Thromb. Vasc. Boil. 2005, 25, 2114–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorburn, A. Death receptor-induced cell killing. Cell. Signal. 2004, 16, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Sata, M.; Suhara, T.; Walsh, K. Vascular endothelial cells and smooth muscle cells differ in expression of Fas and Fas ligand and in sensitivity to Fas ligand-induced cell death: Implications for vascular disease and therapy. Arter. Thromb. Vasc. Boil. 2000, 20, 309–316. [Google Scholar] [CrossRef]
- Kamei, R.; Tanaka, H.Y.; Kawano, T.; Morii, C.; Tanaka, S.; Nishihara, H.; Iwata, C.; Kano, M.R. Regulation of endothelial Fas expression as a mechanism of promotion of vascular integrity by mural cells in tumors. Cancer Sci. 2017, 108, 1080–1088. [Google Scholar] [CrossRef] [PubMed]
- Scheer, J.M.; Romanowski, M.J.; Wells, J.A. A common allosteric site and mechanism in caspases. Proc. Natl. Acad. Sci. USA 2006, 103, 7595–7600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, M.B.; Houbaert, D.; Meçe, O.; Agostinis, P. Autophagy in endothelial cells and tumor angiogenesis. Cell Death Differ. 2019, 26, 665–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boteon, Y.L.; Laing, R.; Mergental, H.; Reynolds, G.M.; Mirza, D.F.; Afford, S.C.; Bhogal, R.H. Mechanisms of autophagy activation in endothelial cell and their targeting during normothermic machine liver perfusion. World J. Gastroenterol. 2017, 23, 8443–8451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, F. Autophagy in vascular endothelial cells. Clin. Exp. Pharmacol. Physiol. 2016, 43, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Hao, Y.; Chong, X.; Zhong, W. Overexpression of microRNA-506-3p aggravates the injury of vascular endothelial cells in patients with hypertension by downregulating Beclin1 expression. Exp. Ther. Med. 2018, 15, 2844–2850. [Google Scholar] [CrossRef]
- Geng, Z.; Xu, F.; Zhang, Y. MiR-129-5p-mediated Beclin-1 suppression inhibits endothelial cell autophagy in atherosclerosis. Am. J. Transl. Res. 2016, 8, 1886–1894. [Google Scholar] [PubMed]
- Rundhaug, J.E. Matrix metalloproteinases and angiogenesis Angiogenesis Review Series. J. Cell. Mol. Med. 2005, 9, 267–285. [Google Scholar] [CrossRef] [PubMed]
- Florence, J.M.; Krupa, A.; Booshehri, L.M.; Allen, T.C.; Kurdowska, A.K. Metalloproteinase-9 contributes to endothelial dysfunction in atherosclerosis via protease activated receptor-1. PLoS ONE 2017, 12, e0171427. [Google Scholar] [CrossRef] [PubMed]
- Papazafiropoulou, A.; Tentolouris, N. Matrix metalloproteinases and cardiovascular diseases. Hippokratia 2009, 13, 76–82. [Google Scholar] [PubMed]
- Nelson, K.K.; Subbaram, S.; Connor, K.M.; Dasgupta, J.; Ha, X.-F.; Meng, T.-C.; Tonks, N.K.; Melendez, J.A. Redox-dependent Matrix Metalloproteinase-1 Expression Is Regulated by JNK through Ets and AP-1 Promoter Motifs. J. Boil. Chem. 2006, 281, 14100–14110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kar, S.; Subbaram, S.; Carrico, P.M.; Melendez, J.A. Redox-control of Matrix Metalloproteinase-1: A critical link between free radicals, matrix remodeling and degenerative disease. Respir. Physiol. Neurobiol. 2010, 174, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Meng, X.P.; Ramasamy, S.; Harrison, D.G.; Galis, Z.S. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J. Clin. Investig. 1996, 98, 2572–2579. [Google Scholar] [CrossRef] [PubMed]
