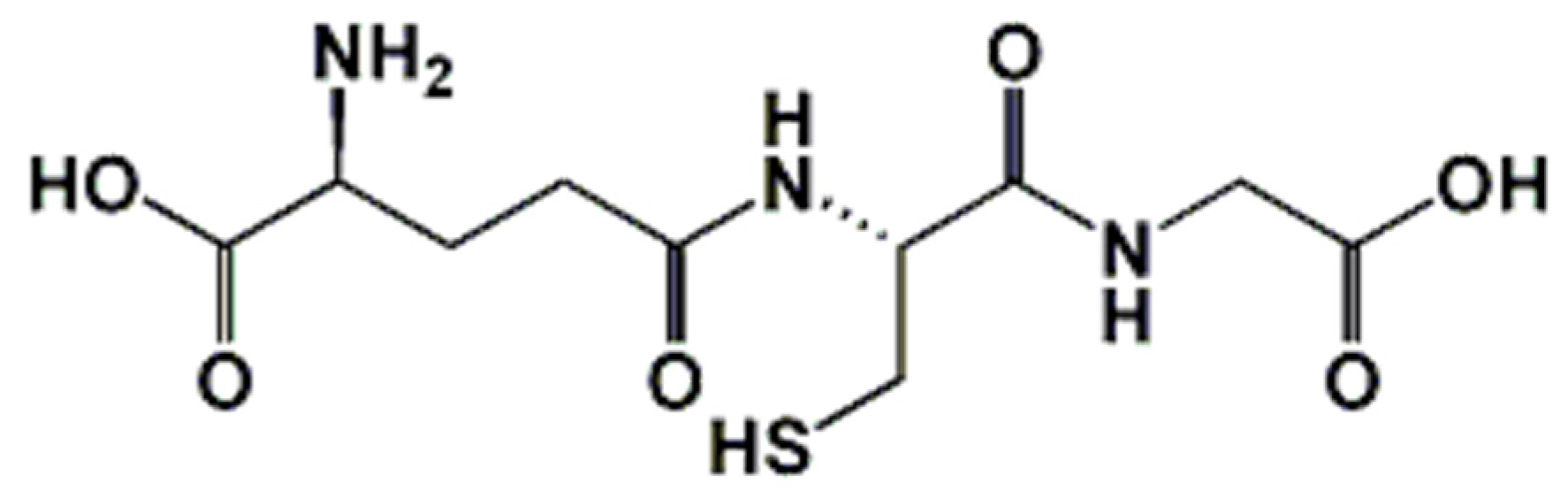
Glutathione: Antioxidant Properties Dedicated to Nanotechnologies

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
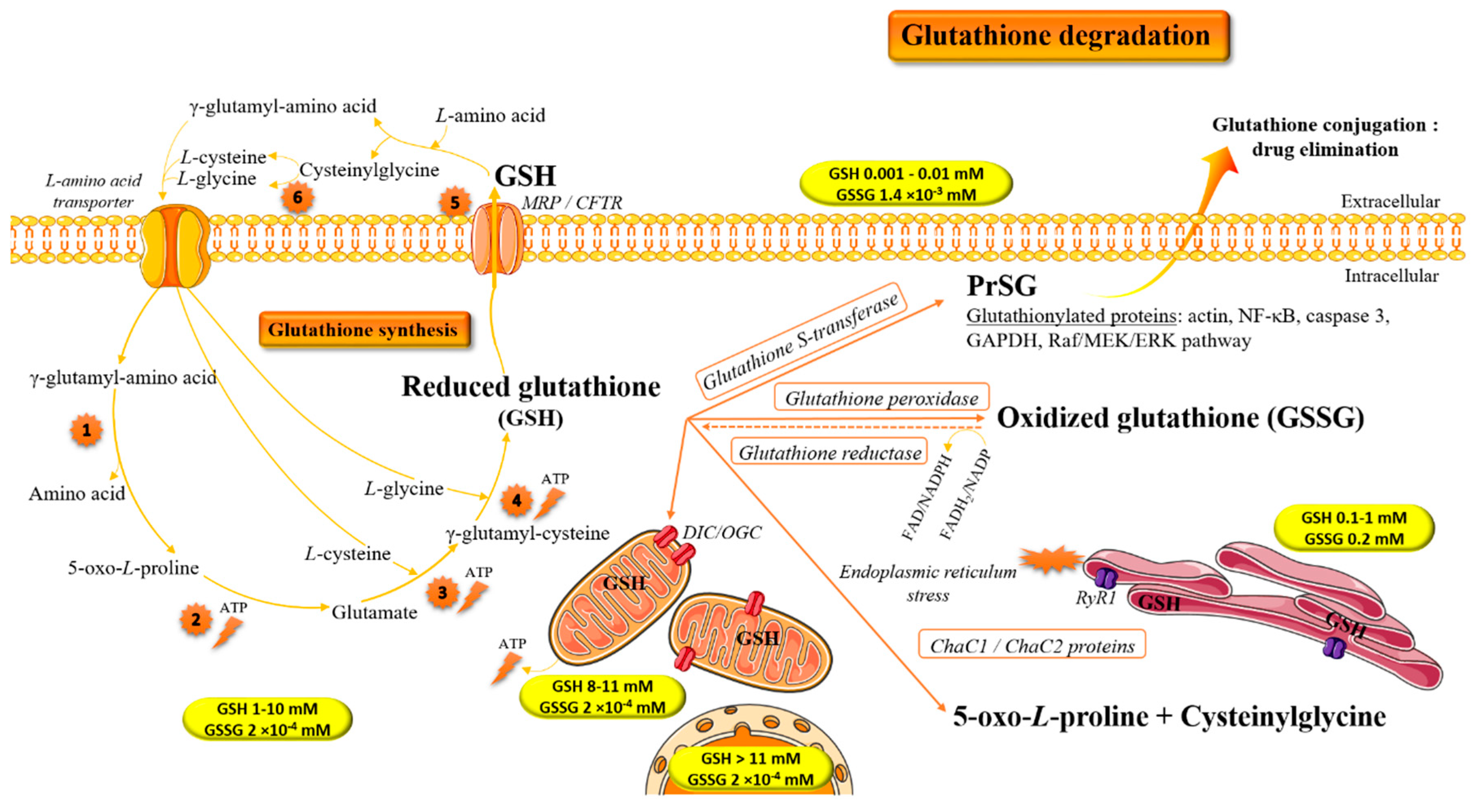
1. Introduction: Biochemical Implication of Glutathione in Redox Homeostasis
2. Glutathione and Antioxidant/Pro-Oxidant Properties
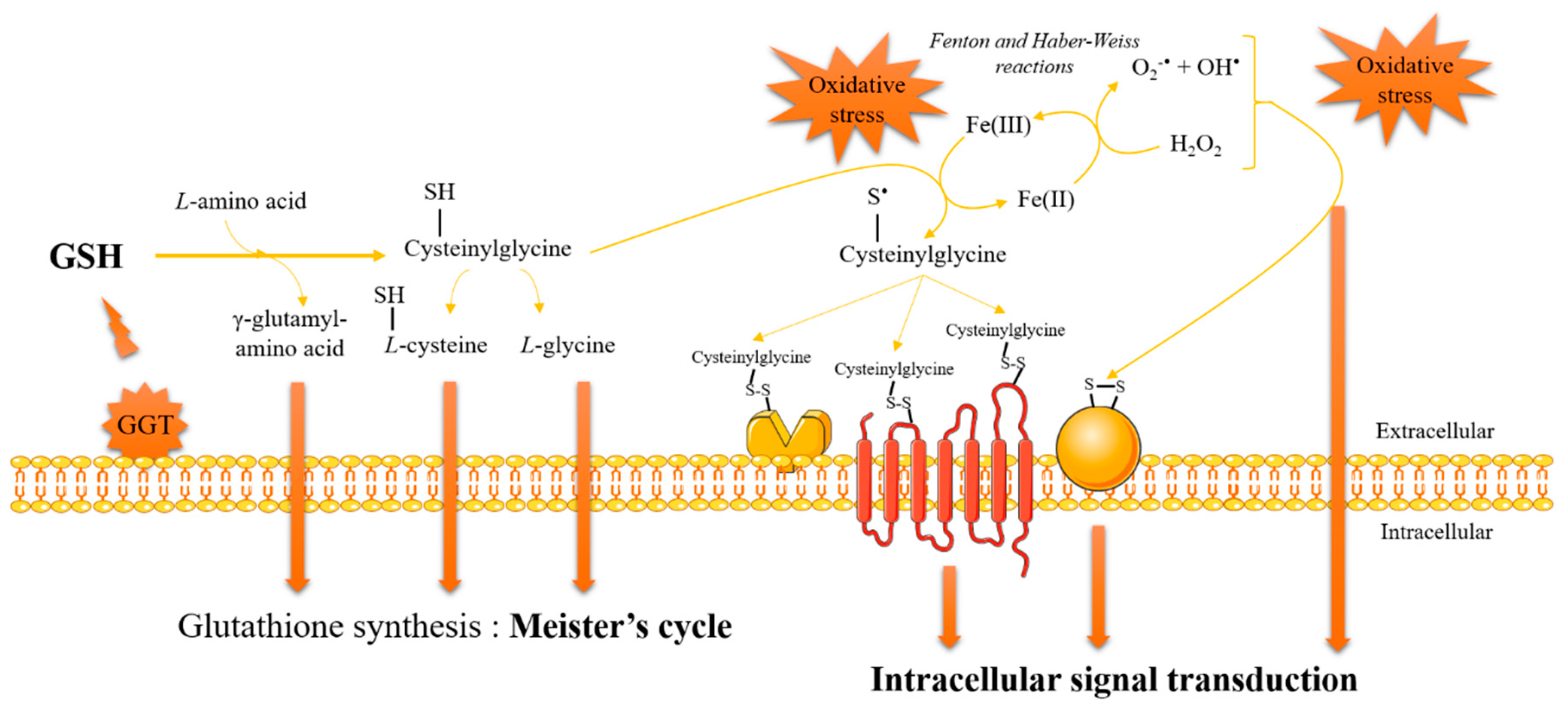
3. Glutathione and Redox Signaling
4. Methodologies for Dosage of Glutathione/Glutathiolated Proteins
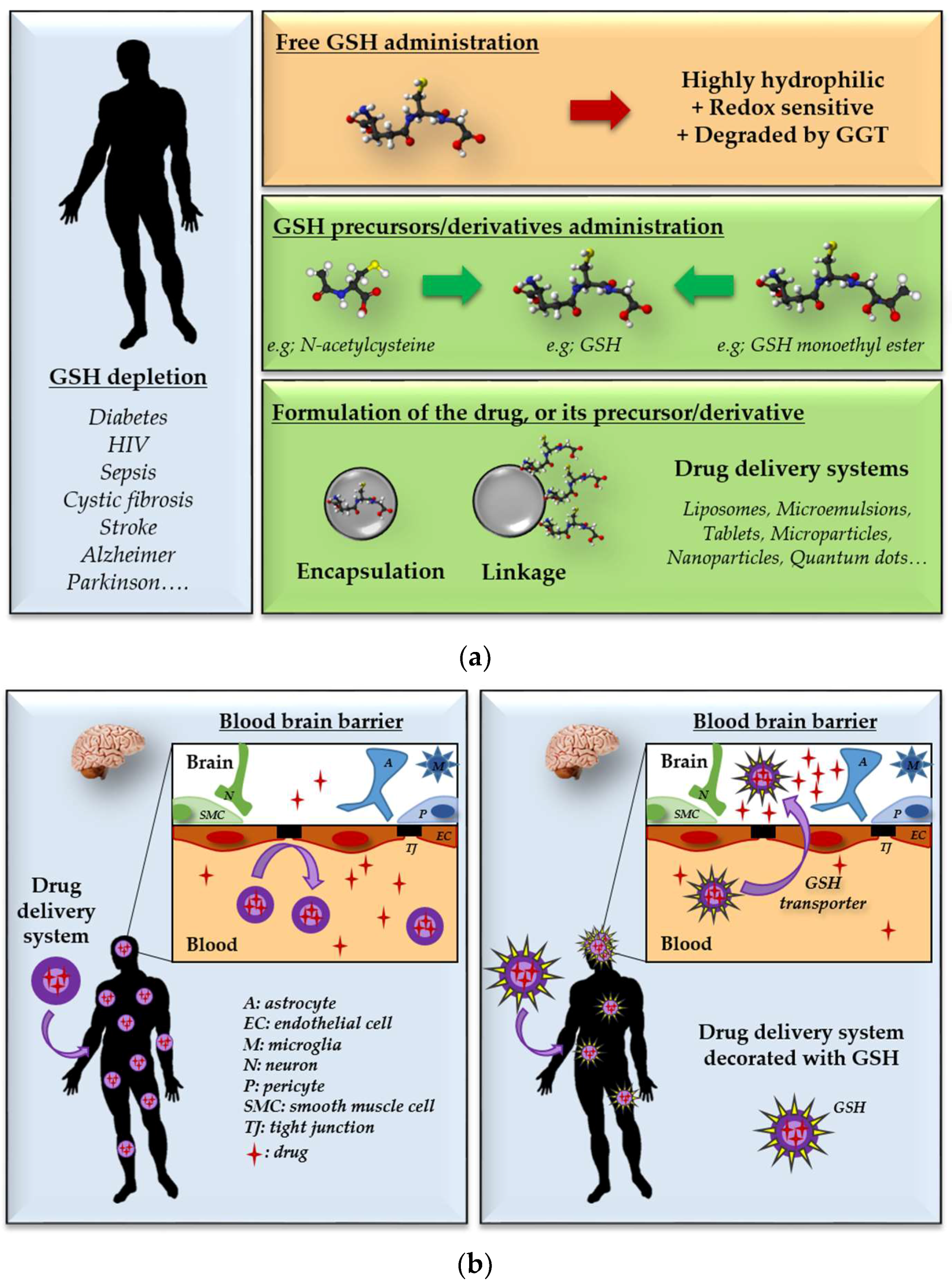
5. Repletion of Glutathione: Therapeutic Opportunities and Challenges
6. GSH Decoration as a Tool for Targeted Drug Delivery Systems
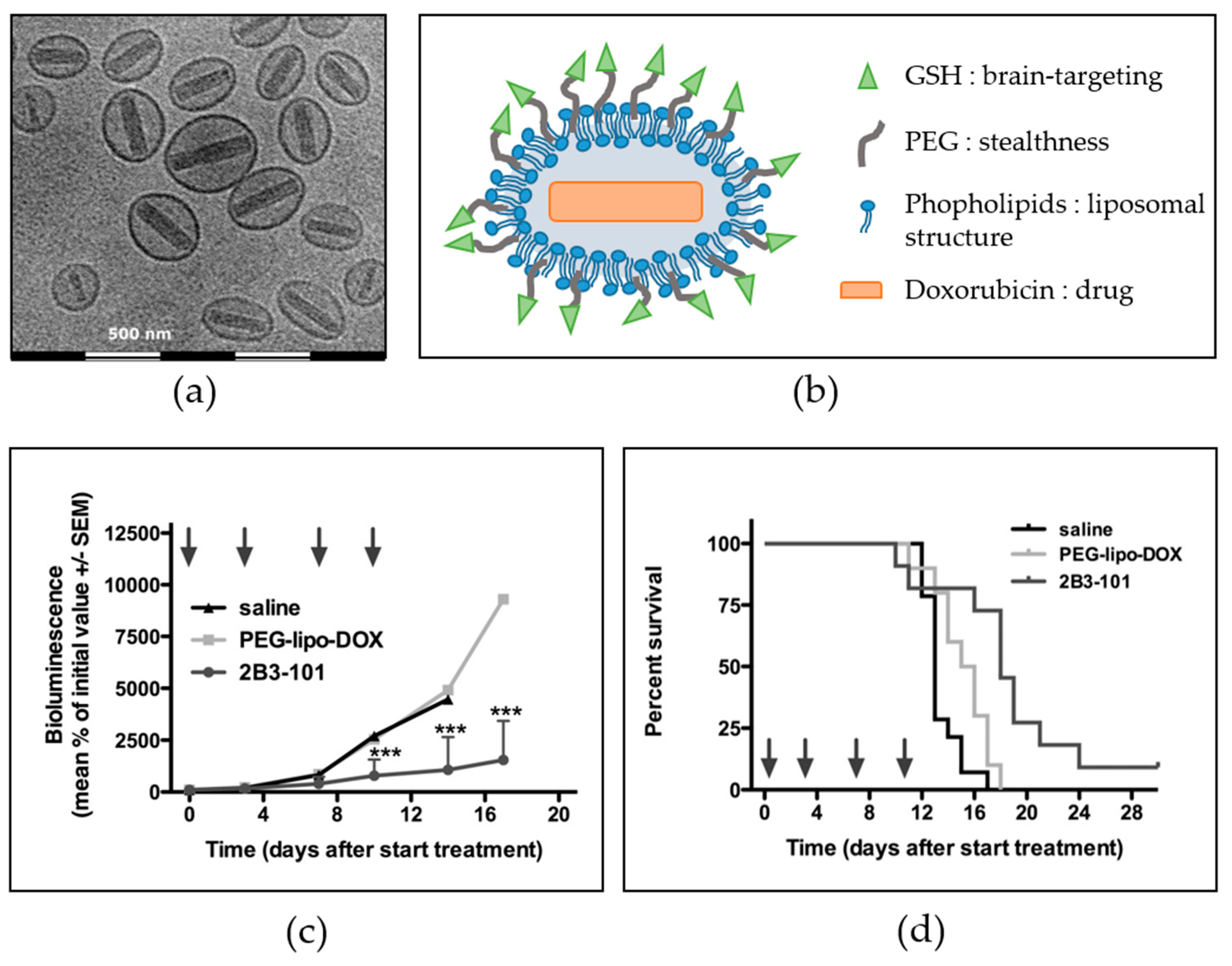
6.1. Brain-Targeted Drug Delivery
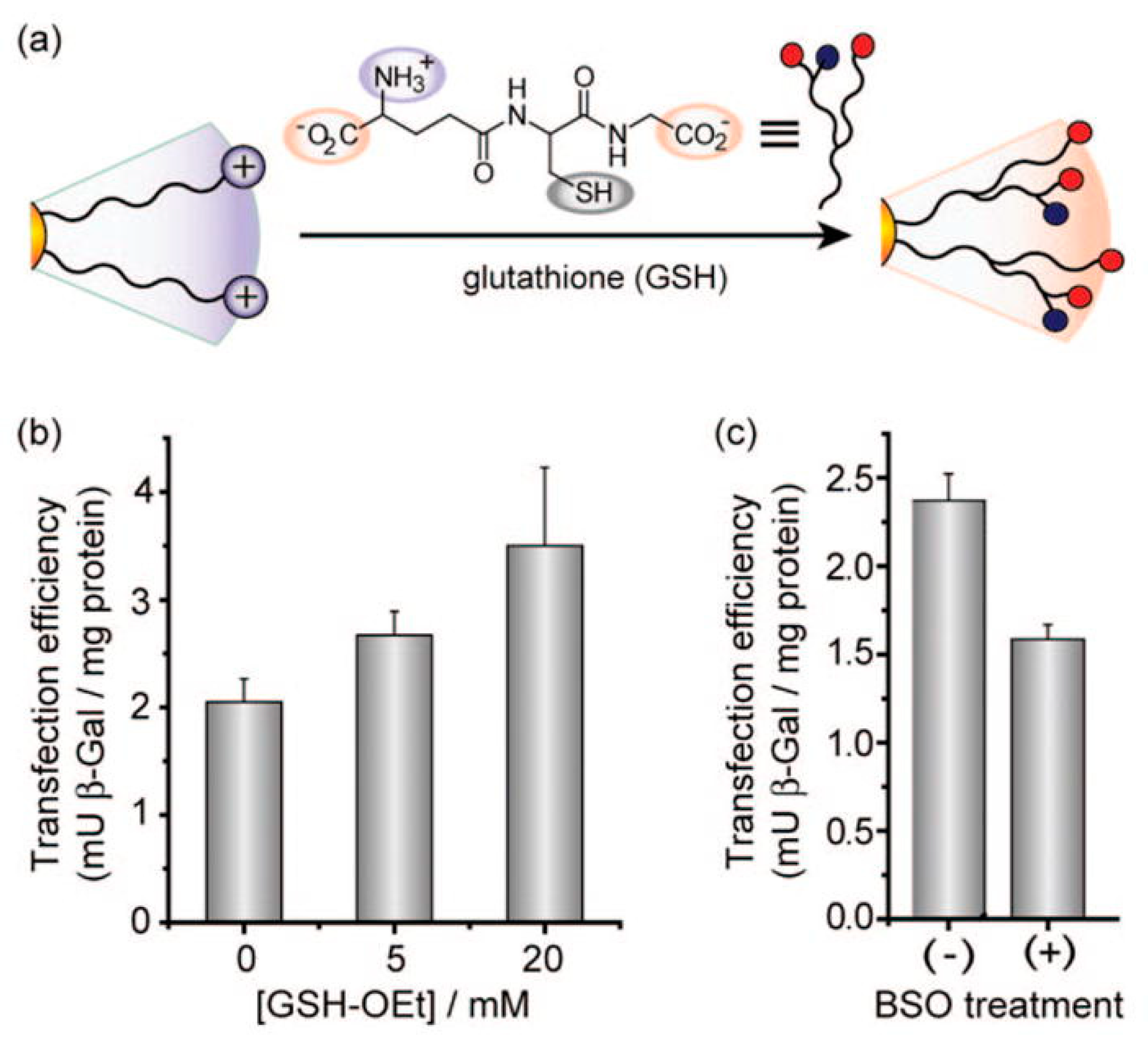
6.2. Intracellular-Targeted Drug Delivery
7. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| ABTS | 2,2′-Azino-bis(3-éthylbenzothiazoline-6-sulphonic) acid |
| AuNP | Gold nanoparticle |
| BSA | Bovine serum albumin |
| BSO | l-Buthionine-[S,R]-sulfoximine |
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| DIC | Dicarboxylate carrier |
| FRAP | Ferric reducing antioxidant power |
| GCL | γ-Glutamylcysteinyl ligase |
| GGT | γ-Glutamyltransferase |
| GPx | Glutathione peroxidase |
| GR | Glutathione disulfide reductase |
| GS | Glutathione synthase |
| GSH | Reduced glutathione |
| GSH-Oet | Glutathione monoester |
| GSNO | S-Nitrosoglutathione |
| GSOH | Glutathione sulfenic acid |
| GSSG | Disulfide glutathione |
| GSTs | Glutathione-S-transferases |
| LDL | Low-density lipoprotein |
| MRP | Multidrug resistance-associated protein |
| NAC | N-Acetylcysteine |
| NFE2L2 | Nuclear factor erythroid-derived 2-like 2 |
| NP | Nanoparticle |
| OGC | Oxoglutarate carrier |
| OPA | O-Phthalaldehyde |
| OTC | l-2-Oxothiazolidine-4-carboxylate |
| PEG | Poly(ethylene)glycol |
| PLGA | Poly(lactic-co-glycolic acid) |
| PSHs | Protein thiol groups |
| PSSGs | S-Glutathiolated proteins |
| PTCA | 2-(RS)-n-Propylthiazolidine-4(R)-carboxylic-acid |
| RyR1 | Ryanodine receptor type 1 |
| SAMe | S-Adenosylmethionine |
References
- Hamilton, C.J.; Arbach, M.; Groom, M. Beyond Glutathione: Different Low Molecular Weight Thiols as Mediators of Redox Regulation and Other Metabolic Functions in Lower Organisms. In Recent Advances in Redox Active Plant and Microbial Products; Jacob, C., Kirsch, G., Slusarenko, A., Winyard, P., Burkholz, T., Eds.; Springer: Dordrecht, The Netherlands, 2014; ISBN 978-94-017-8925-3. [Google Scholar]
- Gowland Hopkins, F.; Dixon, M. On glutathione. II. A thermostable oxidation-reduction system. J. Biol. Chem. 1922, 54, 527–563. [Google Scholar]
- Hunter, G.; Eagles, B.A. Glutathione: A critical study. J. Biol. Chem. 1927, 72, 147–166. [Google Scholar]
- Meister, A.; Tate, S.S. Glutathione and related gamma-glutamyl compounds: Biosynthesis and utilization. Annu. Rev. Biochem. 1976, 45, 559–604. [Google Scholar] [CrossRef] [PubMed]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef] [PubMed]
- Baldelli, S.; Aquilano, K.; Ciriolo, M.R. Punctum on two different transcription factors regulated by PGC-1α: Nuclear factor erythroid-derived 2-like 2 and nuclear respiratory factor 2. Biochim. Biophys. Acta 2013, 1830, 4137–4146. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shertzer, H.G.; Schneider, S.N.; Nebert, D.W.; Dalton, T.P. Glutamate cysteine ligase catalysis: Dependence on ATP and modifier subunit for regulation of tissue glutathione levels. J. Biol. Chem. 2005, 280, 33766–33774. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.G.; Nagy, L.E.; Bray, T.M. Nutritional and hormonal regulation of glutathione homeostasis. Curr. Top. Cell. Regul. 1996, 34, 189–208. [Google Scholar] [PubMed]
