Experimental and Computational Study of the Antioxidative Potential of Novel Nitro and Amino Substituted Benzimidazole/Benzothiazole-2-Carboxamides with Antiproliferative Activity

 , ,
, ,  , and
, and
Abstract
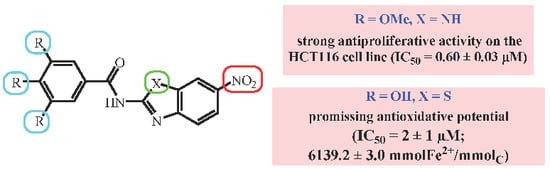
:
1. Introduction
2. Materials and Methods
2.1. Chemistry
2.1.1. General Methods
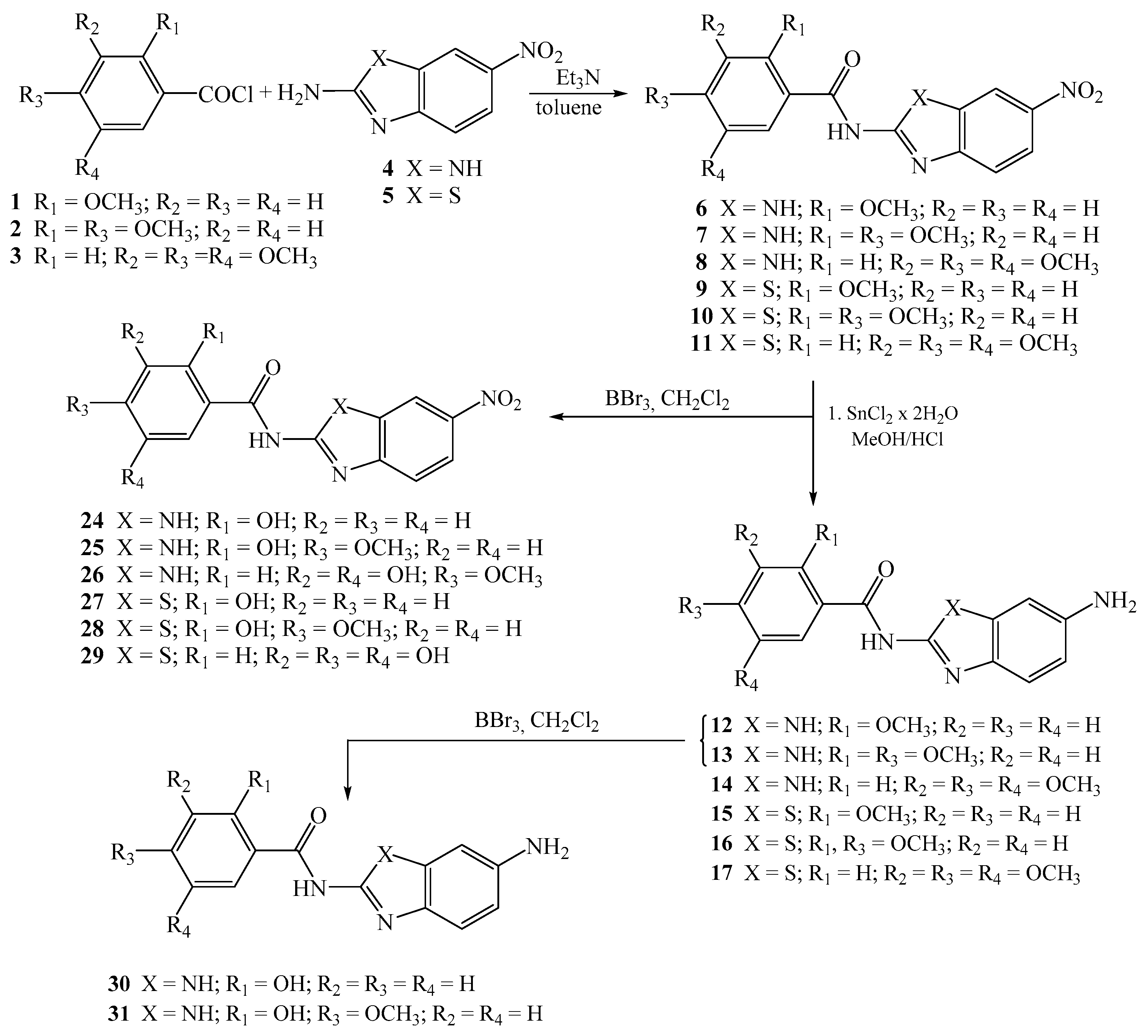
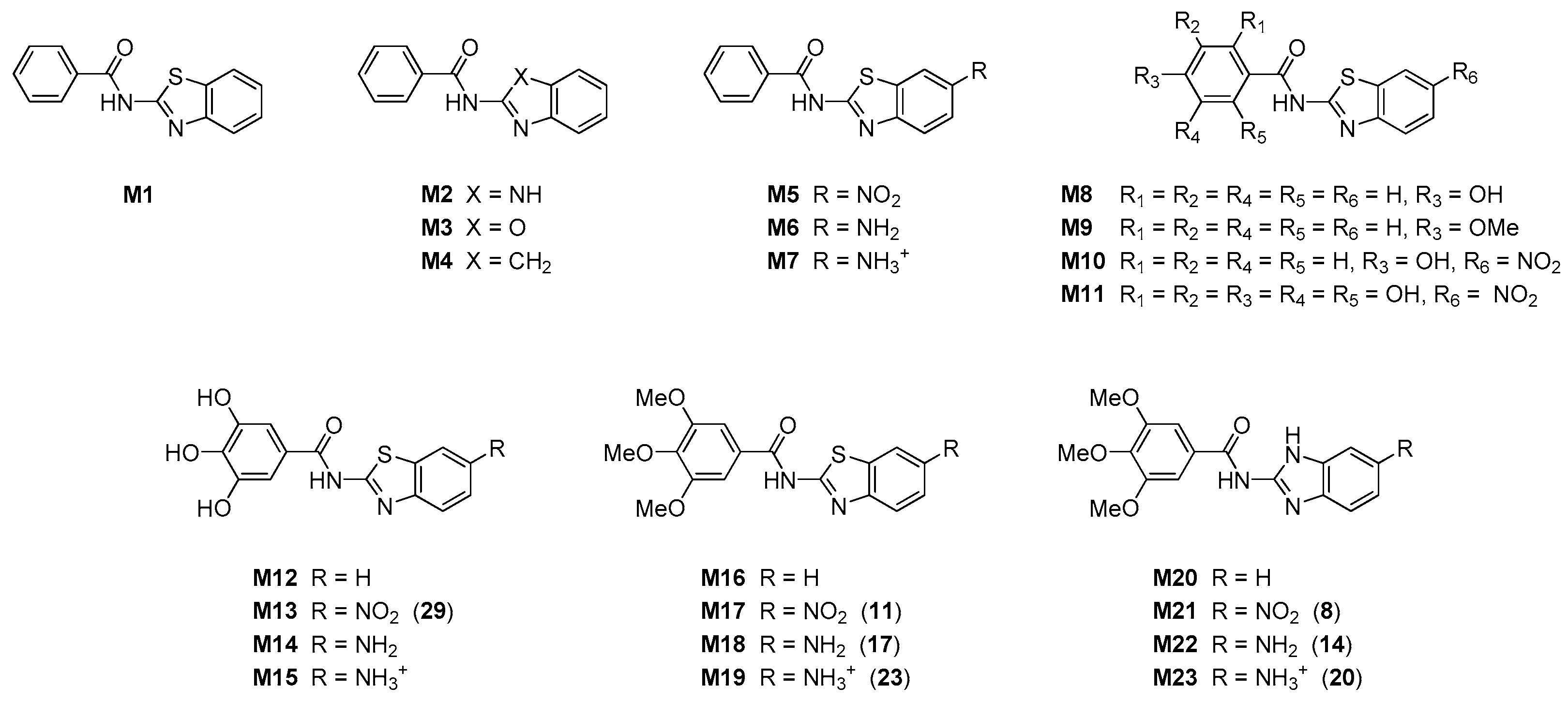
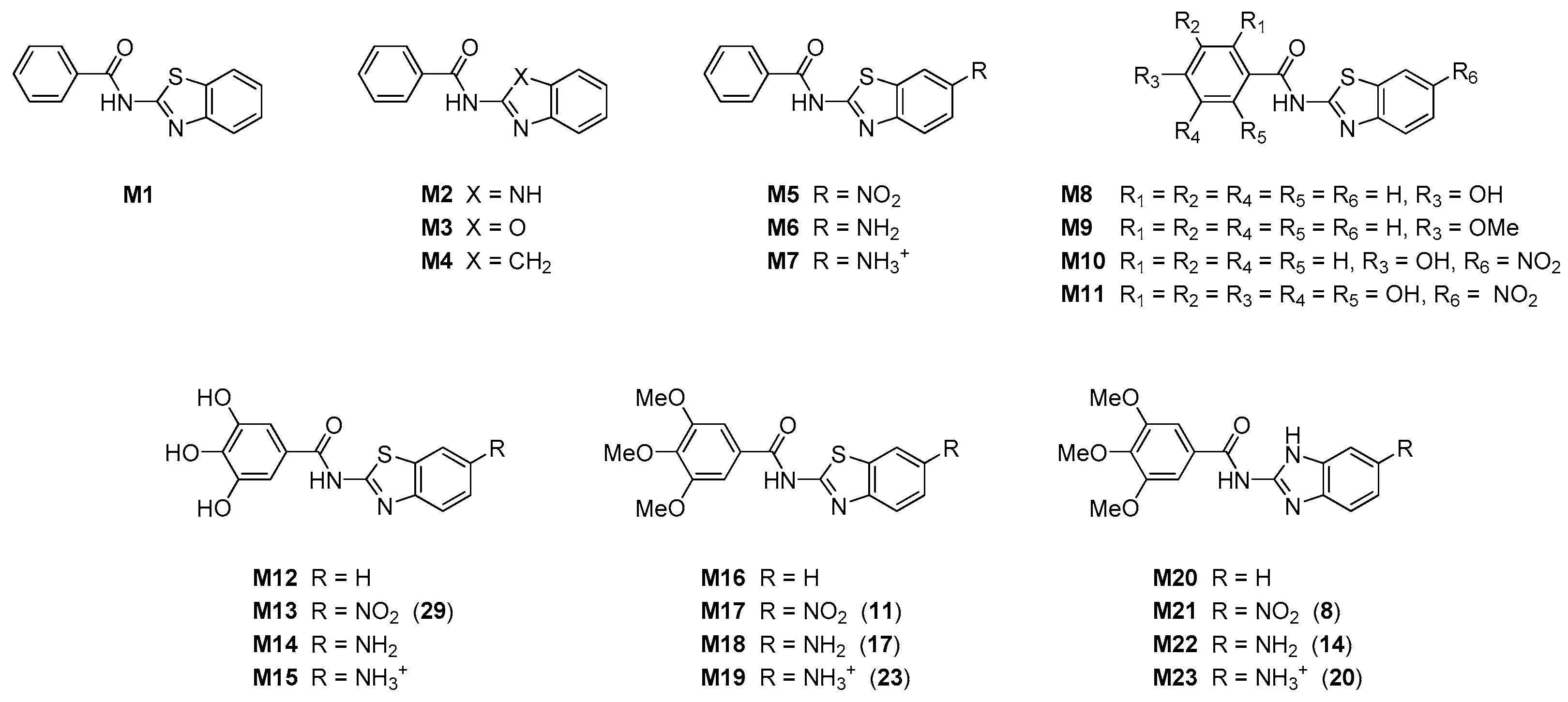
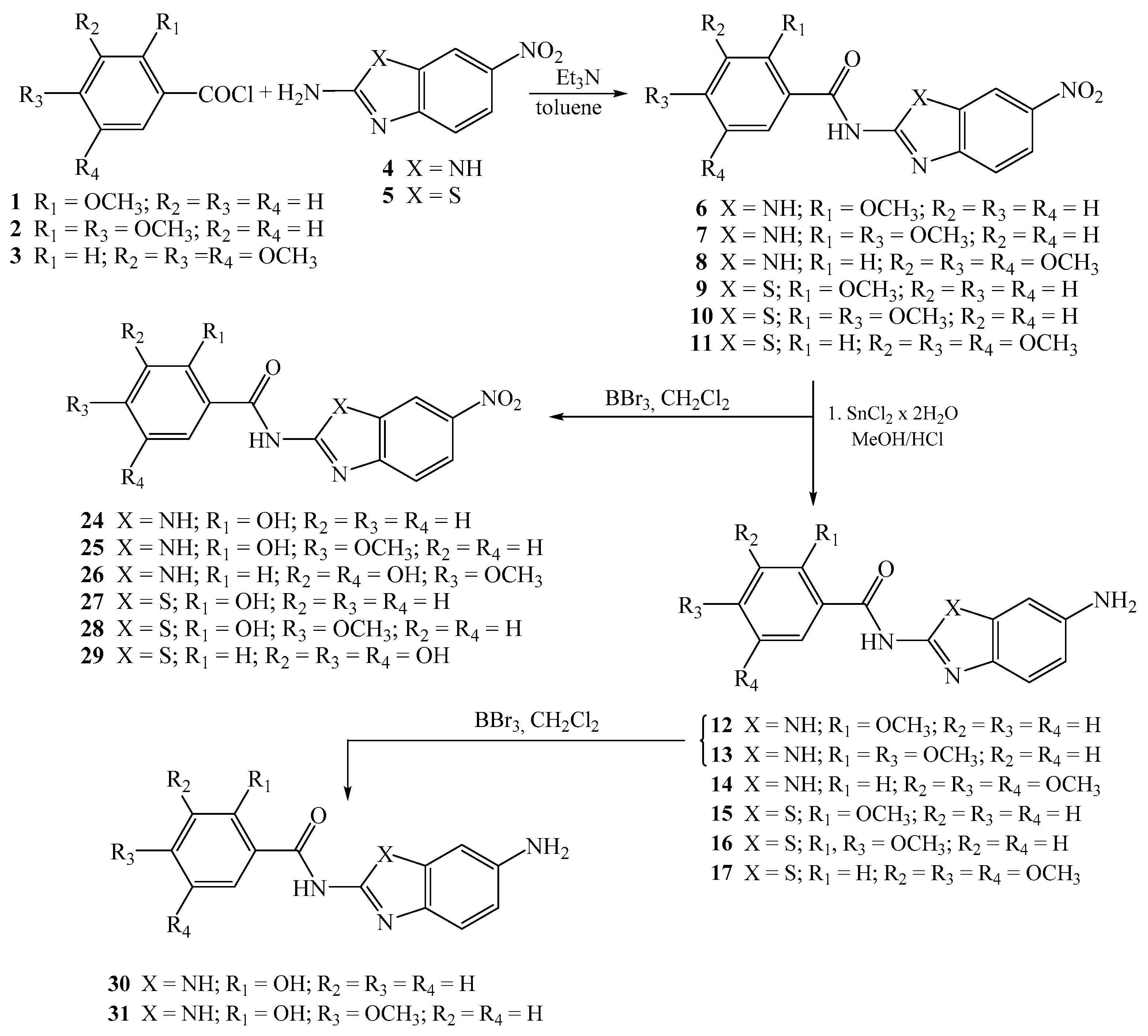
2.1.2. General Method for the Preparation of Methoxy Substituted Nitro Benzamides 6–11
2-methoxy-N-[5(6)-nitrobenzimidazol-2-yl]benzamide 6
2,4-dimethoxy-N-[5(6)-nitrobenzimidazol-2-yl]benzamide 7
3,4,5-trimethoxy-N-[5(6)-nitrobenzimidazol-2-yl]benzamide 8
2-methoxy-N-(6-nitrobenzothiazol-2-yl)benzamide 9
2,4-dimethoxy-N-(6-nitrobenzothiazol-2-yl)benzamide 10
3,4,5-trimethoxy-N-(6-nitrobenzothiazol-2-yl)benzamide 11
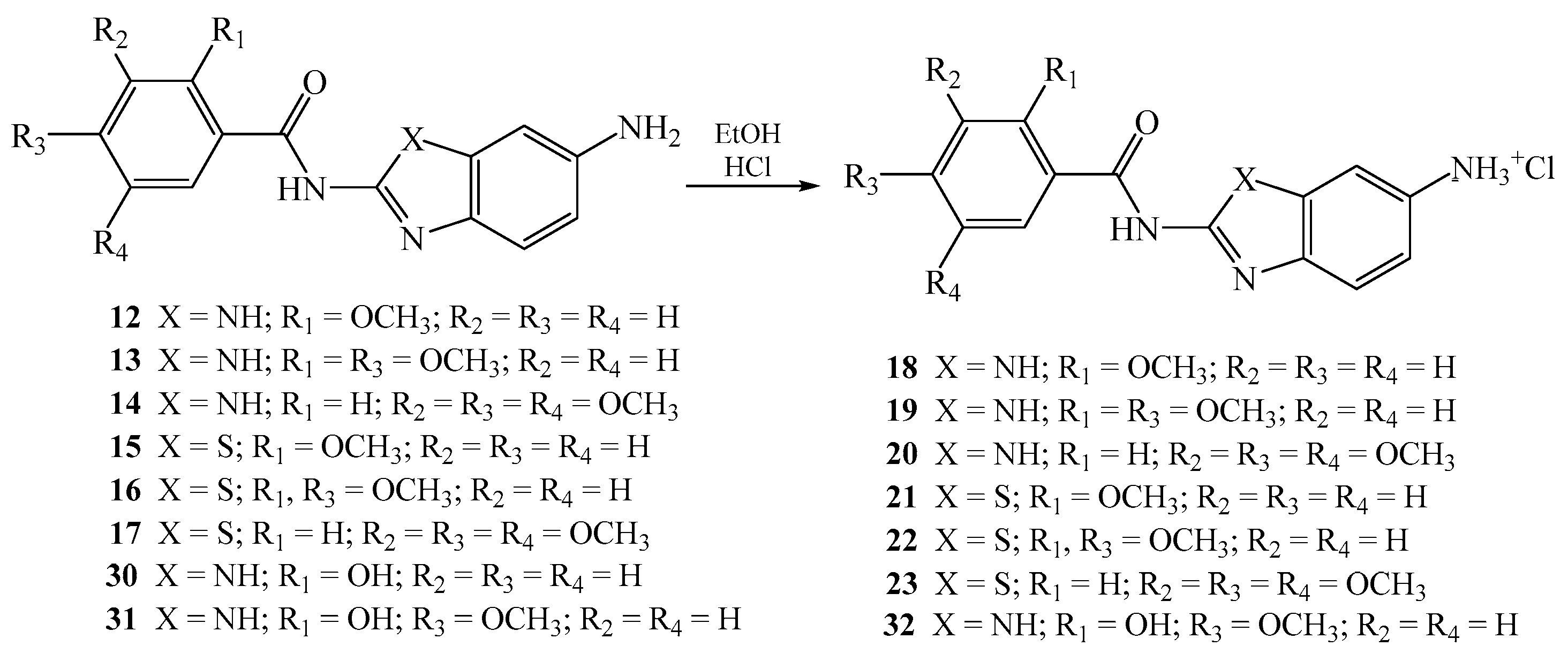
2.1.3. General Method for the Preparation of Methoxy Substituted Amino Benzamides 12–17
N-[5(6)-aminobenzimidazol-2-yl]-2-methoxybenzamide 12
N-[5(6)-aminobenzimidazol-2-yl]-2,4-dimethoxybenzamide 13
N-[5(6)-aminobenzimidazol-2-yl]-3,4,5-trimethoxybenzamide 14
N-(6-aminobenzothiazol-2-yl)-2-methoxybenzamide 15
N-(6-aminobenzothiazol-2-yl)-2,4-dimethoxybenzamide 16
N-(6-aminobenzothiazol-2-yl)-3,4,5-trimethoxybenzamide 17
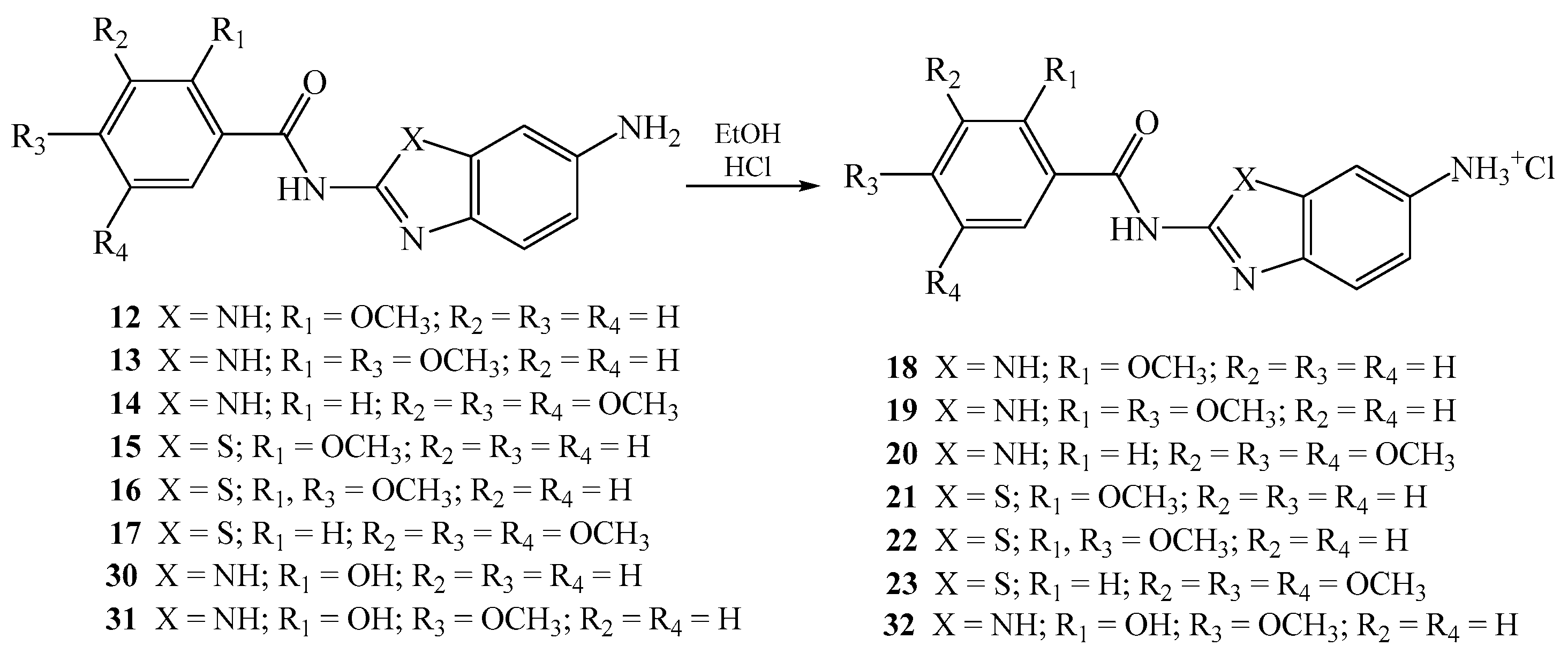
2.1.4. General Method for the Preparation of Amino Substituted Benzamides as Hydrochloride Salts 18–23 and 32
N-[5(6)-aminobenzimidazol-2-yl]-2-methoxybenzamide hydrochloride 18
N-[5(6)-aminobenzimidazol-2-yl]-2,4-dimethoxybenzamide hydrochloride 19
N-(6-aminobenzimidazol-2-yl)-3,4,5-trimethoxybenzamide hydrochloride 20
N-(6-aminobenzothiazol-2-yl)-2-methoxybenzamide hydrochloride 21
N-(6-aminobenzothiazol-2-yl)-2,4-dimethoxybenzamide hydrochloride 22
N-(6-aminobenzothiazol-2-yl)-3,4,5-trimethoxybenzamide hydrochloride 23
N-[5(6)-aminobenzimidazol-2-yl]-2-hydroxy-4-methoxybenzamide 32
2.1.5. General Method for the Preparation of Hydroxy Substituted Amino Benzamides 24–31
2-hydroxy-N-[5(6)-nitrobenzimidazol-2-yl]benzamide 24
2-hydroxy-4-methoxy-N-[5(6)-nitrobenzimidazol-2-yl]benzamide 25
3,5-dihydroxy-4-methoxy-N-[5(6)-nitrobenzimidazol-2-yl]benzamide 26
2-hydroxy-N-(6-nitrobenzothiazol-2-yl)benzamide 27
2-hydroxy-4-methoxy-N-(6-nitrobenzothiazol-2-yl)benzamide 28
3,4,5-trihydroxy-N-(6-nitrobenzothiazol-2-yl)benzamide 29
N-[5(6)-aminobenzimidazol-2-yl]-2-hydroxybenzamide 30
N-[5(6)-aminobenzimidazol-2-yl]-2-hydroxy-4-methoxybenzamide 31
2.2. Biological Activity
2.2.1. Antiproliferative Activity in Vitro
2.2.2. Antioxidative Activity
