Crystal Structure of the Apo-Form of NADPH-Dependent Thioredoxin Reductase from a Methane-Producing Archaeon



Abstract
1. Introduction
2. Materials and Methods
2.1. MmNTR Protein Production and Purification
2.2. Protein Crystallization, Data Collection, and Structure Solution
2.3. SAXS
2.4. The Protein Sequence Analysis and Structure Modeling
3. Results
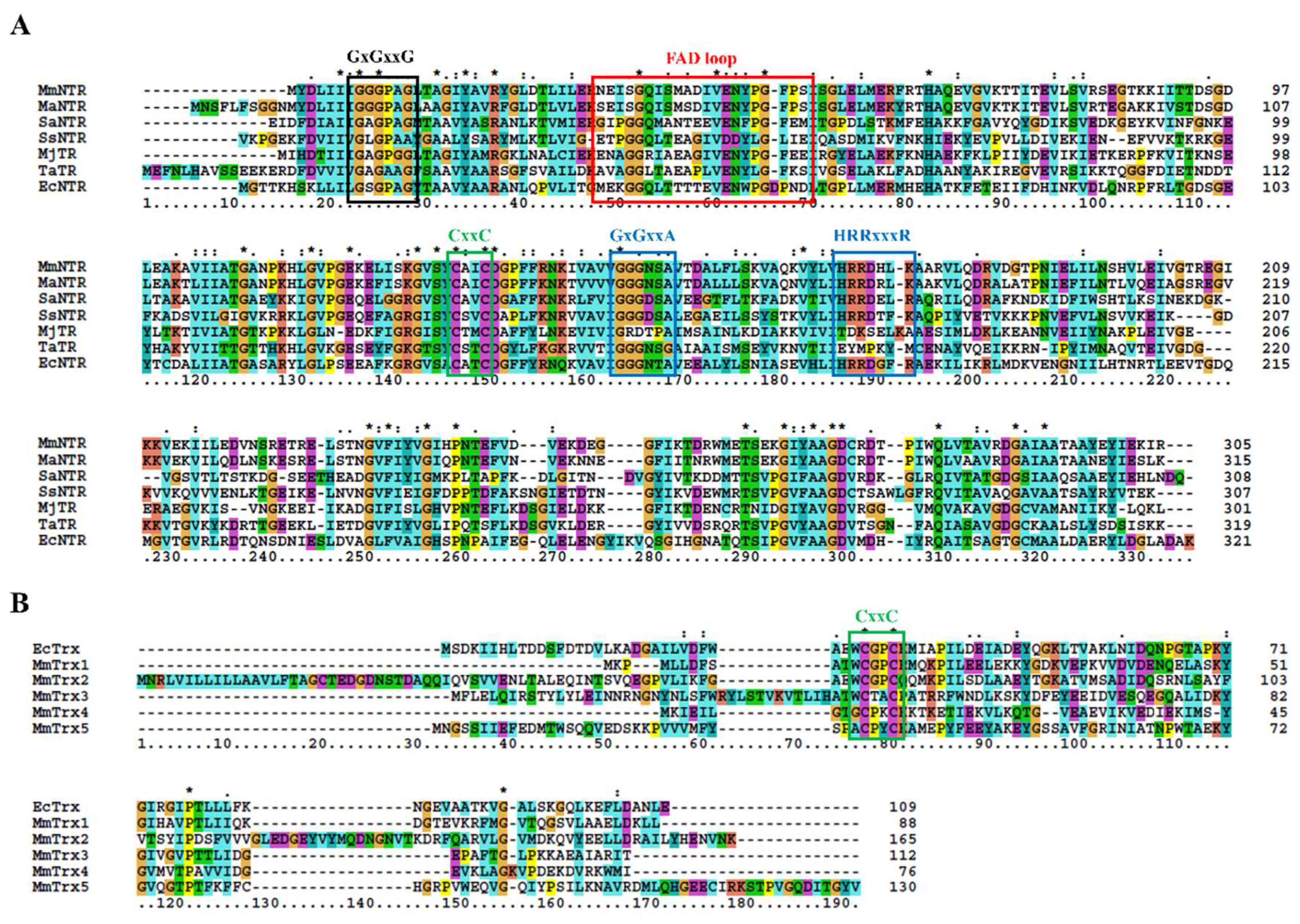
3.1. Description of the M. Mazei Trx System
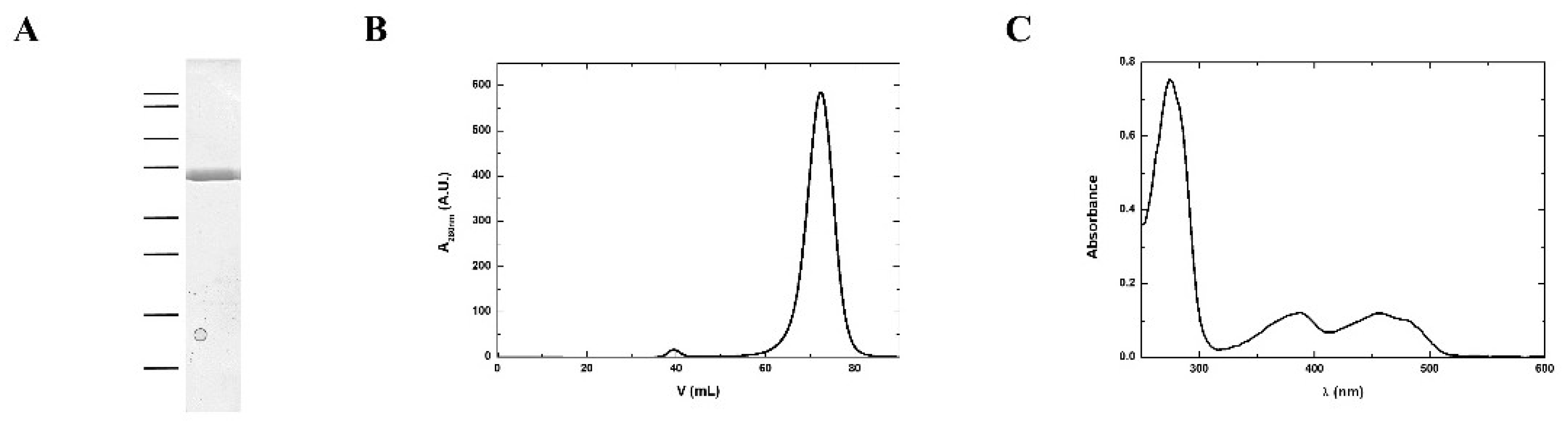
3.2. Purification and Structural Features of MmNTR
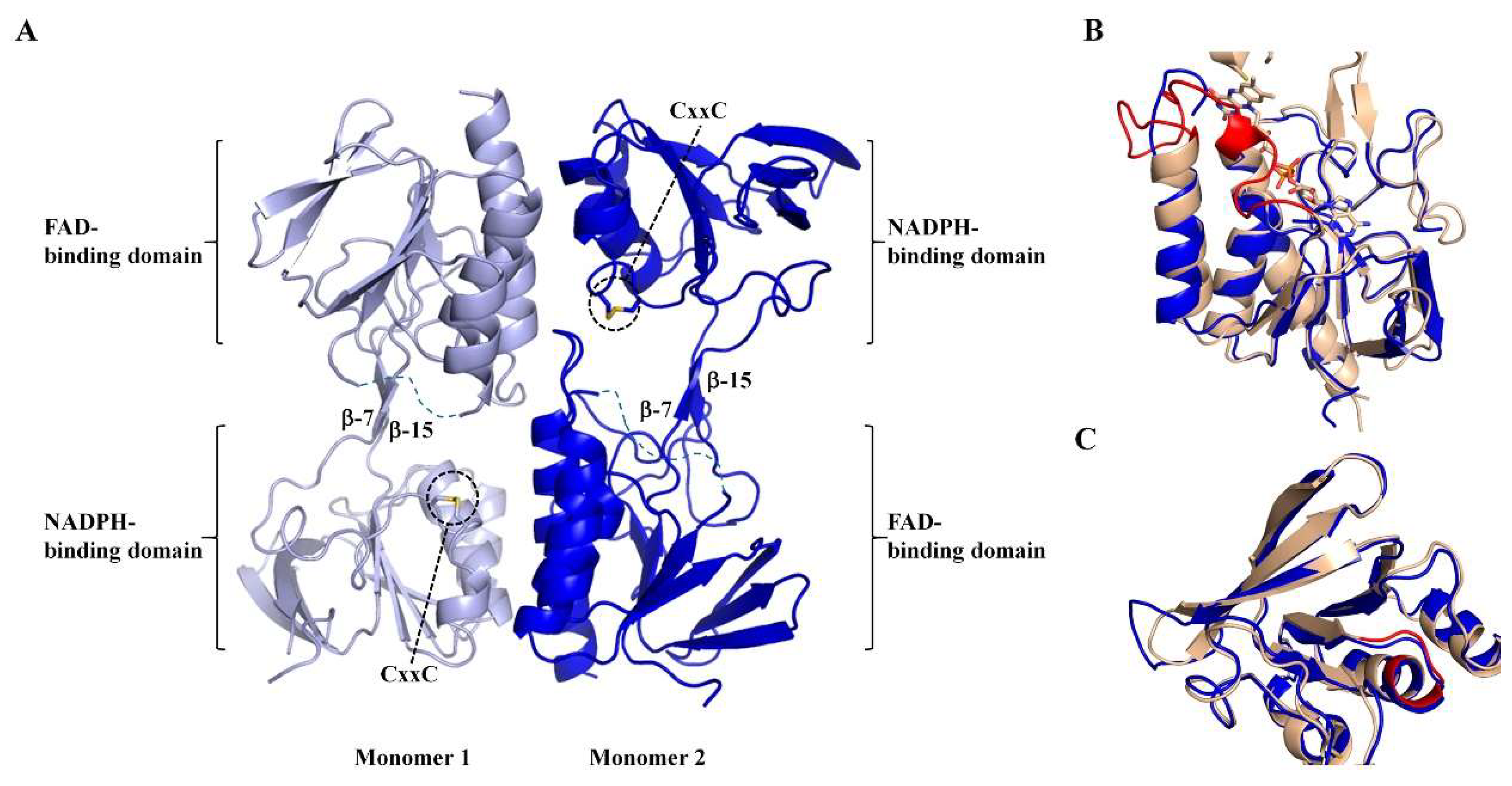
3.3. Apo-MmNTR is a Homodimer Composed of Two Domains
3.4. Apo-MmNTR Monomers Adopt a Flavin-Oxidizing Conformation
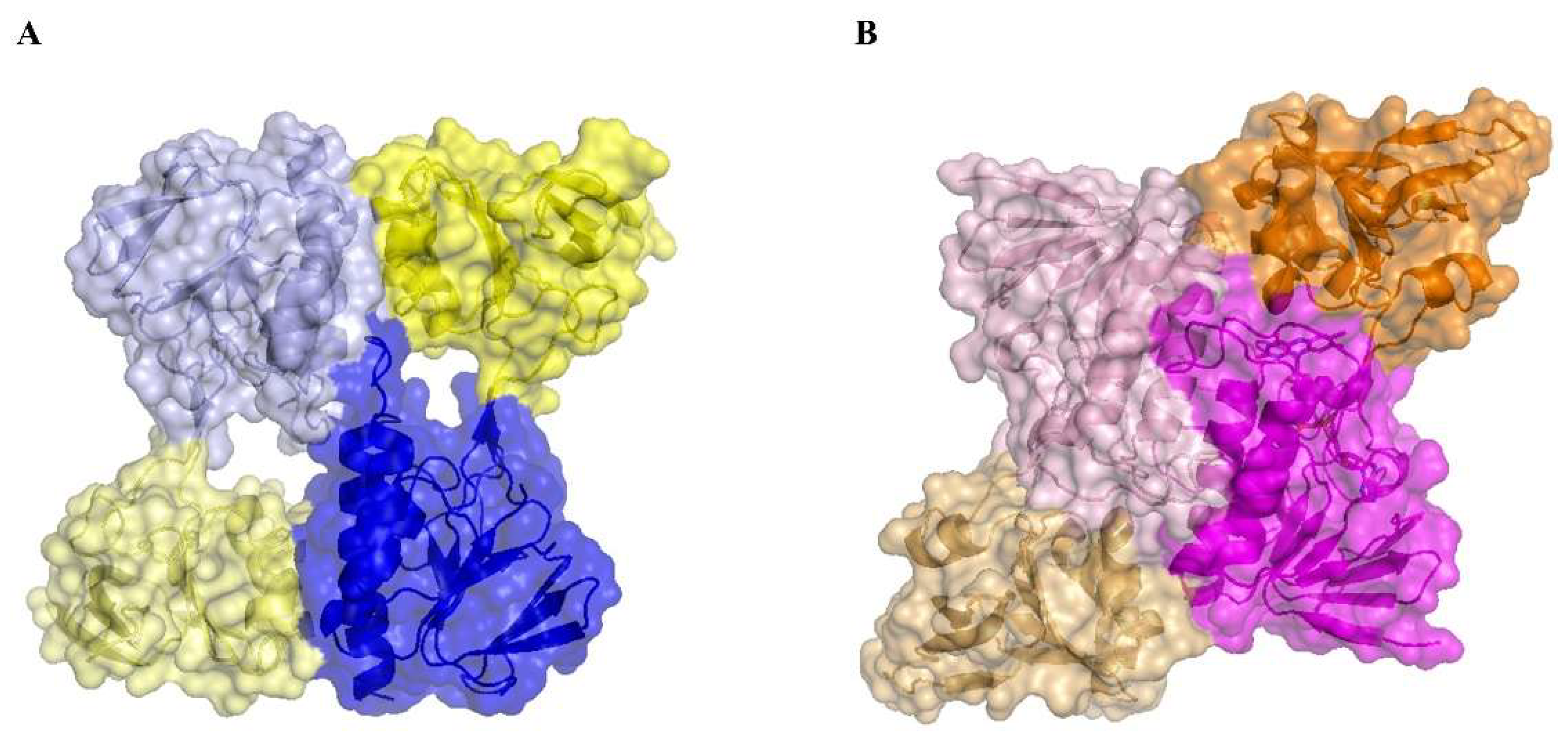
3.5. The Absence of the Flavin Cofactor Compromises Dimer Formation
3.6. SAXS Reveals a Typical LMW-NTR Conformation of MmNTR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jacquot, J.-P.; Eklund, H.; Rouhier, N.; Schürmann, P. Structural and evolutionary aspects of thioredoxin reductases in photosynthetic organisms. Trends Plant Sci. 2009, 14, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Lennon, B.W.; Williams, C.H. Reductive half-reaction of thioredoxin reductase from Escherichia coli. Biochemistry 1997, 36, 9464–9477. [Google Scholar] [CrossRef] [PubMed]
