Redox Biology of Right-Sided Heart Failure

Abstract
1. Introduction
2. Oxidative Modifications in Right-Sided Heart Failure
3. RV-Specific Redox Regulation of GATA4 Gene Expression
4. The RV-Specific Redox Regulation of Serotonin Signaling
5. Metabolomics Analysis to Define the Difference between the RV and LV
6. RV/LV Differences in Oxidative Stress and Antioxidant Defense
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Konstam, M.A.; Kiernan, M.S.; Bernstein, D.; Bozkurt, B.; Jacob, M.; Kapur, N.K.; Kociol, R.D.; Lewis, E.F.; Mehra, M.R.; Pagani, F.D.; et al. Evaluation and Management of Right-Sided Heart Failure: A Scientific Statement from the American Heart Association. Circulation 2018, 137, e578–e622. [Google Scholar] [CrossRef] [PubMed]
- Guimaron, S.; Guihaire, J.; Amsallem, M.; Haddad, F.; Fadel, E.; Mercier, O. Current knowledge and recent advances of right ventricular molecular biology and metabolism from congenital heart disease to chronic pulmonary hypertension. BioMed Res. Int. 2018, 2018, 1981568. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.; Bernstein, D. Molecular mechanisms of right ventricular failure. Circulation 2015, 132, 1734–1742. [Google Scholar] [CrossRef] [PubMed]
- Voelkel, N.F.; Quaife, R.A.; Leinwand, L.A.; Barst, R.J.; McGoon, M.D.; Meldrum, D.R.; Dupuis, J.; Long, C.S.; Rubin, L.J.; Smark, F.W.; et al. Right ventricular function and failure: The need to know more. Report of a National Heart, Lung and Blood Institute Working Group on Cellular and Molecular Mechanisms of Right Heart Failure. Circulation 2006, 114, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
- Boxt, L.M. Radiology of the right ventricle. Radiol. Clin. N. Am. 1999, 37, 379–400. [Google Scholar] [CrossRef]
- Budev, M.M.; Arroliga, A.C.; Wiedemann, H.P.; Matthay, R.A. Cor pulmonale: An overview. Semin. Respir. Crit. Care Med. 2003, 24, 233–244. [Google Scholar] [PubMed]