Determination of the Reducing Activity of the Stable Radical 1,1-diphenyl-picrylhydrazyl (DPPH)
Determination of Ferric Reducing/Antioxidant Power (FRAP assay)
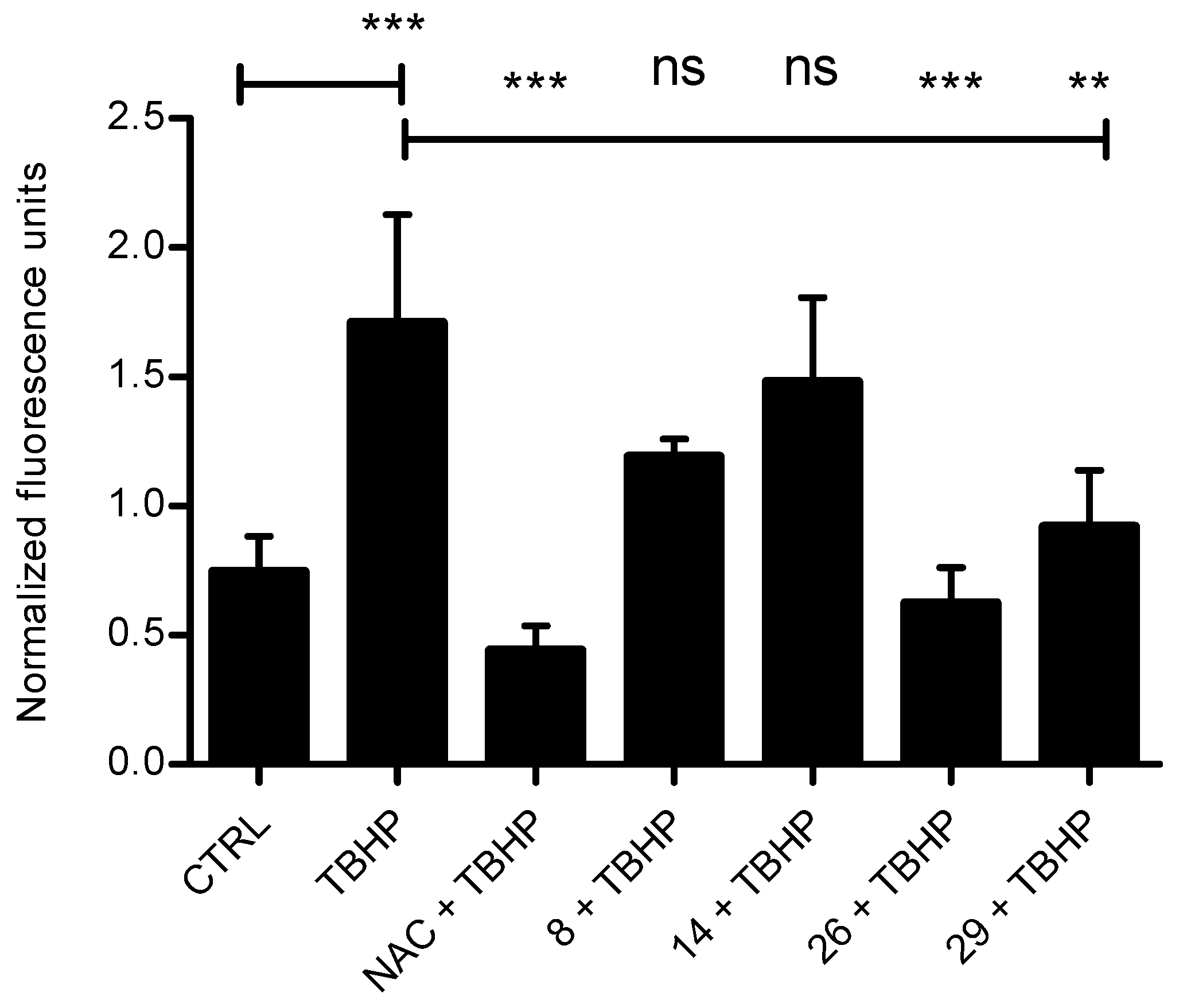
2.2.3. Antioxidative Activity Assay in Cells
2.3. Computational Details
3. Results and Discussion
3.1. Chemistry
3.2. Biological Evaluation
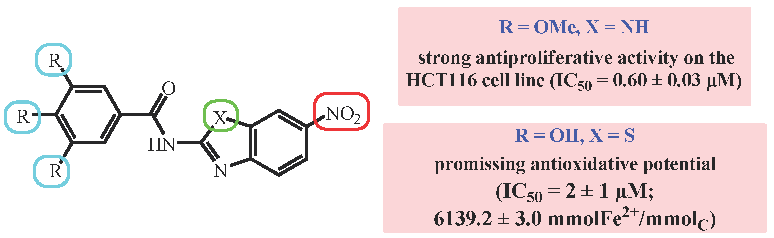
3.2.1. Antiproliferative Activity in Vitro
3.2.2. Antioxidative Capacity of Benzimidazole/Benzothiazole Derivatives
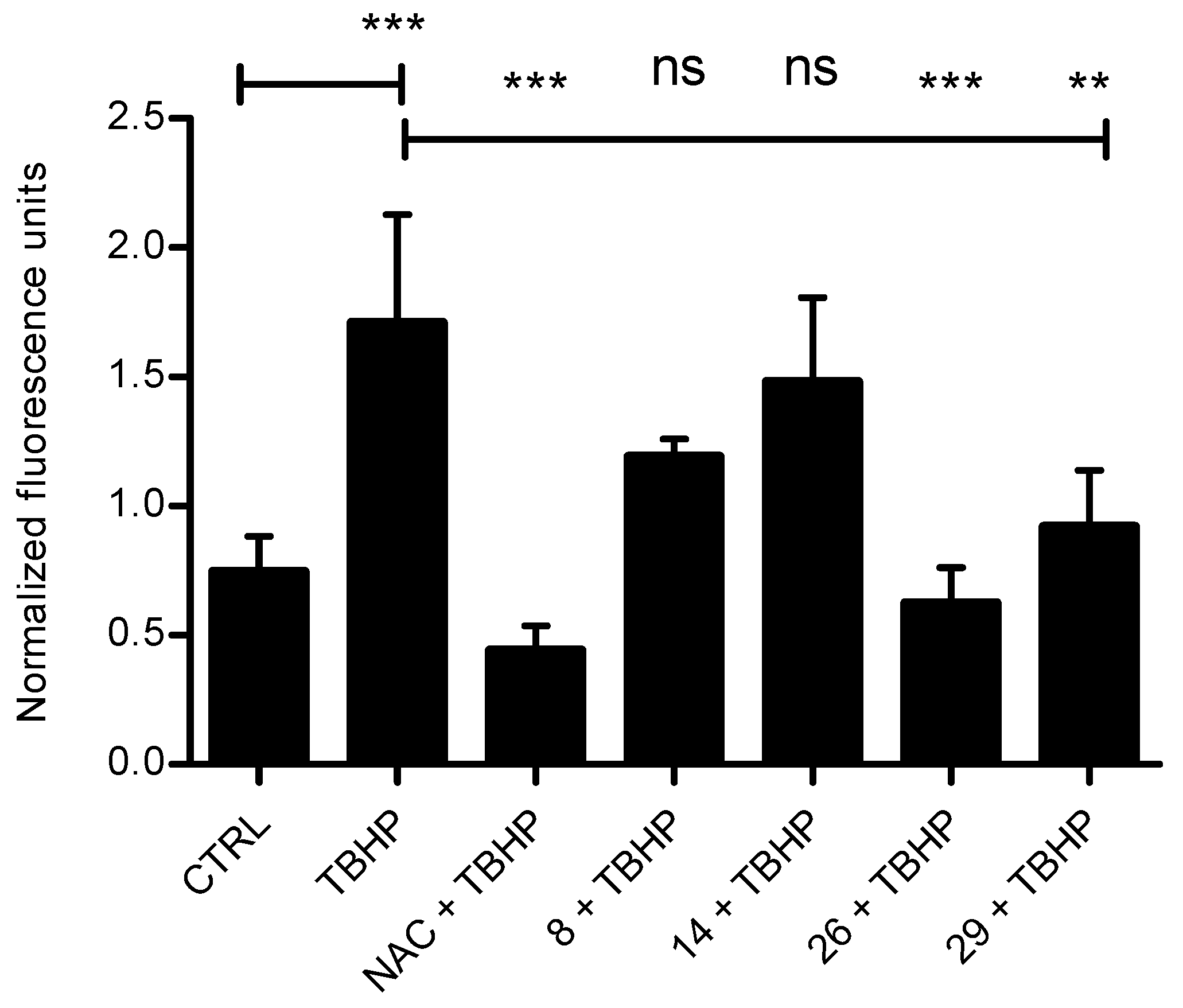
3.2.3. Antioxidant Ability in Cells
3.3. Computational Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bansal, Y.; Silakari, O. The therapeutic journey of benzimidazoles: A review. Bioorg. Med. Chem. 2012, 20, 6208–6236. [Google Scholar] [CrossRef]
- Sharma, P.C.; Sinhmar, A.; Sharma, A.; Rajak, H.; Dharam, P.P. Medicinal significance of benzothiazole scaffold: An insight view. J. Enzyme Inhib. Med. Chem. 2013, 28, 240–266. [Google Scholar] [CrossRef]
- Hranjec, M.; Sović, I.; Ratkaj, I.; Pavlović, G.; Ilić, N.; Valjalo, L.; Pavelić, K.; Kraljević Pavelić, S.; Karminski-Zamola, G. Antiproliferative potency of novel benzofuran-2-carboxamides on tumor cell lines: Cell death mechanisms and determination of crystal structure. Eur. J. Med. Chem. 2013, 59, 111–119. [Google Scholar] [CrossRef]
- Racané, L.; Sedić, M.; Ilić, N.; Aleksić, M.; Kraljević Pavelić, S.; Karminski-Zamola, G. Novel 2-Thienyl- and 2-Benzothienyl-Substituted 6-(2-Imidazolinyl) Benzothiazoles: Synthesis; in vitro Evaluation of Antitumor Effects and Assessment of Mitochondrial Toxicity. Anti-Cancer Ag. Med. Chem. 2017, 17, 57–66. [Google Scholar]
- Gaba, M.; Mohan, C. Development of drugs based on imidazole and benzimidazole bioactive heterocycles: Recent advances and future directions. Med. Chem. Res. 2016, 25, 173–210. [Google Scholar] [CrossRef]
- Rescifina, A.; Zagni, C.; Varrica, M.G.; Pistarà, V.; Corsaro, A. Recent advances in small organic molecules as DNA intercalating agents: Synthesis, activity, and modeling. Eur. J. Med. Chem. 2014, 74, 95–115. [Google Scholar] [CrossRef]
- Shah, K.; Chhabra, S.; Shrivastava, S.K.; Mishra, P. Benzimidazole: A promising pharmacophore. Med. Chem. Res. 2013, 22, 5077–5104. [Google Scholar] [CrossRef]
- Racane, L.; Stojković, R.; Tralić-Kulenović, V.; Cerić, H.; Đaković, M.; Ester, K.; Mišir Krpan, A.; Radić Stojković, M. Interactions with polynucleotides and antitumor activity of amidino and imidazolinyl substituted 2-phenylbenzothiazole mesylates. Eur. J. Med. Chem. 2014, 86, 406–419. [Google Scholar] [CrossRef]
