Targeting Oxidative Stress and Mitochondrial Dysfunction in the Treatment of Impaired Wound Healing: A Systematic Review

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Normal Wound Healing and Redox Regulation
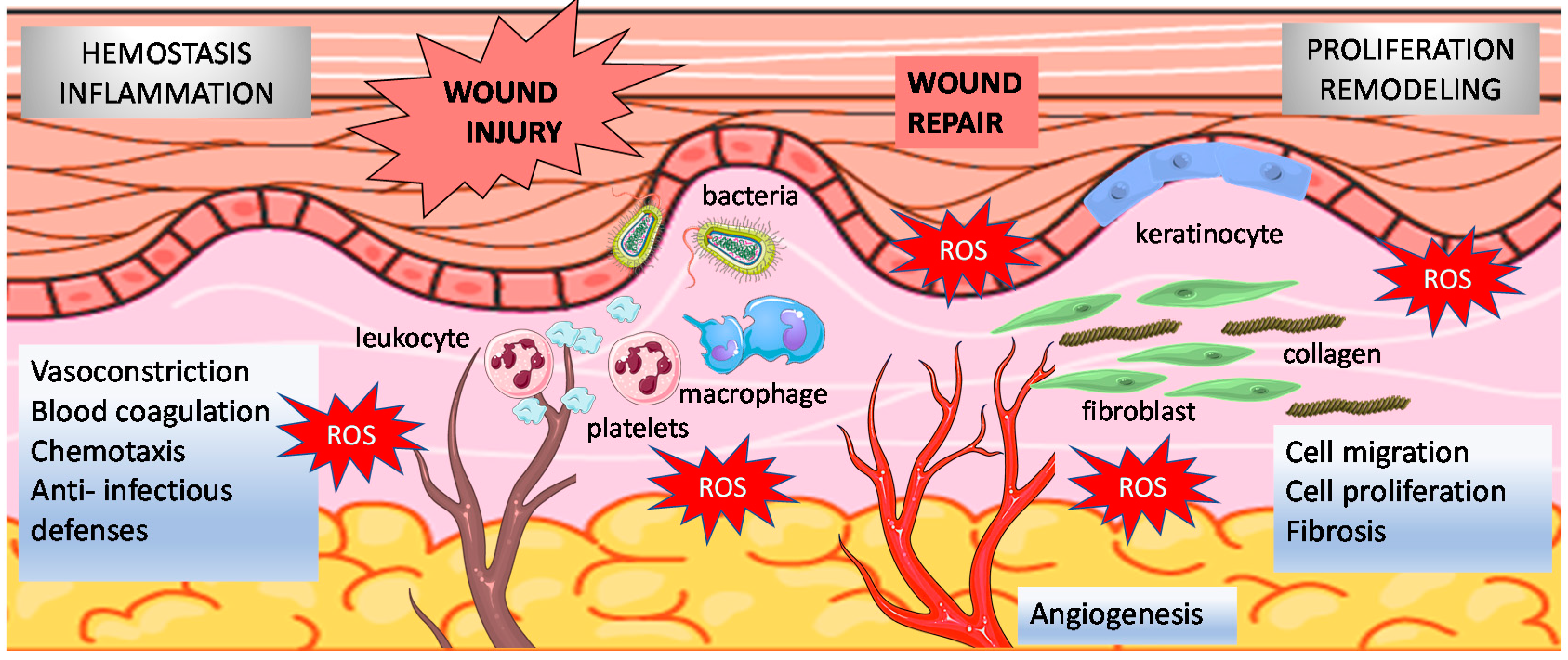
2.1. Wound Healing Phases
2.2. Redox Regulation of Normal Wound Healing
3. Definition of Chronic Wound
4. Oxidative Stress in Chronic Wound
4.1. Hypoxic Wound
4.2. Diabetic Chronic Wound
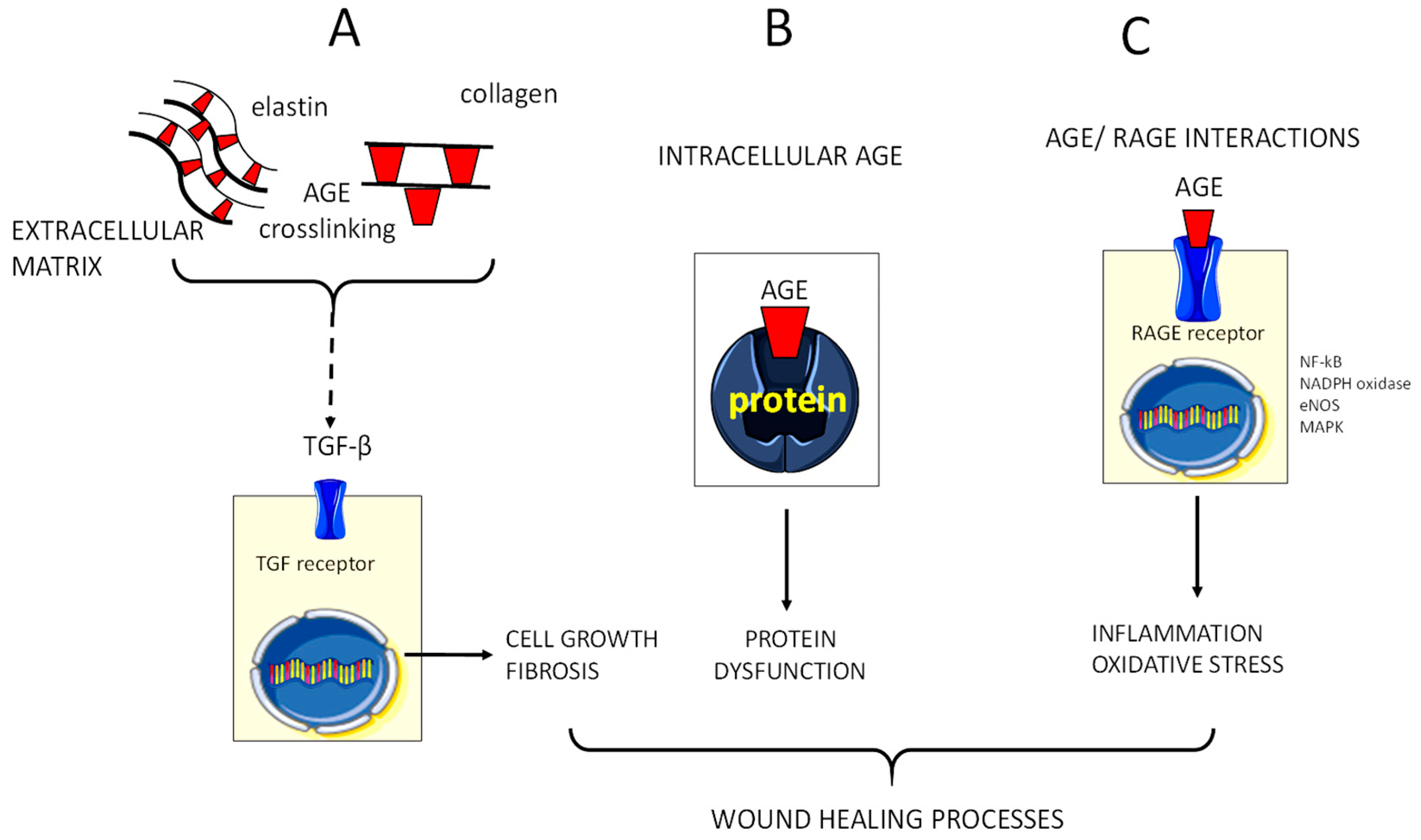
5. Glycoxidation and AGE Formation in the Diabetic Chronic Wound
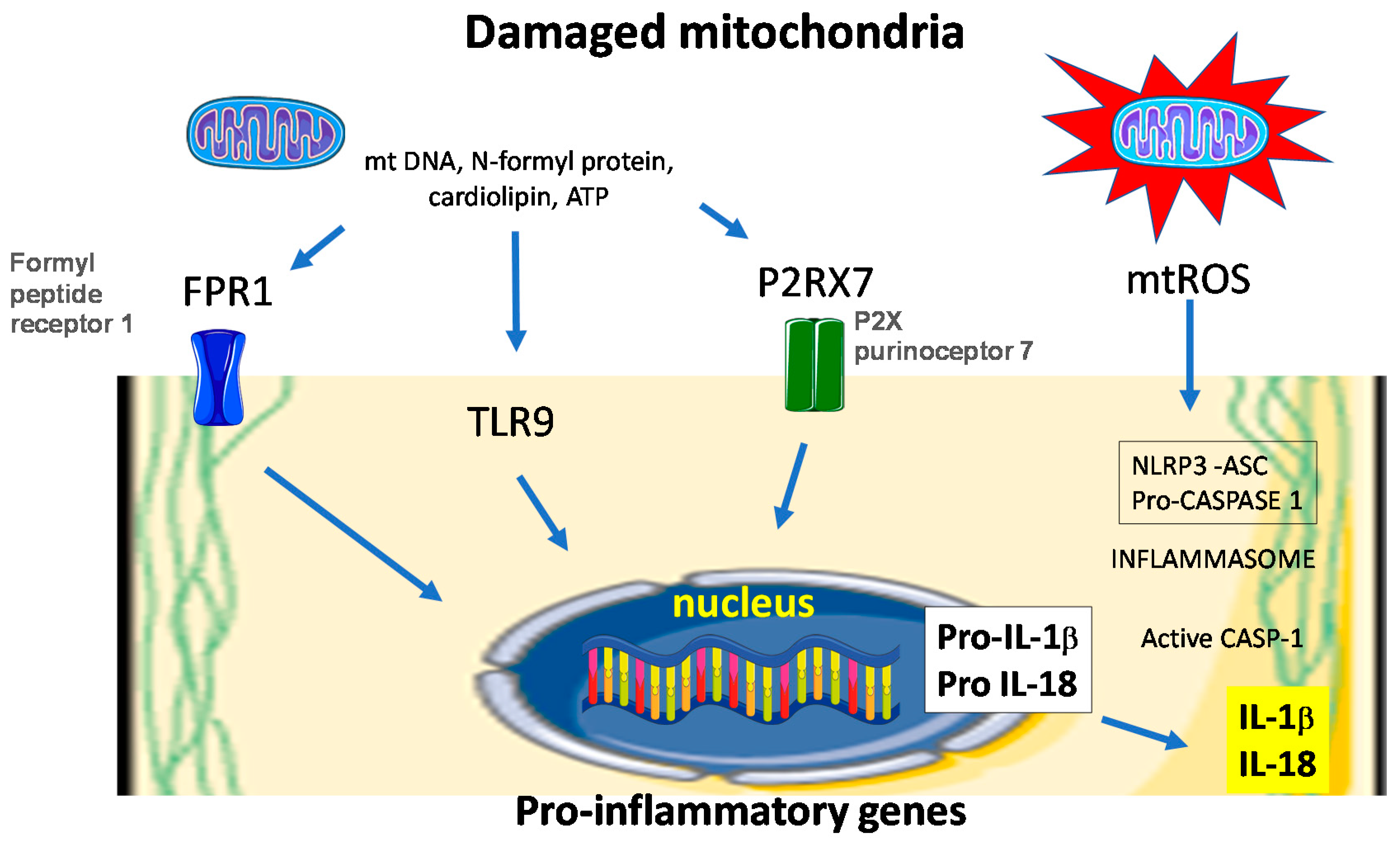
6. Redox Balance Modules Inflammation in the Healing Wound
7. Targeting Oxidative Stress in the Treatment of Chronic Wound
8. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Janis, J.E.; Harrison, B. Wound healing: Part I. Basic science. Plast. Reconstr. Surg. 2016, 138, 9S–17S. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Dipietro, L.A. Factors affecting wound healing. J. Dent. Res. 2010, 89, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Roy, S. Redox signals in wound healing. Biochim. Biophys. Acta 2008, 1780, 1348–1361. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.; Werner, S. Oxidative stress in normal and impaired wound repair. Pharmacol. Res. 2008, 58, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Dunnill, C.; Patton, T.; Brennan, J.; Barrett, J.; Dryden, M.; Cooke, J.; Leaper, D.; Georgopoulos, N.T. Reactive oxygen species (ROS) and wound healing: The functional role of ROS and emerging ROS-modulating technologies for augmentation of the healing process. Int. Wound J. 2017, 14, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K. Wound healing essentials: Let there be oxygen. Wound Repair Regen. 2009, 17, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Bryan, N.; Ahswin, H.; Smart, N.; Bayon, Y.; Wohlert, S.; Hunt, J.A. Reactive oxygen species (ROS)—A family of fate deciding molecules pivotal in constructive inflammation and wound healing. Eur. Cells Mater. 2012, 24, 249–265. [Google Scholar] [CrossRef]
- Fitzmaurice, S.D.; Sivamani, R.K.; Isseroff, R.R. Antioxidant therapies for wound healing: A clinical guide to currently commercially available products. Skin Pharmacol. Physiol. 2011, 24, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Kunkemoeller, B.; Kyriakides, T.R. Redox signaling in diabetic wound healing regulates extracellular matrix deposition. Antioxid. Redox Signal. 2017, 27, 823–838. [Google Scholar] [CrossRef] [PubMed]
