Role of Cytosolic 2-Cys Prx1 and Prx2 in Redox Signaling


Abstract
1. Introduction
2. ROS, Oxidative Stress and Cancer
3. Antioxidant Enzymes; SOD, CAT, GPx, and Prx
3.1. SOD
3.2. CAT
3.3. GPx
3.4. Prx
4. Biological Functions of Prx1 and Prx2
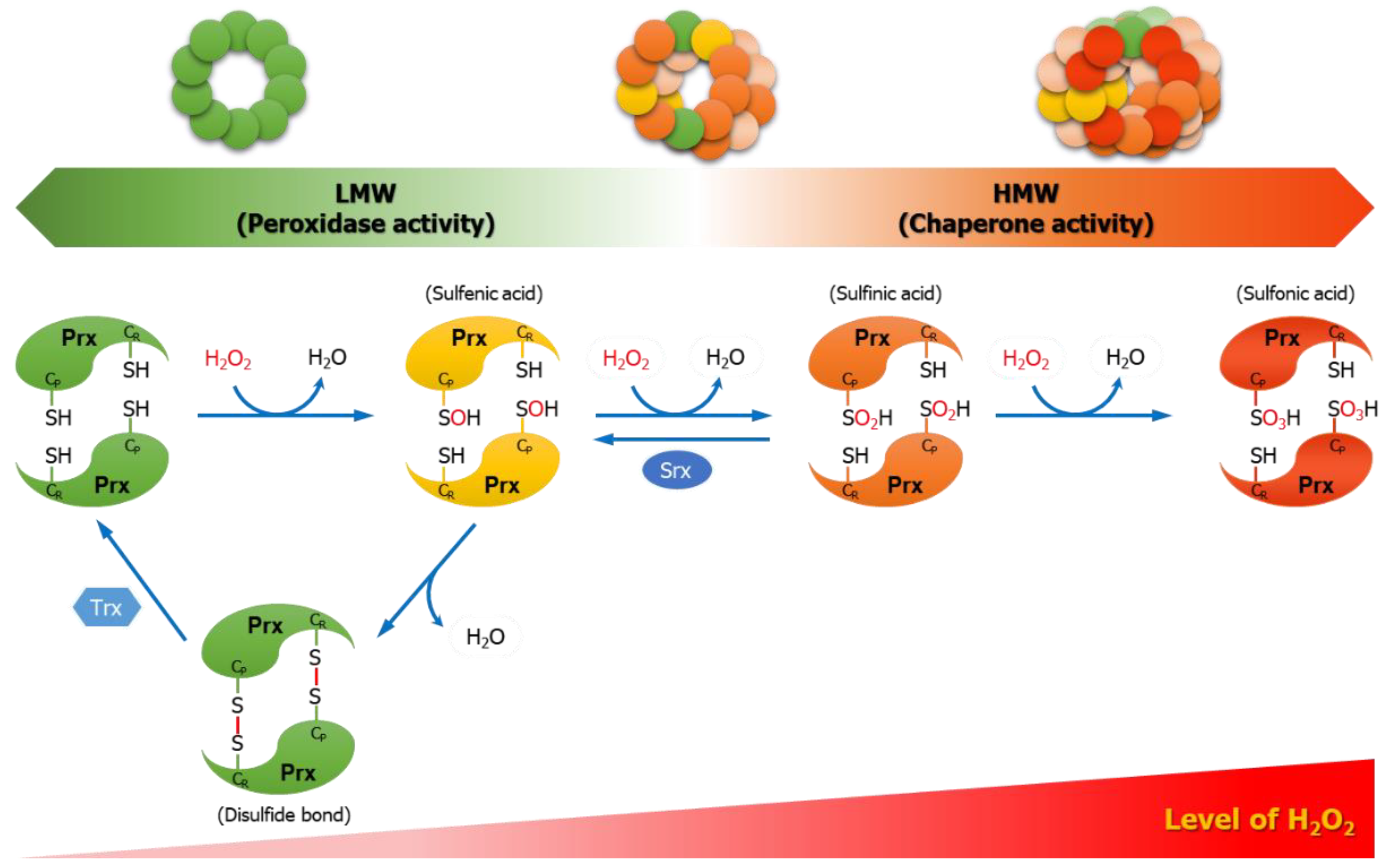
4.1. Peroxidase Function
4.2. Chaperone Function
5. Interaction Partners of Prx1 and/or Prx2
5.1. Interaction Partners of Prx1
5.1.1. Androgen Receptor
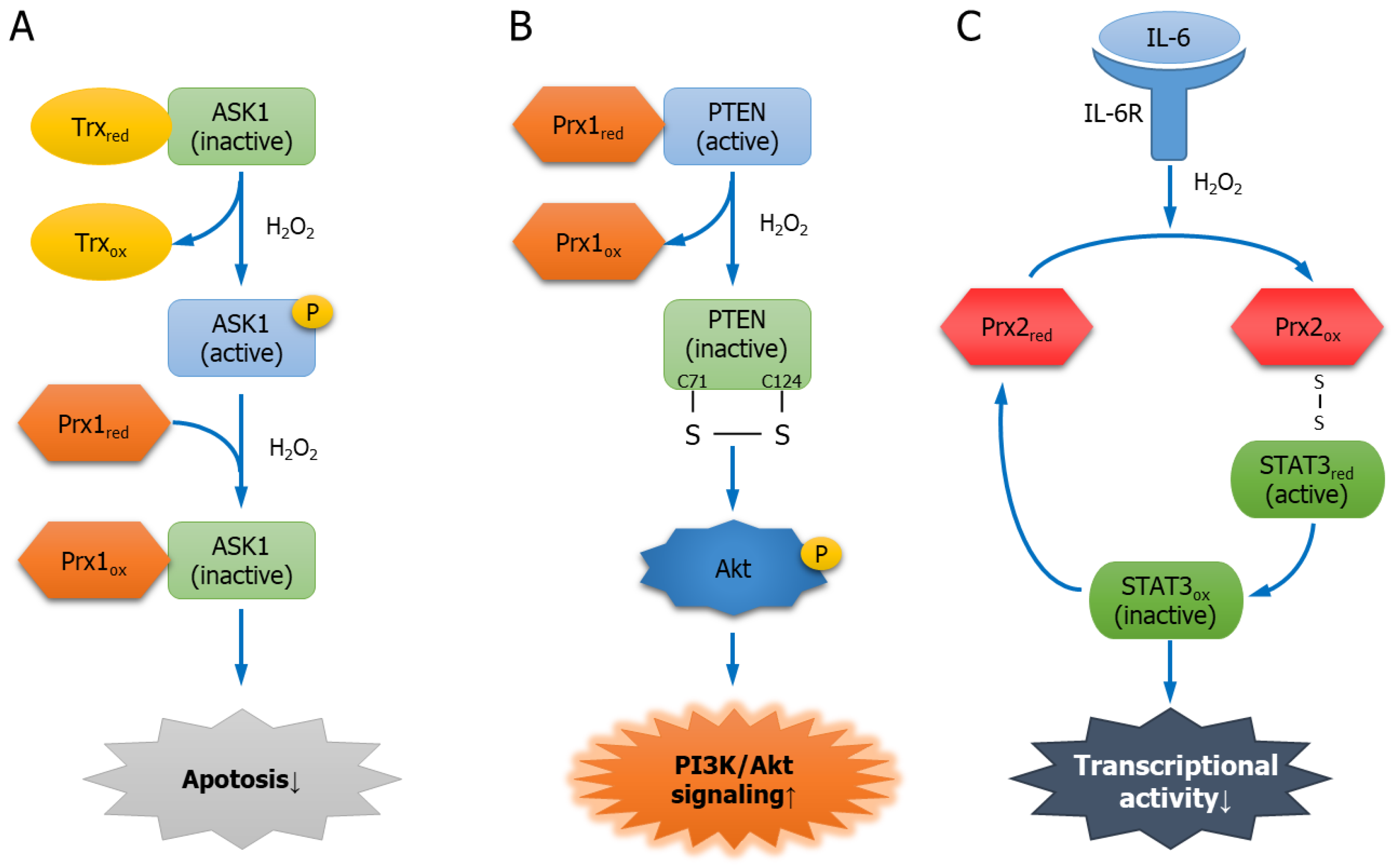
5.1.2. Apoptosis Signal-Regulating Kinase 1
5.1.3. PTEN
5.1.4. Mammalian Ste20-like Kinase-1
5.1.5. Glycerophosphodiester Phosphodiesterase 2
5.1.6. Apurinic/Apyrimidinic Endonuclease 1
5.2. Interaction Partners of Prx2
5.2.1. Platelet-Derived Growth Factor Receptor
5.2.2. Tankyrase
5.2.3. Signal Transducer and Activator of Transcription 3
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxid. Med. Cell. Longev. 2016, 2016, 3164734. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef]
- Mills, G.C. Hemoglobin catabolism. I. Glutathione peroxidase, an erythrocyte enzyme which protects hemoglobin from oxidative breakdown. J. Biol. Chem. 1957, 229, 189–197. [Google Scholar]
- Chae, H.Z.; Chung, S.J.; Rhee, S.G. Thioredoxin-dependent peroxide reductase from yeast. J. Biol. Chem. 1994, 269, 27670–27678. [Google Scholar]
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxiredoxins: Guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 2015, 40, 435–445. [Google Scholar] [CrossRef]
- Rhee, S.G.; Kil, I.S. Multiple Functions and Regulation of Mammalian Peroxiredoxins. Annu. Rev. Biochem. 2017, 86, 749–775. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Warabi, E.; Yanagawa, T. Novel roles of peroxiredoxins in inflammation, cancer and innate immunity. J. Clin. Biochem. Nutr. 2012, 50, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, S.; Lee, S.; Kang, S.W. 2-cys peroxiredoxins: Emerging hubs determining redox dependency of Mammalian signaling networks. Int. J. Cell. Biol. 2014, 2014, 715867. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Jiang, H.; Wu, L.; Chawsheen, H.A.; Wei, Q. The sulfiredoxin-peroxiredoxin (Srx-Prx) axis in cell signal transduction and cancer development. Cancer Lett. 2015, 366, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Phalen, T.J.; Weirather, K.; Deming, P.B.; Anathy, V.; Howe, A.K.; van der Vliet, A.; Jonsson, T.J.; Poole, L.B.; Heintz, N.H. Oxidation state governs structural transitions in peroxiredoxin II that correlate with cell cycle arrest and recovery. J. Cell Biol. 2006, 175, 779–789. [Google Scholar] [CrossRef]
- Jang, H.H.; Lee, K.O.; Chi, Y.H.; Jung, B.G.; Park, S.K.; Park, J.H.; Lee, J.R.; Lee, S.S.; Moon, J.C.; Yun, J.W.; et al. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell 2004, 117, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, T.J.; Lee, K.Y. A novel function of peroxiredoxin 1 (Prx-1) in apoptosis signal-regulating kinase 1 (ASK1)-mediated signaling pathway. FEBS Lett. 2008, 582, 1913–1918. [Google Scholar] [CrossRef]