- Krejsa, C.M.; Franklin, C.C.; White, C.C.; Ledbetter, J.A.; Schieven, G.L.; Kavanagh, T.J. Rapid Activation of Glutamate Cysteine Ligase following Oxidative Stress. J. Biol. Chem. 2010, 285, 16116–16124. [Google Scholar] [CrossRef] [PubMed]
- Lauterburg, B.H.; Adams, J.D.; Mitchell, J.R. Hepatic glutathione homeostasis in the rat: Efflux accounts for glutathione turnover. Hepatology 1984, 4, 586–590. [Google Scholar] [CrossRef] [PubMed]
- DeLeve, L.D.; Kaplowitz, N. Glutathione metabolism and its role in hepatotoxicity. Pharmacol. Ther. 1991, 52, 287–305. [Google Scholar] [CrossRef]
- Cooper, A.J.; Pinto, J.T.; Callery, P.S. Reversible and irreversible protein glutathionylation: Biological and Clinical aspects. Expert Opin. Drug Metab. Toxicol. 2011, 7, 891–910. [Google Scholar] [CrossRef] [PubMed]
- Pineda-Molina, E.; Klatt, P.; Vázquez, J.; Marina, A.; García de Lacoba, M.; Pérez-Sala, D.; Lamas, S. Glutathionylation of the p50 subunit of NF-kappaB: A mechanism for redox-induced inhibition of DNA binding. Biochemistry 2001, 40, 14134–14142. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Pinto, J.T.; Deng, H.; Richie, J.P. Inhibition of caspase-3 activity and activation by protein glutathionylation. Biochem. Pharmacol. 2008, 75, 2234–2244. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Piemonte, F. Protein glutathionylation in cardiovascular diseases. Int. J. Mol. Sci. 2013, 14, 20845–20876. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Aw, T.Y. Glutathione and modulation of cell apoptosis. Biochim. Biophys. Acta 2012, 1823, 1767–1777. [Google Scholar] [CrossRef] [PubMed]
- Deponte, M. The Incomplete Glutathione Puzzle: Just Guessing at Numbers and Figures? Antioxid. Redox Signal. 2017, 27, 1130–1161. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, N.; Ikeda, Y. Gamma-glutamyl transpeptidase: Catalytic mechanism and gene expression. Adv. Enzymol. Relat. Areas Mol. Biol. 1998, 72, 239–278. [Google Scholar] [PubMed]
- Ohkama-Ohtsu, N.; Radwan, S.; Peterson, A.; Zhao, P.; Badr, A.F.; Xiang, C.; Oliver, D.J. Characterization of the extracellular gamma-glutamyl transpeptidases, GGT1 and GGT2, in Arabidposis. Plant J. 2007, 49, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Hanigan, M.H. Gamma-glutamyl transpeptidase: Redox regulation and drug resistance. Adv. Cancer Res. 2014, 122, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Forman, H.J. Redox regulation of gamma-glutamyl transpeptidase. Am. J. Respir. Cell Mol. Biol. 2009, 41, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Tikoo, S.; Maity, S.; Sengupta, S.; Sengupta, S.; Kaur, A.; Bachhawat, A.K. Mammalian proapoptotic factor ChaC1 and its homologues function as γ-glutamyl cyclotransferases acting specifically on glutathione. EMBO Rep. 2012, 13, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Gautam, R.; Srivastava, R.; Chandel, A.; Kumar, A.; Karthikeyan, S.; Bachhawat, A.K. ChaC2, an Enzyme for Slow Turnover of Cytosolic Glutathione. J. Biol. Chem. 2017, 292, 638–651. [Google Scholar] [CrossRef] [PubMed]
