Vitamin E as a Treatment for Nonalcoholic Fatty Liver Disease: Reality or Myth?


Abstract
1. Introduction
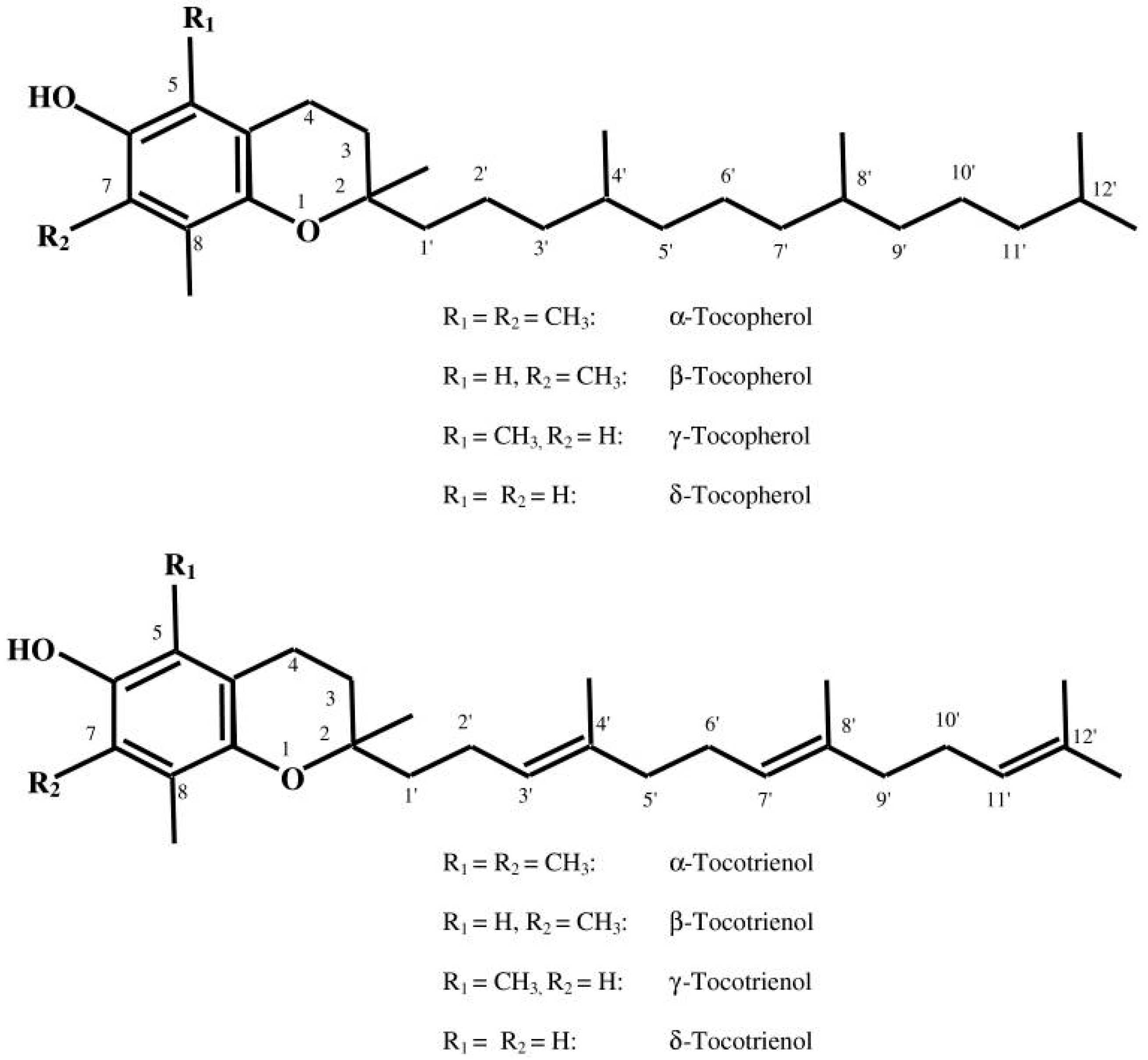
2. Vitamin E: Brief Overview of Structure, Metabolism and Function
2.1. Structural Perspectives
2.2. Insight into Metabolism
2.3. Biological Activity of Vitamin E
2.3.1. Antioxidant Activity
2.3.2. Beyond Vitamin E Antioxidant Activity
3. Vitamin E and NAFLD
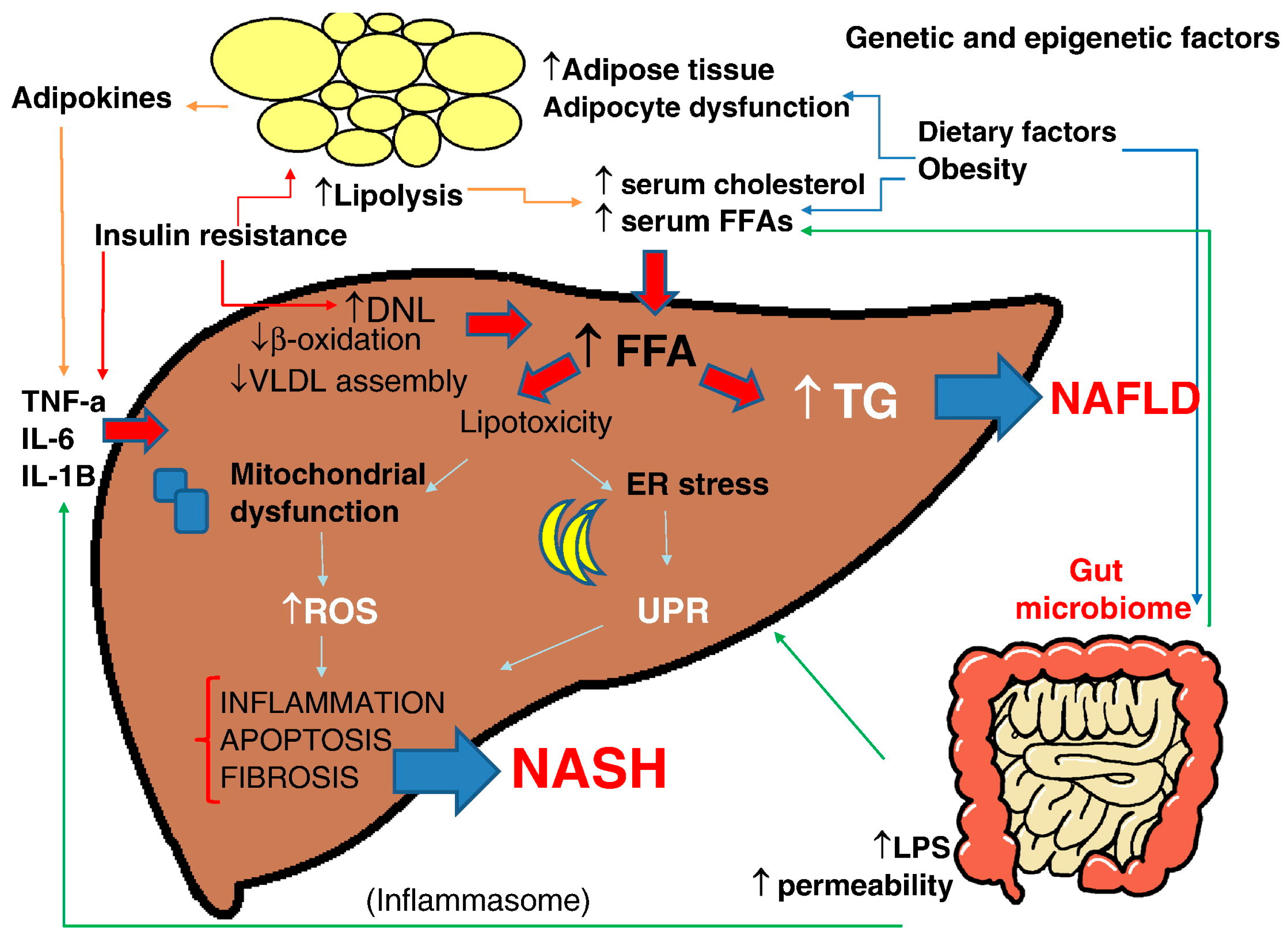
3.1. NAFLD/NASH Pathogenesis
3.2. Vitamin E and NAFLD: Experimental Studies
3.3. Vitamin E and NAFLD: Human Studies
3.4. Vitamin E: Safety Concerns
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Chalasani, N.; Younoss, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Zavos, C. Nonalcoholic fatty liver disease: The pathogenetic roles of insulin resistance and adipocytokines. Curr. Mol. Med. 2009, 72, 299–314. [Google Scholar] [CrossRef]
- Khan, R.S.; Newsome, P.N. Non-alcoholic fatty liver disease and liver transplantation. Metabolism 2016, 65, 1208–1223. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Review, T.; LaBrecque, D.R.; Abbas, Z.; Anania, F.; Ferenci, P.; Khan, A.G.; Goh, K.L.; Hamid, S.S.; Isakov, V.; Lizarzabal, M.; et al. World Gastroenterology Organisation global guidelines: Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. J. Clin. Gastroenterol. 2014, 48, 467–473. [Google Scholar]
- El Hadi, H.; Vettor, R.; Rossato, M. Congenital Vitamin E deficiency. In Handbook of Famine, Starvation, and Nutrient Deprivation; Preedy, V.R., Patel, V.B., Eds.; Springer International Publishing AG: Basel, Switzerland, 2018; pp. 1–18. ISBN 978-3-319-40007-5. [Google Scholar]
- Evans, H.M.; Bishop, K.S. On the existence of a hitherto unrecognized dietary factor essential for reproduction. Science 1922, 56, 650–651. [Google Scholar] [CrossRef] [PubMed]
- Gee, P.T. Unleashing the untold and misunderstood observations on vitamin E. Genes Nutr. 2011, 6, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, A.J.; Pennington, J.A.T.; Weihrauch, J.L. Analysis and distribution of vitamin E in vegetable oils and foods. In Vitamin E in Health and Disease; Packer, L., Fuchs, J., Eds.; Marcel Dekker Inc.: New York, NY, USA, 1993; pp. 9–31. [Google Scholar]
- Karrer, P.; Fritzsche, H.; Ringier, B.; Salomon, H. Synthesis of alpha-tocopherol (vitamin E). Nature 1938, 141, 1057. [Google Scholar] [CrossRef]
- Jensen, S.K.; Lauridsen, C. Alpha-tocopherol stereoisomers. Vitam. Horm. 2007, 76, 281–308. [Google Scholar] [PubMed]