- Fritz-Wolf, K.; Kehr, S.; Stumpf, M.; Rahlfs, S.; Becker, K. Crystal structure of the human thioredoxin reductase–thioredoxin complex. Nat. Commun. 2011, 2, 383. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.-J.; Ishikawa, K. Identification and characterization of thioredoxin and thioredoxin reductase from Aeropyrum pernix K1. Eur. J. Biochem. 2002, 269, 5423–5430. [Google Scholar] [CrossRef] [PubMed]
- Kashima, Y.; Ishikawa, K. A hyperthermostable novel protein-disulfide oxidoreductase is reduced by thioredoxin reductase from hyperthermophilic archaeon Pyrococcus horikoshii. Arch. Biochem. Biophys. 2003, 418, 179–185. [Google Scholar] [CrossRef] [PubMed]
- McCarver, A.C.; Lessner, D.J. Molecular characterization of the thioredoxin system from Methanosarcina acetivorans. FEBS J. 2014, 281, 4598–4611. [Google Scholar] [CrossRef] [PubMed]
- McCarver, A.C.; Lessner, F.H.; Soroeta, J.M.; Lessner, D.J. Methanosarcina acetivorans utilizes a single NADPH-dependent thioredoxin system and contains additional thioredoxin homologues with distinct functions. Microbiology 2017, 163, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, P.; Ruocco, M.R.; Lanzotti, M.A.; Ruggiero, A.; Ruggiero, I.; Arcari, P.; Vitagliano, L.; Masullo, M. Characterisation of the components of the thioredoxin system in the archaeon Sulfolobus solfataricus. Extremophiles 2008, 12, 553. [Google Scholar] [CrossRef] [PubMed]
- Susanti, D.; Loganathan, U.; Mukhopadhyay, B. A novel F420-dependent Thioredoxin Reductase gated by low potential FAD: A tool for redox regulation in an anaerobe. J. Biol. Chem. 2016, 291, 23084–23100. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, H.; Jaquez, O.; Hamill, M.; Elliott, S.; Drennan, C. Thioredoxin reductase from Thermoplasma acidophilum: A new twist on redox regulation. Biochemistry 2008, 47, 9728–9737. [Google Scholar] [CrossRef] [PubMed]