- Golebiewska, E.M.; Poole, A.W. Platelet secretion: From haemostasis to wound healing and beyond. Blood Rev. 2015, 29, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Berna-Erro, A.; Redondo, P.C.; Lopez, E.; Albarran, L.; Rosado, J.A. Molecular interplay between platelets and the vascular wall in thrombosis and hemostasis. Curr. Vasc. Pharmacol. 2013, 11, 409–430. [Google Scholar] [CrossRef] [PubMed]
- Kaltalioglu, K.; Coskun-Cevher, S. A bioactive molecule in a complex wound healing process: Platelet-derived growth factor. Int. J. Dermatol. 2015, 54, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Demidova-Rice, T.N.; Hamblin, M.R.; Herman, I.M. Acute and impaired wound healing: Pathophysiology and current methods for drug delivery, part 2: Role of growth factors in normal and pathological wound healing: Therapeutic potential and methods of delivery. Adv. Skin Wound Care 2012, 25, 349–370. [Google Scholar] [CrossRef] [PubMed]
- Nurden, A.T. The biology of the platelet with special reference to inflammation, wound healing and immunity. Front. Biosci. (Landmark Ed.) 2018, 23, 726–751. [Google Scholar] [CrossRef] [PubMed]
- Portou, M.J.; Baker, D.; Abraham, D.; Tsui, J. The innate immune system, toll-like receptors and dermal wound healing: A review. Vasc. Pharmacol. 2015, 71, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Kasuya, A.; Tokura, Y. Attempts to accelerate wound healing. J. Dermatol. Sci. 2014, 76, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Landen, N.X.; Li, D.; Stahle, M. Transition from inflammation to proliferation: A critical step during wound healing. Cell. Mol. Life Sci. 2016, 73, 3861–3885. [Google Scholar] [CrossRef] [PubMed]
- Tracy, L.E.; Minasian, R.A.; Caterson, E.J. Extracellular matrix and dermal fibroblast function in the healing wound. Adv. Wound Care (New Rochelle) 2016, 5, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, P. Wound healing and the role of fibroblasts. J. Wound Care 2013, 22, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Wang, Q.; Lu, S.; Niu, Y. Hydrogen peroxide: A potential wound therapeutic target? Med. Princ. Pract. 