- Traber, M.G. Vitamin E regulatory mechanisms. Annu. Rev. Nutr. 2007, 27, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Kayden, H.J.; Traber, M.G. Absorption, lipoprotein transport, and regulation of plasma concentrations of vitamin E in humans. J. Lipid Res. 1993, 34, 343–358. [Google Scholar] [PubMed]
- Kaempf-Rotzoll, D.E.; Traber, M.G.; Arai, H. Vitamin E and transfer proteins. Curr. Opin. Lipidol. 2003, 14, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q. Natural forms of vitamin E: Metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free Radic. Biol. Med. 2014, 72, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Bardowell, S.A.; Duan, F.; Manor, D.; Swanson, J.E.; Parker, R.S. Disruption of mouse cytochrome p4504f14 (Cyp4f14 gene) causes severe perturbations in vitamin E metabolism. J. Biol. Chem. 2012, 287, 26077–26086. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Peh, H.Y.; Tan, W.S.; Liao, W.; Wong, W.S. Vitamin E therapy beyond cancer: Tocopherol versus tocotrienol. Pharmacol. Ther. 2016, 162, 152–169. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Saito, Y.; Jones, L.S.; Shigeri, Y. Chemical reactivities and physical effects in comparison between tocopherols and tocotrienols: Physiological significance and prospects as antioxidants. J. Biosci. Bioeng. 2007, 104, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Cooney, R.V.; Harwood, P.J.; Franke, A.A.; Narala, K.; Sundström, A.K.; Berggren, P.O.; Mordan, L.J. Products of gamma-tocopherol reaction with NO2 and their formation in rat insulinoma (RINm5F) cells. Free Radic. Biol. Med. 1995, 19, 259–269. [Google Scholar] [CrossRef]
- Christen, S.; Jiang, Q.; Shigenaga, M.K.; Ames, B.N. Analysis of plasma tocopherols α, γ, and 5-nitro-γ in rats with inflammation by HPLC coulometric detection. J. Lipid Res. 2002, 43, 1978–1985. [Google Scholar] [CrossRef] [PubMed]
- Berbée, M.; Fu, Q.; Boerma, M.; Wang, J.; Kumar, K.S.; Hauer-Jensen, M. gamma-Tocotrienol ameliorates intestinal radiation injury and reduces vascular oxidative stress after total-body irradiation by an HMG-CoA reductase-dependent mechanism. Radiat. Res. 2009, 171, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Upston, J.M.; Terentis, A.C.; Stocker, R. Tocopherol-mediated peroxidation of lipoproteins: Implications for vitamin E as a potential antiatherogenic supplement. FASEB J. 1999, 13, 977–994. [Google Scholar] [PubMed]
