H2O2 Metabolism in Normal Thyroid Cells and in Thyroid Tumorigenesis: Focus on NADPH Oxidases




Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
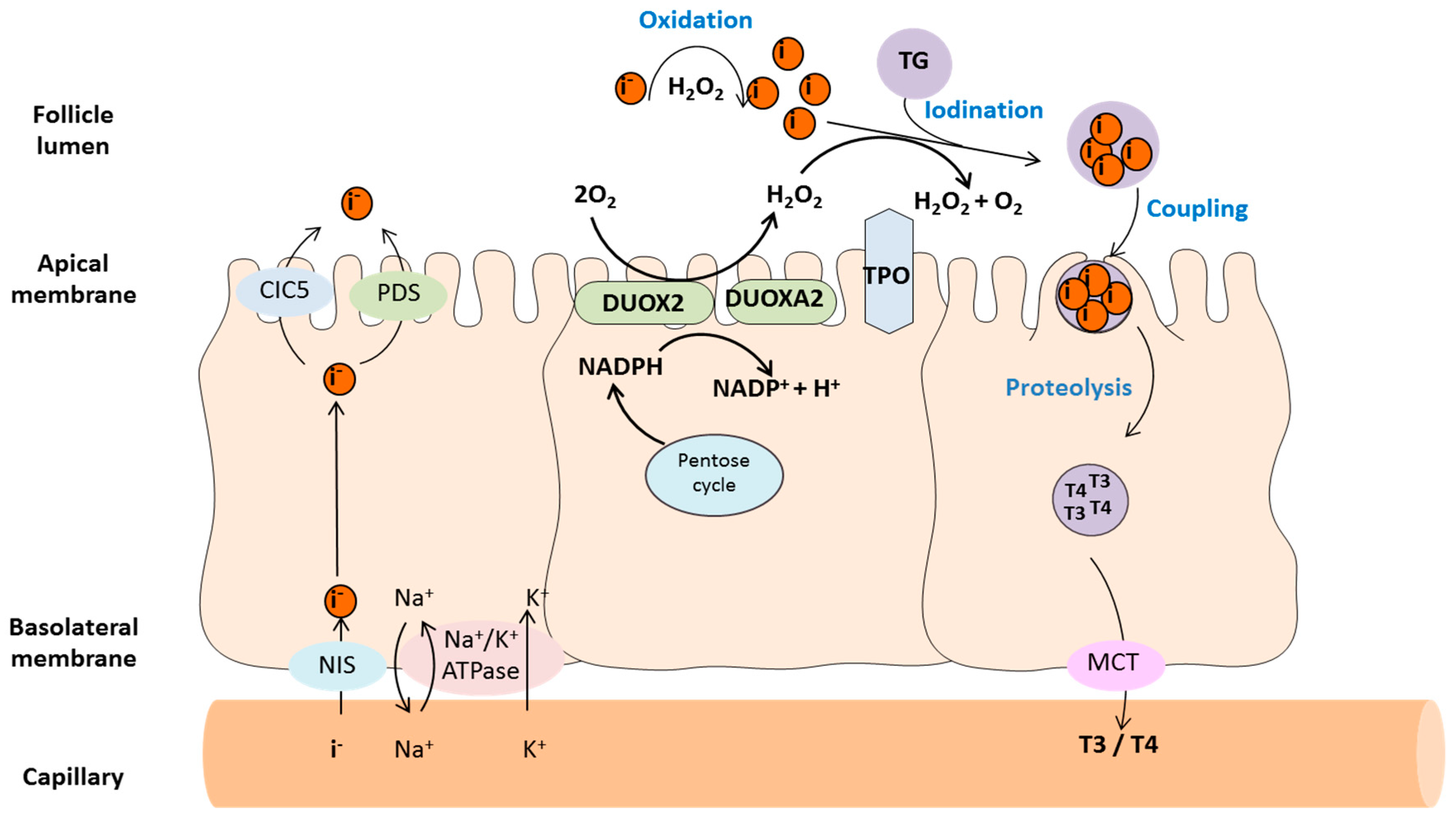
2. Redox Enzymes in Thyroid Hormone Synthesis
2.1. Thyroid Peroxidase (TPO)
2.2. Dual Oxidases (DUOX1 and DUOX2)
3. NADPH Oxidases in Thyroid Pathologies
3.1. DUOX1/2 in Thyroid Dyshormonogenesis
3.2. DUOX1/2 in Thyroid Tumorigenesis
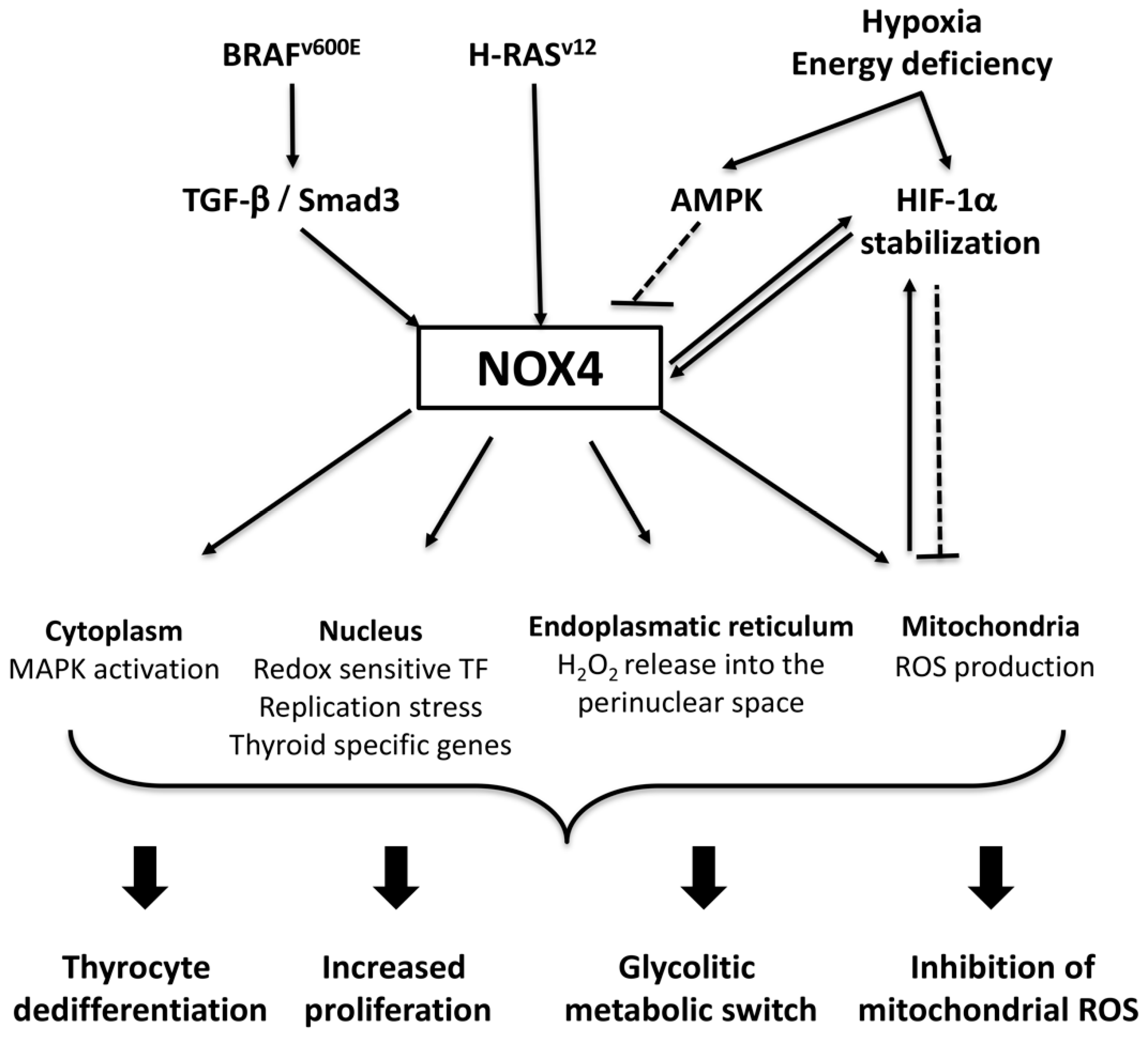
3.3. NADPH Oxidase 4 (NOX4)
NOX4 in Thyroid Tumorigenesis
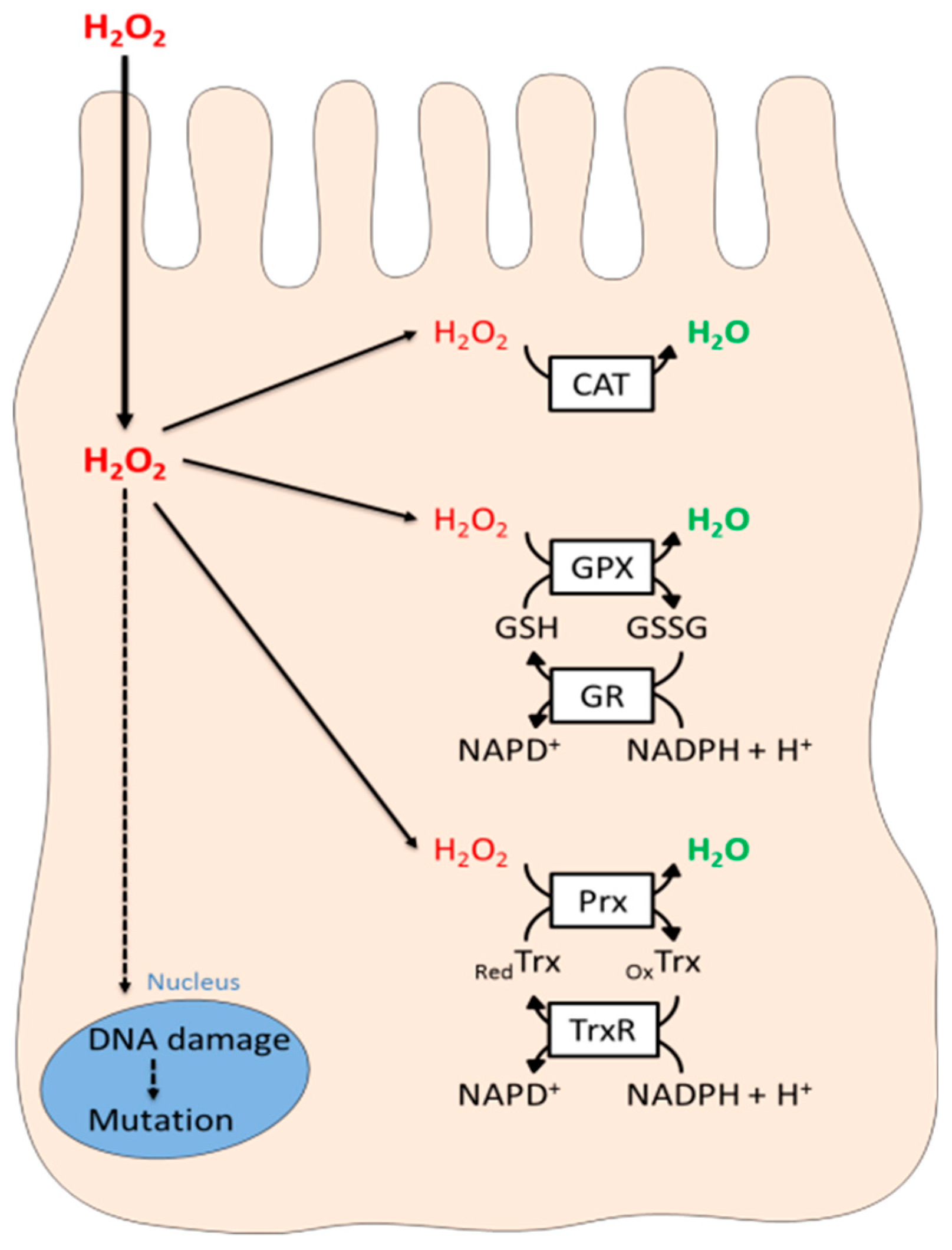
4. The Thyrocyte Anti-Oxidant Defense System
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Demeester-Mirkine, N.; Van Sande, J.; Corvilain, J.; Dumont, J.E. Benign thyroid nodule with normal iodide trap and defective organification. J. Clin. Endocrinol. Metab. 1975, 41, 1169–1171. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, U.; Ekholm, R. Generation of H2O2 in isolated porcine thyroid follicles. Endocrinology 1984, 115, 392–398. [Google Scholar] [CrossRef]
- Deme, D.; Virion, A.; Hammou, N.A.; Pommier, J. NADPH-dependent generation of H2O2 in a thyroid particulate fraction requires Ca2+. FEBS Lett. 1985, 186, 107–110. [Google Scholar] [CrossRef]
- Virion, A.; Michot, J.L.; Deme, D.; Kaniewski, J.; Pommier, J. NADPH-dependent H2O2 generation and peroxidase activity in thyroid particular fraction. Mol. Cell. Endocrinol. 1984, 36, 95–105. [Google Scholar] [CrossRef]
- De Deken, X.; Wang, D.; Many, M.C.; Costagliola, S.; Libert, F.; Vassart, G.; Dumont, J.E.; Miot, F. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J. Biol. Chem. 2000, 275, 23227–23233. [Google Scholar] [CrossRef]
- Dupuy, C.; Ohayon, R.; Valent, A.; Noel-Hudson, M.S.; Deme, D.; Virion, A. Purification of a novel flavoprotein involved in the thyroid NADPH oxidase. Cloning of the porcine and human cdnas. J. Biol. Chem. 1999, 274, 37265–37269. [Google Scholar] [CrossRef]
- Ameziane-El-Hassani, R.; Morand, S.; Boucher, J.L.; Frapart, Y.M.; Apostolou, D.; Agnandji, D.; Gnidehou, S.; Ohayon, R.; Noel-Hudson, M.S.; Francon, J.; et al. Dual oxidase-2 has an intrinsic Ca2+-dependent H2O2-generating activity. J. Biol. Chem. 2005, 280, 30046–30054. [Google Scholar] [CrossRef]
- Brandes, R.P.; Weissmann, N.; Schroder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Redox-dependent signal transduction. FEBS Lett. 2000, 476, 52–54. [Google Scholar] [CrossRef]
- Alexandrova, A.Y.; Kopnin, P.B.; Vasiliev, J.M.; Kopnin, B.P. ROS up-regulation mediates Ras-induced changes of cell morphology and motility. Exp. Cell Res. 2006, 312, 2066–2073. [Google Scholar] [CrossRef]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Weyemi, U.; Lagente-Chevallier, O.; Boufraqech, M.; Prenois, F.; Courtin, F.; Caillou, B.; Talbot, M.; Dardalhon, M.; Al Ghuzlan, A.; Bidart, J.M.; et al. ROS-generating NADPH oxidase NOX4 is a critical mediator in oncogenic H-Ras-induced DNA damage and subsequent senescence. Oncogene 2012, 31, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Azouzi, N.; Cailloux, J.; Cazarin, J.M.; Knauf, J.A.; Cracchiolo, J.; Al Ghuzlan, A.; Hartl, D.; Polak, M.; Carre, A.; El Mzibri, M.; et al. NADPH Oxidase NOX4 Is a Critical Mediator of BRAF(V600E)-Induced Downregulation of the Sodium/Iodide Symporter in Papillary Thyroid Carcinomas. Antioxid. Redox Signal. 2017, 26, 864–877. [Google Scholar] [CrossRef] [PubMed]
- Godlewska, M.; Gora, M.; Buckle, A.M.; Porebski, B.T.; Kemp, E.H.; Sutton, B.J.; Czarnocka, B.; Banga, J.P. A redundant role of human thyroid peroxidase propeptide for cellular, enzymatic, and immunological activity. Thyroid 2014, 24, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Fayadat, L.; Niccoli-Sire, P.; Lanet, J.; Franc, J.L. Role of heme in intracellular trafficking of thyroperoxidase and involvement of H2O2 generated at the apical surface of thyroid cells in autocatalytic covalent heme binding. J. Biol. Chem. 1999, 274, 10533–10538. [Google Scholar] [CrossRef] [PubMed]
- Taurog, A. Molecular evolution of thyroid peroxidase. Biochimie 1999, 81, 557–562. [Google Scholar] [CrossRef]
- Morand, S.; Chaaraoui, M.; Kaniewski, J.; Deme, D.; Ohayon, R.; Noel-Hudson, M.S.; Virion, A.; Dupuy, C. Effect of iodide on nicotinamide adenine dinucleotide phosphate oxidase activity and Duox2 protein expression in isolated porcine thyroid follicles. Endocrinology 2003, 144, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Uyttersprot, N.; Pelgrims, N.; Carrasco, N.; Gervy, C.; Maenhaut, C.; Dumont, J.E.; Miot, F. Moderate doses of iodide in vivo inhibit cell proliferation and the expression of thyroperoxidase and Na+/I- symporter mRNAs in dog thyroid. Mol. Cell. Endocrinol. 1997, 131, 195–203. [Google Scholar] [CrossRef]
- Lambeth, J.D.; Neish, A.S. Nox enzymes and new thinking on reactive oxygen: A double-edged sword revisited. Annu. Rev. Pathol. 2014, 9, 119–145. [Google Scholar] [CrossRef]
- Skonieczna, M.; Hejmo, T.; Poterala-Hejmo, A.; Cieslar-Pobuda, A.; Buldak, R.J. NADPH Oxidases: Insights into Selected Functions and Mechanisms of Action in Cancer and Stem Cells. Oxid. Med. Cell. Longev. 2017, 2017, 9420539. [Google Scholar] [CrossRef]
- Ambasta, R.K.; Kumar, P.; Griendling, K.K.; Schmidt, H.H.; Busse, R.; Brandes, R.P. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J. Biol. Chem. 2004, 279, 45935–45941. [Google Scholar] [CrossRef]
- Geiszt, M.; Kopp, J.B.; Varnai, P.; Leto, T.L. Identification of renox, an NAD(P)H oxidase in kidney. Proc. Natl. Acad. Sci. USA 2000, 97, 8010–8014. [Google Scholar] [CrossRef]