- Yan, Y.; Sabharwal, P.; Rao, M.; Sockanathan, S. The antioxidant enzyme Prdx1 controls neuronal differentiation by thiol-redox-dependent activation of GDE2. Cell 2009, 138, 1209–1221. [Google Scholar] [CrossRef]
- Riddell, J.R.; Maier, P.; Sass, S.N.; Moser, M.T.; Foster, B.A.; Gollnick, S.O. Peroxiredoxin 1 stimulates endothelial cell expression of VEGF via TLR4 dependent activation of HIF-1alpha. PLoS ONE 2012, 7, e50394. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Agarwal, A.; Gupta, S.; Sikka, S. The role of free radicals and antioxidants in reproduction. Curr. Opin. Obstet. Gynecol. 2006, 18, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: An essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 2014, 94, 329–354. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014, 2, 17. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P.; Cruzat, V.F.; Keane, K.N.; Carlessi, R.; de Bittencourt, P.I., Jr. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem. J. 2016, 473, 4527–4550. [Google Scholar] [CrossRef]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Miki, H.; Funato, Y. Regulation of intracellular signalling through cysteine oxidation by reactive oxygen species. J. Biochem. 2012, 151, 255–261. [Google Scholar] [CrossRef]
- Hohn, A.; Jung, T.; Grune, T. Pathophysiological importance of aggregated damaged proteins. Free Radic. Biol. Med. 2014, 71, 70–89. [Google Scholar] [CrossRef]
- Berlett, B.S.; Stadtman, E.R. Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 1997, 272, 20313–20316. [Google Scholar] [CrossRef]
- Wu, Q.; Ni, X. ROS-mediated DNA methylation pattern alterations in carcinogenesis. Curr. Drug Targets 2015, 16, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Wagner, J.R. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb. Perspect. Biol. 2013, 5, a012559. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Badana, A.K.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.W.; Byzova, T.V. Oxidative stress in angiogenesis and vascular disease. Blood 2014, 123, 625–631. [Google Scholar] [CrossRef]
- Havens, C.G.; Ho, A.; Yoshioka, N.; Dowdy, S.F. Regulation of late G1/S phase transition and APC Cdh1 by reactive oxygen species. Mol. Cell. Biol. 2006, 26, 4701–4711. [Google Scholar] [CrossRef]
- Nishikawa, M. Reactive oxygen species in tumor metastasis. Cancer Lett. 2008, 266, 53–59. [Google Scholar] [CrossRef]
- Robinson, C.M.; Ohh, M. The multifaceted von Hippel-Lindau tumour suppressor protein. FEBS Lett. 2014, 588, 2704–2711. [Google Scholar] [CrossRef]
- Qutub, A.A.; Popel, A.S. Reactive oxygen species regulate hypoxia-inducible factor 1alpha differentially in cancer and ischemia. Mol. Cell. Biol. 2008, 28, 5106–5119. [Google Scholar] [CrossRef]
- Liu, D.; Xu, Y. p53, oxidative stress, and aging. Antioxid. Redox Signal. 2011, 15, 1669–1678. [Google Scholar] [CrossRef]