- Mungrue, I.N.; Pagnon, J.; Kohannim, O.; Gargalovic, P.S.; Lusis, A.J. CHAC1/MGC4504 is a novel proapoptotic component of the unfolded protein response, downstream of the ATF4-ATF3-CHOP cascade. J. Immunol. 2009, 182, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Lash, L.H. Mitochondrial glutathione transport: Physiological, pathological and toxicological implications. Chem. Biol. Interact. 2006, 163, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Bánhegyi, G.; Csala, M.; Nagy, G.; Sorrentino, V.; Fulceri, R.; Benedetti, A. Evidence for the transport of glutathione through ryanodine receptor channel type 1. Biochem. J. 2003, 376, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Csala, M.; Fulceri, R.; Mandl, J.; Benedetti, A.; Bánhegyi, G. Ryanodine receptor channel-dependent glutathione transport in the sarcoplasmic reticulum of skeletal muscle. Biochem. Biophys. Res. Commun. 2001, 287, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Belcastro, E.; Wu, W.; Fries-Raeth, I.; Corti, A.; Pompella, A.; Leroy, P.; Lartaud, I.; Gaucher, C. Oxidative stress enhances and modulates protein S-nitrosation in smooth muscle cells exposed to S-nitrosoglutathione. Nitric Oxide Biol. Chem. 2017, 69, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Ballatori, N.; Krance, S.M.; Marchan, R.; Hammond, C.L. Plasma membrane glutathione transporters and their roles in cell physiology and pathophysiology. Mol. Asp. Med. 2009, 30, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Muanprasat, C.; Wongborisuth, C.; Pathomthongtaweechai, N.; Satitsri, S.; Hongeng, S. Protection against oxidative stress in beta thalassemia/hemoglobin E erythrocytes by inhibitors of glutathione efflux transporters. PLoS ONE 2013, 8, e55685. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, D.A.; Forman, H.J. Cellular glutathione and thiols metabolism. Biochem. Pharmacol. 2002, 64, 1019–1026. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Nagy, P. Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol-oxidation pathways. Antioxid. Redox Signal. 2013, 18, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Paolicchi, A.; Dominici, S.; Pieri, L.; Maellaro, E.; Pompella, A. Glutathione catabolism as a signaling mechanism. Biochem. Pharmacol. 2002, 64, 1027–1035. [Google Scholar] [CrossRef]
- Dominici, S.; Paolicchi, A.; Corti, A.; Maellaro, E.; Pompella, A. Prooxidant reactions promoted by soluble and cell-bound gamma-glutamyltransferase activity. Methods Enzymol. 2005, 401, 484–501. [Google Scholar] [CrossRef] [PubMed]
- Drozdz, R.; Parmentier, C.; Hachad, H.; Leroy, P.; Siest, G.; Wellman, M. gamma-Glutamyltransferase dependent generation of reactive oxygen species from a glutathione/transferrin system. Free Radic. Biol. Med. 1998, 25, 786–792. [Google Scholar] [CrossRef]
- Dominici, S.; Pieri, L.; Comporti, M.; Pompella, A. Possible role of membrane gamma-glutamyltransferase activity in the facilitation of transferrin-dependent and -independent iron uptake by cancer cells. Cancer Cell Int. 2003, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Glass, G.A.; Stark, A.A. Promotion of glutathione-gamma-glutamyl transpeptidase-dependent lipid peroxidation by copper and ceruloplasmin: The requirement for iron and the effects of antioxidants and antioxidant enzymes. Environ. Mol. Mutagen. 1997, 29, 73–80. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Hori, O.; Brett, J.; Yan, S.D.; Wautier, J.L.; Stern, D. Cellular receptors for advanced glycation end products. Implications for induction of oxidant stress and cellular dysfunction in the pathogenesis of vascular lesions. Arterioscler. Thromb. J. Vasc. Biol. 1994, 14, 1521–1528. [Google Scholar] [CrossRef]
- Gimbrone, M.A. Vascular endothelium: An integrator of pathophysiologic stimuli in atherosclerosis. Am. J. Cardiol. 1995, 75, 67B–70B. [Google Scholar] [CrossRef]