- Susanti, D.; Loganathan, U.; Compton, A.; Mukhopadhyay, B. A reexamination of thioredoxin reductase from Thermoplasma acidophilum, a thermoacidophilic euryarchaeon, identifies it as an NADH-dependent enzyme. ACS Omega 2017, 2, 4180–4187. [Google Scholar] [CrossRef] [PubMed]
- Deppenmeier, U.; Johann, A.; Hartsch, T.; Merkl, R.; Schmitz, R.A.; Martinez-Arias, R.; Henne, A.; Wiezer, A.; Baumer, S.; Jacobi, C.; et al. The genome of Methanosarcina mazei: Evidence for lateral gene transfer between bacteria and archaea. J. Mol. Microbiol. Biotechnol. 2002, 4, 453–461. [Google Scholar] [PubMed]
- Thauer, R.K.; Kaster, A.-K.; Seedorf, H.; Buckel, W.; Hedderich, R. Methanogenic archaea: Ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 2008, 6, 579. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Cryst. D 2010, 66, 133–144. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Cryst. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Rychlewski, L.; Jaroszewski, L.; Li, W.; Godzik, A. Comparison of sequence profiles. Strategies for structural predictions using sequence information. Protein Sci. 2000, 9, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Cryst. D 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Cryst. D 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System; Version 1.8; Schrödinger, LLC: New York, NY, USA, 2015.