- Zaffran, S.; Kelly, R.G.; Meilhac, S.M.; Buckingham, M.E.; Brown, N.A. Right ventricular myocardium derives from the anterior heart field. Circ. Res. 2004, 95, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Verzi, M.P.; McCulley, D.J.; De Val, S.; Dodou, E.; Black, B.L. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev. Biol. 2005, 287, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, D. Making or breaking the heart: From lineage determination to morphogenesis. Cell 2006, 126, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.J. Molecular basis of right ventricular hypertrophy and failure in pulmonary vascular disease. In Textbook of Pulmonary Vascular Disease; Yuan, J.X.J., Garcia, J.G.N., Hales, C.A., Archer, S.L., Rich, S., West, J.B., Eds.; Springer: New York, NY, USA, 2011; pp. 1305–1312. [Google Scholar]
- Liu, L.; Marcocci, L.; Wong, C.M.; Park, A.M.; Suzuki, Y.J. Serotonin-mediated protein carbonylation in the right heart. Free Radic. Biol. Med. 2008, 45, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Park, A.; Wong, C.; Jelinkova, L.; Liu, L.; Nagase, H.; Suzuki, Y.J. Pulmonary hypertension-induced GATA4 activation in the right ventricle. Hypertension 2010, 56, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Zungu-Edmondson, M.; Suzuki, Y.J. Differential stress response mechanisms in right and left ventricles. J. Rare Dis. Res. Treat. 2016, 1, 39–45. [Google Scholar] [PubMed]
- Taraseviciene-Stewart, L.; Kasahara, Y.; Alger, L.; Hirth, P.; Mc Mahon, G.; Waltenberger, J.; Voelkel, N.F.; Tuder, R.M. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001, 15, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, S.; Farkas, L.; Al Husseini, A.; Farkas, D.; Gomez-Arroyo, J.; Kraskauskas, D.; Nicolls, M.R.; Cool, C.D.; Bogaard, H.J.; Voelkel, N.F. Severe pulmonary arterial hypertension induced by SU5416 and ovalbumin immunization. Am. J. Respir. Cell Mol. Biol. 2012, 47, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Bogaard, H.J.; Natarajan, R.; Henderson, S.C.; Long, C.S.; Kraskauskas, D.; Smithson, L.; Ockaili, R.; McCord, J.M.; Voelkel, N.F. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation 2009, 120, 1951–1960. [Google Scholar] [CrossRef] [PubMed]
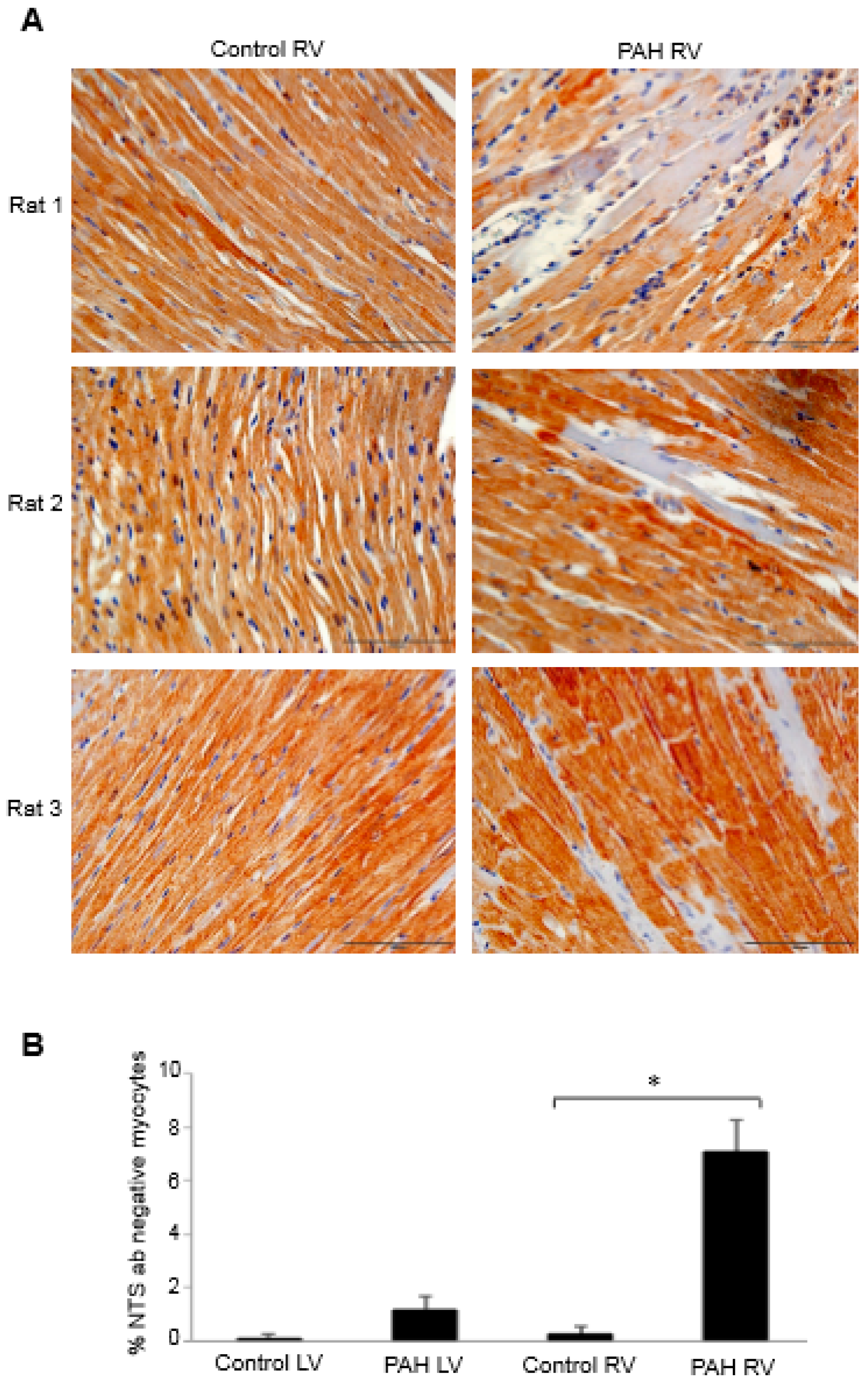
- Wang, X.; Shults, N.V.; Suzuki, Y.J. Oxidative profiling of the failing right heart in rats with pulmonary hypertension. PLoS ONE 2017, 12, e0176887. [Google Scholar] [CrossRef] [PubMed]