- Budanov, A.V. The role of tumor suppressor p53 in the antioxidant defense and metabolism. Subcell. Biochem. 2014, 85, 337–358. [Google Scholar]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Kalo, E.; Kogan-Sakin, I.; Solomon, H.; Bar-Nathan, E.; Shay, M.; Shetzer, Y.; Dekel, E.; Goldfinger, N.; Buganim, Y.; Stambolsky, P.; et al. Mutant p53R273H attenuates the expression of phase 2 detoxifying enzymes and promotes the survival of cells with high levels of reactive oxygen species. J. Cell Sci. 2012, 125, 5578–5586. [Google Scholar] [CrossRef] [PubMed]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [PubMed]
- Carnero, A.; Blanco-Aparicio, C.; Renner, O.; Link, W.; Leal, J.F. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr. Cancer Drug Targets 2008, 8, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Shen, W.H. PTEN: A new guardian of the genome. Oncogene 2008, 27, 5443–5453. [Google Scholar] [CrossRef]
- Poljsak, B.; Suput, D.; Milisav, I. Achieving the balance between ROS and antioxidants: When to use the synthetic antioxidants. Oxid. Med. Cell. Longev. 2013, 2013, 956792. [Google Scholar] [CrossRef] [PubMed]
- Dietz, K.J. Peroxiredoxins in plants and cyanobacteria. Antioxid. Redox Signal. 2011, 15, 1129–1159. [Google Scholar] [CrossRef]
- Bayliak, M.; Semchyshyn, H.; Lushchak, V. Effect of hydrogen peroxide on antioxidant enzyme activities in Saccharomyces cerevisiae is strain-specific. Biochemistry 2006, 71, 1013–1020. [Google Scholar] [CrossRef]
- Nystrom, T.; Yang, J.; Molin, M. Peroxiredoxins, gerontogenes linking aging to genome instability and cancer. Genes Dev. 2012, 26, 2001–2008. [Google Scholar] [CrossRef]
- Brown, D.I.; Griendling, K.K. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ. Res. 2015, 116, 531–549. [Google Scholar] [CrossRef]
- Azadmanesh, J.; Borgstahl, G.E.O. A Review of the Catalytic Mechanism of Human Manganese Superoxide Dismutase. Antioxidants 2018, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef]
- Culotta, V.C.; Yang, M.; O’Halloran, T.V. Activation of superoxide dismutases: Putting the metal to the pedal. Biochim. Biophys. Acta 2006, 1763, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.R.; Cotter, T.G. Hydrogen peroxide: A Jekyll and Hyde signalling molecule. Cell Death Dis. 2011, 2, e213. [Google Scholar] [CrossRef]
- Goyal, M.M.; Basak, A. Human catalase: Looking for complete identity. Protein Cell 2010, 1, 888–897. [Google Scholar] [CrossRef]
- Hoffschir, F.; Daya-Grosjean, L.; Petit, P.X.; Nocentini, S.; Dutrillaux, B.; Sarasin, A.; Vuillaume, M. Low catalase activity in xeroderma pigmentosum fibroblasts and SV40-transformed human cell lines is directly related to decreased intracellular levels of the cofactor, NADPH. Free Radic. Biol. Med. 1998, 24, 809–816. [Google Scholar] [CrossRef]
- Hojo, Y.; Saito, Y.; Tanimoto, T.; Hoefen, R.J.; Baines, C.P.; Yamamoto, K.; Haendeler, J.; Asmis, R.; Berk, B.C. Fluid shear stress attenuates hydrogen peroxide-induced c-Jun NH2-terminal kinase activation via a glutathione reductase-mediated mechanism. Circ. Res. 2002, 91, 712–718. [Google Scholar] [CrossRef]