- Pearson, P.; Lewis, S.A.; Britton, J.; Young, I.S.; Fogarty, A. The pro-oxidant activity of high-dose vitamin E supplements in vivo. BioDrugs 2006, 20, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.B.; VanderWerken, B.S.; Anderson, R.A.; Stegner, J.E.; Thomas, M.J. Pro-oxidant effect of vitamin E in cigarette smokers consuming a high polyunsaturated fat diet. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Zingg, J.M.; Azzi, A. Non-antioxidant activities of vitamin E. Curr. Med. Chem. 2004, 11, 1113–1133. [Google Scholar] [CrossRef] [PubMed]
- Rimbach, G.; Minihane, A.M.; Majewicz, J.; Fischer, A.; Pallauf, J.; Virgli, F.; Weinberg, P.D. Regulation of cell signalling by vitamin E. Proc. Nutr. Soc. 2002, 61, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Boscoboinik, D.; Szewczyk, A.; Hensey, C.; Azzi, A. Inhibition of cell proliferation by alpha-tocopherol. Role of protein kinase C. J. Biol. Chem. 1991, 266, 6188–6194. [Google Scholar] [PubMed]
- Cook-Mills, J.M. Isoforms of Vitamin E Differentially Regulate PKC α and Inflammation: A Review. J. Clin. Cell. Immunol. 2013, 4, 1000137. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, S.K.; Devaraj, S.; Yang, T.; Jialal, I. Alpha-tocopherol decreases superoxide anion release in human monocytes under hyperglycemic conditions via inhibition of protein kinase C-alpha. Diabetes 2002, 51, 3049–3054. [Google Scholar] [CrossRef] [PubMed]
- Martin-Nizard, F.; Boullier, A.; Fruchart, J.C.; Duriez, P. Alpha-tocopherol but not beta-tocopherol inhibits thrombin-induced PKC activation and endothelin secretion in endothelial cells. J. Cardiovasc. Risk 1998, 5, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Rimbach, G.; Moehring, J.; Huebbe, P.; Lodge, J.K. Gene-regulatory activity of alpha-tocopherol. Molecules 2010, 15, 1746–1761. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, H.; Ahad, A.; Iqbal, J.; Siddiqui, W.A. Pharmacological potential of tocotrienols: A review. Nutr. Metab. 2014, 11, 52. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Hassan, K.; Bhalla, V.; El Regal, M.E.; A-Kader, H.H. Nonalcoholic fatty liver disease: A comprehensive review of a growing epidemic. World J. Gastroenterol. 2014, 20, 12082–12101. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Mohammed, B.S.; Magkos, F.; Korenblat, K.M.; Patterson, B.W.; Klein, S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, R.N.; Fisher, C.D.; Canet, M.J.; Lake, A.D.; Cherrington, N.J. Diversity in antioxidant response enzymes in progressive stages of human nonalcoholic fatty liver disease. Drug Metab. Dispos. 2010, 38, 2293–2301. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P. Pathogenesis of steatohepatitis. Best Prac. Res. Clin. Gastroenterol. 2002, 16, 663–678. [Google Scholar] [CrossRef]