2017, 26, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Andre-Levigne, D.; Modarressi, A.; Pepper, M.S.; Pittet-Cuenod, B. Reactive oxygen species and nox enzymes are emerging as key players in cutaneous wound repair. Int. J. Mol. Sci. 2017, 18, 2149. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.H.; Griffiths, H.R. The dual role of ROS in autoimmune and inflammatory diseases: Evidence from preclinical models. Free Radic. Biol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Zhang, Y.; Dusting, G.J. Nadph oxidase-mediated redox signaling: Roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev. 2011, 63, 218–242. [Google Scholar] [CrossRef] [PubMed]
- Levigne, D.; Modarressi, A.; Krause, K.H.; Pittet-Cuenod, B. Nadph oxidase 4 deficiency leads to impaired wound repair and reduced dityrosine-crosslinking, but does not affect myofibroblast formation. Free Radic. Biol. Med. 2016, 96, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Hesketh, M.; Sahin, K.B.; West, Z.E.; Murray, R.Z. Macrophage phenotypes regulate scar formation and chronic wound healing. Int. J. Mol. Sci. 2017, 18, 1545. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Maheshwari, A.; Chandra, A. Biomarkers for wound healing and their evaluation. J. Wound Care 2016, 25, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.A.; Waypa, G.B.; Schumacker, P.T. Redox signaling during hypoxia in mammalian cells. Redox Biol. 2017, 13, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Waypa, G.B.; Smith, K.A.; Schumacker, P.T. O2 sensing, mitochondria and ROS signaling: The fog is lifting. Mol. Asp. Med. 2016, 47–48, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, D.C.; Brune, B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. 2017, 12, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Wautier, M.P.; Guillausseau, P.J.; Wautier, J.L. Activation of the receptor for advanced glycation end products and consequences on health. Diabetes Metab. Syndr. 2017, 11, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.S.; Brownlee, M. Molecular and cellular mechanisms of cardiovascular disorders in diabetes. Circ. Res. 2016, 118, 1808–1829. [Google Scholar] [CrossRef] [PubMed]