- Matouskova, P.; Hanouskova, B.; Skalova, L. MicroRNAs as Potential Regulators of Glutathione Peroxidases Expression and Their Role in Obesity and Related Pathologies. Int. J. Mol. Sci. 2018, 19, 1199. [Google Scholar] [CrossRef]
- Chae, H.Z.; Oubrahim, H.; Park, J.W.; Rhee, S.G.; Chock, P.B. Protein glutathionylation in the regulation of peroxiredoxins: A family of thiol-specific peroxidases that function as antioxidants, molecular chaperones, and signal modulators. Antioxid. Redox Signal. 2012, 16, 506–523. [Google Scholar] [CrossRef]
- Rhee, S.G.; Woo, H.A.; Kil, I.S.; Bae, S.H. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. 2012, 287, 4403–4410. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 1–5. [Google Scholar] [PubMed]
- Hopkins, B.L.; Nadler, M.; Skoko, J.J.; Bertomeu, T.; Pelosi, A.; Shafaei, P.M.; Levine, K.; Schempf, A.; Pennarun, B.; Yang, B.; et al. A Peroxidase Peroxiredoxin 1-Specific Redox Regulation of the Novel FOXO3 microRNA Target let-7. Antioxid. Redox Signal. 2018, 28, 62–77. [Google Scholar] [CrossRef] [PubMed]
- Mullen, L.; Hanschmann, E.M.; Lillig, C.H.; Herzenberg, L.A.; Ghezzi, P. Cysteine Oxidation Targets Peroxiredoxins 1 and 2 for Exosomal Release through a Novel Mechanism of Redox-Dependent Secretion. Mol. Med. 2015, 21, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Cunniff, B.; Newick, K.; Nelson, K.J.; Wozniak, A.N.; Beuschel, S.; Leavitt, B.; Bhave, A.; Butnor, K.; Koenig, A.; Chouchani, E.T.; et al. Disabling Mitochondrial Peroxide Metabolism via Combinatorial Targeting of Peroxiredoxin 3 as an Effective Therapeutic Approach for Malignant Mesothelioma. PLoS ONE 2015, 10, e0127310. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Li, D.F.; Wang, X.; Liang, J.; Sitia, R.; Wang, C.C.; Wang, X. Crystal Structure of the ERp44-Peroxiredoxin 4 Complex Reveals the Molecular Mechanisms of Thiol-Mediated Protein Retention. Structure 2016, 24, 1755–1765. [Google Scholar] [CrossRef] [PubMed]
- Knoops, B.; Goemaere, J.; Van der Eecken, V.; Declercq, J.P. Peroxiredoxin 5: Structure, mechanism, and function of the mammalian atypical 2-Cys peroxiredoxin. Antioxid. Redox Signal. 2011, 15, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.B. Peroxiredoxin 6: A bifunctional enzyme with glutathione peroxidase and phospholipase A(2) activities. Antioxid. Redox Signal. 2011, 15, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Netto, L.E.; Antunes, F. The Roles of Peroxiredoxin and Thioredoxin in Hydrogen Peroxide Sensing and in Signal Transduction. Mol. Cells 2016, 39, 65–71. [Google Scholar]
- Barranco-Medina, S.; Lazaro, J.J.; Dietz, K.J. The oligomeric conformation of peroxiredoxins links redox state to function. FEBS Lett. 2009, 583, 1809–1816. [Google Scholar] [CrossRef]