- Shiose, A.; Kuroda, J.; Tsuruya, K.; Hirai, M.; Hirakata, H.; Naito, S.; Hattori, M.; Sakaki, Y.; Sumimoto, H. A novel superoxide-producing NAD(P)H oxidase in kidney. J. Biol. Chem. 2001, 276, 1417–1423. [Google Scholar] [CrossRef]
- Serrander, L.; Cartier, L.; Bedard, K.; Banfi, B.; Lardy, B.; Plastre, O.; Sienkiewicz, A.; Forro, L.; Schlegel, W.; Krause, K.H. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem. J. 2007, 406, 105–114. [Google Scholar] [CrossRef]
- Nakano, Y.; Banfi, B.; Jesaitis, A.J.; Dinauer, M.C.; Allen, L.A.; Nauseef, W.M. Critical roles for p22phox in the structural maturation and subcellular targeting of Nox3. Biochem. J. 2007, 403, 97–108. [Google Scholar] [CrossRef]
- Zhu, Y.; Marchal, C.C.; Casbon, A.J.; Stull, N.; von Lohneysen, K.; Knaus, U.G.; Jesaitis, A.J.; McCormick, S.; Nauseef, W.M.; Dinauer, M.C. Deletion mutagenesis of p22phox subunit of flavocytochrome b558: Identification of regions critical for gp91phox maturation and NADPH oxidase activity. J. Biol. Chem. 2006, 281, 30336–30346. [Google Scholar] [CrossRef]
- Ameziane-El-Hassani, R.; Schlumberger, M.; Dupuy, C. NADPH oxidases: New actors in thyroid cancer? Nat. Rev. Endocrinol. 2016, 12, 485–494. [Google Scholar] [CrossRef]
- Rigutto, S.; Hoste, C.; Grasberger, H.; Milenkovic, M.; Communi, D.; Dumont, J.E.; Corvilain, B.; Miot, F.; De Deken, X. Activation of dual oxidases Duox1 and Duox2: Differential regulation mediated by camp-dependent protein kinase and protein kinase C-dependent phosphorylation. J. Biol. Chem. 2009, 284, 6725–6734. [Google Scholar] [CrossRef]
- Song, Y.; Ruf, J.; Lothaire, P.; Dequanter, D.; Andry, G.; Willemse, E.; Dumont, J.E.; Van Sande, J.; De Deken, X. Association of duoxes with thyroid peroxidase and its regulation in thyrocytes. J. Clin. Endocrinol. Metab. 2010, 95, 375–382. [Google Scholar] [CrossRef]
- Caillou, B.; Dupuy, C.; Lacroix, L.; Nocera, M.; Talbot, M.; Ohayon, R.; Deme, D.; Bidart, J.M.; Schlumberger, M.; Virion, A. Expression of reduced nicotinamide adenine dinucleotide phosphate oxidase (ThoX, LNOX, Duox) genes and proteins in human thyroid tissues. J. Clin. Endocrinol. Metab. 2001, 86, 3351–3358. [Google Scholar] [CrossRef]
- Grasberger, H.; Refetoff, S. Identification of the maturation factor for dual oxidase. Evolution of an eukaryotic operon equivalent. J. Biol. Chem. 2006, 281, 18269–18272. [Google Scholar] [CrossRef] [PubMed]
- Morand, S.; Ueyama, T.; Tsujibe, S.; Saito, N.; Korzeniowska, A.; Leto, T.L. Duox maturation factors form cell surface complexes with Duox affecting the specificity of reactive oxygen species generation. FASEB J. 2009, 23, 1205–1218. [Google Scholar] [CrossRef] [PubMed]
- Luxen, S.; Noack, D.; Frausto, M.; Davanture, S.; Torbett, B.E.; Knaus, U.G. Heterodimerization controls localization of Duox-DuoxA NADPH oxidases in airway cells. J. Cell Sci. 2009, 122, 1238–1247. [Google Scholar] [CrossRef]
- Pachucki, J.; Wang, D.; Christophe, D.; Miot, F. Structural and functional characterization of the two human ThOX/Duox genes and their 5′-flanking regions. Mol. Cell. Endocrinol. 2004, 214, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Forteza, R.; Salathe, M.; Miot, F.; Forteza, R.; Conner, G.E. Regulated hydrogen peroxide production by Duox in human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2005, 32, 462–469. [Google Scholar] [CrossRef]
- Geiszt, M.; Witta, J.; Baffi, J.; Lekstrom, K.; Leto, T.L. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J. 2003, 17, 1502–1504. [Google Scholar] [CrossRef]
- Schwarzer, C.; Machen, T.E.; Illek, B.; Fischer, H. NADPH oxidase-dependent acid production in airway epithelial cells. J. Biol. Chem. 2004, 279, 36454–36461. [Google Scholar] [CrossRef]
- El Hassani, R.A.; Benfares, N.; Caillou, B.; Talbot, M.; Sabourin, J.C.; Belotte, V.; Morand, S.; Gnidehou, S.; Agnandji, D.; Ohayon, R.; et al. Dual oxidase2 is expressed all along the digestive tract. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G933–G942. [Google Scholar] [CrossRef]
- Ryu, J.H.; Yoo, J.Y.; Kim, M.J.; Hwang, S.G.; Ahn, K.C.; Ryu, J.C.; Choi, M.K.; Joo, J.H.; Kim, C.H.; Lee, S.N.; et al. Distinct TLR-mediated pathways regulate house dust mite-induced allergic disease in the upper and lower airways. J. Allergy Clin. Immunol. 2013, 131, 549–561. [Google Scholar] [CrossRef]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of chaperone-mediated autophagy during oxidative stress. Mol. Biol. Cell 2004, 15, 4829–4840. [Google Scholar] [CrossRef]
- Corvilain, B.; Collyn, L.; van Sande, J.; Dumont, J.E. Stimulation by iodide of H(2)O(2) generation in thyroid slices from several species. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E692–E699. [Google Scholar] [CrossRef]
- Carre, A.; Louzada, R.A.; Fortunato, R.S.; Ameziane-El-Hassani, R.; Morand, S.; Ogryzko, V.; de Carvalho, D.P.; Grasberger, H.; Leto, T.L.; Dupuy, C. When an Intramolecular Disulfide Bridge Governs the Interaction of DUOX2 with Its Partner DUOXA2. Antioxid. Redox Signal. 2015, 23, 724–733. [Google Scholar] [CrossRef]
- Grasberger, H.; De Deken, X.; Miot, F.; Pohlenz, J.; Refetoff, S. Missense mutations of dual oxidase 2 (DUOX2) implicated in congenital hypothyroidism have impaired trafficking in cells reconstituted with DUOX2 maturation factor. Mol. Endocrinol. 2007, 21, 1408–1421. [Google Scholar] [CrossRef]
- Fischer, H.; Gonzales, L.K.; Kolla, V.; Schwarzer, C.; Miot, F.; Illek, B.; Ballard, P.L. Developmental regulation of DUOX1 expression and function in human fetal lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L1506–L1514. [Google Scholar] [CrossRef]