- Schramm, A.; Matusik, P.; Osmenda, G.; Guzik, T.J. Targeting nadph oxidases in vascular pharmacology. Vasc. Pharmacol. 2012, 56, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Kurosaka, M.; Suzuki, T.; Hosono, K.; Kamata, Y.; Fukamizu, A.; Kitasato, H.; Fujita, Y.; Majima, M. Reduced angiogenesis and delay in wound healing in angiotensin ii type 1a receptor-deficient mice. Biomed. Pharmacother. 2009, 63, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.L.; Stupar, D.; Croll, T.; Leavesley, D.; Upton, Z. Xanthine oxidoreductase: A novel therapeutic target for the treatment of chronic wounds? Adv. Wound Care (New Rochelle) 2018, 7, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive oxygen species in metabolic and inflammatory signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef] [PubMed]
- Zinkevich, N.S.; Gutterman, D.D. Ros-induced ROS release in vascular biology: Redox-redox signaling. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H647–H653. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, M.; Vats, P. Reactive metabolites and antioxidant gene polymorphisms in type 2 diabetes mellitus. Redox Biol. 2014, 2, 170–177. [Google Scholar] [CrossRef] [PubMed]
- David, J.A.; Rifkin, W.J.; Rabbani, P.S.; Ceradini, D.J. The Nrf2/Keap1/ARE pathway and oxidative stress as a therapeutic target in type ii diabetes mellitus. J. Diabetes Res. 2017, 2017, 4826724. [Google Scholar] [CrossRef] [PubMed]
- Bitar, M.S.; Al-Mulla, F. A defect in Nrf2 signaling constitutes a mechanism for cellular stress hypersensitivity in a genetic rat model of type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E1119–H1129. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.A.; Cohen, O.D.; Low, Y.C.; Sartor, R.A.; Ellison, T.; Anil, U.; Anzai, L.; Chang, J.B.; Saadeh, P.B.; Rabbani, P.S.; et al. Restoration of Nrf2 signaling normalizes the regenerative niche. Diabetes 2016, 65, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Ambrozova, N.; Ulrichova, J.; Galandakova, A. Models for the study of skin wound healing. The role of Nrf2 and NF-kappaB. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2017, 161, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; He, R.; Zhao, J.; Mei, J.C.; Shao, M.Z.; Pan, Y.; Zhang, J.; Wu, H.S.; Yu, M.; Yan, W.C.; et al. Negative pressure wound therapy inhibits inflammation and upregulates activating transcription factor-3 and downregulates nuclear factor-kappab in diabetic patients with foot ulcerations. Diabetes Metab. Res. Rev. 2017, 33. [Google Scholar] [CrossRef] [PubMed]