- Wood, Z.A.; Poole, L.B.; Karplus, P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 2003, 300, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.Q.; Zhou, W.S.; Li, M.; Ren, F.; Han, Y.W. Characterization and Expression Analysis of Peroxiredoxin Genes in NNK-induced V79 Cells. Biomed. Environ. Sci. 2017, 30, 224–228. [Google Scholar] [PubMed]
- Seo, J.H.; Lim, J.C.; Lee, D.Y.; Kim, K.S.; Piszczek, G.; Nam, H.W.; Kim, Y.S.; Ahn, T.; Yun, C.H.; Kim, K.; et al. Novel protective mechanism against irreversible hyperoxidation of peroxiredoxin: Nalpha-terminal acetylation of human peroxiredoxin II. J. Biol. Chem. 2009, 284, 13455–13465. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, J.A.; Nelson, K.J.; Haynes, A.C.; Lee, J.; Reisz, J.A.; Graff, A.H.; Clodfelter, J.E.; Parsonage, D.; Poole, L.B.; Furdui, C.M.; et al. Novel hyperoxidation resistance motifs in 2-Cys peroxiredoxins. J. Biol. Chem. 2018, 293, 11901–11912. [Google Scholar] [CrossRef] [PubMed]
- Dalla Rizza, J.; Randall, L.M.; Santos, J.; Ferrer-Sueta, G.; Denicola, A. Differential parameters between cytosolic 2-Cys peroxiredoxins, PRDX1 and PRDX2. Protein Sci. 2019, 28, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.G.; Pearson, A.G.; Pullar, J.M.; Jonsson, T.J.; Lowther, W.T.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial peroxiredoxin 3 is more resilient to hyperoxidation than cytoplasmic peroxiredoxins. Biochem. J. 2009, 421, 51–58. [Google Scholar] [CrossRef]
- Pascual, M.B.; Mata-Cabana, A.; Florencio, F.J.; Lindahl, M.; Cejudo, F.J. Overoxidation of 2-Cys peroxiredoxin in prokaryotes: Cyanobacterial 2-Cys peroxiredoxins sensitive to oxidative stress. J. Biol. Chem. 2010, 285, 34485–34492. [Google Scholar] [CrossRef]
- Bertoldi, M. Human Peroxiredoxins 1 and 2 and Their Interacting Protein Partners; Through Structure Toward Functions of Biological Complexes. Protein Pept. Lett. 2016, 23, 69–77. [Google Scholar] [CrossRef]
- Hampton, M.B.; Vick, K.A.; Skoko, J.J.; Neumann, C.A. Peroxiredoxin Involvement in the Initiation and Progression of Human Cancer. Antioxid. Redox Signal. 2018, 28, 591–608. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, Y.J.; Gao, A.C.; Mohler, J.L.; Onate, S.A.; Hidalgo, A.A.; Ip, C.; Park, E.M.; Yoon, S.Y.; Park, Y.M. Hypoxia increases androgen receptor activity in prostate cancer cells. Cancer Res. 2006, 66, 5121–5129. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Ahn, J.Y.; Liang, P.; Ip, C.; Zhang, Y.; Park, Y.M. Human prx1 gene is a target of Nrf2 and is up-regulated by hypoxia/reoxygenation: Implication to tumor biology. Cancer Res. 2007, 67, 546–554. [Google Scholar] [CrossRef]
- Zhang, M.; Hou, M.; Ge, L.; Miao, C.; Zhang, J.; Jing, X.; Shi, N.; Chen, T.; Tang, X. Induction of peroxiredoxin 1 by hypoxia regulates heme oxygenase-1 via NF-kappaB in oral cancer. PLoS ONE 2014, 9, e105994. [Google Scholar]
- Park, S.Y.; Yu, X.; Ip, C.; Mohler, J.L.; Bogner, P.N.; Park, Y.M. Peroxiredoxin 1 interacts with androgen receptor and enhances its transactivation. Cancer Res. 2007, 67, 9294–9303. [Google Scholar] [CrossRef] [PubMed]