- Stupack, D.G.; Cheresh, D.A. Integrins and Angiogenesis. Curr. Top. Dev. Biol. 2004, 64, 207–238. [Google Scholar] [PubMed]
- Eble, J.A.; de Rezende, F.F. Redox-Relevant Aspects of the Extracellular Matrix and Its Cellular Contacts via Integrins. Antioxid. Redox Signal. 2013, 20, 1977–1993. [Google Scholar] [CrossRef]
- Liu, S.-Y.; Tsai, M.-Y.; Chuang, K.-P.; Huang, Y.-F.; Shieh, C.-C. Ligand binding of leukocyte integrin very late antigen-4 involves exposure of sulfhydryl groups and is subject to redox modulation. Eur. J. Immunol. 2008, 38, 410–423. [Google Scholar] [CrossRef]
- You, Y.; Chen, J.; Zhu, F.; Xu, Q.; Han, L.; Gao, X.; Zhang, X.; Luo, H.R.; Miao, J.; Sun, X.; et al. Glutaredoxin 1 up-regulates deglutathionylation of α4 integrin and thereby restricts neutrophil mobilization from bone marrow. J. Biol. Chem. 2019, 294, 2616–2627. [Google Scholar] [CrossRef] [PubMed]
- Deem, T.L.; Cook-Mills, J.M. Vascular cell adhesion molecule 1 (VCAM-1) activation of endothelial cell matrix metalloproteinases: Role of reactive oxygen species. Blood 2004, 104, 2385–2393. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.-H.; Kim, Y.K.; Kim, M.R.; Jang, J.H.; Lee, S. Emerging Roles of Vascular Cell Adhesion Molecule-1 (VCAM-1) in Immunological Disorders and Cancer. Int. J. Mol. Sci. 2018, 19, 1057. [Google Scholar] [CrossRef] [PubMed]
- Fearnley, G.W.; Odell, A.F.; Latham, A.M.; Mughal, N.A.; Bruns, A.F.; Burgoyne, N.J.; Homer-Vanniasinkam, S.; Zachary, I.C.; Hollstein, M.C.; Wheatcroft, S.B.; et al. VEGF-A isoforms differentially regulate ATF-2–dependent VCAM-1 gene expression and endothelial–leukocyte interactions. Mol. Boil. Cell 2014, 25, 2509–2521. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Gonzalez-Billault, C. Regulation of cytoskeletal dynamics by redox signaling and oxidative stress: Implications for neuronal development and trafficking. Front. Cell. Neurosci. 2015, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Protein Name | Protein Type | Glutathionylated Cysteine(s) | Direct Effect(s) on Protein | Physiological Effect(s) in ECs | Reference(s) |
|---|---|---|---|---|---|
| 4.1. Epigenetics regulators | |||||
| Histone H3 | Nucleosomal | C110 | Not confirmed | Regulation of gene expression via modulating chromatin structure | [13,14] |
| Sirtuin1 | Histone deacetylase | C67 C268 C623 | Inhibition of enzymatic activity | Apoptosis - Senescence | [15,16,17] |
| 4.2. Transcription factors | |||||
| p65 | Transcription factor | Unknown | Inhibition of nuclear translocation | Angiogenesis & cell survival | [18] |
| p50 | C62 | Inhibition of DNA-binding activity | [19] | ||
| c-jun | C269 | Unknown | [20,21] | ||
| p53 | C124 C141 C182 | Inhibition of DNA-binding and protein dimerization | Angiogenesis & cell survival (supposed) | [22] | |
| HIF-1a | C520 | Protein stabilization | Angiogenesis & ischemic revascularisation | [23,24] | |
| STAT3 | C328 C542 | Inhibition of phosphorylation and activity | Anti-angiogenesis and reduced inflammation | [25,26,27,28] | |
| STAT1 | C324 C492 | Protein activation | Unknown | [28] | |
| Keap1 | Nrf2 inhibitor | C434 | Inhibition of Nrf2 binding | Antioxidant and anti-inflammatory response via Nrf2 signalling | [29,30] |