- Luxen, S.; Belinsky, S.A.; Knaus, U.G. Silencing of DUOX NADPH oxidases by promoter hypermethylation in lung cancer. Cancer Res. 2008, 68, 1037–1045. [Google Scholar] [CrossRef]
- Yoshihara, A.; Hara, T.; Kawashima, A.; Akama, T.; Tanigawa, K.; Wu, H.; Sue, M.; Ishido, Y.; Hiroi, N.; Ishii, N.; et al. Regulation of dual oxidase expression and H2O2 production by thyroglobulin. Thyroid 2012, 22, 1054–1062. [Google Scholar] [CrossRef]
- Cardoso, L.C.; Martins, D.C.; Figueiredo, M.D.; Rosenthal, D.; Vaisman, M.; Violante, A.H.; Carvalho, D.P. Ca(2+)/nicotinamide adenine dinucleotide phosphate-dependent H(2)O(2) generation is inhibited by iodide in human thyroids. J. Clin. Endocrinol. Metab. 2001, 86, 4339–4343. [Google Scholar] [CrossRef]
- De Deken, X.; Wang, D.; Dumont, J.E.; Miot, F. Characterization of ThOX proteins as components of the thyroid H(2)O(2)-generating system. Exp. Cell Res. 2002, 273, 187–196. [Google Scholar] [CrossRef]
- Corvilain, B.; Van Sande, J.; Dumont, J.E. Inhibition by iodide of iodide binding to proteins: The “Wolff-Chaikoff” effect is caused by inhibition of H2O2 generation. Biochem. Biophys. Res. Commun. 1988, 154, 1287–1292. [Google Scholar] [CrossRef]
- Carvalho, D.P.; Dupuy, C.; Gorin, Y.; Legue, O.; Pommier, J.; Haye, B.; Virion, A. The Ca2+- and reduced nicotinamide adenine dinucleotide phosphate-dependent hydrogen peroxide generating system is induced by thyrotropin in porcine thyroid cells. Endocrinology 1996, 137, 1007–1012. [Google Scholar] [CrossRef]
- Wolff, J.; Chaikoff, I.L. Plasma inorganic iodide as a homeostatic regulator of thyroid function. J. Biol. Chem. 1948, 174, 555–564. [Google Scholar]
- Harper, R.W.; Xu, C.; Eiserich, J.P.; Chen, Y.; Kao, C.Y.; Thai, P.; Setiadi, H.; Wu, R. Differential regulation of dual NADPH oxidases/peroxidases, Duox1 and Duox2, by Th1 and Th2 cytokines in respiratory tract epithelium. FEBS Lett. 2005, 579, 4911–4917. [Google Scholar] [CrossRef] [PubMed]
- Raad, H.; Eskalli, Z.; Corvilain, B.; Miot, F.; De Deken, X. Thyroid hydrogen peroxide production is enhanced by the Th2 cytokines, IL-4 and IL-13, through increased expression of the dual oxidase 2 and its maturation factor DUOXA2. Free Radic. Biol. Med. 2013, 56, 216–225. [Google Scholar] [CrossRef]
- Eskalli, Z.; Achouri, Y.; Hahn, S.; Many, M.C.; Craps, J.; Refetoff, S.; Liao, X.H.; Dumont, J.E.; Van Sande, J.; Corvilain, B.; et al. Overexpression of Interleukin-4 in the Thyroid of Transgenic Mice Upregulates the Expression of Duox1 and the Anion Transporter Pendrin. Thyroid 2016, 26, 1499–1512. [Google Scholar] [CrossRef] [PubMed]
- Bliddal, S.; Nielsen, C.H.; Feldt-Rasmussen, U. Recent advances in understanding autoimmune thyroid disease: The tallest tree in the forest of polyautoimmunity. F1000Res 2017, 6, 1776. [Google Scholar] [CrossRef]
- Mancini, A.; Di Segni, C.; Raimondo, S.; Olivieri, G.; Silvestrini, A.; Meucci, E.; Curro, D. Thyroid Hormones, Oxidative Stress, and Inflammation. Mediat. Inflamm. 2016, 2016, 6757154. [Google Scholar] [CrossRef] [PubMed]
- Hanley, P.; Lord, K.; Bauer, A.J. Thyroid Disorders in Children and Adolescents: A Review. JAMA Pediatr. 2016, 170, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.R.; Marden, C.C.; Ward-Bailey, P.; Gagnon, L.H.; Bronson, R.T.; Donahue, L.R. Congenital hypothyroidism, dwarfism, and hearing impairment caused by a missense mutation in the mouse dual oxidase 2 gene, Duox2. Mol. Endocrinol. 2007, 21, 1593–1602. [Google Scholar] [CrossRef]
- Grasberger, H. Defects of thyroidal hydrogen peroxide generation in congenital hypothyroidism. Mol. Cell. Endocrinol. 2010, 322, 99–106. [Google Scholar] [CrossRef]
- Moreno, J.C.; Bikker, H.; Kempers, M.J.; van Trotsenburg, A.S.; Baas, F.; de Vijlder, J.J.; Vulsma, T.; Ris-Stalpers, C. Inactivating mutations in the gene for thyroid oxidase 2 (THOX2) and congenital hypothyroidism. N. Engl. J. Med. 2002, 347, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Hoste, C.; Rigutto, S.; Van Vliet, G.; Miot, F.; De Deken, X. Compound heterozygosity for a novel hemizygous missense mutation and a partial deletion affecting the catalytic core of the H2O2-generating enzyme DUOX2 associated with transient congenital hypothyroidism. Hum. Mutat. 2010, 31, E1304–E1319. [Google Scholar] [CrossRef] [PubMed]
- Maruo, Y.; Takahashi, H.; Soeda, I.; Nishikura, N.; Matsui, K.; Ota, Y.; Mimura, Y.; Mori, A.; Sato, H.; Takeuchi, Y. Transient congenital hypothyroidism caused by biallelic mutations of the dual oxidase 2 gene in Japanese patients detected by a neonatal screening program. J. Clin. Endocrinol. Metab. 2008, 93, 4261–4267. [Google Scholar] [CrossRef] [PubMed]
- Grasberger, H.; De Deken, X.; Mayo, O.B.; Raad, H.; Weiss, M.; Liao, X.H.; Refetoff, S. Mice deficient in dual oxidase maturation factors are severely hypothyroid. Mol. Endocrinol. 2012, 26, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Chopra, K.; Ishibashi, S.; Amaya, E. Zebrafish duox mutations provide a model for human congenital hypothyroidism. Biol. Open 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, M.D.; Cardoso, L.C.; Ferreira, A.C.; Campos, D.V.; da Cruz Domingos, M.; Corbo, R.; Nasciutti, L.E.; Vaisman, M.; Carvalho, D.P. Goiter and hypothyroidism in two siblings due to impaired Ca(+2)/NAD(P)H-dependent H(2)O(2)-generating activity. J. Clin. Endocrinol. Metab. 2001, 86, 4843–4848. [Google Scholar] [CrossRef]
- Vigone, M.C.; Fugazzola, L.; Zamproni, I.; Passoni, A.; Di Candia, S.; Chiumello, G.; Persani, L.; Weber, G. Persistent mild hypothyroidism associated with novel sequence variants of the DUOX2 gene in two siblings. Hum. Mutat. 2005, 26, 395. [Google Scholar] [CrossRef]