- Hatai, T.; Matsuzawa, A.; Inoshita, S.; Mochida, Y.; Kuroda, T.; Sakamaki, K.; Kuida, K.; Yonehara, S.; Ichijo, H.; Takeda, K. Execution of apoptosis signal-regulating kinase 1 (ASK1)-induced apoptosis by the mitochondria-dependent caspase activation. J. Biol. Chem. 2000, 275, 26576–26581. [Google Scholar] [CrossRef] [PubMed]
- Fujino, G.; Noguchi, T.; Matsuzawa, A.; Yamauchi, S.; Saitoh, M.; Takeda, K.; Ichijo, H. Thioredoxin and TRAF family proteins regulate reactive oxygen species-dependent activation of ASK1 through reciprocal modulation of the N-terminal homophilic interaction of ASK1. Mol. Cell. Biol. 2007, 27, 8152–8163. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef]
- Hsieh, C.C.; Papaconstantinou, J. Thioredoxin-ASK1 complex levels regulate ROS-mediated p38 MAPK pathway activity in livers of aged and long-lived Snell dwarf mice. FASEB J. 2006, 20, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Soga, M.; Matsuzawa, A.; Ichijo, H. Oxidative Stress-Induced Diseases via the ASK1 Signaling Pathway. Int. J. Cell Biol. 2012, 2012, 439587. [Google Scholar] [CrossRef]
- Jarvis, R.M.; Hughes, S.M.; Ledgerwood, E.C. Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radic. Biol. Med. 2012, 53, 1522–1530. [Google Scholar] [CrossRef]
- Cao, J.; Schulte, J.; Knight, A.; Leslie, N.R.; Zagozdzon, A.; Bronson, R.; Manevich, Y.; Beeson, C.; Neumann, C.A. Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. EMBO J. 2009, 28, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- Neumann, C.A.; Cao, J.; Manevich, Y. Peroxiredoxin 1 and its role in cell signaling. Cell Cycle 2009, 8, 4072–4078. [Google Scholar] [CrossRef] [PubMed]
- Rawat, S.J.; Creasy, C.L.; Peterson, J.R.; Chernoff, J. The tumor suppressor Mst1 promotes changes in the cellular redox state by phosphorylation and inactivation of peroxiredoxin-1 protein. J. Biol. Chem. 2013, 288, 8762–8771. [Google Scholar] [CrossRef] [PubMed]
- Del Re, D.P.; Matsuda, T.; Zhai, P.; Maejima, Y.; Jain, M.R.; Liu, T.; Li, H.; Hsu, C.P.; Sadoshima, J. Mst1 promotes cardiac myocyte apoptosis through phosphorylation and inhibition of Bcl-xL. Mol. Cell 2014, 54, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Morinaka, A.; Funato, Y.; Uesugi, K.; Miki, H. Oligomeric peroxiredoxin-I is an essential intermediate for p53 to activate MST1 kinase and apoptosis. Oncogene 2011, 30, 4208–4218. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Sockanathan, S. Transmembrane protein GDE2 induces motor neuron differentiation in vivo. Science 2005, 309, 2212–2215. [Google Scholar] [CrossRef]
- Izumi, T.; Hazra, T.K.; Boldogh, I.; Tomkinson, A.E.; Park, M.S.; Ikeda, S.; Mitra, S. Requirement for human AP endonuclease 1 for repair of 3′-blocking damage at DNA single-strand breaks induced by reactive oxygen species. Carcinogenesis 2000, 21, 1329–1334. [Google Scholar] [CrossRef][Green Version]