- Berlett, B.S.; Stadtman, E.R. Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 1997, 272, 20313–20316. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.J.; Shults, N.V. Redox Signaling in the Right Ventricle. Adv. Exp. Med. Biol. 2017, 967, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D.; Olson, E.N. GATA4: A novel transcriptional regulator of cardiac hypertrophy? Circulation 1997, 96, 3833–3835. [Google Scholar] [PubMed]
- Van Berlo, J.H.; Elrod, J.W.; Aronow, B.J.; Pu, W.T.; Molkentin, J.D. Serine 105 phosphorylation of transcription factor GATA4 is necessary for stress-induced cardiac hypertrophy in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 12331–12336. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.J.; Nagase, H.; Wong, C.M.; Kumar, S.V.; Jain, V.; Park, A.M.; Day, R.M. Regulation of Bcl-xL expression in lung vascular smooth muscle. Am. J. Respir. Cell Mol. Biol. 2007, 36, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.M.; Cheema, A.K.; Zhang, L.; Suzuki, Y.J. Protein carbonylation as a novel mechanism in redox signaling. Circ. Res. 2008, 102, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Kitta, K.; Day, R.M.; Kim, Y.; Torregroza, I.; Evans, T.; Suzuki, Y.J. Hepatocyte growth factor induces GATA-4 phosphorylation and cell survival in cardiac muscle cells. J. Biol. Chem. 2003, 278, 4705–4712. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Ma, A.G.; Kitta, K.; Fitch, S.N.; Ikeda, T.; Ihara, Y.; Simon, A.R.; Evans, T.; Suzuki, Y.J. Anthracycline-induced suppression of GATA-4 transcription factor: Implication in the regulation of cardiac myocyte apoptosis. Mol. Pharmacol. 2003, 63, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, Y.F.; Wong, C.M.; Pavlickova, L.; Liu, L.; Trasar, L.; Bansal, G.; Suzuki, Y.J. Mechanism of the susceptibility of remodeled pulmonary vessels to drug-induced cell killing. J. Am. Heart Assoc. 2014, 3, e000520. [Google Scholar] [CrossRef] [PubMed]
- Zungu-Edmondson, M.; Shults, N.V.; Melnyk, O.; Suzuki, Y.J. Natural reversal of pulmonary vascular remodeling and right ventricular remodeling in SU5416/hypoxia-treated Sprague-Dawley rats. PLoS ONE 2017, 12, e0182551. [Google Scholar] [CrossRef] [PubMed]