- Ross, R. The pathogenesis of atherosclerosis: A perspective for the 1990s. Nature 1993, 362, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.R. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Paolicchi, A.; Minotti, G.; Tonarelli, P.; Tongiani, R.; De Cesare, D.; Mezzetti, A.; Dominici, S.; Comporti, M.; Pompella, A. Gamma-glutamyl transpeptidase-dependent iron reduction and LDL oxidation—A potential mechanism in atherosclerosis. J. Investig. Med. 1999, 47, 151–160. [Google Scholar] [PubMed]
- Berliner, J.A.; Heinecke, J.W. The role of oxidized lipoproteins in atherogenesis. Free Radic. Biol. Med. 1996, 20, 707–727. [Google Scholar] [CrossRef]
- Jones, D.P. Redox potential of GSH/GSSG couple: Assay and biological significance. Methods Enzymol. 2002, 348, 93–112. [Google Scholar] [PubMed]
- Sen, C.K. Cellular thiols and redox-regulated signal transduction. Curr. Top. Cell. Regul. 2000, 36, 1–30. [Google Scholar] [PubMed]
- Conour, J.E.; Graham, W.V.; Gaskins, H.R. A combined in vitro/bioinformatic investigation of redox regulatory mechanisms governing cell cycle progression. Physiol. Genom. 2004, 18, 196–205. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Menon, S.G.; Sarsour, E.H.; Spitz, D.R.; Higashikubo, R.; Sturm, M.; Zhang, H.; Goswami, P.C. Redox regulation of the G1 to S phase transition in the mouse embryo fibroblast cell cycle. Cancer Res. 2003, 63, 2109–2117. [Google Scholar] [PubMed]
- Menon, S.G.; Sarsour, E.H.; Kalen, A.L.; Venkataraman, S.; Hitchler, M.J.; Domann, F.E.; Oberley, L.W.; Goswami, P.C. Superoxide signaling mediates N-acetyl-L-cysteine-induced G1 arrest: Regulatory role of cyclin D1 and manganese superoxide dismutase. Cancer Res. 2007, 67, 6392–6399. [Google Scholar] [CrossRef] [PubMed]
- Belcastro, E.; Gaucher, C.; Corti, A.; Leroy, P.; Lartaud, I.; Pompella, A. Regulation of protein function by S-nitrosation and S-glutathionylation: Processes and targets in cardiovascular pathophysiology. Biol. Chem. 2017, 398, 1267–1293. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A. Redox regulation by thioredoxin and thioredoxin reductase. BioFactors 2000, 11, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Federici, G.; Bertini, E.; Piemonte, F. Analysis of glutathione: Implication in redox and detoxification. Clin. Chim. Acta 2003, 333, 19–39. [Google Scholar] [CrossRef]
- Schulz, J.B.; Lindenau, J.; Seyfried, J.; Dichgans, J. Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem. 2000, 267, 4904–4911. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.F.; Nilsson, M.; Eriksson, P.S.; Sims, N.R. Glutathione monoethyl ester provides neuroprotection in a rat model of stroke. Neurosci. Lett. 2004, 354, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Vargas, F.; Rodríguez-Gómez, I.; Pérez-Abud, R.; Vargas Tendero, P.; Baca, Y.; Wangensteen, R. Cardiovascular and renal manifestations of glutathione depletion induced by buthionine sulfoximine. Am. J. Hypertens. 2012, 25, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Beutler, E.; Duron, O.; Kelly, B.M. Improved method for the determination of blood glutathione. J. Lab. Clin. Med. 1963, 61, 882–888. [Google Scholar] [PubMed]
- Mansoor, M.A.; Svardal, A.M.; Ueland, P.M. Determination of the in vivo redox status of cysteine, cysteinylglycine, homocysteine, and glutathione in human plasma. Anal. Biochem. 1992, 200, 218–229. [Google Scholar] [CrossRef]