- Varela, V.; Rivolta, C.M.; Esperante, S.A.; Gruneiro-Papendieck, L.; Chiesa, A.; Targovnik, H.M. Three mutations (p.Q36H, p.G418fsX482, and g.IVS19-2A>C) in the dual oxidase 2 gene responsible for congenital goiter and iodide organification defect. Clin. Chem. 2006, 52, 182–191. [Google Scholar] [CrossRef]
- Pfarr, N.; Korsch, E.; Kaspers, S.; Herbst, A.; Stach, A.; Zimmer, C.; Pohlenz, J. Congenital hypothyroidism caused by new mutations in the thyroid oxidase 2 (THOX2) gene. Clin. Endocrinol. 2006, 65, 810–815. [Google Scholar] [CrossRef]
- De Marco, G.; Agretti, P.; Montanelli, L.; Di Cosmo, C.; Bagattini, B.; De Servi, M.; Ferrarini, E.; Dimida, A.; Freitas Ferreira, A.C.; Molinaro, A.; et al. Identification and functional analysis of novel dual oxidase 2 (DUOX2) mutations in children with congenital or subclinical hypothyroidism. J. Clin. Endocrinol. Metab. 2011, 96, E1335–E1339. [Google Scholar] [CrossRef]
- Lu, Z.P.; Li, G.H.; Li, W.J.; Liu, S.G. [DUOX2 gene mutation in patients with congenital goiter with hypothyroidism]. Zhonghua Er Ke Za Zhi 2011, 49, 943–946. [Google Scholar]
- Muzza, M.; Rabbiosi, S.; Vigone, M.C.; Zamproni, I.; Cirello, V.; Maffini, M.A.; Maruca, K.; Schoenmakers, N.; Beccaria, L.; Gallo, F.; et al. The clinical and molecular characterization of patients with dyshormonogenic congenital hypothyroidism reveals specific diagnostic clues for DUOX2 defects. J. Clin. Endocrinol. Metab. 2014, 99, E544–E553. [Google Scholar] [CrossRef]
- Wang, F.; Lu, K.; Yang, Z.; Zhang, S.; Lu, W.; Zhang, L.; Liu, S.; Yan, S. Genotypes and phenotypes of congenital goitre and hypothyroidism caused by mutations in dual oxidase 2 genes. Clin. Endocrinol. 2014, 81, 452–457. [Google Scholar] [CrossRef]
- Jin, H.Y.; Heo, S.H.; Kim, Y.M.; Kim, G.H.; Choi, J.H.; Lee, B.H.; Yoo, H.W. High frequency of DUOX2 mutations in transient or permanent congenital hypothyroidism with eutopic thyroid glands. Horm. Res. Paediatr. 2014, 82, 252–260. [Google Scholar] [CrossRef]
- Muzza, M.; Fugazzola, L. Disorders of H2O2 generation. Best Pract. Res. Clin. Endocrinol. Metab. 2017, 31, 225–240. [Google Scholar] [CrossRef]
- Moreno, J.C.; Visser, T.J. New phenotypes in thyroid dyshormonogenesis: Hypothyroidism due to DUOX2 mutations. Endocr. Dev. 2007, 10, 99–117. [Google Scholar] [CrossRef]
- Makretskaya, N.; Bezlepkina, O.; Kolodkina, A.; Kiyaev, A.; Vasilyev, E.V.; Petrov, V.; Kalinenkova, S.; Malievsky, O.; Dedov, I.I.; Tiulpakov, A. High frequency of mutations in ‘dyshormonogenesis genes’ in severe congenital hypothyroidism. PLoS ONE 2018, 13, e0204323. [Google Scholar] [CrossRef]
- Donko, A.; Ruisanchez, E.; Orient, A.; Enyedi, B.; Kapui, R.; Peterfi, Z.; de Deken, X.; Benyo, Z.; Geiszt, M. Urothelial cells produce hydrogen peroxide through the activation of Duox1. Free Radic. Biol. Med. 2010, 49, 2040–2048. [Google Scholar] [CrossRef]
- Xing, M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat. Rev. Cancer 2013, 13, 184–199. [Google Scholar] [CrossRef]
- Coclet, J.; Foureau, F.; Ketelbant, P.; Galand, P.; Dumont, J.E. Cell population kinetics in dog and human adult thyroid. Clin. Endocrinol. 1989, 31, 655–665. [Google Scholar] [CrossRef]
- Saad, A.G.; Kumar, S.; Ron, E.; Lubin, J.H.; Stanek, J.; Bove, K.E.; Nikiforov, Y.E. Proliferative activity of human thyroid cells in various age groups and its correlation with the risk of thyroid cancer after radiation exposure. J. Clin. Endocrinol. Metab. 2006, 91, 2672–2677. [Google Scholar] [CrossRef]
- Block, K.; Gorin, Y. Aiding and abetting roles of NOX oxidases in cellular transformation. Nat. Rev. Cancer 2012, 12, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Ling, Q.; Shi, W.; Huang, C.; Zheng, J.; Cheng, Q.; Yu, K.; Chen, S.; Zhang, H.; Li, N.; Chen, M. Epigenetic silencing of dual oxidase 1 by promoter hypermethylation in human hepatocellular carcinoma. Am. J. Cancer Res. 2014, 4, 508–517. [Google Scholar] [PubMed]
- Wu, Y.; Antony, S.; Hewitt, S.M.; Jiang, G.; Yang, S.X.; Meitzler, J.L.; Juhasz, A.; Lu, J.; Liu, H.; Doroshow, J.H.; et al. Functional activity and tumor-specific expression of dual oxidase 2 in pancreatic cancer cells and human malignancies characterized with a novel monoclonal antibody. Int. J. Oncol. 2013, 42, 1229–1238. [Google Scholar] [CrossRef]
- Ramsey, M.R.; Sharpless, N.E. ROS as a tumour suppressor? Nat. Cell Biol. 2006, 8, 1213–1215. [Google Scholar] [CrossRef]
- Ameziane-El-Hassani, R.; Talbot, M.; de Souza Dos Santos, M.C.; Al Ghuzlan, A.; Hartl, D.; Bidart, J.M.; De Deken, X.; Miot, F.; Diallo, I.; de Vathaire, F.; et al. NADPH oxidase DUOX1 promotes long-term persistence of oxidative stress after an exposure to irradiation. Proc. Natl. Acad. Sci. USA 2015, 112, 5051–5056. [Google Scholar] [CrossRef]
- Ameziane El Hassani, R.; Buffet, C.; Leboulleux, S.; Dupuy, C. Oxidative stress in thyroid carcinomas: Biological and clinical significance. Endocr. Relat. Cancer 2019, 26, R131–R143. [Google Scholar] [CrossRef]
- Lacroix, L.; Nocera, M.; Mian, C.; Caillou, B.; Virion, A.; Dupuy, C.; Filetti, S.; Bidart, J.M.; Schlumberger, M. Expression of nicotinamide adenine dinucleotide phosphate oxidase flavoprotein DUOX genes and proteins in human papillary and follicular thyroid carcinomas. Thyroid 2001, 11, 1017–1023. [Google Scholar] [CrossRef]
- Weyemi, U.; Redon, C.E.; Parekh, P.R.; Dupuy, C.; Bonner, W.M. NADPH Oxidases NOXs and DUOXs as putative targets for cancer therapy. Anticancer Agents Med. Chem. 2013, 13, 502–514. [Google Scholar]