- Vistoli, G.; De Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced glycoxidation and lipoxidation end products (ages and ales): An overview of their mechanisms of formation. Free Radic. Res. 2013, 47 (Suppl. 1), 3–27. [Google Scholar] [CrossRef] [PubMed]
- Frimat, M.; Daroux, M.; Litke, R.; Neviere, R.; Tessier, F.J.; Boulanger, E. Kidney, heart and brain: Three organs targeted by ageing and glycation. Clin. Sci. (Lond.) 2017, 131, 1069–1092. [Google Scholar] [CrossRef] [PubMed]
- Neviere, R.; Yu, Y.; Wang, L.; Tessier, F.; Boulanger, E. Implication of advanced glycation end products (AGES) and their receptor (RAGE) on myocardial contractile and mitochondrial functions. Glycoconj. J. 2016, 33, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Van Putte, L.; De Schrijver, S.; Moortgat, P. The effects of advanced glycation end products (ages) on dermal wound healing and scar formation: A systematic review. Scars Burn Heal. 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Huijberts, M.S.; Schaper, N.C.; Schalkwijk, C.G. Advanced glycation end products and diabetic foot disease. Diabetes Metab. Res. Rev. 2008, 24 (Suppl. 1), S19–S24. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.I.; Nakamura, N.; Matsui, T. Glycation and cardiovascular disease in diabetes: A perspective on the concept of metabolic memory. J. Diabetes 2017, 9, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Peppa, M.; Stavroulakis, P.; Raptis, S.A. Advanced glycoxidation products and impaired diabetic wound healing. Wound Repair Regen. 2009, 17, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Koulis, C.; Watson, A.M.; Gray, S.P.; Jandeleit-Dahm, K.A. Linking rage and nox in diabetic micro- and macrovascular complications. Diabetes Metab. 2015, 41, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Xie, T.; Ge, K.; Lin, Y.; Lu, S. Effects of extracellular matrix glycosylation on proliferation and apoptosis of human dermal fibroblasts via the receptor for advanced glycosylated end products. Am. J. Dermatopathol. 2008, 30, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Peppa, M.; Brem, H.; Ehrlich, P.; Zhang, J.G.; Cai, W.; Li, Z.; Croitoru, A.; Thung, S.; Vlassara, H. Adverse effects of dietary glycotoxins on wound healing in genetically diabetic mice. Diabetes 2003, 52, 2805–2813. [Google Scholar] [CrossRef] [PubMed]