- Tietze, F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: Applications to mammalian blood and other tissues. Anal. Biochem. 1969, 27, 502–522. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, Z.; Chen, J.; Wang, S.; Huang, X. Sensitive and selective detection of glutathione based on resonance light scattering using sensitive gold nanoparticles as colorimetric probes. Analyst 2012, 137, 3132–3137. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Hepel, M. “Molecular beacon”—Based fluorescent assay for selective detection of glutathione and cysteine. Anal. Chem. 2011, 83, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, G.; Fiorani, M.; Biagiarelli, B.; Palma, F.; Potenza, L.; Amicucci, A.; Stocchi, V. Simultaneous high-performance capillary electrophoretic determination of reduced and oxidized glutathione in red blood cells in the femtomole range. J. Chromatogr. A 1994, 676, 239–246. [Google Scholar] [CrossRef]
- Park, S.K.; Boulton, R.B.; Noble, A.C. Automated HPLC analysis of glutathione and thiol-containing compounds in grape juice and wine using pre-column derivatization with fluorescence detection. Food Chem. 2000, 68, 475–480. [Google Scholar] [CrossRef]
- Parmentier, C.; Leroy, P.; Wellman, M.; Nicolas, A. Determination of cellular thiols and glutathione-related enzyme activities: Versatility of high-performance liquid chromatography-spectrofluorimetric detection. J. Chromatogr. B. Biomed. Sci. Appl. 1998, 719, 37–46. [Google Scholar] [CrossRef]
- Parmentier, C.; Wellman, M.; Nicolas, A.; Siest, G.; Leroy, P. Simultaneous measurement of reactive oxygen species and reduced glutathione using capillary electrophoresis and laser-induced fluorescence detection in cultured cell lines. Electrophoresis 1999, 20, 2938–2944. [Google Scholar] [CrossRef]
- Lewicki, K.; Marchand, S.; Matoub, L.; Lulek, J.; Coulon, J.; Leroy, P. Development of a fluorescence-based microtiter plate method for the measurement of glutathione in yeast. Talanta 2006, 70, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, B.; Khosropour, H.; Ensafi, A.A.; Hadadzadeh, H.; Farrokhpour, H. A Differential Pulse Voltammetric Sensor for Determination of Glutathione in Real Samples Using a Trichloro(terpyridine)ruthenium(III)/Multiwall Carbon Nanotubes Modified Paste Electrode. IEEE Sens. J. 2015, 15, 483–490. [Google Scholar] [CrossRef]
- Burford, N.; Eelman, M.D.; Mahony, D.E.; Morash, M. Definitive identification of cysteine and glutathione complexes of bismuth by mass spectrometry: Assessing the biochemical fate of bismuth pharmaceutical agents. Chem. Commun. 2003, 1, 146–147. [Google Scholar] [CrossRef]
- Han, H.-Y.; He, Z.-K.; Zeng, Y.-E. Chemiluminescence method for the determination of glutathione in human serum using the Ru(phen)32+-KMnO4 system. Microchim. Acta 2006, 155, 431–434. [Google Scholar] [CrossRef]
- Mandal, P.K.; Tripathi, M.; Sugunan, S. Brain oxidative stress: Detection and mapping of anti-oxidant marker “Glutathione” in different brain regions of healthy male/female, MCI and Alzheimer patients using non-invasive magnetic resonance spectroscopy. Biochem. Biophys. Res. Commun. 2012, 417, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.G.; Hossain, M.K.; Han, X.X.; Ozaki, Y. A novel reversed reporting agent method for surface-enhanced Raman scattering; highly sensitive detection of glutathione in aqueous solutions. Analyst 2009, 134, 2468–2474. [Google Scholar] [CrossRef] [PubMed]
- Vallverdú-Queralt, A.; Verbaere, A.; Meudec, E.; Cheynier, V.; Sommerer, N. Straightforward method to quantify GSH, GSSG, GRP, and hydroxycinnamic acids in wines by UPLC-MRM-MS. J. Agric. Food Chem. 2015, 63, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Parent, M.; Dahboul, F.; Schneider, R.; Clarot, I.; Maincent, P.; Leroy, P.; Boudier, A. A Complete Physicochemical Identity Card of S-Nitrosoglutathione. Available online: http://www.eurekaselect.com/105996/article (accessed on 22 March 2018).