- Mouche, S.; Mkaddem, S.B.; Wang, W.; Katic, M.; Tseng, Y.H.; Carnesecchi, S.; Steger, K.; Foti, M.; Meier, C.A.; Muzzin, P.; et al. Reduced expression of the NADPH oxidase NOX4 is a hallmark of adipocyte differentiation. Biochim. Biophys. Acta 2007, 1773, 1015–1027. [Google Scholar] [CrossRef]
- Mahadev, K.; Wu, X.; Zilbering, A.; Zhu, L.; Lawrence, J.T.; Goldstein, B.J. Hydrogen peroxide generated during cellular insulin stimulation is integral to activation of the distal insulin signaling cascade in 3T3-L1 adipocytes. J. Biol. Chem. 2001, 276, 48662–48669. [Google Scholar] [CrossRef]
- Carmona-Cuenca, I.; Herrera, B.; Ventura, J.J.; Roncero, C.; Fernandez, M.; Fabregat, I. EGF blocks NADPH oxidase activation by TGF-beta in fetal rat hepatocytes, impairing oxidative stress, and cell death. J. Cell. Physiol. 2006, 207, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.F.; Qiao, M.; Schroder, K.; Zhao, Q.; Asmis, R. Nox4 is a novel inducible source of reactive oxygen species in monocytes and macrophages and mediates oxidized low density lipoprotein-induced macrophage death. Circ. Res. 2010, 106, 1489–1497. [Google Scholar] [CrossRef] [PubMed]
- Vallet, P.; Charnay, Y.; Steger, K.; Ogier-Denis, E.; Kovari, E.; Herrmann, F.; Michel, J.P.; Szanto, I. Neuronal expression of the NADPH oxidase NOX4, and its regulation in mouse experimental brain ischemia. Neuroscience 2005, 132, 233–238. [Google Scholar] [CrossRef]
- Nisimoto, Y.; Diebold, B.A.; Cosentino-Gomes, D.; Lambeth, J.D. Nox4: A hydrogen peroxide-generating oxygen sensor. Biochemistry 2014, 53, 5111–5120. [Google Scholar] [CrossRef] [PubMed]
- Takac, I.; Schroder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef]
- Block, K.; Gorin, Y.; Abboud, H.E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 14385–14390. [Google Scholar] [CrossRef] [PubMed]
- Lyle, A.N.; Deshpande, N.N.; Taniyama, Y.; Seidel-Rogol, B.; Pounkova, L.; Du, P.; Papaharalambus, C.; Lassegue, B.; Griendling, K.K. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ. Res. 2009, 105, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Shanmugasundaram, K.; Nayak, B.K.; Friedrichs, W.E.; Kaushik, D.; Rodriguez, R.; Block, K. NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nat. Commun. 2017, 8, 997. [Google Scholar] [CrossRef]
- Kuroda, J.; Sadoshima, J. NADPH oxidase and cardiac failure. J. Cardiovasc. Transl. Res. 2010, 3, 314–320. [Google Scholar] [CrossRef]
- Kuroda, J.; Nakagawa, K.; Yamasaki, T.; Nakamura, K.; Takeya, R.; Kuribayashi, F.; Imajoh-Ohmi, S.; Igarashi, K.; Shibata, Y.; Sueishi, K.; et al. The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes Cells 2005, 10, 1139–1151. [Google Scholar] [CrossRef]
- Leto, T.L.; Morand, S.; Hurt, D.; Ueyama, T. Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid. Redox Signal. 2009, 11, 2607–2619. [Google Scholar] [CrossRef] [PubMed]
- Martyn, K.D.; Frederick, L.M.; von Loehneysen, K.; Dinauer, M.C.; Knaus, U.G. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal. 2006, 18, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Vukelic, S.; Xu, Q.; Seidel-Rogol, B.; Faidley, E.A.; Dikalova, A.E.; Hilenski, L.L.; Jorde, U.; Poole, L.B.; Lassegue, B.; Zhang, G.; et al. NOX4 (NADPH Oxidase 4) and Poldip2 (Polymerase delta-Interacting Protein 2) Induce Filamentous Actin Oxidation and Promote Its Interaction With Vinculin During Integrin-Mediated Cell Adhesion. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2423–2434. [Google Scholar] [CrossRef]
- Fortunato, R.S.; Braga, W.M.; Ortenzi, V.H.; Rodrigues, D.C.; Andrade, B.M.; Miranda-Alves, L.; Rondinelli, E.; Dupuy, C.; Ferreira, A.C.; Carvalho, D.P. Sexual dimorphism of thyroid reactive oxygen species production due to higher NADPH oxidase 4 expression in female thyroid glands. Thyroid 2013, 23, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Weyemi, U.; Caillou, B.; Talbot, M.; Ameziane-El-Hassani, R.; Lacroix, L.; Lagent-Chevallier, O.; Al Ghuzlan, A.; Roos, D.; Bidart, J.M.; Virion, A.; et al. Intracellular expression of reactive oxygen species-generating NADPH oxidase NOX4 in normal and cancer thyroid tissues. Endocr. Relat. Cancer 2010, 17, 27–37. [Google Scholar] [CrossRef]
- Santos, M.C.; Louzada, R.A.; Souza, E.C.; Fortunato, R.S.; Vasconcelos, A.L.; Souza, K.L.; Castro, J.P.; Carvalho, D.P.; Ferreira, A.C. Diabetes mellitus increases reactive oxygen species production in the thyroid of male rats. Endocrinology 2013, 154, 1361–1372. [Google Scholar] [CrossRef]
- Li, Y.; Mouche, S.; Sajic, T.; Veyrat-Durebex, C.; Supale, R.; Pierroz, D.; Ferrari, S.; Negro, F.; Hasler, U.; Feraille, E.; et al. Deficiency in the NADPH oxidase 4 predisposes towards diet-induced obesity. Int. J. Obes. 2012, 36, 1503–1513. [Google Scholar] [CrossRef] [PubMed]
- Matos, L.P.L.; Penha, R.C.C.; Cardoso-Weide, L.C.; Freitas, M.L.; Silva, D.; Ferreira, A.C.F. Regulation of thyroid sodium-iodide symporter in different stages of goiter: Possible involvement of reactive oxygen species. Clin. Exp. Pharmacol. Physiol. 2018, 45, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Oglio, R.; Salvarredi, L.; Rossich, L.; Copelli, S.; Pisarev, M.; Juvenal, G.; Thomasz, L. Participation of NADPH 4 oxidase in thyroid regulation. Mol. Cell. Endocrinol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.T.; Lin, X.L.; Wu, S.; Liang, Q.; Yang, L.; Gao, Y.J.; Ge, Z.Z. NOX4-driven ROS formation regulates proliferation and apoptosis of gastric cancer cells through the GLI1 pathway. Cell Signal. 2018, 46, 52–63. [Google Scholar] [CrossRef]