- Racane, L.; Tralić-Kulenović, V.; Kraljević Pavelić, S.; Ratkaj, I.; Peixoto, P.; Nhili, R.; Depauw, S.; Hildebrand, M.P.; David-Cordonnier, M.H.; Pavelić, K.; et al. Novel Diamidino-Substituted Derivatives of Phenyl Benzothiazolyl and Dibenzothiazolyl Furans and Thiophenes: Synthesis, Antiproliferative and DNA Binding Properties. J. Med. Chem. 2010, 53, 2418–2432. [Google Scholar] [CrossRef]
- Hranjec, M.; Kralj, M.; Piantanida, I.; Sedić, M.; Šuman, L.; Pavelić, K.; Karminski-Zamola, G. Novel cyano- and amidino-substituted derivatives of styryl-2-benzimidazoles and benzimidazo[1,2-a]quinolines. Synthesis, photochemical synthesis, DNA binding and antitumor evaluation, Part 3. J. Med. Chem. 2007, 50, 5696–5711. [Google Scholar] [CrossRef]
- Klaunig, J.E.; Kamendulis, L.M.; Hocevar, B.A. Oxidative stress and oxidative damage in carcinogenesis. Toxicol. Pathol. 2010, 38, 96–109. [Google Scholar]
- Tailor, N.; Sharma, M. Antioxidant hybrid compounds: A promising therapeutic intervention in oxidative stress induced diseases. Mini Rev. Med. Chem. 2013, 13, 280–297. [Google Scholar]
- Lu, J.M.; Lin, P.H.; Yao, Q.; Chen, C. Chemical and molecular mechanisms of antioxidants: Experimental approaches and model systems. J. Cell. Mol. Med. 2010, 14, 840–860. [Google Scholar] [CrossRef]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Asp. Med. 2011, 32, 234–246. [Google Scholar] [CrossRef]
- Weisiger, R.A.; Fridovich, I. Superoxide dismutase. Organelle specificity. J. Biol. Chem. 1973, 248, 3582–3592. [Google Scholar]
- Pavlin, M.; Repič, M.; Vianello, R.; Mavri, J. The Chemistry of Neurodegeneration: Kinetic Data and Their Implications. Mol. Neurobiol. 2016, 53, 3400–3415. [Google Scholar] [CrossRef]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef]
- Lu, A.L.; Li, X.; Gu, Y.; Wright, P.M.; Chang, D.Y. Repair of oxidative DNA damage: Mechanisms and functions. Cell. Biochem. Biophys. 2001, 35, 141–170. [Google Scholar] [CrossRef]
- Zhou, B.; Li, B.; Yi, W.; Bu, X.; Ma, L. Synthesis, antioxidant, and antimicrobial evaluation of some 2-arylbenzimidazole derivatives. Bioorg. Med. Chem. Lett. 2013, 23, 3759–3763. [Google Scholar] [CrossRef]
- Likhar, R.; Perumal, P.; Kolhe, N.; Bhaskar, V.H.; Daroi, P. Synthesis and antioxidant activity novel 2-aryl are substituted benzothiazole derivatives. Int. J. Curr. Pharm. Res. 2015, 7, 34–37. [Google Scholar]
- Cindrić, M.; Sović, I.; Martin-Kleiner, I.; Kralj, M.; Mašek, T.; Hranjec, M.; Starčević, K. Synthesis, antioxidative and antiproliferative activity of methoxy amidino substituted benzamides and benzimidazoles. Med. Chem. Res. 2017, 26, 2024–2037. [Google Scholar] [CrossRef]
- Racané, L.; Cindrić, M.; Perin, N.; Roškarić, P.; Starčević, K.; Mašek, T.; Maurić, M.; Dogan, J.; Karminski-Zamola, G. Synthesis and Antioxidative Potency of Novel Amidino Substituted Benzimidazole and Benzothiazole Derivatives. Croat. Chem. Acta 2017, 90, 187–195. [Google Scholar] [CrossRef]