- Caldwell, S.H.; Chang, C.Y.; Nakamoto, R.K.; Krugner-Higby, L. Mitochondria in nonalcoholic fatty liver disease. Clin. Liver Dis. 2004, 8, 595–617. [Google Scholar] [CrossRef] [PubMed]
- Aubert, J.; Begriche, K.; Knockaert, L.; Robin, M.A.; Fromenty, B. Increased expression of cytochrome P450 2E1 in nonalcoholic fatty liver disease: Mechanisms and pathophysiological role. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, I.A.; Farrell, G.C.; Field, J.; Bell, D.R.; Gonzalez, F.J.; Robertson, G.R. CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J. Clin. Investig. 2000, 105, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, S.; Zhou, Y.; Su, W.; Ruan, X.; Wang, B.; Zheng, F.; Warner, M.; Gustafsson, J.Å.; Guan, Y. Ablation of cytochrome P450 omega-hydroxylase 4A14 gene attenuates hepatic steatosis and fibrosis. Proc. Natl. Acad. Sci. USA 2017, 114, 3181–3185. [Google Scholar] [CrossRef] [PubMed]
- Demeilliers, C.; Maisonneuve, C.; Grodet, A.; Mansouri, A.; Nguyen, R.; Tinel, M.; Lettéron, P.; Degott, C.; Feldmann, G.; Pessayre, D.; et al. Impaired adaptive resynthesis and prolonged depletion of hepatic mitochondrial DNA after repeated alcohol binges in mice. Gastroenterology 2002, 123, 1278–1290. [Google Scholar] [CrossRef] [PubMed]
- Dara, L.; Ji, C.; Kaplowitz, N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology 2011, 53, 1752–1763. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Larter, C.Z.; Yeh, M.M.; Williams, J.; Bell-Anderson, K.S.; Farrell, G.C. MCD-induced steatohepatitis is associated with hepatic adiponectin resistance and adipogenic transformation of hepatocytes. J. Hepatol. 2008, 49, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Phung, N.; Pera, N.; Farrell, G.; Leclercq, I.; Hou, J.Y.; George, J. Pro-oxidant-mediated hepatic fibrosis and effects of antioxidant intervention in murine dietary steatohepatitis. Int. J. Mol. Med. 2009, 24, 171–180. [Google Scholar] [PubMed]
- Nan, Y.M.; Wu, W.J.; Fu, N.; Liang, B.L.; Wang, R.Q.; Li, L.X.; Zhao, S.X.; Zhao, J.M.; Yu, J. Antioxidants vitamin E and 1-aminobenzotriazole prevent experimental non-alcoholic steatohepatitis in mice. Scand. J. Gastroenterol. 2009, 44, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Karimian, G.; Kirschbaum, M.; Veldhuis, Z.J.; Bomfati, F.; Porte, R.J.; Lisman, T. Vitamin E Attenuates the Progression of Non-Alcoholic Fatty Liver Disease Caused by Partial Hepatectomy in Mice. PLoS ONE 2015, 10, e0143121. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.Y.; Yeung, S.F.; Park, H.J.; Volek, J.S.; Bruno, R.S. Dietary α- and γ-tocopherol supplementation attenuates lipopolysaccharide-induced oxidative stress and inflammatory-related responses in an obese mouse model of nonalcoholic steatohepatitis. J. Nutr. Biochem. 