- Hanley, C.J.; Mellone, M.; Ford, K.; Thirdborough, S.M.; Mellows, T.; Frampton, S.J.; Smith, D.M.; Harden, E.; Szyndralewiez, C.; Bullock, M.; et al. Targeting the Myofibroblastic Cancer-Associated Fibroblast Phenotype Through Inhibition of NOX4. J. Natl. Cancer Inst. 2018, 110. [Google Scholar] [CrossRef] [PubMed]
- Salmeen, A.; Park, B.O.; Meyer, T. The NADPH oxidases NOX4 and DUOX2 regulate cell cycle entry via a p53-dependent pathway. Oncogene 2010, 29, 4473–4484. [Google Scholar] [CrossRef]
- Meng, T.C.; Fukada, T.; Tonks, N.K. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol. Cell 2002, 9, 387–399. [Google Scholar] [CrossRef]
- Santos, C.X.; Hafstad, A.D.; Beretta, M.; Zhang, M.; Molenaar, C.; Kopec, J.; Fotinou, D.; Murray, T.V.; Cobb, A.M.; Martin, D.; et al. Targeted redox inhibition of protein phosphatase 1 by Nox4 regulates eIF2alpha-mediated stress signaling. EMBO J. 2016, 35, 319–334. [Google Scholar] [CrossRef]
- Chen, K.; Kirber, M.T.; Xiao, H.; Yang, Y.; Keaney, J.F., Jr. Regulation of ROS signal transduction by NADPH oxidase 4 localization. J. Cell Biol. 2008, 181, 1129–1139. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef]
- Thomasz, L.; Oglio, R.; Salvarredi, L.; Perona, M.; Rossich, L.; Copelli, S.; Pisarev, M.; Juvenal, G. Regulation of NADPH oxidase NOX4 by delta iodolactone (IL-delta) in thyroid cancer cells. Mol. Cell. Endocrinol. 2018, 470, 115–126. [Google Scholar] [CrossRef]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef]
- Boudreau, H.E.; Casterline, B.W.; Rada, B.; Korzeniowska, A.; Leto, T.L. Nox4 involvement in TGF-beta and SMAD3-driven induction of the epithelial-to-mesenchymal transition and migration of breast epithelial cells. Free Radic. Biol. Med. 2012, 53, 1489–1499. [Google Scholar] [CrossRef]
- Carmona-Cuenca, I.; Roncero, C.; Sancho, P.; Caja, L.; Fausto, N.; Fernandez, M.; Fabregat, I. Upregulation of the NADPH oxidase NOX4 by TGF-beta in hepatocytes is required for its pro-apoptotic activity. J. Hepatol. 2008, 49, 965–976. [Google Scholar] [CrossRef]
- Yan, F.; Wang, Y.; Wu, X.; Peshavariya, H.M.; Dusting, G.J.; Zhang, M.; Jiang, F. Nox4 and redox signaling mediate TGF-beta-induced endothelial cell apoptosis and phenotypic switch. Cell Death Dis. 2014, 5, e1010. [Google Scholar] [CrossRef]
- Colletta, G.; Cirafici, A.M.; Imbriaco, M.; Vecchio, G. Inhibitory action of transforming growth factor beta on thyroid cells. Ann. N. Y. Acad. Sci. 1988, 551, 372–373. [Google Scholar] [CrossRef]
- Taton, M.; Lamy, F.; Roger, P.P.; Dumont, J.E. General inhibition by transforming growth factor beta 1 of thyrotropin and cAMP responses in human thyroid cells in primary culture. Mol. Cell. Endocrinol. 1993, 95, 13–21. [Google Scholar] [CrossRef]
- Ely, K.A.; Bischoff, L.A.; Weiss, V.L. Wnt Signaling in Thyroid Homeostasis and Carcinogenesis. Genes 2018, 9, 204. [Google Scholar] [CrossRef]
- Pisarev, M.A.; Thomasz, L.; Juvenal, G.J. Role of transforming growth factor beta in the regulation of thyroid function and growth. Thyroid 2009, 19, 881–892. [Google Scholar] [CrossRef]
- Oglio, R.; Thomasz, L.; Salvarredi, L.; Juvenal, G.; Pisarev, M. Comparative effects of transforming growth factor beta isoforms on redox metabolism in thyroid cells. Mol. Cell. Endocrinol. 2018, 470, 168–178. [Google Scholar] [CrossRef]
- Fagin, J.A.; Wells, S.A., Jr. Biologic and Clinical Perspectives on Thyroid Cancer. N. Engl. J. Med. 2016, 375, 1054–1067. [Google Scholar] [CrossRef]
- Kimura, E.T.; Nikiforova, M.N.; Zhu, Z.; Knauf, J.A.; Nikiforov, Y.E.; Fagin, J.A. High prevalence of BRAF mutations in thyroid cancer: Genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003, 63, 1454–1457. [Google Scholar]
- Li, X.; Abdel-Mageed, A.B.; Kandil, E. BRAF mutation in papillary thyroid carcinoma. Int. J. Clin. Exp. Med. 2012, 5, 310–315. [Google Scholar]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Kebebew, E.; Weng, J.; Bauer, J.; Ranvier, G.; Clark, O.H.; Duh, Q.Y.; Shibru, D.; Bastian, B.; Griffin, A. The prevalence and prognostic value of BRAF mutation in thyroid cancer. Ann. Surg. 2007, 246, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, D.; Murugan, A.K.; Liu, Z.; Xing, M. Histone deacetylation of NIS promoter underlies BRAF V600E-promoted NIS silencing in thyroid cancer. Endocr. Relat. Cancer 2014, 21, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Nath, M.C.; Erickson, L.A. Aggressive Variants of Papillary Thyroid Carcinoma: Hobnail, Tall Cell, Columnar, and Solid. Adv. Anat. Pathol. 2018, 25, 172–179. [Google Scholar] [CrossRef]
- Boudreau, H.E.; Casterline, B.W.; Burke, D.J.; Leto, T.L. Wild-type and mutant p53 differentially regulate NADPH oxidase 4 in TGF-beta-mediated migration of human lung and breast epithelial cells. Br. J. Cancer 2014, 110, 2569–2582. [Google Scholar] [CrossRef]
- Tang, P.; Dang, H.; Huang, J.; Xu, T.; Yuan, P.; Hu, J.; Sheng, J.F. NADPH oxidase NOX4 is a glycolytic regulator through mROS-HIF1alpha axis in thyroid carcinomas. Sci. Rep. 2018, 8, 15897. [Google Scholar] [CrossRef]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef]
- Jiang, B. Aerobic glycolysis and high level of lactate in cancer metabolism and microenvironment. Genes Dis. 2017, 4, 25–27. [Google Scholar] [CrossRef]
- Coelho, R.G.; Cazarin, J.M.; Cavalcanti de Albuquerque, J.P.; de Andrade, B.M.; Carvalho, D.P. Differential glycolytic profile and Warburg effect in papillary thyroid carcinoma cell lines. Oncol. Rep. 2016, 36, 3673–3681. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Mehrmohamadi, M.; Liu, X.; Shestov, A.A.; Locasale, J.W. Characterization of the usage of the serine metabolic network in human cancer. Cell Rep. 2014, 9, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid. Redox Signal. 2008, 10, 179–206. [Google Scholar] [CrossRef] [PubMed]