- Lauriane, N.T.R.; Bouali, J.; Najih, R.; Khouili, M.; Hafid, A.; Chtaini, A. Synthesis and electrochemical evaluation of some organic molecules as an antioxidant agents. Pharm. Anal. Acta 2014, 5, 1–5. [Google Scholar]
- Perin, N.; Roškarić, P.; Sović, I.; Boček, I.; Starčević, K.; Hranjec, M.; Vianello, R. Amino-Substituted Benzamide Derivatives as Promising Antioxidant Agents: A Combined Experimental and Computational Study. Chem. Res. Toxicol. 2018, 31, 974–984. [Google Scholar] [CrossRef]
- Foti, M.C. Use and abuse of the DPPH• radical. J. Agric. Food Chem. 2015, 62, 8765–8776. [Google Scholar] [CrossRef]
- Tireli, M.; Starčević, K.; Martinović, T.; Kraljević Pavelić, S.; Karminski-Zamola, G.; Hranjec, M. Antioxidative and antiproliferative activities of novel pyrido[1,2-a]benzimidazoles. Mol. Divers. 2017, 21, 201–210. [Google Scholar] [CrossRef]
- Uzelac, L.; Škalamera, Đ.; Mlinarić-Majerski, K.; Basarić, N.; Kralj, M. Selective photocytotoxicity of anthrols on cancer stem-like cells: The effect of quinone methides or reactive oxygen species. Eur. J. Med. Chem. 2017, 8, 558–574. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A. 03; Gaussian. Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Leopoldini, M.; Russo, N.; Toscano, M. The molecular basis of working mechanism of natural polyphenolic antioxidants. Food Chem. 2011, 125, 288–306. [Google Scholar] [CrossRef]
- Tandarić, T.; Vianello, R. Computational insight into the mechanism of the irreversible inhibition of monoamine oxidase enzymes by the anti-parkinsonian propargylamine inhibitors rasagiline and selegiline. ACS Chem. Neurosci. 2019, 10, 3532–3542. [Google Scholar] [CrossRef]
- Gregorić, T.; Sedić, M.; Grbčić, P.; Tomljenović Paravić, A.; Kraljević Pavelić, S.; Cetina, M.; Vianello, R.; Raić-Malić, S. Novel pyrimidine-2,4-dione-1,2,3-triazole and furo[2,3-d]pyrimidine-2-one-1,2,3-triazole hybrids as potential anti-cancer agents: Synthesis, computational and X-ray analysis and biological evaluation. Eur. J. Med. Chem. 2017, 125, 1247–1267. [Google Scholar] [CrossRef]
- Saftić, D.; Vianello, R.; Žinić, B. 5-Triazolyluracils and their N1-sulfonyl derivatives: Intriguing reactivity differences in the sulfonation of triazole N1′-substituted and N1′-unsubstituted uracil molecules. Eur. J. Org. Chem. 2015, 35, 7695–7704. [Google Scholar] [CrossRef]
- Huang, D.; Ou, B.; Prior, R.L. The chemistry behind antioxidant capacity assays. J. Agric. Food Chem. 2005, 53, 1841–1856. [Google Scholar] [CrossRef] [PubMed]