- Goova, M.T.; Li, J.; Kislinger, T.; Qu, W.; Lu, Y.; Bucciarelli, L.G.; Nowygrod, S.; Wolf, B.M.; Caliste, X.; Yan, S.F.; et al. Blockade of receptor for advanced glycation end-products restores effective wound healing in diabetic mice. Am. J. Pathol. 2001, 159, 513–525. [Google Scholar] [CrossRef]
- Wear-Maggitti, K.; Lee, J.; Conejero, A.; Schmidt, A.M.; Grant, R.; Breitbart, A. Use of topical srage in diabetic wounds increases neovascularization and granulation tissue formation. Ann. Plast. Surg. 2004, 52, 519–521. [Google Scholar] [CrossRef] [PubMed]
- Bigot, N.; Beauchef, G.; Hervieu, M.; Oddos, T.; Demoor, M.; Boumediene, K.; Galera, P. Nf-kappab accumulation associated with col1a1 transactivators defects during chronological aging represses type I collagen expression through a -112/-61-bp region of the col1a1 promoter in human skin fibroblasts. J. Investig. Dermatol. 2012, 132, 2360–2367. [Google Scholar] [CrossRef] [PubMed]
- Artlett, C.M. Inflammasomes in wound healing and fibrosis. J. Pathol. 2013, 229, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Kanbe, A.; Sakai, H.; Seishima, M. Activation of NLRP3 signalling accelerates skin wound healing. Exp. Dermatol. 2018, 27, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Weinheimer-Haus, E.M.; Mirza, R.E.; Koh, T.J. Nod-like receptor protein-3 inflammasome plays an important role during early stages of wound healing. PLoS ONE 2015, 10, e0119106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dai, J.; Li, L.; Chen, H.; Chai, Y. Nlrp3 inflammasome expression and signaling in human diabetic wounds and in high glucose induced macrophages. J. Diabetes Res. 2017, 2017, 5281358. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Laverny, G.; Bernardi, L.; Charles, A.L.; Alsaleh, G.; Pottecher, J.; Sibilia, J.; Geny, B. Mitochondria: An organelle of bacterial origin controlling inflammation. Front. Immunol. 2018, 9, 536. [Google Scholar] [CrossRef] [PubMed]
- Demyanenko, I.A.; Zakharova, V.V.; Ilyinskaya, O.P.; Vasilieva, T.V.; Fedorov, A.V.; Manskikh, V.N.; Zinovkin, R.A.; Pletjushkina, O.Y.; Chernyak, B.V.; Skulachev, V.P.; et al. Mitochondria-targeted antioxidant SkQ1 improves dermal wound healing in genetically diabetic mice. Oxid. Med. Cell. Longev. 2017, 2017, 6408278. [Google Scholar] [CrossRef] [PubMed]