- Panjkovich, A.; Svergun, D.I. CHROMIXS: Automatic and interactive analysis of chromatography-coupled small-angle X-ray scattering data. Bioinformatics 2018, 34, 1944–1946. [Google Scholar] [CrossRef] [PubMed]
- Svergun, D.; Barberato, C.; Koch, M.H.J. CRYSOL—A program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Cryst. 1995, 28, 768–773. [Google Scholar] [CrossRef]
- Konarev, P.V.; Petoukhov, M.V.; Volkov, V.V.; Svergun, D.I. ATSAS 2.1, a program package for small-angle scattering data analysis. J. Appl. Cryst. 2006, 39, 277–286. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, T.; Sandalova, T.; Lu, J.; Holmgren, A.; Schneider, G. High-resolution structures of oxidized and reduced thioredoxin reductase from Helicobacter pylori. Acta Cryst. D 2007, 63, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A. Thioredoxin. Annu. Rev. Biochem. 1985, 54, 237–271. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Collet, J.F. Many roles of the bacterial envelope reducing pathways. Antioxid. Redox Signal. 2013, 18, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Prakash, D.; Walters, K.A.; Martinie, R.J.; McCarver, A.C.; Kumar, A.K.; Lessner, D.J.; Krebs, C.; Golbeck, J.H.; Ferry, J.G. Toward a mechanistic and physiological understanding of a ferredoxin:disulfide reductase from the domains Archaea and Bacteria. J. Biol. Chem. 2018, 293, 9198–9209. [Google Scholar] [CrossRef] [PubMed]