- Metere, A.; Frezzotti, F.; Graves, C.E.; Vergine, M.; De Luca, A.; Pietraforte, D.; Giacomelli, L. A possible role for selenoprotein glutathione peroxidase (GPx1) and thioredoxin reductases (TrxR1) in thyroid cancer: Our experience in thyroid surgery. Cancer Cell Int. 2018, 18, 7. [Google Scholar] [CrossRef]
- Yi, J.W.; Park, J.Y.; Sung, J.Y.; Kwak, S.H.; Yu, J.; Chang, J.H.; Kim, J.H.; Ha, S.Y.; Paik, E.K.; Lee, W.S.; et al. Genomic evidence of reactive oxygen species elevation in papillary thyroid carcinoma with Hashimoto thyroiditis. Endocr. J. 2015, 62, 857–877. [Google Scholar] [CrossRef]
- Diebold, I.; Petry, A.; Hess, J.; Gorlach, A. The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Mol. Biol. Cell 2010, 21, 2087–2096. [Google Scholar] [CrossRef]
- Cazarin, J.M.; Coelho, R.G.; Hecht, F.; Andrade, B.M.; Carvalho, D.P. 5′-AMP-Activated Protein Kinase Regulates Papillary (TPC-1 and BCPAP) Thyroid Cancer Cell Survival, Migration, Invasion, and Epithelial-to-Mesenchymal Transition. Thyroid 2016, 26, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.P.; Andrade, B.M.; Vaisman, F.; Cazarin, J.; Pinto, L.F.; Breitenbach, M.M.; Corbo, R.; Caroli-Bottino, A.; Soares, F.; Vaisman, M.; et al. AMP-activated protein kinase signaling is upregulated in papillary thyroid cancer. Eur. J. Endocrinol. 2013, 169, 521–528. [Google Scholar] [CrossRef]
- Rabinovitch, R.C.; Samborska, B.; Faubert, B.; Ma, E.H.; Gravel, S.P.; Andrzejewski, S.; Raissi, T.C.; Pause, A.; St-Pierre, J.; Jones, R.G. AMPK Maintains Cellular Metabolic Homeostasis through Regulation of Mitochondrial Reactive Oxygen Species. Cell Rep. 2017, 21, 1–9. [Google Scholar] [CrossRef]
- Coelho, R.G.; Fortunato, R.S.; Carvalho, D.P. Metabolic Reprogramming in Thyroid Carcinoma. Front. Oncol. 2018, 8, 82. [Google Scholar] [CrossRef]
- Hardie, D.G.; Lin, S.C. AMP-activated protein kinase—Not just an energy sensor. F1000Res 2017, 6, 1724. [Google Scholar] [CrossRef]
- Eid, A.A.; Ford, B.M.; Block, K.; Kasinath, B.S.; Gorin, Y.; Ghosh-Choudhury, G.; Barnes, J.L.; Abboud, H.E. AMP-activated protein kinase (AMPK) negatively regulates Nox4-dependent activation of p53 and epithelial cell apoptosis in diabetes. J. Biol. Chem. 2010, 285, 37503–37512. [Google Scholar] [CrossRef]
- Sato, N.; Takasaka, N.; Yoshida, M.; Tsubouchi, K.; Minagawa, S.; Araya, J.; Saito, N.; Fujita, Y.; Kurita, Y.; Kobayashi, K.; et al. Metformin attenuates lung fibrosis development via NOX4 suppression. Respir. Res. 2016, 17, 107. [Google Scholar] [CrossRef]
- Eid, A.A.; Lee, D.Y.; Roman, L.J.; Khazim, K.; Gorin, Y. Sestrin 2 and AMPK connect hyperglycemia to Nox4-dependent endothelial nitric oxide synthase uncoupling and matrix protein expression. Mol. Cell. Biol. 2013, 33, 3439–3460. [Google Scholar] [CrossRef]
- Ryder, M.; Ghossein, R.A.; Ricarte-Filho, J.C.; Knauf, J.A.; Fagin, J.A. Increased density of tumor-associated macrophages is associated with decreased survival in advanced thyroid cancer. Endocr. Relat. Cancer 2008, 15, 1069–1074. [Google Scholar] [CrossRef]
- Caillou, B.; Talbot, M.; Weyemi, U.; Pioche-Durieu, C.; Al Ghuzlan, A.; Bidart, J.M.; Chouaib, S.; Schlumberger, M.; Dupuy, C. Tumor-associated macrophages (TAMs) form an interconnected cellular supportive network in anaplastic thyroid carcinoma. PLoS ONE 2011, 6, e22567. [Google Scholar] [CrossRef]
- Zhang, J.; Li, H.; Wu, Q.; Chen, Y.; Deng, Y.; Yang, Z.; Zhang, L.; Liu, B. Tumoral NOX4 recruits M2 tumor-associated macrophages via ROS/PI3K signaling-dependent various cytokine production to promote NSCLC growth. Redox Biol. 2019, 22, 101116. [Google Scholar] [CrossRef]
- Burdon, R.H. Superoxide and hydrogen peroxide in relation to mammalian cell proliferation. Free Radic. Biol. Med. 1995, 18, 775–794. [Google Scholar] [CrossRef]
- Hansberg, W.; Aguirre, J. Hyperoxidant states cause microbial cell differentiation by cell isolation from dioxygen. J. Theor. Biol. 1990, 142, 201–221. [Google Scholar] [CrossRef]
- Toledo, I.; Aguirre, J.; Hansberg, W. Enzyme inactivation related to a hyperoxidant state during conidiation of Neurospora crassa. Microbiology 1994, 140 Pt 9, 2391–2397. [Google Scholar] [CrossRef][Green Version]
- Demelash, A.; Karlsson, J.O.; Nilsson, M.; Bjorkman, U. Selenium has a protective role in caspase-3-dependent apoptosis induced by H2O2 in primary cultured pig thyrocytes. Eur. J. Endocrinol. 2004, 150, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Buttke, T.M.; Sandstrom, P.A. Oxidative stress as a mediator of apoptosis. Immunol. Today 1994, 15, 7–10. [Google Scholar] [CrossRef]
- Riou, C.; Tonoli, H.; Bernier-Valentin, F.; Rabilloud, R.; Fonlupt, P.; Rousset, B. Susceptibility of differentiated thyrocytes in primary culture to undergo apoptosis after exposure to hydrogen peroxide: Relation with the level of expression of apoptosis regulatory proteins, Bcl-2 and Bax. Endocrinology 1999, 140, 1990–1997. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Song, Y.; Driessens, N.; Costa, M.; De Deken, X.; Detours, V.; Corvilain, B.; Maenhaut, C.; Miot, F.; Van Sande, J.; Many, M.C.; et al. Roles of hydrogen peroxide in thyroid physiology and disease. J. Clin. Endocrinol. Metab. 2007, 92, 3764–3773. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Oshino, N. Kinetics and mechanisms of catalase in peroxisomes of the mitochondrial fraction. Biochem. J. 1971, 122, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, U.; Ekholm, R. Hydrogen peroxide degradation and glutathione peroxidase activity in cultures of thyroid cells. Mol. Cell. Endocrinol. 1995, 111, 99–107. [Google Scholar] [CrossRef]
