Effects of Bcl-2/Bcl-xL Inhibitors on Pulmonary Artery Smooth Muscle Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture Experiments
2.2. Animal Experiments
2.3. Histological Measurements
2.4. Western Blotting
2.5. Fluorescence-Based Cell Assays for Apoptosis and Autophagy
2.6. Statistical Analysis
3. Results
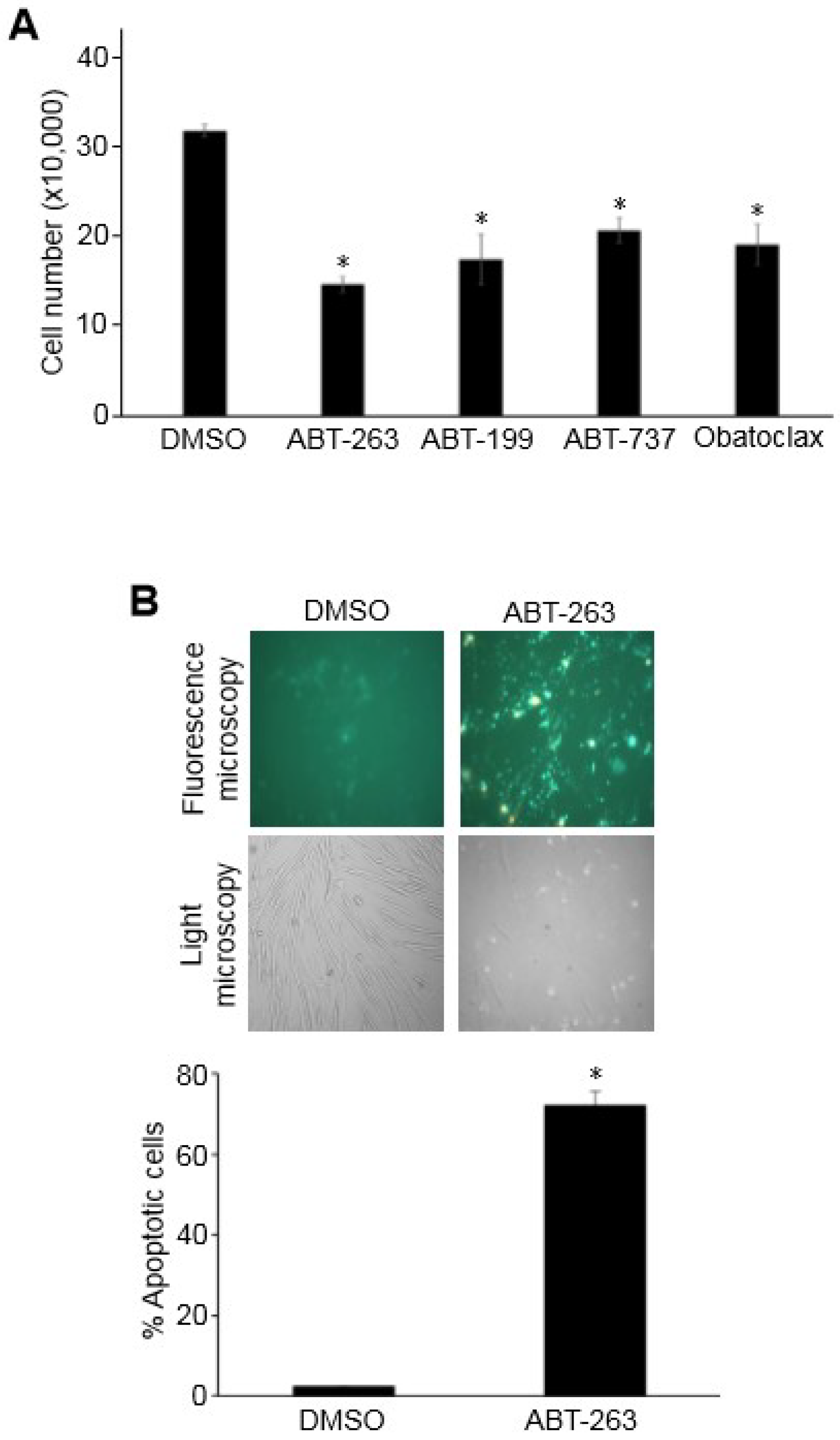
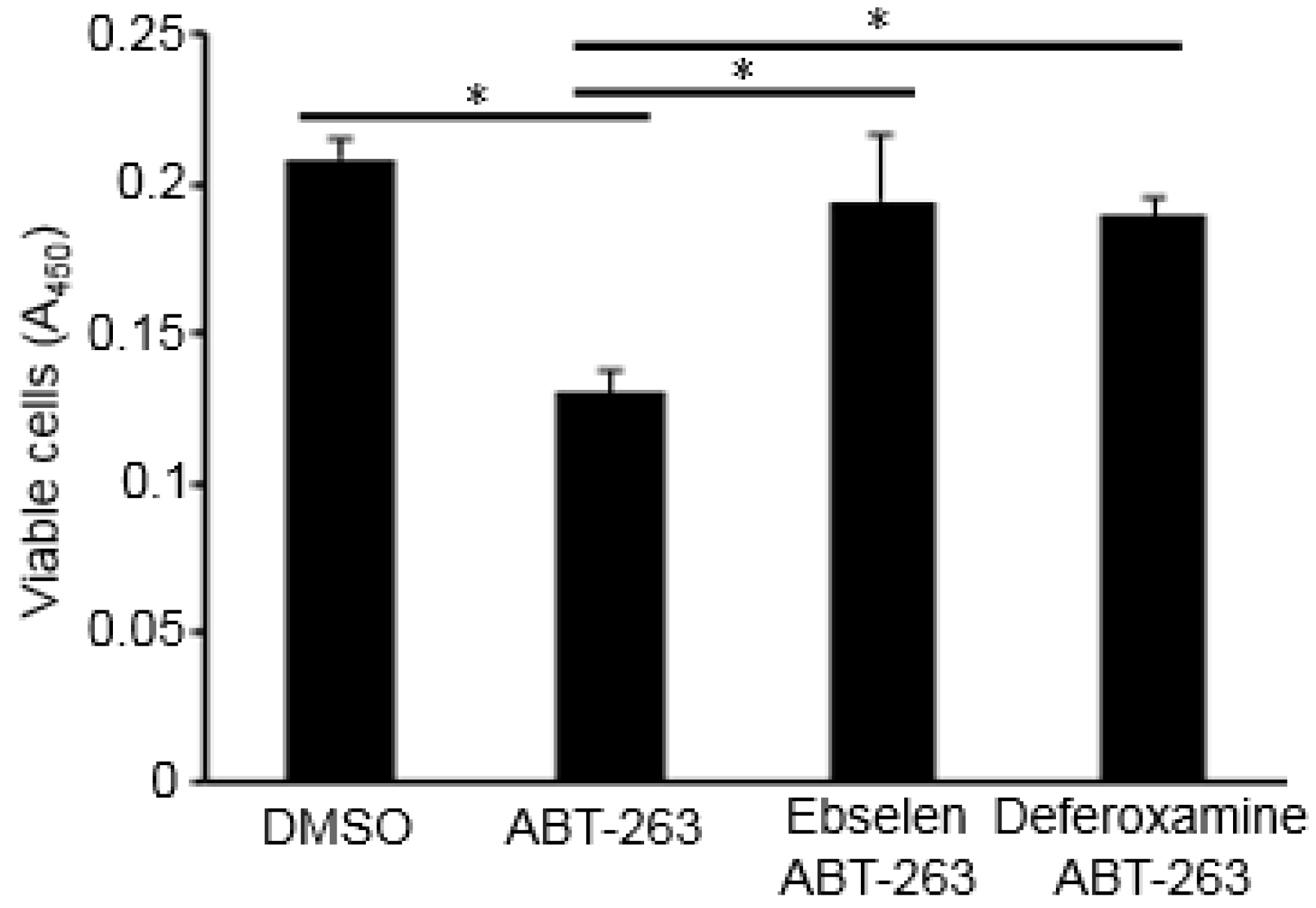
3.1. Bcl-2/Bcl-xL Inhibitors Promote the Death of PASMCs
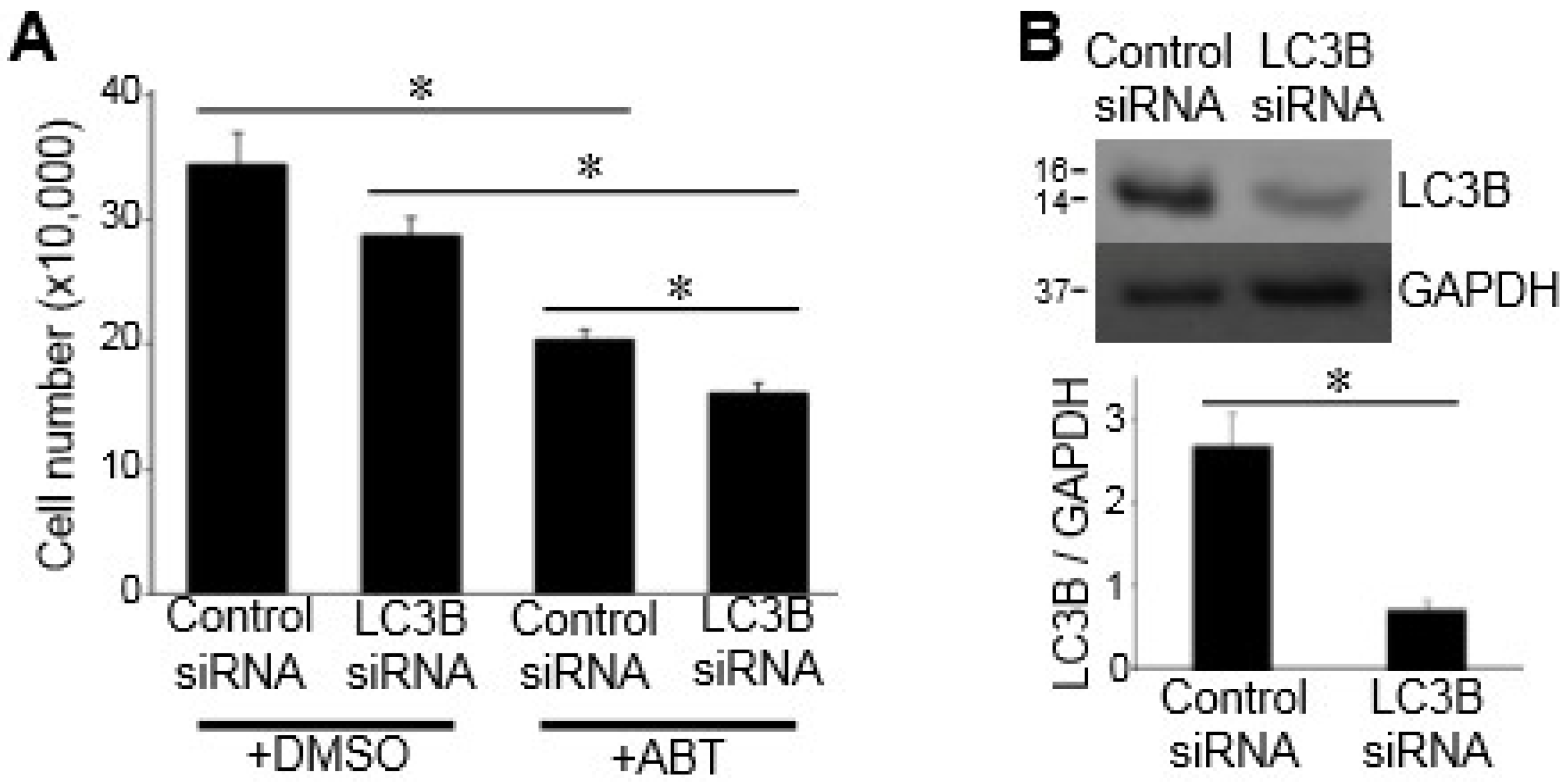
3.2. Autophagy Does Not Mediate Bcl-2/Bcl-xL-Inhibitor-Induced Cell Death
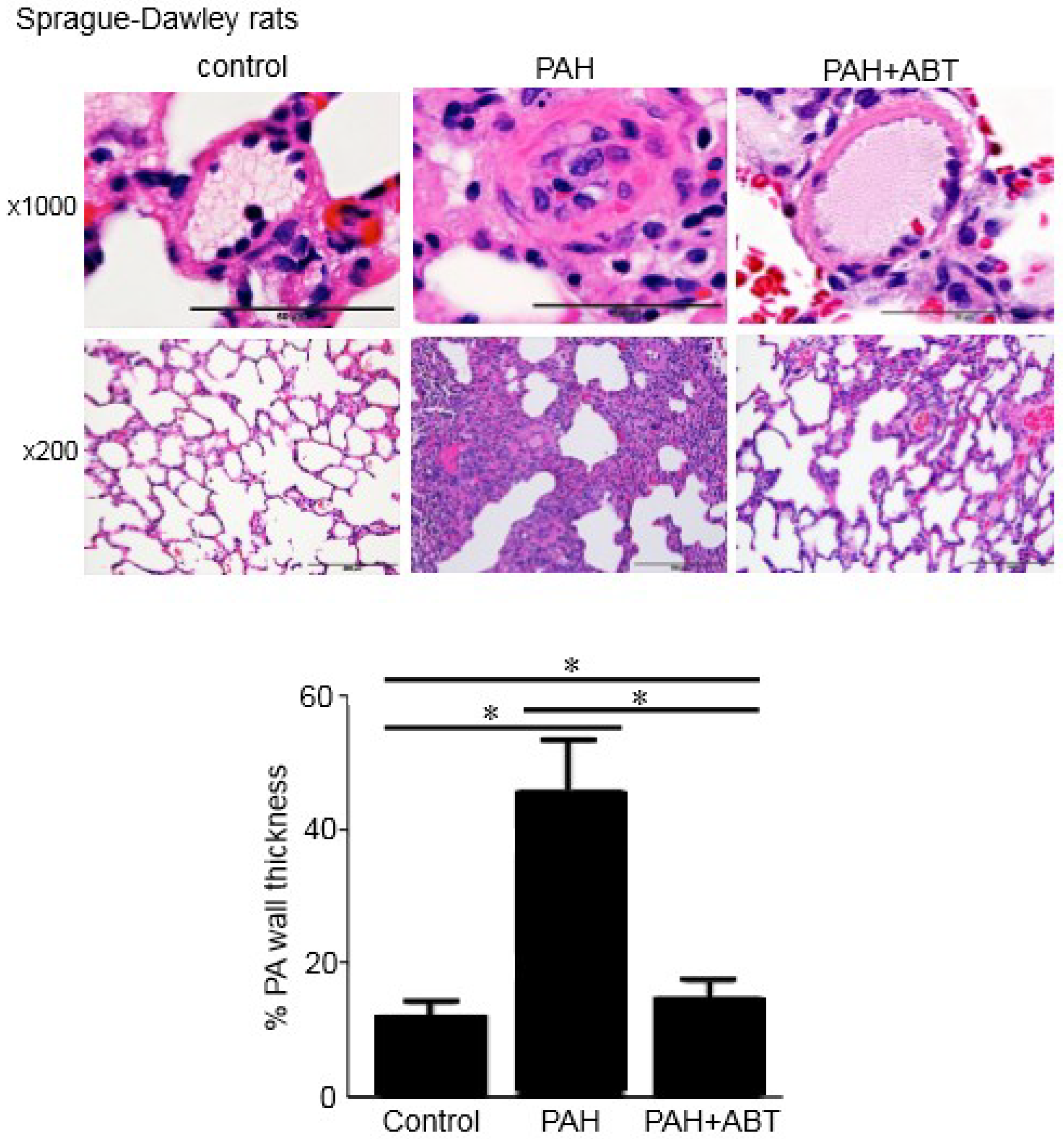
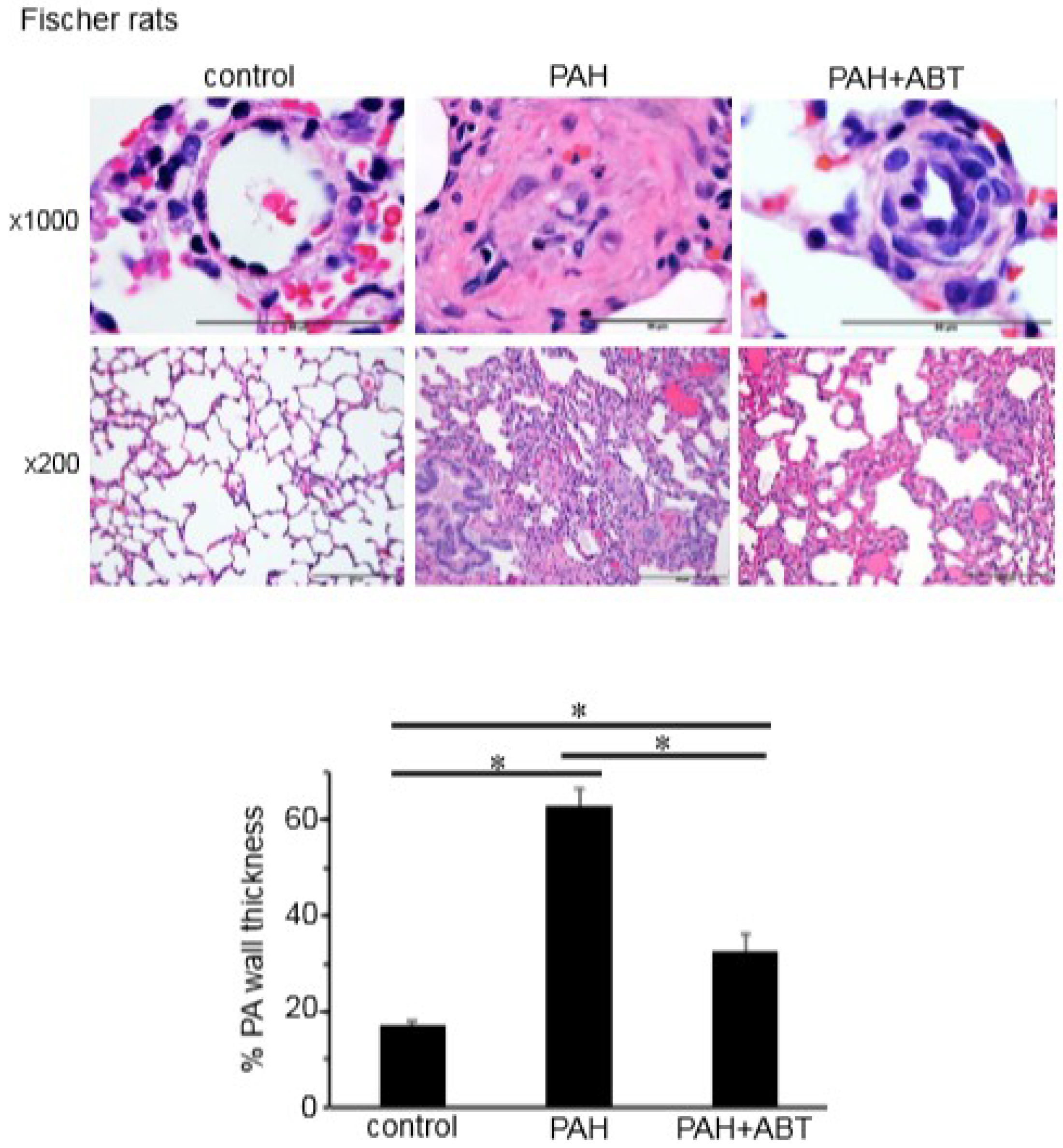
3.3. Bcl-2/Bcl-xL-Inhibition Reverses Pulmonary Vascular Remodeling in Rats
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fallah, F. Recent strategies in treatment of pulmonary arterial hypertension, a review. Glob. J. Health Sci. 2015, 7, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, S. Pulmonary hypertension 2015: Current definitions, terminology, and novel treatment options. Clin. Res. Cardiol. 