- Balsera, M.; Uberegui, E.; Susanti, D.; Schmitz, R.; Mukhopadhyay, B.; Schürmann, P.; Buchanan, B. Ferredoxin:thioredoxin reductase (FTR) links the regulation of oxygenic photosynthesis to deeply rooted bacteria. Planta 2013, 237, 619–635. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.K.; Kumar, R.S.; Yennawar, N.H.; Yennawar, H.P.; Ferry, J.G. Structural and biochemical characterization of a Ferredoxin:Thioredoxin reductase-like enzyme from Methanosarcina acetivorans. Biochemistry 2015, 54, 3122–3128. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Li, B.; Prakash, D.; Ferry, J.G.; Elliott, S.J.; Stubbe, J. A ferredoxin disulfide reductase delivers electrons to the Methanosarcina barkeri class III Ribonucleotide Reductase. Biochemistry 2015, 54, 7019–7028. [Google Scholar] [CrossRef] [PubMed]
- Kuriyan, J.; Krishna, T.S.R.; Wong, L.; Guenther, B.; Pahler, A.; Williams, C.H.; Model, P. Convergent evolution of similar function in two structurally divergent enzymes. Nature 1991, 352, 172–174. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Laakso, L.M. Dali server update. Nucleic Acids Res. 2016, 44, W351–W355. [Google Scholar] [CrossRef] [PubMed]
- Lennon, B.W.; Williams, C.H., Jr.; Ludwig, M.L. Twists in catalysis: Alternating conformations of Escherichia coli thioredoxin reductase. Science 2000, 289, 1190–1194. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.H., Jr. Mechanism and structure of thioredoxin reductase from Escherichia coli. FASEB J. 1995, 9, 1267–1276. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M. Mazei Trx | Gene ID | Catalytic Motif | M. Acetivorans Homolog [6] | Reductant | Reduction of Insulin [7] |