- Kelley, M.R.; Georgiadis, M.M.; Fishel, M.L. APE1/Ref-1 role in redox signaling: Translational applications of targeting the redox function of the DNA repair/redox protein APE1/Ref-1. Curr. Mol. Pharmacol. 2012, 5, 36–53. [Google Scholar] [CrossRef]
- Nassour, H.; Wang, Z.; Saad, A.; Papaluca, A.; Brosseau, N.; Affar, E.B.; Alaoui-Jamali, M.A.; Ramotar, D. Peroxiredoxin 1 interacts with and blocks the redox factor APE1 from activating interleukin-8 expression. Sci. Rep. 2016, 6, 29389. [Google Scholar] [CrossRef]
- Kim, H.K.; Kim, J.W.; Zilberstein, A.; Margolis, B.; Kim, J.G.; Schlessinger, J.; Rhee, S.G. PDGF stimulation of inositol phospholipid hydrolysis requires PLC-gamma 1 phosphorylation on tyrosine residues 783 and 1254. Cell 1991, 65, 435–441. [Google Scholar] [CrossRef]
- Reyhani, V.; Tsioumpekou, M.; van Wieringen, T.; Rask, L.; Lennartsson, J.; Rubin, K. PDGF-BB enhances collagen gel contraction through a PI3K-PLCgamma-PKC-cofilin pathway. Sci. Rep. 2017, 7, 8924. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Huang, H.J.; Kazlauskas, A.; Cavenee, W.K. Induction of vascular endothelial growth factor expression in endothelial cells by platelet-derived growth factor through the activation of phosphatidylinositol 3-kinase. Cancer Res. 1999, 59, 1464–1472. [Google Scholar] [PubMed]
- Bae, Y.S.; Sung, J.Y.; Kim, O.S.; Kim, Y.J.; Hur, K.C.; Kazlauskas, A.; Rhee, S.G. Platelet-derived growth factor-induced H2O2 production requires the activation of phosphatidylinositol 3-kinase. J. Biol. Chem. 2000, 275, 10527–10531. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.H.; Lee, I.K.; Kim, G.W.; Kim, B.U.; Han, Y.H.; Yu, D.Y.; Park, H.S.; Kim, K.Y.; Lee, J.S.; Choi, C.; et al. Regulation of PDGF signalling and vascular remodelling by peroxiredoxin II. Nature 2005, 435, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Li, V.S.; Ng, S.S.; Boersema, P.J.; Low, T.Y.; Karthaus, W.R.; Gerlach, J.P.; Mohammed, S.; Heck, A.J.; Maurice, M.M.; Mahmoudi, T.; et al. Wnt signaling through inhibition of beta-catenin degradation in an intact Axin1 complex. Cell 2012, 149, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Amit, S.; Hatzubai, A.; Birman, Y.; Andersen, J.S.; Ben-Shushan, E.; Mann, M.; Ben-Neriah, Y.; Alkalay, I. Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: A molecular switch for the Wnt pathway. Genes Dev. 2002, 16, 1066–1076. [Google Scholar] [CrossRef] [PubMed]
- Stamos, J.L.; Weis, W.I. The beta-catenin destruction complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef]
- Riffell, J.L.; Lord, C.J.; Ashworth, A. Tankyrase-targeted therapeutics: Expanding opportunities in the PARP family. Nat. Rev. Drug Discov. 2012, 11, 923–936. [Google Scholar] [CrossRef]
- Li, X.; Han, H.; Zhou, M.T.; Yang, B.; Ta, A.P.; Li, N.; Chen, J.; Wang, W. Proteomic Analysis of the Human Tankyrase Protein Interaction Network Reveals Its Role in Pexophagy. Cell Rep. 2017, 20, 737–749. [Google Scholar] [CrossRef]