| IKKb | Kinase | C179 | Inhibition of kinase activity | Angiogenesis & neovascularisation | [31] |
| 5. Kinases & phosphatases | |||||
| LMW PTP | Phosphatase | Unknown | Inhibition of activity | Cell migration and angiogenesis | [32] |
| PTP1B | C215 | Not confirmed | [33,34] | ||
| Rac1 | Small Rho GTPase | C81 C157 | Altered actin structure and barrier function | [35,36] | |
| Rac2 | C157 | Increased GTP-binding activity | Unknown | [36,37] | |
| Ras | GTPase | C118 | Not confirmed | Unknown | [36,38,39] |
| PKA | Kinase | C199 | Inhibition of activity | Alteration of barrier function and blood pressure regulation (supposed) | [40,41] |
| PKB | Unknown | [40,42,43] | |||
| MEKK1 | C1238 | [40,44] | |||
| PKC | Unknown | [40,45] | |||
| 6. RONS production | |||||
| p47 phox | NADPH oxidase | C98 C111 C196 | Enhanced protein function | Sustained superoxide production Endothelial dysfunction (supposed) | [46] |
| Complex I | NADH-ubiquinone oxidoreductase | Unknown | [47] | ||
| eNOS | Oxide synthase | C689 C908 | Protein uncoupling | Sustained superoxide production Impaired vasodilation and endothelial dysfunction | [48,49,50,51,52] |
| 7. Calcium-dependent channels | |||||
| IP3R | Ca2+ channel | Unknown (C34 C42 C65?) | Protein activation | Increased [Ca2+]i Regulation of Ca2+ homeostasis | [53,54,55] |
| PMCA | Ca2+ ATPase pump | Unknown | Protein inhibition | [56] | |
| SERCA2b | C674 | Protein activation | Increased Ca2+ uptake in ER stores Cell migration and angiogenesis | [57,58,59,60,61,62] | |
| STIM1 | Ca2+ sensor | C56 | Protein oligomerization | Increased [Ca]i via Orai1 activation Mitochondrial dysfunction | [63] |
| 8. Apoptosis and autophagy | |||||
| Fas | Death receptor | C294 | Enhanced activity | Cell death | [64] |
| Caspase-3 | Protease | C45 (p12) C135 (p17) | Inhibition of proteolytic activity | Cell survival | [65,66] |
| Caspase-8 | Unknown | [67] | |||
| Beclin-1 | Autophagy-related protein | Unknown | Upregulation of protein activity | Not confirmed | [68] |
| 9. Cell structure and dynamics | |||||
| ProMMPs | Metalloprotease | PRCGVPD motif on inhibitory domain | Activation | Angiogenesis and vascular permeability | [69] |
| ICAM-1 | Adhesion receptor | Unknown | Protein degradation | Cell junction disassembly | [70] |
| Actin | Cytoskeletal | C374 | Inhibition of polymerization | Inhibition of cell motility | [71,72,73] |
| Microtubules | Unknown | Cell growth arrest and apoptosis | [74,75] | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lermant, A.; Murdoch, C.E. Cysteine Glutathionylation Acts as a Redox Switch in Endothelial Cells. Antioxidants 2019, 8, 315. https://doi.org/10.3390/antiox8080315
Lermant A, Murdoch CE. Cysteine Glutathionylation Acts as a Redox Switch in Endothelial Cells. Antioxidants. 2019; 8(8):315. https://doi.org/10.3390/antiox8080315
Chicago/Turabian StyleLermant, Agathe, and Colin E. Murdoch. 2019. "Cysteine Glutathionylation Acts as a Redox Switch in Endothelial Cells" Antioxidants 8, no. 8: 315. https://doi.org/10.3390/antiox8080315
APA StyleLermant, A., & Murdoch, C. E. (2019). Cysteine Glutathionylation Acts as a Redox Switch in Endothelial Cells. Antioxidants, 8(8), 315. https://doi.org/10.3390/antiox8080315