2010, 21, 1200–1206. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Harper, A.F.; Zhao, J.; Corl, B.A.; LeRoith, T.; Dalloul, R.A. Effects of a dietary antioxidant blend and vitamin E on fatty acid profile, liver function, and inflammatory response in broiler chickens fed a diet high in oxidants. Poult. Sci. 2014, 93, 1658–1666. [Google Scholar] [CrossRef] [PubMed]
- Podszun, M.C.; Grebenstein, N.; Spruss, A.; Schlueter, T.; Kremoser, C.; Bergheim, I.; Frank, J. Dietary alpha-tocopherol and atorvastatin reduce high-fat-induced lipid accumulation and down-regulate CD36 protein in the liver of guinea pigs. J. Nutr. Biochem. 2014, 25, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Lavine, J.E. Vitamin E treatment of nonalcoholic steatohepatitis in children: A pilot study. J. Pediatr. 2000, 136, 734–738. [Google Scholar] [CrossRef]
- Hasegawa, T.; Yoneda, M.; Nakamura, K.; Makino, I.; Terano, A. Plasma transforming growth factor-beta1 level and efficacy of alpha-tocopherol in patients with non-alcoholic steatohepatitis: A pilot study. Aliment. Pharmacol. Ther. 2001, 15, 1667–1672. [Google Scholar] [CrossRef] [PubMed]
- Kugelmas, M.; Hill, D.B.; Vivian, B.; Marsano, L.; McClain, C.J. Cytokines and NASH: A pilot study of the effects of lifestyle modification and vitamin E. Hepatology 2003, 38, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Yakaryilmaz, F.; Guliter, S.; Savas, B.; Erdem, O.; Ersoy, R.; Erden, E.; Akyol, G.; Bozkaya, H.; Ozenirler, S. Effects of vitamin E treatment on peroxisome proliferator-activated receptor-alpha expression and insulin resistance in patients with non-alcoholic steatohepatitis: Results of a pilot study. Intern. Med. J. 2007, 37, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; Gentilcore, E.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; David, E.; Rizzetto, M.; Marchesini, G. A randomized controlled trial of metformin versus vitamin E or prescriptive diet in nonalcoholic fatty liver disease. Am. J. Gastroenterol. 2005, 100, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Vajro, P.; Mandato, C.; Franzese, A.; Ciccimarra, E.; Lucariello, S.; Savoia, M.; Capuano, G.; Migliaro, F. Vitamin E treatment in pediatric obesity-related liver disease: A randomized study. J. Pediatr. Gastroenterol. Nutr. 2004, 38, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [PubMed]