- Mariotti, L.; Pollock, K.; Guettler, S. Regulation of Wnt/beta-catenin signalling by tankyrase-dependent poly(ADP-ribosyl)ation and scaffolding. Br. J. Pharmacol. 2017, 174, 4611–4636. [Google Scholar] [CrossRef]
- Kang, D.H.; Lee, D.J.; Lee, S.; Lee, S.Y.; Jun, Y.; Kim, Y.; Kim, Y.; Lee, J.S.; Lee, D.K.; Lee, S.; et al. Interaction of tankyrase and peroxiredoxin II is indispensable for the survival of colorectal cancer cells. Nat. Commun. 2017, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.R.; Rai, U.; Fanburg, B.L.; Cochran, B.H. Activation of the JAK-STAT pathway by reactive oxygen species. Am. J. Physiol. 1998, 275, C1640–C1652. [Google Scholar] [CrossRef] [PubMed]
- Mamoon, N.M.; Smith, J.K.; Chatti, K.; Lee, S.; Kundrapu, K.; Duhe, R.J. Multiple cysteine residues are implicated in Janus kinase 2-mediated catalysis. Biochemistry 2007, 46, 14810–14818. [Google Scholar] [CrossRef] [PubMed]
- Sobotta, M.C.; Liou, W.; Stocker, S.; Talwar, D.; Oehler, M.; Ruppert, T.; Scharf, A.N.; Dick, T.P. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 2015, 11, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Choi, K.S.; Riddell, J.; Ip, C.; Ghosh, D.; Park, J.H.; Park, Y.M. Human peroxiredoxin 1 and 2 are not duplicate proteins: The unique presence of CYS83 in Prx1 underscores the structural and functional differences between Prx1 and Prx2. J. Biol. Chem. 2007, 282, 22011–22022. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Isoform | Localization | Catalytic Residue | Ref. |
|---|---|---|---|
| GPX1 | Cytosolic | Sec | [52,59] |
| GPX2 | Gastrointestinal | Sec | [52,59] |
| GPX3 | Plasma | Sec | [52,59] |
| GPX4 | PH 1 | Sec | [52,59] |
| GPX5 | EL 2 | Cys | [52,59] |
| GPX6 | Olfactory | Sec | [52,59] |
| GPX7 | ER 3 | Cys | [52,59] |
| GPX8 | ER 3 (putative) | Cys | [52,59] |
| Class | Isoform | CP | CR | Non-Catalytic | Localization | Ref. |
|---|---|---|---|---|---|---|
| Typical 2-Cys | Prx1 | 52 | 173 | 71, 83 | Cytosol, nucleus, PM 1 | [11,63] |
| Prx2 | 51 | 172 | 70 | Cytosol, nucleus, PM 1 | [11,64] | |
| Prx3 | 108 | 229 | 127 | Mitochondria | [11,65] | |
| Prx4 | 87 | 208 | 14, 111 | Cytosol, secretion, ER 2 | [11,66] | |
| Atypical 2-Cys | Prx5 | 47 | 151 | 72 | Cytosol, peroxisome mitochondria | [11,67] |
| 1-Cys | Prx6 | 47 | − | 91 | Cytosol, secretion lysosome | [11,68] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Jang, H.H. Role of Cytosolic 2-Cys Prx1 and Prx2 in Redox Signaling. Antioxidants 2019, 8, 169. https://doi.org/10.3390/antiox8060169
Kim Y, Jang HH. Role of Cytosolic 2-Cys Prx1 and Prx2 in Redox Signaling. Antioxidants. 2019; 8(6):169. https://doi.org/10.3390/antiox8060169
Chicago/Turabian StyleKim, Yosup, and Ho Hee Jang. 2019. "Role of Cytosolic 2-Cys Prx1 and Prx2 in Redox Signaling" Antioxidants 8, no. 6: 169. https://doi.org/10.3390/antiox8060169
APA StyleKim, Y., & Jang, H. H. (2019). Role of Cytosolic 2-Cys Prx1 and Prx2 in Redox Signaling. Antioxidants, 8(6), 169. https://doi.org/10.3390/antiox8060169