2015, 104, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Delcroix, M.; Naeije, R. Optimising the management of pulmonary arterial hypertension patients: Emergency treatments. Eur. Respir. Rev. 2010, 19, 204–211. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, V.V.; Shah, S.J.; Souza, R.; Humbert, M. Management of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2015, 65, 1976–1997. [Google Scholar] [CrossRef] [PubMed]
- D’Alonzo, G.E.; Barst, R.J.; Ayres, S.M.; Bergofsky, E.H.; Brundage, B.H.; Detre, K.M.; Fishman, A.P.; Goldring, R.M.; Groves, B.M.; Kernis, J.T.; et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann. Intern. Med. 1991, 115, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Runo, J.R.; Loyd, J.E. Primary pulmonary hypertension. Lancet 2003, 361, 1533–1544. [Google Scholar] [CrossRef]
- Benza, R.L.; Miller, D.P.; Frost, A.; Barst, R.J.; Krichman, A.M.; McGoon, M.D. Analysis of the lung allocation score estimation of risk of death in patients with pulmonary arterial hypertension using data from the REVEAL Registry. Transplantation 2010, 90, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Sitbon, O.; Yaïci, A.; Montani, D.; O’Callaghan, D.S.; Jaïs, X.; Parent, F.; Savale, L.; Natali, D.; Günther, S.; et al. French Pulmonary Arterial Hypertension Network. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur. Respir. J. 2010, 36, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Thenappan, T.; Shah, S.J.; Rich, S.; Tian, L.; Archer, S.L.; Gomberg-Maitland, M. Survival in pulmonary arterial hypertension: A reappraisal of the NIH risk stratification equation. Eur. Respir. J. 2010, 35, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Olsson, K.M.; Delcroix, M.; Ghofrani, H.A.; Tiede, H.; Huscher, D.; Speich, R.; Grünig, E.; Staehler, G.; Rosenkranz, S.; Halank, M.; et al. Anticoagulation and survival in pulmonary arterial hypertension: Results from the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA). Circulation 2014, 129, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.J.; Nagase, H.; Wong, C.M.; Kumar, S.V.; Jain, V.; Park, A.M.; Day, R.M. Regulation of Bcl-xL expression in lung vascular smooth muscle. Am. J. Respir. Cell. Mol. Biol. 2007, 36, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, Y.F.; Wong, C.M.; Pavlickova, L.; Liu, L.; Trasar, L.; Bansal, G.; Suzuki, Y.J. Mechanism of the susceptibility of remodeled pulmonary vessels to drug-induced cell killing. J. Am. Heart Assoc. 2014, 3, e000520. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ibrahim, Y.F.; Das, D.; Zungu-Edmondson, M.; Shults, N.V.; Suzuki, Y.J. Carfilzomib reverses pulmonary arterial hypertension. Cardiovasc. Res. 2016, 110, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, Y.F.; Shults, N.V.; Rybka, V.; Suzuki, Y.J. Docetaxel reverses pulmonary vascular remodeling by decreasing autophagy and resolves right ventricular fibrosis. J. Pharmacol. Exp. Ther. 2017, 363, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Hockenbery, D.M.; Oltvai, Z.N.; Yin, X.M.; Milliman, C.L.; Korsmeyer, S.J. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell 1993, 75, 241–251. [Google Scholar] [CrossRef]
- Perini, G.F.; Ribeiro, G.N.; Pinto Neto, J.V.; Campos, L.T.; Hamerschlak, N. BCL-2 as therapeutic target for hematological malignancies. J. Hematol. Oncol. 2018, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Taraseviciene-Stewart, L.; Kasahara, Y.; Alger, L.; Hirth, P.; Mc Mahon, G.; Waltenberger, J.; Voelkel, N.F.; Tuder, R.M. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001, 15, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Oka, M.; Homma, N.; Taraseviciene-Stewart, L.; Morris, K.G.; Kraskauskas, D.; Burns, N.; Voelkel, N.F.; McMurtry, I.F. Rho kinase-mediated vasoconstriction is important in severe occlusive pulmonary arterial hypertension in rats. Circ. Res. 2007, 100, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Toba, M.; Alzoubi, A.; Ito, M.; Fagan, K.A.; Cool, C.D.; Voelkel, N.F.; McMurtry, I.F.; Oka, M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 2010, 121, 2747–2754. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Deng, Y.; Suen, C.; Taha, M.; Chaudhary, K.R.; Courtman, D.W.; Stewart, D.J. Marked strain-specific differences in the SU5416 rat model of severe pulmonary arterial hypertension. Am. J. Respir. Cell. Mol. Biol. 2016, 54, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Park, A.; Wong, C.; Jelinkova, L.; Liu, L.; Nagase, H.; Suzuki, Y.J. Pulmonary hypertension-induced GATA4 activation in the right ventricle. Hypertension 2010, 56, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Billard, C. BH3 mimetics: Status of the field and new developments. Mol. Cancer Ther. 2013, 12, 1691–1700. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.; Strasser, A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell. Death Differ. 2015, 22, 1071–1080. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rybka, V.; Suzuki, Y.J.; Shults, N.V. Effects of Bcl-2/Bcl-xL Inhibitors on Pulmonary Artery Smooth Muscle Cells. Antioxidants 2018, 7, 150. https://doi.org/10.3390/antiox7110150
Rybka V, Suzuki YJ, Shults NV. Effects of Bcl-2/Bcl-xL Inhibitors on Pulmonary Artery Smooth Muscle Cells. Antioxidants. 2018; 7(11):150. https://doi.org/10.3390/antiox7110150
Chicago/Turabian StyleRybka, Vladyslava, Yuichiro J. Suzuki, and Nataliia V. Shults. 2018. "Effects of Bcl-2/Bcl-xL Inhibitors on Pulmonary Artery Smooth Muscle Cells" Antioxidants 7, no. 11: 150. https://doi.org/10.3390/antiox7110150
APA StyleRybka, V., Suzuki, Y. J., & Shults, N. V. (2018). Effects of Bcl-2/Bcl-xL Inhibitors on Pulmonary Artery Smooth Muscle Cells. Antioxidants, 7(11), 150. https://doi.org/10.3390/antiox7110150