- Cindrić, M.; Jambon, S.; Harej, A.; Depauw, S.; David-Cordonnier, M.H.; Kraljević Pavelić, S.; Karminski-Zamola, G.; Hranjec, M. Novel amidino substituted benzimidazole and benzothiazole benzo[b]thieno-2-carboxamides exert strong antiproliferative and DNA binding properties. Eur. J. Med. Chem. 2017, 136, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Lautraite, S.; Bigot-Lasserre, D.; Bars, R.; Carmichael, N. Optimisation of cell-based assays for medium throughput screening of oxidative stress. Toxicol. In Vitro 2003, 17, 207–220. [Google Scholar] [CrossRef]
- Huyut, Z.; Beydemir, Ş.; Gülçin, İ. Antioxidant and Antiradical Properties of Selected Flavonoids and Phenolic Compounds. Biochem. Res. Int. 2017, 2017, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Marteau, C.; Nardello-Rataj, V.D.; Favier, D.; Aubry, J.M. Dual role of phenols as fragrances and antioxidants: Mechanism, kinetics and drastic solvent effect. Flavour Fragr. J. 2013, 28, 30–38. [Google Scholar] [CrossRef]
- Foti, M.C.C.; Daquino, C.; Mackie, I.D.; DiLabio, G.A.; Ingold, K.U. Reaction of phenols with the 2,2-diphenyl-1-picrylhydrazyl radical. Kinetics and DFT calculations applied to determine ArO-H bond dissociation enthalpies and reaction mechanism. J. Org. Chem. 2008, 73, 9270–9282. [Google Scholar] [CrossRef]
- Liu, W.Z.; Bordwell, F.G. Gas-phase and solution-phase homolytic bond dissociation energies of H−N+ bonds in the conjugate acids of nitrogen bases. J. Org. Chem. 1996, 61, 4778–4783. [Google Scholar] [CrossRef]
- Wright, J.S.; Johnson, E.R.; DiLabio, G.A. Predicting the activity of phenolic antioxidants: Theoretical method, analysis of substituent effects, and application to major families of antioxidants. J. Am. Chem. Soc. 2001, 123, 1173–1183. [Google Scholar] [CrossRef]
- Fox, T.; Kollman, P.A. Calculation of ionization potentials and C−H bond dissociation energies of toluene derivatives. J. Phys. Chem. 1996, 100, 2950–2956. [Google Scholar] [CrossRef]
- Wright, J.S.; Carpenter, D.J.; McKay, D.J.; Ingold, K.U.J. Theoretical calculation of substituent effects on the O−H bond strength of phenolic antioxidants related to vitamin E. J. Am. Chem. Soc. 1997, 119, 4245–4252. [Google Scholar] [CrossRef]
- DiLabio, G.A.; Pratt, D.A.; LoFaro, A.D.; Wright, J.S. Theoretical study of X−H bond energetics (X = C, N, O, S): Application to substituent effects, gas phase acidities, and redox potentials. J. Phys. Chem. A 1999, 103, 1653–1661. [Google Scholar] [CrossRef]
- Lucarini, M.; Pedulli, G.F. Free radical intermediates in the inhibition of the autoxidation reaction. Chem. Soc. Rev. 2010, 39, 2106–2119. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.J.; Tronic, T.A.; Mayer, J.M. Thermochemistry of proton-coupled electron transfer reagents and its implications. Chem. Rev. 2010, 110, 6961–7001. [Google Scholar] [CrossRef]