- Neuschwander-Tetri, B.A.; Roll, F.J. Glutathione measurement by high-performance liquid chromatography separation and fluorometric detection of the glutathione-orthophthalaldehyde adduct. Anal. Biochem. 1989, 179, 236–241. [Google Scholar] [CrossRef]
- Serru, V.; Baudin, B.; Ziegler, F.; David, J.P.; Cals, M.J.; Vaubourdolle, M.; Mario, N. Quantification of reduced and oxidized glutathione in whole blood samples by capillary electrophoresis. Clin. Chem. 2001, 47, 1321–1324. [Google Scholar] [PubMed]
- European Department for the Quality of Medicines. Glutathione, Monograph 01/2017: 1670, European Pharmacopoeia; European Department for the Quality of Medicines: Strasbourg, France, 2018. [Google Scholar]
- Pensa, E.; Cortés, E.; Corthey, G.; Carro, P.; Vericat, C.; Fonticelli, M.H.; Benítez, G.; Rubert, A.A.; Salvarezza, R.C. The chemistry of the sulfur-gold interface: In search of a unified model. Acc. Chem. Res. 2012, 45, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Monteiro-Riviere, N.A.; Riviere, J.E. Pharmacokinetics of metallic nanoparticles. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2015, 7, 189–217. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, A.; Chakraborty, K.; Shukla, A. Cellular copper homeostasis: Current concepts on its interplay with glutathione homeostasis and its implication in physiology and human diseases. Metallomics 2017, 9, 1376–1388. [Google Scholar] [CrossRef] [PubMed]
- Reimers, J.R.; Ford, M.J.; Marcuccio, S.M.; Ulstrup, J.; Hush, N.S. Competition of van der Waals and chemical forces on gold–sulfur surfaces and nanoparticles. Nat. Rev. Chem. 2017, 1, 17. [Google Scholar] [CrossRef]
- Tsogas, G.Z.; Kappi, F.A.; Vlessidis, A.G.; Giokas, D.L. Recent Advances in Nanomaterial Probes for Optical Biothiol Sensing: A Review. Anal. Lett. 2018, 51, 443–468. [Google Scholar] [CrossRef]
- Li, Z.-J.; Zheng, X.-J.; Zhang, L.; Liang, R.-P.; Li, Z.-M.; Qiu, J.-D. Label-free colorimetric detection of biothiols utilizing SAM and unmodified Au nanoparticles. Biosens. Bioelectron. 2015, 68, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-F.; Huang, P.-C.; Wu, F.-Y. Highly selective and sensitive detection of glutathione based on anti-aggregation of gold nanoparticles via pH regulation. Sens. Actuators B Chem. 2017, 240, 553–559. [Google Scholar] [CrossRef]
- Shen, C.-C.; Tseng, W.-L.; Hsieh, M.-M. Selective enrichment of aminothiols using polysorbate 20-capped gold nanoparticles followed by capillary electrophoresis with laser-induced fluorescence. J. Chromatogr. A 2009, 1216, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.-M.; Chen, Q.; Sun, Z.-Y.; Chen, X.-W.; Wang, J.-H. Assay of biothiols by regulating the growth of silver nanoparticles with C-dots as reducing agent. Anal. Chem. 2014, 86, 5002–5008. [Google Scholar] [CrossRef] [PubMed]
- Kappi, F.A.; Papadopoulos, G.A.; Tsogas, G.Z.; Giokas, D.L. Low-cost colorimetric assay of biothiols based on the photochemical reduction of silver halides and consumer electronic imaging devices. Talanta 2017, 172, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Koehler, Y.; Derr, L.; Tomba, G.; Schmidt, M.M.; Treccani, L.; Colombi Ciacchi, L.; Rezwan, K. Adsorption and reduction of glutathione disulfide on α-Al2O3 nanoparticles: Experiments and modeling. Langmuir 2011, 27, 9449–9457. [Google Scholar] [CrossRef] [PubMed]
- Barman, U.; Mukhopadhyay, G.; Goswami, N.; Ghosh, S.S.; Paily, R.P. Detection of Glutathione by Glutathione-S-Transferase-Nanoconjugate Ensemble Electrochemical Device. IEEE Trans. Nanobiosci. 2017, 16, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Estrela, J.; Obrador, E.; Navarro, J.; Delavega, M.; Pellicer, J. Elimination of Ehrlich Tumors by ATP-Induced Growth-Inhibition, Glutathione Depletion and X-Rays. Nat. Med. 1995, 1, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Mena, S.; Benlloch, M.; Ortega, A.; Carretero, J.; Obrador, E.; Asensi, M.; Petschen, I.; Brown, B.D.; Estrela, J.M. Bcl-2 and glutathione depletion sensitizes B16 melanoma to combination therapy and eliminates metastatic disease. Clin. Cancer Res. 2007, 13, 2658–2666. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.R.; Garcia, C.C.M.; Vieira, D.B.; Quinet, A.; de Andrade-Lima, L.C.; Munford, V.; Belizario, J.E.; Menck, C.F.M. Glutathione depletion sensitizes cisplatin- and temozolomide-resistant glioma cells in vitro and in vivo. Cell Death Dis. 2014, 5, e1505. [Google Scholar] [CrossRef] [PubMed]
- Lagman, M.; Ly, J.; Saing, T.; Kaur Singh, M.; Vera Tudela, E.; Morris, D.; Chi, P.-T.; Ochoa, C.; Sathananthan, A.; Venketaraman, V. Investigating the causes for decreased levels of glutathione in individuals with type II diabetes. PLoS ONE 2015, 10, e0118436. [Google Scholar] [CrossRef] [PubMed]
- Herzenberg, L.A.; DeRosa, S.C.; Dubs, J.G.; Roederer, M.; Anderson, M.T.; Ela, S.W.; Deresinski, S.C.; Herzenberg, L.A. Glutathione deficiency is associated with impaired survival in HIV disease. Proc. Natl. Acad. Sci. USA 1997, 94, 1967–1972. [Google Scholar] [CrossRef] [PubMed]
- Biolo, G.; Antonione, R.; De Cicco, M. Glutathione metabolism in sepsis. Crit. Care Med. 2007, 35, S591–S595. [Google Scholar] [CrossRef] [PubMed]
- Day, B.J. Glutathione—A radical treatment for cystic fibrosis lung disease? Chest 2005, 127, 12–14. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.L.; Teismann, P. Glutathione—A review on its role and significance in Parkinson’s disease. FASEB J. 2009, 23, 3263–3272. [Google Scholar] [CrossRef] [PubMed]
- Pocernich, C.B.; Butterfield, D.A. Elevation of glutathione as a therapeutic strategy in Alzheimer disease. Biochim. Biophys. Acta 2012, 1822, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Chauhan, V.; Chauhan, A. Glutathione redox imbalance in brain disorders. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P.; Mody, V.C.; Carlson, J.L.; Lynn, M.J.; Sternberg, P. Redox analysis of human plasma allows separation of pro-oxidant events of aging from decline in antioxidant defenses. Free Radic. Biol. Med. 2002, 33, 1290–1300. [Google Scholar] [CrossRef]
- Witschi, A.; Reddy, S.; Stofer, B.; Lauterburg, B.H. The systemic availability of oral glutathione. Eur. J. Clin. Pharmacol. 1992, 43, 667–669. [Google Scholar] [CrossRef] [PubMed]
- Shibui, Y.; Sakai, R.; Manabe, Y.; Masuyama, T. Comparisons of l-cysteine and d-cysteine toxicity in 4-week repeated-dose toxicity studies of rats receiving daily oral administration. J. Toxicol. Pathol. 2017, 30, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Greene, S.C.; Noonan, P.K.; Sanabria, C.; Peacock, W.F. Effervescent N-Acetylcysteine Tablets versus Oral Solution N-Acetylcysteine in Fasting Healthy Adults: An Open-Label, Randomized, Single-Dose, Crossover, Relative Bioavailability Study. Curr. Ther. Res. Clin. Exp. 2016, 83, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W.; Breitkreutz, R. N-acetyl-cysteine in the therapy of HIV-positive patients. Curr. Opin. Clin. Nutr. Metab. Care 1999, 2, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Borgström, L.; Kågedal, B.; Paulsen, O. Pharmacokinetics of N-acetylcysteine in man. Eur. J. Clin. Pharmacol. 1986, 31, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Grinberg, L.; Fibach, E.; Amer, J.; Atlas, D. N-acetylcysteine amide, a novel cell-permeating thiol, restores cellular glutathione and protects human red blood cells from oxidative stress. Free Radic. Biol. Med. 2005, 38, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.P.; Sullivan, P.G.; Pandya, J.D.; Goldstein, G.A.; VanRooyen, J.L.; Yonutas, H.M.; Eldahan, K.C.; Morehouse, J.; Magnuson, D.S.K.; Rabchevsky, A.G. N-acetylcysteine amide preserves mitochondrial bioenergetics and improves functional recovery following spinal trauma. Exp. Neurol. 2014, 257, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Giustarini, D.; Milzani, A.; Dalle-Donne, I.; Tsikas, D.; Rossi, R. N-Acetylcysteine ethyl ester (NACET): A novel lipophilic cell-permeable cysteine derivative with an unusual pharmacokinetic feature and remarkable antioxidant potential. Biochem. Pharmacol. 2012, 84, 1522–1533. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.E.; Powrie, F.; Puri, R.N.; Meister, A. Glutathione monoethyl ester: Preparation, uptake by tissues, and conversion to glutathione. Arch. Biochem. Biophys. 1985, 239, 538–548. [Google Scholar] [CrossRef]