- Wang, D.H.; Tsutsui, K.; Sano, K.; Masuoka, N.; Kira, S. cDNA cloning and expression of mutant catalase from the hypocatalasemic mouse: Comparison with the acatalasemic mutant. Biochim. Biophys. Acta 2001, 1522, 217–220. [Google Scholar] [CrossRef]
- Ho, Y.S.; Xiong, Y.; Ma, W.; Spector, A.; Ho, D.S. Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J. Biol. Chem. 2004, 279, 32804–32812. [Google Scholar] [CrossRef]
- Goth, L.; Nagy, T. Inherited catalase deficiency: Is it benign or a factor in various age related disorders? Mutat. Res. 2013, 753, 147–154. [Google Scholar] [CrossRef]
- Howie, A.F.; Arthur, J.R.; Nicol, F.; Walker, S.W.; Beech, S.G.; Beckett, G.J. Identification of a 57-kilodalton selenoprotein in human thyrocytes as thioredoxin reductase and evidence that its expression is regulated through the calcium-phosphoinositol signaling pathway. J. Clin. Endocrinol. Metab. 1998, 83, 2052–2058. [Google Scholar] [CrossRef]
- Howie, A.F.; Walker, S.W.; Akesson, B.; Arthur, J.R.; Beckett, G.J. Thyroidal extracellular glutathione peroxidase: A potential regulator of thyroid-hormone synthesis. Biochem. J. 1995, 308 Pt 3, 713–717. [Google Scholar] [CrossRef]
- Kim, H.; Lee, T.H.; Park, E.S.; Suh, J.M.; Park, S.J.; Chung, H.K.; Kwon, O.Y.; Kim, Y.K.; Ro, H.K.; Shong, M. Role of peroxiredoxins in regulating intracellular hydrogen peroxide and hydrogen peroxide-induced apoptosis in thyroid cells. J. Biol. Chem. 2000, 275, 18266–18270. [Google Scholar] [CrossRef]
- Arriagada, A.A.; Albornoz, E.; Opazo, M.C.; Becerra, A.; Vidal, G.; Fardella, C.; Michea, L.; Carrasco, N.; Simon, F.; Elorza, A.A.; et al. Excess iodide induces an acute inhibition of the sodium/iodide symporter in thyroid male rat cells by increasing reactive oxygen species. Endocrinology 2015, 156, 1540–1551. [Google Scholar] [CrossRef]
- Leoni, S.G.; Kimura, E.T.; Santisteban, P.; De la Vieja, A. Regulation of thyroid oxidative state by thioredoxin reductase has a crucial role in thyroid responses to iodide excess. Mol. Endocrinol. 2011, 25, 1924–1935. [Google Scholar] [CrossRef]
- Xing, M. Oxidative stress: A new risk factor for thyroid cancer. Endocr. Relat. Cancer 2012, 19, C7-11. [Google Scholar] [CrossRef]
- Maier, J.; van Steeg, H.; van Oostrom, C.; Karger, S.; Paschke, R.; Krohn, K. Deoxyribonucleic acid damage and spontaneous mutagenesis in the thyroid gland of rats and mice. Endocrinology 2006, 147, 3391–3397. [Google Scholar] [CrossRef]
- Halliwell, B. Can oxidative DNA damage be used as a biomarker of cancer risk in humans? Problems, resolutions and preliminary results from nutritional supplementation studies. Free Radic. Res. 1998, 29, 469–486. [Google Scholar] [CrossRef]
- Laatikainen, L.E.; Castellone, M.D.; Hebrant, A.; Hoste, C.; Cantisani, M.C.; Laurila, J.P.; Salvatore, G.; Salerno, P.; Basolo, F.; Nasman, J.; et al. Extracellular superoxide dismutase is a thyroid differentiation marker down-regulated in cancer. Endocr. Relat. Cancer 2010, 17, 785–796. [Google Scholar] [CrossRef]
- Howell, G.M.; Hodak, S.P.; Yip, L. RAS mutations in thyroid cancer. Oncologist 2013, 18, 926–932. [Google Scholar] [CrossRef]
- Cammarota, F.; de Vita, G.; Salvatore, M.; Laukkanen, M.O. Ras oncogene-mediated progressive silencing of extracellular superoxide dismutase in tumorigenesis. Biomed. Res. Int. 2015, 2015, 780409. [Google Scholar] [CrossRef]
- Ashtekar, A.; Huk, D.; Magner, A.; La Perle, K.M.D.; Boucai, L.; Kirschner, L.S. Alterations in Sod2-Induced Oxidative Stress Affect Endocrine Cancer Progression. J. Clin. Endocrinol. Metab. 2018, 103, 4135–4145. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Feng, J.F.; Zeng, P.; Yang, Y.H.; Luo, J.; Yang, Y.W. Total oxidant/antioxidant status in sera of patients with thyroid cancers. Endocr. Relat. Cancer 2011, 18, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Drummond, G.R.; Selemidis, S.; Griendling, K.K.; Sobey, C.G. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 453–471. [Google Scholar] [CrossRef]
- Schmidt, H.H.; Stocker, R.; Vollbracht, C.; Paulsen, G.; Riley, D.; Daiber, A.; Cuadrado, A. Antioxidants in Translational Medicine. Antioxid. Redox Signal. 2015, 23, 1130–1143. [Google Scholar] [CrossRef]
- Cifuentes-Pagano, M.E.; Meijles, D.N.; Pagano, P.J. Nox Inhibitors & Therapies: Rational Design of Peptidic and Small Molecule Inhibitors. Curr. Pharm. Des. 2015, 21, 6023–6035. [Google Scholar]
- Cifuentes-Pagano, E.; Meijles, D.N.; Pagano, P.J. The quest for selective nox inhibitors and therapeutics: challenges, triumphs and pitfalls. Antioxid. Redox Signal. 2014, 20, 2741–2754. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szanto, I.; Pusztaszeri, M.; Mavromati, M. H2O2 Metabolism in Normal Thyroid Cells and in Thyroid Tumorigenesis: Focus on NADPH Oxidases. Antioxidants 2019, 8, 126. https://doi.org/10.3390/antiox8050126
Szanto I, Pusztaszeri M, Mavromati M. H2O2 Metabolism in Normal Thyroid Cells and in Thyroid Tumorigenesis: Focus on NADPH Oxidases. Antioxidants. 2019; 8(5):126. https://doi.org/10.3390/antiox8050126
Chicago/Turabian StyleSzanto, Ildiko, Marc Pusztaszeri, and Maria Mavromati. 2019. "H2O2 Metabolism in Normal Thyroid Cells and in Thyroid Tumorigenesis: Focus on NADPH Oxidases" Antioxidants 8, no. 5: 126. https://doi.org/10.3390/antiox8050126
APA StyleSzanto, I., Pusztaszeri, M., & Mavromati, M. (2019). H2O2 Metabolism in Normal Thyroid Cells and in Thyroid Tumorigenesis: Focus on NADPH Oxidases. Antioxidants, 8(5), 126. https://doi.org/10.3390/antiox8050126