- Guitard, R.; Nardello-Rataj, V.; Aubry, J.M. Theoretical and kinetic tools for selecting effective antioxidants: Application to the protection of omega-3 oils with natural and synthetic phenols. Int. J. Mol. Sci. 2016, 17, 1220. [Google Scholar] [CrossRef] [PubMed]
- Ingold, K.U.; Pratt, D.A. Advances in radical-trapping antioxidant chemistry in the 21st century: A kinetics and mechanisms perspective. Chem. Rev. 2014, 114, 9022–9046. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, M.; Lind, J.; Merenyi, G.; Eriksen, T.E. N–H bond dissociation energies, reduction potentials and pKas of multisubstituted anilines and aniline radical cations. J. Chem. Soc. Perkin Trans. 2 1995, 1, 61–65. [Google Scholar] [CrossRef]
- Benzie, I.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef]
- Chen, Y.; Xiao, H.; Zheng, J.; Liang, G. Structure-thermodynamics-antioxidant activity relationships of selected natural phenolic acids and derivatives: An experimental and theoretical evaluation. PLoS ONE 2015, 10, e0121276. [Google Scholar] [CrossRef]
- Litwinienko, G.; Ingold, K.U. Solvent effects on the rates and mechanisms of reaction of phenols with free radicals. Acc. Chem. Res. 2007, 40, 222–230. [Google Scholar] [CrossRef]
- Lucarini, M.; Mugnaini, V.; Pedulli, G.F. Bond dissociation enthalpies of polyphenols: The importance of cooperative effects. J. Org. Chem. 2002, 67, 928–931. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd | R | R1 | R2 | R3 | R4 | X | IC50 (µM) | |||
|---|---|---|---|---|---|---|---|---|---|---|
| HCT116 | MCF–7 | H 460 | HEK 293 | |||||||
| 6 | NO2 | OMe | H | H | H | NH | 6 ± 1 | 4.0 ± 0.3 | 6 ± 5 | 3.0 ± 0.6 |
| 7 | NO2 | OMe | H | OMe | H | NH | 3.0 ± 0.2 | 2.0 ± 0.9 | 4 ± 2 | 2.0 ± 0.3 |
| 8 | NO2 | H | OMe | OMe | OMe | NH | 0.60 ± 0.03 | 1.0 ± 0.2 | 2.0 ± 0.2 | 2.0 ± 0.5 |
| 9 | NO2 | OMe | H | H | H | S | 44 ± 25 | 9 ± 1 | 46 ± 3 | n.t. b |
| 10 | NO2 | OMe | H | OMe | H | S | 9 ± 7 | 5 ± 2 | 20 ± 3 | 5.0 ± 1.0 |
| 11 | NO2 | H | OMe | OMe | OMe | S | 37.0 ± 0.8 | 17 ± 9 | 23 ± 4 | n.t. |
| 12 | NH2 | OMe | H | H | H | NH | 48 ± 4 | 53 ± 9 | 39 ± 11 | n.t. |
| 13 | NH2 | OMe | H | OMe | H | NH | 17 ± 8 | 20 ± 4 | 24 ± 8 | n.t. |
| 14 | NH2 | H | OMe | OMe | OMe | NH | 76 ± 24 | 43 ± 15 | >100 | 43.0 ± 2.0 |
| 15 | NH2 | OMe | H | H | H | S | 6 ± 4 | 3 ± 2 | 3.0 ± 0.9 | n.t. |
| 16 | NH2 | OMe | H | OMe | H | S | 6 ± 1 | 5.0 ± 0.3 | 43 ± 29 | 11.0 ± 8.0 |
| 17 | NH2 | H | OMe | OMe | OMe | S | 28 ± 4 | 5 ± 3 | ≥100 | n.t. |
| 18 | NH3+Cl− | OMe | H | H | H | NH | 33 ± 7 | 23 ± 2 | 36 ± 8 | n.t. |
| 19 | NH3+Cl− | OMe | H | OMe | H | NH | ≥100 | 34 ± 24 | >100 | n.t. |
| 20 | NH3+Cl− | H | OMe | OMe | OMe | NH | ≥100 | 34 ± 30 | >100 | n.t. |