- Grattagliano, I.; Wieland, P.; Schranz, C.; Lauterburg, B.H. Effect of oral glutathione monoethyl ester and glutathione on circulating and hepatic sulfhydrils in the rat. Pharmacol. Toxicol. 1994, 75, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Minhas, H.S.; Thornalley, P.J. Comparison of the delivery of reduced glutathione into P388D1 cells by reduced glutathione and its mono- and diethyl ester derivatives. Biochem. Pharmacol. 1995, 49, 1475–1482. [Google Scholar] [CrossRef]
- Zampagni, M.; Wright, D.; Cascella, R.; D’Adamio, G.; Casamenti, F.; Evangelisti, E.; Cardona, F.; Goti, A.; Nacmias, B.; Sorbi, S.; et al. Novel S-acyl glutathione derivatives prevent amyloid oxidative stress and cholinergic dysfunction in Alzheimer disease models. Free Radic. Biol. Med. 2012, 52, 1362–1371. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. S-Adenosyl-L-methionine and alcoholic liver disease in animal models: Implications for early intervention in human beings. Alcohol 2002, 27, 173–177. [Google Scholar] [CrossRef]
- Fawcett, J.P.; Schiller, B.; Jiang, R.; Moran, J.; Walker, R.J. Supplementation with L-2-oxothiazolidine-4-carboxylic acid, a cysteine precursor, does not protect against lipid peroxidation in puromycin aminonucleoside-induced nephropathy. Exp. Nephrol. 1996, 4, 248–252. [Google Scholar] [PubMed]
- Oz, H.S.; Chen, T.S.; Nagasawa, H. Comparative efficacies of 2 cysteine prodrugs and a glutathione delivery agent in a colitis model. Transl. Res. J. Lab. Clin. Med. 2007, 150, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Zarka, M.H.; Bridge, W.J. Oral administration of γ-glutamylcysteine increases intracellular glutathione levels above homeostasis in a randomised human trial pilot study. Redox Biol. 2017, 11, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Rosenblat, M.; Volkova, N.; Coleman, R.; Aviram, M. Anti-oxidant and anti-atherogenic properties of liposomal glutathione: Studies in vitro, and in the atherosclerotic apolipoprotein E-deficient mice. Atherosclerosis 2007, 195, e61–e68. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Du, Y.; Li, D.; Alany, R. Development of water-in-oil microemulsions with the potential of prolonged release for oral delivery of L-glutathione. Pharm. Dev. Technol. 2013, 18, 1424–1429. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; Choy, J.-H.; Choi, S.-J. Montmorillonite intercalated with glutathione for antioxidant delivery: Synthesis, characterization, and bioavailability evaluation. Int. J. Pharm. 2012, 425, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Trapani, A.; Laquintana, V.; Denora, N.; Lopedota, A.; Cutrignelli, A.; Franco, M.; Trapani, G.; Liso, G. Eudragit RS 100 microparticles containing 2-hydroxypropyl-β-cyclodextrin and glutathione: Physicochemical characterization, drug release and transport studies. Eur. J. Pharm. Sci. 2007, 30, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Trapani, A.; Lopedota, A.; Franco, M.; Cioffi, N.; Ieva, E.; Garcia-Fuentes, M.; Alonso, M.J. A comparative study of chitosan and chitosan/cyclodextrin nanoparticles as potential carriers for the oral delivery of small peptides. Eur. J. Pharm. Biopharm. 2010, 75, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Naji-Tabasi, S.; Razavi, S.M.A.; Mehditabar, H. Fabrication of basil seed gum nanoparticles as a novel oral delivery system of glutathione. Carbohydr. Polym. 2017, 157, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Mandracchia, D.; Denora, N.; Franco, M.; Pitarresi, G.; Giammona, G.; Trapani, G. New Biodegradable Hydrogels Based on Inulin and alpha,beta-Polyaspartylhydrazide Designed for Colonic Drug Delivery: In Vitro Release of Glutathione and Oxytocin. J. Biomater. Sci. Polym. Ed. 2011, 22, 313–328. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, G.; Bunt, C.; Wen, J. Mucoadhesive polymers-based film as a carrier system for sublingual delivery of glutathione. J. Pharm. Pharmacol. 2015, 67, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Buonocore, D.; Grosini, M.; Giardina, S.; Michelotti, A.; Carrabetta, M.; Seneci, A.; Verri, M.; Dossena, M.; Marzatico, F. Bioavailability Study of an Innovative Orobuccal Formulation of Glutathione. Oxid. Med. Cell. Longev. 2016, 2016, 3286365. [Google Scholar] [CrossRef] [PubMed]
- Beqa, L.; Singh, A.K.; Khan, S.A.; Senapati, D.; Arumugam, S.R.; Ray, P.C. Gold nanoparticle-based simple colorimetric and ultrasensitive dynamic light scattering assay for the selective detection of Pb(II) from paints, plastics, and water samples. ACS Appl. Mater. Interfaces 2011, 3, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Jia, J.; Lai, Y.; Ma, Y.; Weng, J.; Sun, L. Conjugating folic acid to gold nanoparticles through glutathione for targeting and detecting cancer cells. Bioorg. Med. Chem. 2010, 18, 5528–5534. [Google Scholar] [CrossRef] [PubMed]
- Valusová, E.; Svec, P.; Antalík, M. Structural and thermodynamic behavior of cytochrome c assembled with glutathione-covered gold nanoparticles. J. Biol. Inorg. Chem. 2009, 14, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Polavarapu, L.; Manna, M.; Xu, Q.-H. Biocompatible glutathione capped gold clusters as one- and two-photon excitation fluorescence contrast agents for live cells imaging. Nanoscale 2011, 3, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Simpson, C.A.; Salleng, K.J.; Cliffel, D.E.; Feldheim, D.L. In vivo toxicity, biodistribution, and clearance of glutathione-coated gold nanoparticles. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.H.; Lee, J.-S.; Kim, G.-H.; Lee, H.G. Preparation, Characteristics, and Stability of Glutathione-Loaded Nanoparticles. J. Agric. Food Chem. 2011, 59, 11264–11269. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.R.; Lepene, B.S.; Thatcher, C.D.; Long, T.E. Synthesis and characterization of poly(ethylene glycol)-glutathione conjugate self-assembled nanoparticles for antioxidant delivery. Biomacromolecules 2009, 10, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Tortiglione, C.; Quarta, A.; Tino, A.; Manna, L.; Cingolani, R.; Pellegrino, T. Synthesis and biological assay of GSH functionalized fluorescent quantum dots for staining Hydra vulgaris. Bioconjug. Chem. 2007, 18, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Tournebize, J.; Boudier, A.; Sapin-Minet, A.; Maincent, P.; Leroy, P.; Schneider, R. Role of gold nanoparticles capping density on stability and surface reactivity to design drug delivery platforms. ACS Appl. Mater. Interfaces 2012, 4, 5790–5799. [Google Scholar] [CrossRef] [PubMed]
- Tournebize, J.; Boudier, A.; Joubert, O.; Eidi, H.; Bartosz, G.; Maincent, P.; Leroy, P.; Sapin-Minet, A. Impact of gold nanoparticle coating on redox homeostasis. Int. J. Pharm. 2012, 438, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Boudier, A.; Clarot, I.; Maincent, P.; Schneider, R.; Leroy, P. Gold Nanoparticles Grafted by Reduced Glutathione With Thiol Function Preservation. Colloid Interface Sci. Commun. 2016, 14, 8–12. [Google Scholar] [CrossRef]
- Levin, V.A. Relationship of octanol/water partition coefficient and molecular weight to rat brain capillary permeability. J. Med. Chem. 1980, 23, 682–684. [Google Scholar] [CrossRef] [PubMed]
- Kannan, R.; Kuhlenkamp, J.F.; Jeandidier, E.; Trinh, H.; Ookhtens, M.; Kaplowitz, N. Evidence for carrier-mediated transport of glutathione across the blood-brain barrier in the rat. J. Clin. Investig. 1990, 85, 2009–2013. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V.; Mackic, J.B.; McComb, J.G.; Weiss, M.H.; Kaplowitz, N.; Kannan, R. Evidence for transcapillary transport of reduced glutathione in vascular perfused guinea-pig brain. Biochem. Biophys. Res. Commun. 1994, 201, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Kannan, R.; Chakrabarti, R.; Tang, D.; Kim, K.J.; Kaplowitz, N. GSH transport in human cerebrovascular endothelial cells and human astrocytes: Evidence for luminal localization of Na+-dependent GSH transport in HCEC. Brain Res. 2000, 852, 374–382. [Google Scholar] [CrossRef]