- Zungu-Edmondson, M.; Shults, N.V.; Wong, C.M.; Suzuki, Y.J. Modulators of right ventricular apoptosis and contractility in a rat model of pulmonary hypertension. Cardiovasc. Res. 2016, 110, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Schreckenberg, R.; Rebelo, M.; Deten, A.; Weber, M.; Rohrbach, S.; Pipicz, M.; Csonka, C.; Ferdinandy, P.; Schulz, R.; Schlüter, K.D. Specific Mechanisms Underlying Right Heart Failure: The Missing upregulation of superoxide dismutase-2 and its decisive role in antioxidative defense. Antioxid. Redox Signal. 2015, 23, 1220–1232. [Google Scholar] [CrossRef] [PubMed]
- Borchi, E.; Bargelli, V.; Stillitano, F.; Giordano, C.; Sebastiani, M.; Nassi, P.A.; d’Amati, G.; Cerbai, E.; Nediani, C. Enhanced ROS production by NADPH oxidase is correlated to changes in antioxidant enzyme activity in human heart failure. Biochim. Biophys. Acta 2010, 1802, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, R.R.; Shore, D.F.; Lincoln, C.; Mumby, S.; Kemp, M.; Brierly, J.; Petros, A.; Gutteridge, J.M.; Hooper, J.; Redington, A.N. Acute right ventricular restrictive physiology after repair of tetralogy of Fallot: Association with myocardial injury and oxidative stress. Circulation 1999, 100, 1540–1547. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.; Osorio, J.C.; Duque, A.M.; Kaufman, B.D.; Phillips, A.B.; Chen, J.M.; Quaegebeur, J.; Mosca, R.S.; Mital, S. Failure of right ventricular adaptation in children with tetralogy of Fallot. Circulation 2006, 114, I-37–I-42. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Peptides | Fold Difference (Control/PAH) | Total # of AAs | Carbonylation Susceptible AAs | |
|---|---|---|---|---|
| # | % | |||
| Glu Ile Lys Pro | 4.8 | 4 | 2 | 50 |
| Asp Lys Lys Pro | 2.5 | 4 | 3 | 75 |
| Lys Arg Thr Thr | 2.2 | 4 | 4 | 100 |
| Phe Gly Arg Arg | 4.5 | 4 | 2 | 50 |
| Ser Val Lys Arg | 2.5 | 4 | 2 | 50 |
| Lys Trp Lys | 2.0 | 3 | 2 | 67 |
| Lys Tyr Ile Glu | 2.7 | 4 | 1 | 25 |
| Ser Leu Leu Ser Phe | 2.2 | 5 | 0 | 0 |
| Asp Leu Phe Arg | 2.4 | 4 | 1 | 25 |