| 21 | NH3+Cl− | OMe | H | H | H | S | 10 ± 8 | 3.0 ± 0.7 | 4.0 ± 0.1 | 25.0 ± 1.5 |
| 22 | NH3+Cl− | OMe | H | OMe | H | S | 8 ± 2 | 3.0 ± 0.2 | 50 ± 8 | 11.0 ± 1.0 |
| 23 | NH3+Cl− | H | OMe | OMe | OMe | S | 50 ± 11 | 12 ± 24 | 34 ± 9 | n.t. |
| 24 | NO2 | OH | H | H | H | NH | 61.0 ± 0.1 | 42 ± 7 | 42 ± 20 | n.t. |
| 25 | NO2 | OH | H | OMe | H | NH | 31 ± 4 | 28 ± 6 | 18.00 ± 0.01 | n.t. |
| 26 | NO2 | H | OH | OMe | OH | NH | 4.0 ± 0.1 | 3 ± 1 | 3.0 ± 0.5 | 3.0 ± 0.7 |
| 27 | NO2 | OH | H | H | H | S | 6 ± 1 | 2.0 ± 0.6 | 3.0 ± 0.2 | 3.0 ± 1.0 |
| 28 | NO2 | OH | H | OMe | H | S | 7.0 ± 0.4 | 10 ± 8 | 8 ± 1 | 4.0 ± 2.0 |
| 29 | NO2 | H | OH | OH | OH | S | 7 ± 1 | 4.0 ± 0.7 | 2 ± 1 | 6.0 ± 4.0 |
| 30 | NH2 | OH | H | H | H | NH | 46.0 ± 0.2 | 30.0 ± 0.8 | >100 | n.t. |
| 31 | NH2 | OH | H | OMe | H | NH | 37 ± 7 | ≥100 | 75 ± 27 | n.t. |
| 32 | NH3+Cl− | OH | H | OMe | H | NH | 50 ± 13 | 80 ± 4 | ≥100 | n.t. |
| Etoposide | 5 ± 2 | 1.0 ± 0.7 | 0.10 ± 0.04 | 1.6 ± 0.4 | ||||||
| Cpd | R | R1 | R2 | R3 | R4 | X | FRAP a mmolFe2+/mmolC | DPPH b IC50/μM |
|---|---|---|---|---|---|---|---|---|
| 6 | NO2 | OCH3 | H | H | H | NH | 123.5 ± 2.8 | – c |
| 7 | NO2 | OCH3 | H | OCH3 | H | NH | 139.6 ± 16.9 | – |
| 8 | NO2 | H | OCH3 | OCH3 | OCH3 | NH | 89.7 ± 6.2 | – |
| 9 | NO2 | OCH3 | H | H | H | S | 6.7 ± 0.6 | – |
| 10 | NO2 | OCH3 | H | OCH3 | H | S | – | – |
| 11 | NO2 | H | OCH3 | OCH3 | OCH3 | S | 155.5 ± 3.0 | – |
| 12 | NH2 | OCH3 | H | H | H | NH | 256.3 ± 2.8 | 28.90 ± 1.03 |
| 13 | NH2 | OCH3 | H | OCH3 | H | NH | 238.4 ± 2.1 | 17.71 ± 2.81 |
| 14 | NH2 | H | OCH3 | OCH3 | OCH3 | NH | 1102.5 ± 14.1 | 10.70 ± 0.23 |
| 15 | NH2 | OCH3 | H | H | H | S | 163.4 ± 4.7 | – |
| 16 | NH2 | OCH3 | H | OCH3 | H | S | 203.6 ± 5.8 | – |
| 17 | NH2 | H | OCH3 | OCH3 | OCH3 | S | 235.4 ± 2.8 | 40.4 ± 0.4 |
| 18 | NH3+Cl− | OCH3 | H | H | H | NH | 267.2 ± 5.0 | 9.850 ± 0.003 |
| 19 | NH3+Cl− | OCH3 | H | OCH3 | H | NH | 259.1 ± 0.6 | 13.3 ± 0.8 |
| 20 | NH3+Cl− | H | OCH3 | OCH3 | OCH3 | NH | 267.4 ± 1.4 | 8.06 ± 0.03 |
| 21 | NH3+Cl− | OCH3 | H | H | H | S | 214.3 ± 3.4 | 12.0 ± 1.1 |
| 22 | NH3+Cl− | OCH3 | H | OCH3 | H | S | 244.2 ± 3.0 | 4.00 ± 0.04 |
| 23 | NH3+Cl− | H | OCH3 | OCH3 | OCH3 | S | 241.3 ± 1.6 | 1.5 ± 0.5 |
| 24 | NO2 | OH | H | H | H | NH | 39.1 ± 2.0 | – |
| 25 | NO2 | OH | H | OCH3 | H | NH | 150.4 ± 3.4 | – |
| 26 | NO2 | H | OH | OCH3 | OH | NH | 244.9 ± 4.8 | 8.1 ± 1.9 |
| 27 | NO2 | OH | H | H | H | S | – | – |
| 28 | NO2 | OH | H | OCH3 | H | S | 39.8 ± 1.6 | – |
| 29 | NO2 | H | OH | OH | OH | S | 6139.2 ± 3.0 | 2.00 ± 0.15 |
| 30 | NH2 | OH | H | H | H | NH | 272.6 ± 7.1 | 42.5 ± 0.5 |
| 31 | NH2 | OH | H | OCH3 | H | NH | 258.4 ± 6.4 | 9.2 ± 0.6 |