- Rip, J.; Chen, L.; Hartman, R.; van den Heuvel, A.; Reijerkerk, A.; van Kregten, J.; van der Boom, B.; Appeldoorn, C.; de Boer, M.; Maussang, D.; et al. Glutathione PEGylated liposomes: Pharmacokinetics and delivery of cargo across the blood-brain barrier in rats. J. Drug Target. 2014, 22, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Rotman, M.; Welling, M.M.; Bunschoten, A.; de Backer, M.E.; Rip, J.; Nabuurs, R.J.A.; Gaillard, P.J.; van Buchem, M.A.; van der Maarel, S.M.; van der Weerd, L. Enhanced glutathione PEGylated liposomal brain delivery of an anti-amyloid single domain antibody fragment in a mouse model for Alzheimer’s disease. J. Control. Release 2015, 203, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Maussang, D.; Rip, J.; van Kregten, J.; van den Heuvel, A.; van der Pol, S.; van der Boom, B.; Reijerkerk, A.; Chen, L.; de Boer, M.; Gaillard, P.; et al. Glutathione conjugation dose-dependently increases brain-specific liposomal drug delivery in vitro and in vivo. Drug Discov. Today Technol. 2016, 20, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Birngruber, T.; Raml, R.; Gladdines, W.; Gatschelhofer, C.; Gander, E.; Ghosh, A.; Kroath, T.; Gaillard, P.J.; Pieber, T.R.; Sinner, F. Enhanced doxorubicin delivery to the brain administered through glutathione PEGylated liposomal doxorubicin (2B3-101) as compared with generic Caelyx,(®)/Doxil(®)—A cerebral open flow microperfusion pilot study. J. Pharm. Sci. 2014, 103, 1945–1948. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, P.J.; Appeldoorn, C.C.M.; Dorland, R.; van Kregten, J.; Manca, F.; Vugts, D.J.; Windhorst, B.; van Dongen, G.A.M.S.; de Vries, H.E.; Maussang, D.; et al. Pharmacokinetics, brain delivery, and efficacy in brain tumor-bearing mice of glutathione pegylated liposomal doxorubicin (2B3-101). PLoS ONE 2014, 9, e82331. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, P.J.; Appeldoorn, C.C.M.; Rip, J.; Dorland, R.; van der Pol, S.M.A.; Kooij, G.; de Vries, H.E.; Reijerkerk, A. Enhanced brain delivery of liposomal methylprednisolone improved therapeutic efficacy in a model of neuroinflammation. J. Control. Release 2012, 164, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Kanhai, K.M.S.; Zuiker, R.G.J.A.; Stavrakaki, I.; Gladdines, W.; Gaillard, P.J.; Klaassen, E.S.; Groeneveld, G.J. Glutathione-PEGylated liposomal methylprednisolone in comparison to free methylprednisolone: Slow release characteristics and prolonged lymphocyte depression in a first-in-human study. Br. J. Clin. Pharmacol. 2018. [CrossRef] [PubMed]
- Gaillard, P.J.; Kerklaan, B.M.; Aftimos, P.; Altintas, S.; Jager, A.; Gladdines, W.; Lonnqvist, F.; Soetekouw, P.; Verheul, H.; Awada, A.; et al. Abstract CT216: Phase I dose escalating study of 2B3-101, glutathione PEGylated liposomal doxorubicin, in patients with solid tumors and brain metastases or recurrent malignant glioma. Cancer Res. 2014, 74, CT216. [Google Scholar] [CrossRef]
- Brandsma, D.; Kerklaan, B.M.; Diéras, V.; Altintas, S.; Anders, C.K.; Ballester, M.A.; Gelderblom, H.; Soetekouw, P.M.M.B.; Gladdines, W.; Lonnqvist, F.; et al. Phase 1/2a study of glutathione pegylated liposomal doxorubicin (2b3-101) in patients with brain metastases (BM) from solid tumors or recurrent high grade gliomas (HGG). Ann. Oncol. 2014, 25, iv157–iv158. [Google Scholar] [CrossRef]
- Veszelka, S.; Meszaros, M.; Kiss, L.; Kota, Z.; Pali, T.; Hoyk, Z.; Bozso, Z.; Fulop, L.; Toth, A.; Rakhely, G.; et al. Biotin and Glutathione Targeting of Solid Nanoparticles to Cross Human Brain Endothelial Cells. Curr. Pharm. Des. 2017, 23, 4198–4205. [Google Scholar] [CrossRef] [PubMed]
- Grover, A.; Hirani, A.; Pathak, Y.; Sutariya, V. Brain-targeted delivery of docetaxel by glutathione-coated nanoparticles for brain cancer. AAPS PharmSciTech 2014, 15, 1562–1568. [Google Scholar] [CrossRef] [PubMed]
- Geldenhuys, W.; Wehrung, D.; Groshev, A.; Hirani, A.; Sutariya, V. Brain-targeted delivery of doxorubicin using glutathione-coated nanoparticles for brain cancers. Pharm. Dev. Technol. 2015, 20, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Geldenhuys, W.; Mbimba, T.; Bui, T.; Harrison, K.; Sutariya, V. Brain-targeted delivery of paclitaxel using glutathione-coated nanoparticles for brain cancers. J. Drug Target. 2011, 19, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Raval, N.; Mistry, T.; Acharya, N.; Acharya, S. Development of glutathione-conjugated asiatic acid-loaded bovine serum albumin nanoparticles for brain-targeted drug delivery. J. Pharm. Pharmacol. 2015, 67, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Acharya, N.; Acharya, S. Development and characterization of glutathione-conjugated albumin nanoparticles for improved brain delivery of hydrophilic fluorescent marker. Drug Deliv. 2013, 20, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Mdzinarishvili, A.; Sutariya, V.; Talasila, P.K.; Geldenhuys, W.J.; Sadana, P. Engineering triiodothyronine (T3) nanoparticle for use in ischemic brain stroke. Drug Deliv. Transl. Res. 2013, 3, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Englert, C.; Trützschler, A.-K.; Raasch, M.; Bus, T.; Borchers, P.; Mosig, A.S.; Traeger, A.; Schubert, U.S. Crossing the blood-brain barrier: Glutathione-conjugated poly(ethylene imine) for gene delivery. J. Control. Release 2016, 241, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Chen, Y.; Zhang, Z.-H.; Han, X. Stimuli-Responsive Block Copolymer-Based Assemblies for Cargo Delivery and Theranostic Applications. Polymers 2016, 8, 268. [Google Scholar] [CrossRef]
- Liu, X.; Yang, Y.; Urban, M.W. Stimuli-Responsive Polymeric Nanoparticles. Macromol. Rapid Commun. 2017, 38, 13. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Meng, F.; Deng, C.; Klok, H.-A.; Zhong, Z. Dual and multi-stimuli responsive polymeric nanoparticles for programmed site-specific drug delivery. Biomaterials 2013, 34, 3647–3657. [Google Scholar] [CrossRef] [PubMed]
- Son, S.; Namgung, R.; Kim, J.; Singha, K.; Kim, W.J. Bioreducible polymers for gene silencing and delivery. Acc. Chem. Res. 2012, 45, 1100–1112. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Li, Y.; Xu, H.; Wang, Z.; Zhang, X. Dual redox responsive assemblies formed from diselenide block copolymers. J. Am. Chem. Soc. 2010, 132, 442–443. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Zhang, X.-Y.; Wang, Y.-L.; Li, X.; Yang, X.-H.; Huang, M.; Hu, K.; Li, L.-H.; Wei, Y. Redox-responsive theranostic nanoplatforms based on inorganic nanomaterials. J. Control. Release 2017, 259, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.S.; Kim, C.-K.; Han, G.; Forbes, N.S.; Rotello, V.M. Efficient Gene Delivery Vectors by Tuning the Surface Charge Density of Amino Acid-Functionalized Gold Nanoparticles. ACS Nano 2008, 2, 2213–2218. [Google Scholar] [CrossRef] [PubMed]
- Tournebize, J.; Sapin-Minet, A.; Bartosz, G.; Leroy, P.; Boudier, A. Pitfalls of assays devoted to evaluation of oxidative stress induced by inorganic nanoparticles. Talanta 2013, 116, 753–763. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaucher, C.; Boudier, A.; Bonetti, J.; Clarot, I.; Leroy, P.; Parent, M. Glutathione: Antioxidant Properties Dedicated to Nanotechnologies. Antioxidants 2018, 7, 62. https://doi.org/10.3390/antiox7050062
Gaucher C, Boudier A, Bonetti J, Clarot I, Leroy P, Parent M. Glutathione: Antioxidant Properties Dedicated to Nanotechnologies. Antioxidants. 2018; 7(5):62. https://doi.org/10.3390/antiox7050062
Chicago/Turabian StyleGaucher, Caroline, Ariane Boudier, Justine Bonetti, Igor Clarot, Pierre Leroy, and Marianne Parent. 2018. "Glutathione: Antioxidant Properties Dedicated to Nanotechnologies" Antioxidants 7, no. 5: 62. https://doi.org/10.3390/antiox7050062
APA StyleGaucher, C., Boudier, A., Bonetti, J., Clarot, I., Leroy, P., & Parent, M. (2018). Glutathione: Antioxidant Properties Dedicated to Nanotechnologies. Antioxidants, 7(5), 62. https://doi.org/10.3390/antiox7050062