- Lavine, J.E.; Schwimmer, J.B.; Van Natta, M.L.; Molleston, J.P.; Murray, K.F.; Rosenthal, P.; Abrams, S.H.; Scheimann, A.O.; Sanyal, A.J.; Chalasani, N.; et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: The TONIC randomized controlled trial. JAMA 2011, 305, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Pietu, F.; Guillaud, O.; Walter, T.; Vallin, M.; Hervieu, V.; Scoazec, J.Y.; Dumortier, J. Ursodeoxycholic acid with vitamin E in patients with nonalcoholic steatohepatitis: Long-term results. Clin. Res. Hepatol. Gastroenterol. 2012, 36, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.; Budoff, M.J.; Saab, S.; Ahmadi, N.; Gordon, C.; Guerci, A.D. Atorvastatin and antioxidants for the treatment of nonalcoholic fatty liver disease: The St Francis Heart Study randomized clinical trial. Am. J. Gastroenterol. 2011, 106, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Dufour, J.F.; Oneta, C.M.; Gonvers, J.J.; Bihl, F.; Cerny, A.; Cereda, J.M.; Zala, J.F.; Helbling, B.; Steuerwald, M.; Zimmermann, A. Swiss Association for the Study of the Liver. Randomized placebo-controlled trial of ursodeoxycholic acid with vitamin E in nonalcoholic steatohepatitis. Clin. Gastroenterol. Hepatol. 2006, 4, 1537–1543. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Manco, M.; Devito, R.; Ciampalini, P.; Piemonte, F.; Marcellini, M. Effect of vitamin E on aminotransferase levels and insulin resistance in children with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2006, 24, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Manco, M.; Devito, R.; Di Ciommo, V.; Comparcola, D.; Sartorelli, M.R.; Piemonte, F.; Marcellini, M.; Angulo, P. Lifestyle intervention and antioxidant therapy in children with nonalcoholic fatty liver disease: A randomized, controlled trial. Hepatology 2008, 48, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Mofrad, P.S.; Contos, M.J.; Sargeant, C.; Luketic, V.A.; Sterling, R.K.; Stravitz, R.T.; Shiffman, M.L.; Clore, J.; Mills, A.S. A pilot study of vitamin E versus vitamin E and pioglitazone for the treatment of nonalcoholic steatohepatitis. Clin. Gastroenterol. Hepatol. 2004, 2, 1107–1115. [Google Scholar] [CrossRef]
- Harrison, S.A.; Torgerson, S.; Hayashi, P.; Ward, J.; Schenker, S. Vitamin E and vitamin C treatment improves fibrosis in patients with nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2003, 98, 2485–2490. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar]
- Miller, E.R.; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 2005, 142, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Gerss, J.; Köpcke, W. The questionable association of vitamin E supplementation and mortality--inconsistent results of different meta-analytic approaches. Cell. Mol. Biol. 2009, 55 (Suppl. OL), 1111–1120. [Google Scholar]