|---|---|---|---|---|---|
| MmTrx1 | MM_RS02345 | WCGPC | MaTrx2 | unknown | + |
| MmTrx2 | MM_RS05160 | WCGPC | MaTrx6 | unknown | + |
| MmTrx3 | MM_RS12205 | WCTAC | MaTrx7 | NTR [6] | + |
| MmTrx4 | MM_RS11655 | GCPKC | MaTrx5 | unknown | - |
| MmTrx5 | MM_RS10780 | ACPYC | MaTrx1 | FDR [29] | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buey, R.M.; Schmitz, R.A.; Buchanan, B.B.; Balsera, M. Crystal Structure of the Apo-Form of NADPH-Dependent Thioredoxin Reductase from a Methane-Producing Archaeon. Antioxidants 2018, 7, 166. https://doi.org/10.3390/antiox7110166
Buey RM, Schmitz RA, Buchanan BB, Balsera M. Crystal Structure of the Apo-Form of NADPH-Dependent Thioredoxin Reductase from a Methane-Producing Archaeon. Antioxidants. 2018; 7(11):166. https://doi.org/10.3390/antiox7110166
Chicago/Turabian StyleBuey, Rubén M., Ruth A. Schmitz, Bob B. Buchanan, and Monica Balsera. 2018. "Crystal Structure of the Apo-Form of NADPH-Dependent Thioredoxin Reductase from a Methane-Producing Archaeon" Antioxidants 7, no. 11: 166. https://doi.org/10.3390/antiox7110166
APA StyleBuey, R. M., Schmitz, R. A., Buchanan, B. B., & Balsera, M. (2018). Crystal Structure of the Apo-Form of NADPH-Dependent Thioredoxin Reductase from a Methane-Producing Archaeon. Antioxidants, 7(11), 166. https://doi.org/10.3390/antiox7110166