- Dashdorj, A.; Jyothi, K.R.; Lim, S.; Jo, A.; Nguyen, M.N.; Ha, J.; Yoon, K.S.; Kim, H.J.; Park, J.H.; Murphy, M.P.; et al. Mitochondria-targeted antioxidant mitoq ameliorates experimental mouse colitis by suppressing nlrp3 inflammasome-mediated inflammatory cytokines. BMC Med. 2013, 11, 178. [Google Scholar] [CrossRef] [PubMed]
- Jabaut, J.; Ather, J.L.; Taracanova, A.; Poynter, M.E.; Ckless, K. Mitochondria-targeted drugs enhance Nlrp3 inflammasome-dependent IL-1beta secretion in association with alterations in cellular redox and energy status. Free Radic. Biol. Med. 2013, 60, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Zielins, E.R.; Brett, E.A.; Luan, A.; Hu, M.S.; Walmsley, G.G.; Paik, K.; Senarath-Yapa, K.; Atashroo, D.A.; Wearda, T.; Lorenz, H.P.; et al. Emerging drugs for the treatment of wound healing. Expert Opin. Emerg. Drugs 2015, 20, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Iuchi, Y.; Roy, D.; Okada, F.; Kibe, N.; Tsunoda, S.; Suzuki, S.; Takahashi, M.; Yokoyama, H.; Yoshitake, J.; Kondo, S.; et al. Spontaneous skin damage and delayed wound healing in sod1-deficient mice. Mol. Cell. Biochem. 2010, 341, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Deshane, J.; Chen, S.; Caballero, S.; Grochot-Przeczek, A.; Was, H.; Li Calzi, S.; Lach, R.; Hock, T.D.; Chen, B.; Hill-Kapturczak, N.; et al. Stromal cell-derived factor 1 promotes angiogenesis via a heme oxygenase 1-dependent mechanism. J. Exp. Med. 2007, 204, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, A.L.; Lalezarzadeh, F.D.; Soares, M.A.; Saadeh, P.B.; Ceradini, D.J. Normalizing dysfunctional purine metabolism accelerates diabetic wound healing. Wound Repair Regen. 2015, 23, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Albiero, M.; Menegazzo, L.; Boscaro, E.; Pagnin, E.; Iori, E.; Cosma, C.; Lapolla, A.; Pengo, V.; Stendardo, M.; et al. The redox enzyme p66Shc contributes to diabetes and ischemia-induced delay in cutaneous wound healing. Diabetes 2010, 59, 2306–2314. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.D.; Wang, Y.Y.; Fu, W.L.; Wu, J.; Chen, A.F. Gene therapy of endothelial nitric oxide synthase and manganese superoxide dismutase restores delayed wound healing in type 1 diabetic mice. Circulation 2004, 110, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Justiniano, R.; Zhang, D.D.; Wondrak, G.T. The Nrf2-inducers tanshinone I and dihydrotanshinone protect human skin cells and reconstructed human skin against solar simulated Uv. Redox Biol. 2013, 1, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Han, J. Mitochondria-targeted antioxidants for the treatment of cardiovascular disorders. Adv. Exp. Med. Biol. 2017, 982, 621–646. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.P.; Reddy, P.H. Mitochondria-targeted molecules as potential drugs to treat patients with alzheimer’s disease. Prog. Mol. Biol. Transl. Sci. 2017, 146, 173–201. [Google Scholar] [CrossRef] [PubMed]
- Karaa, A.; Haas, R.; Goldstein, A.; Vockley, J.; Weaver, W.D.; Cohen, B.H. Randomized dose-escalation trial of elamipretide in adults with primary mitochondrial myopathy. Neurology 2018, 90, e1212–e1221. [Google Scholar] [CrossRef] [PubMed]
- Daubert, M.A.; Yow, E.; Dunn, G.; Marchev, S.; Barnhart, H.; Douglas, P.S.; O’Connor, C.; Goldstein, S.; Udelson, J.E.; Sabbah, H.N. Novel mitochondria-targeting peptide in heart failure treatment: A randomized, placebo-controlled trial of elamipretide. Circ. Heart Fail. 2017, 10, e004389. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cano Sanchez, M.; Lancel, S.; Boulanger, E.; Neviere, R. Targeting Oxidative Stress and Mitochondrial Dysfunction in the Treatment of Impaired Wound Healing: A Systematic Review. Antioxidants 2018, 7, 98. https://doi.org/10.3390/antiox7080098
Cano Sanchez M, Lancel S, Boulanger E, Neviere R. Targeting Oxidative Stress and Mitochondrial Dysfunction in the Treatment of Impaired Wound Healing: A Systematic Review. Antioxidants. 2018; 7(8):98. https://doi.org/10.3390/antiox7080098
Chicago/Turabian StyleCano Sanchez, Mariola, Steve Lancel, Eric Boulanger, and Remi Neviere. 2018. "Targeting Oxidative Stress and Mitochondrial Dysfunction in the Treatment of Impaired Wound Healing: A Systematic Review" Antioxidants 7, no. 8: 98. https://doi.org/10.3390/antiox7080098
APA StyleCano Sanchez, M., Lancel, S., Boulanger, E., & Neviere, R. (2018). Targeting Oxidative Stress and Mitochondrial Dysfunction in the Treatment of Impaired Wound Healing: A Systematic Review. Antioxidants, 7(8), 98. https://doi.org/10.3390/antiox7080098