| Thr Thr Gly Leu Ile | 2.8 | 5 | 2 | 40 |
| Lys Tyr Thr Arg | 2.5 | 4 | 3 | 75 |
| Arg Ser Lys Arg | 3.0 | 4 | 3 | 75 |
| Trp Phe Trp | 2.3 | 3 | 0 | 0 |
| Asn Arg Phe Lys | 2.8 | 4 | 2 | 50 |
| His Ile Ile Val | 3.1 | 4 | 0 | 0 |
| Arg Lys Lys Cys | 3.0 | 4 | 3 | 75 |
| Asn Arg Phe Lys | 3.2 | 4 | 2 | 50 |
| Phe Ile Gln Lys | 3.0 | 4 | 1 | 25 |
| Ala Arg Tyr Arg | 2.6 | 4 | 2 | 50 |
| Ala Ala Ile Lys | 4.8 | 4 | 1 | 25 |
| Glu Phe Pro Trp | 2.3 | 4 | 1 | 25 |
| Phe Thr Thr Thr | 2.2 | 4 | 3 | 75 |
| Val Arg His Arg | 2.6 | 4 | 2 | 50 |
| Ile Ile Val Tyr | 2.2 | 4 | 0 | 0 |
| Pro Gln Arg Thr | 3.0 | 4 | 3 | 75 |
| Phe Lys Lys | 41.7 | 4 | 2 | 50 |
| Thr Thr Gly Leu Ile | 2.5 | 4 | 2 | 50 |
| Glu Lys Ala Arg | 2.1 | 4 | 2 | 50 |
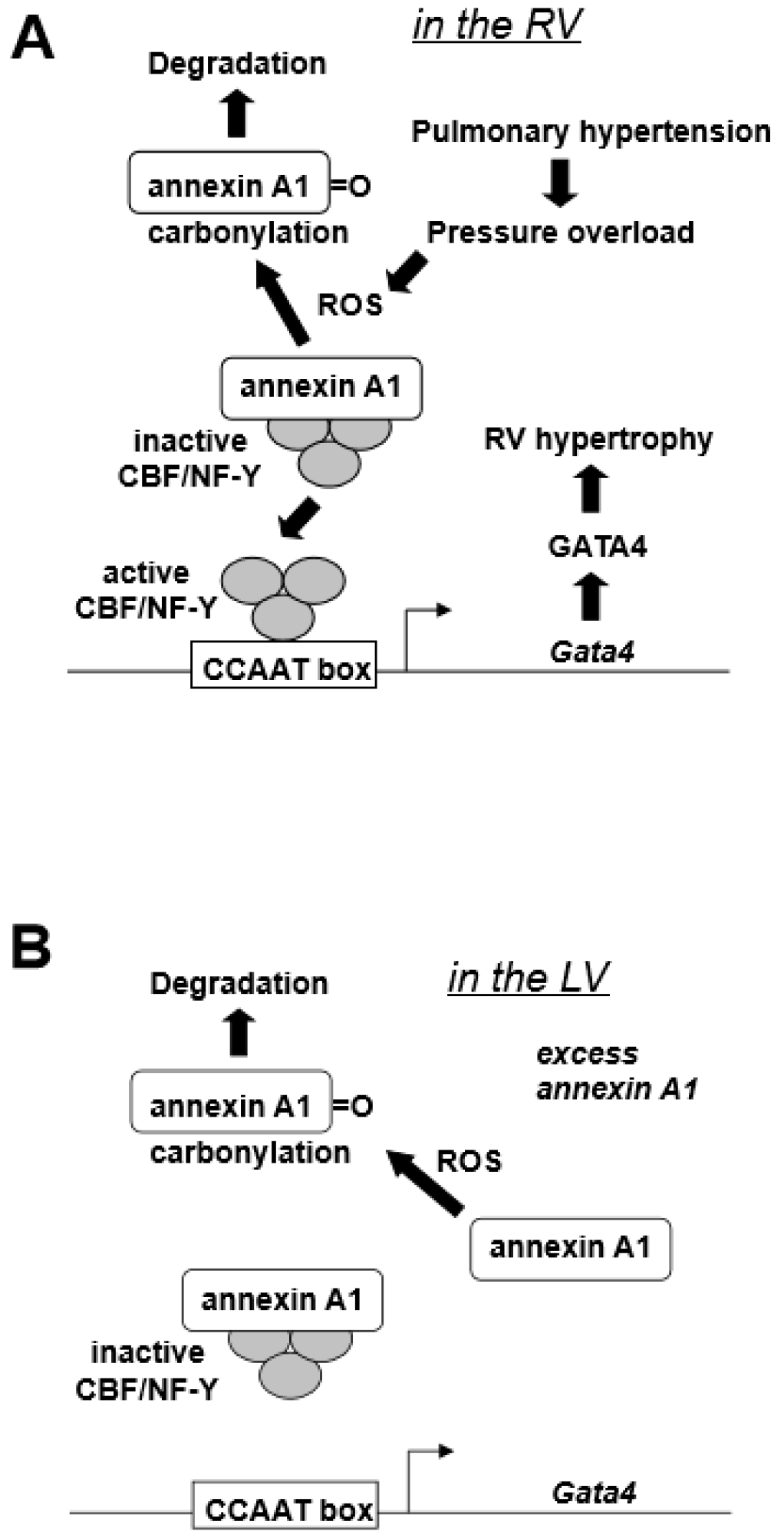
| Pulmonary hypertension activates GATA4 DNA binding activity in the RV. |
| Pulmonary hypertension increases GATA4 protein expression in the RV. |
| Pulmonary hypertension increases Gata4 mRNA expression in the RV. |
| The Gata4 promoter contains a functionally important CCAAT box. |
| The CBF/NF-Y transcription factor regulates CCAAT box of the Gata4 promoter. |
| CBF/NF-Y binds to annexin A1. |
| Pulmonary hypertension promotes the degradation of annexin A1. |
| The degradation of annexin A1 is regulated by metal-catalyzed oxidation of annexin A1. |
| The RV has higher CBF/NF-Y-to-annexin A1 ratio than the left ventricle (LV) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shults, N.V.; Melnyk, O.; Suzuki, D.I.; Suzuki, Y.J. Redox Biology of Right-Sided Heart Failure. Antioxidants 2018, 7, 106. https://doi.org/10.3390/antiox7080106
Shults NV, Melnyk O, Suzuki DI, Suzuki YJ. Redox Biology of Right-Sided Heart Failure. Antioxidants. 2018; 7(8):106. https://doi.org/10.3390/antiox7080106
Chicago/Turabian StyleShults, Nataliia V., Oleksiy Melnyk, Dante I. Suzuki, and Yuichiro J. Suzuki. 2018. "Redox Biology of Right-Sided Heart Failure" Antioxidants 7, no. 8: 106. https://doi.org/10.3390/antiox7080106
APA StyleShults, N. V., Melnyk, O., Suzuki, D. I., & Suzuki, Y. J. (2018). Redox Biology of Right-Sided Heart Failure. Antioxidants, 7(8), 106. https://doi.org/10.3390/antiox7080106