| 32 | NH3+Cl− | OH | H | OCH3 | H | NH | 304.3 ± 6.4 | 29.5 ± 0.1 |
| BHT | 2089 ± 60 | 25 ± 4 | ||||||
| System | BDE | Site of the M–H Cleavage | IE |
|---|---|---|---|
| M1 | 82.7 | amide N–H | 174.3 |
| M2 | 81.0 | amide N–H | 169.3 |
| M3 | 79.4 | amide N–H | 177.3 |
| M4 | 72.6 | C3-indole C–H | 170.3 |
| M5 | 86.0 | amide N–H | 186.8 |
| M6 | 75.6 | amide N–H | 154.8 |
| M7 | 48.3 | benzothiazole N–H | 247.3 |
| M8 | 77.9 | C4-phenolic O–H | 171.8 |
| M9 | 82.2 | amide N–H | 170.3 |
| M10 | 79.0 | C4-phenolic O–H | 183.6 |
| M11 | 70.4 | C3-phenolic O–H | 172.6 |
| M12 | 64.3 | C4-phenolic O–H | 170.4 |
| M13 | 65.1 | C4-phenolic O–H | 181.2 |
| M14 | 63.8 | C4-phenolic O–H | 151.7 |
| M15 | 46.4 | benzothiazole N–H | 227.6 |
| M16 | 83.3 | amide N–H | 166.7 |
| M17 | 86.5 | amide N–H | 173.2 |
| M18 | 75.1 | amide N–H | 152.0 |
| M19 | 45.9 | benzothiazole N–H | 212.3 |
| M20 | 82.9 | amide N–H | 162.3 |
| M21 | 86.6 | amide N–H | 171.8 |
| M22 | 71.8 | amide N–H | 146.2 |
| M23 | 45.5 | benzene N–H | 213.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cindrić, M.; Sović, I.; Mioč, M.; Hok, L.; Boček, I.; Roškarić, P.; Butković, K.; Martin-Kleiner, I.; Starčević, K.; Vianello, R.; et al. Experimental and Computational Study of the Antioxidative Potential of Novel Nitro and Amino Substituted Benzimidazole/Benzothiazole-2-Carboxamides with Antiproliferative Activity. Antioxidants 2019, 8, 477. https://doi.org/10.3390/antiox8100477
Cindrić M, Sović I, Mioč M, Hok L, Boček I, Roškarić P, Butković K, Martin-Kleiner I, Starčević K, Vianello R, et al. Experimental and Computational Study of the Antioxidative Potential of Novel Nitro and Amino Substituted Benzimidazole/Benzothiazole-2-Carboxamides with Antiproliferative Activity. Antioxidants. 2019; 8(10):477. https://doi.org/10.3390/antiox8100477
Chicago/Turabian StyleCindrić, Maja, Irena Sović, Marija Mioč, Lucija Hok, Ida Boček, Petra Roškarić, Kristina Butković, Irena Martin-Kleiner, Kristina Starčević, Robert Vianello, and et al. 2019. "Experimental and Computational Study of the Antioxidative Potential of Novel Nitro and Amino Substituted Benzimidazole/Benzothiazole-2-Carboxamides with Antiproliferative Activity" Antioxidants 8, no. 10: 477. https://doi.org/10.3390/antiox8100477
APA StyleCindrić, M., Sović, I., Mioč, M., Hok, L., Boček, I., Roškarić, P., Butković, K., Martin-Kleiner, I., Starčević, K., Vianello, R., Kralj, M., & Hranjec, M. (2019). Experimental and Computational Study of the Antioxidative Potential of Novel Nitro and Amino Substituted Benzimidazole/Benzothiazole-2-Carboxamides with Antiproliferative Activity. Antioxidants, 8(10), 477. https://doi.org/10.3390/antiox8100477