- Abner, E.L.; Schmitt, F.A.; Mendiondo, M.S.; Marcum, J.L.; Kryscio, R.J. Vitamin E and all-cause mortality: A meta-analysis. Curr. Aging Sci. 2011, 4, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Schurks, M.; Glynn, R.J.; Rist, P.M.; Tzourio, C.; Kurth, T. Effects of vitamin E on stroke subtypes: Meta-analysis of randomised controlled trials. BMJ 2010, 341, c5702. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.A.; Thompson, I.M.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the risk of prostate cancer: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Author | Study Design | Duration | Vitamin E Dosage | Effect of Vitamin E on NAFLD/NASH Histology and Biochemistry Compared to Placebo | ||||
|---|---|---|---|---|---|---|---|---|
| ALT | Steatosis | Inflammation | Hepatocytes Ballooning | Fibrosis | ||||
| Dufour et al., 2006 [64] | 48 NASH patients randomized to vitamin E plus UDCA, UDCA versus placebo | 2 years | 400 IU twice daily | ↓ | ↓ | ns | NA | ns |
| Nobili et al., 2008 [66] | 53 NAFLD patients randomized to vitamin E plus ascorbic and life style intervention versus placebo | 2 years | 600 IU/day | ns | ns | ns | ns | ns |
| Sanyal et al., 2010 [60] | 247 NASH patients randomized to vitamin E, pioglitazone versus placebo | 96 weeks | 800 IU daily | ↓ | ↓ | ↓ | ns | |
| Foster et al., 2011 [63] | 1005 randomized to vitamin E combined with ascorbic acid and atorvastatin versus placebo. At baseline 80 had CT-diagnosed NAFLD | 4 years | 1000 IU daily | ns | ↓ (Based on abdominal CT scan) | NA | NA | NA |
| Lavine et al., 2011 [61] | 173 NAFLD patients randomized to vitamin E, metformin versus placebo | 96 weeks | 400 IU twice daily | ns | ns | ns | ↓ | ns |
| Pietu et al., 2012 [62] | 101 patients treated with a combination of vitamin E with UDCA. 10 patients had a second liver biopsy during follow-up | 4 years | 500 IU daily | ↓ | ↓ 3/10 ↑ 1/10 patients | ↓ 3/10 ↑ 2/10 patients | ↓ 3/10 ↑ 1/10patients | ↓ 4/10 ↑ 1/10 patients |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hadi, H.E.; Vettor, R.; Rossato, M. Vitamin E as a Treatment for Nonalcoholic Fatty Liver Disease: Reality or Myth? Antioxidants 2018, 7, 12. https://doi.org/10.3390/antiox7010012
Hadi HE, Vettor R, Rossato M. Vitamin E as a Treatment for Nonalcoholic Fatty Liver Disease: Reality or Myth? Antioxidants. 2018; 7(1):12. https://doi.org/10.3390/antiox7010012
Chicago/Turabian StyleHadi, Hamza El, Roberto Vettor, and Marco Rossato. 2018. "Vitamin E as a Treatment for Nonalcoholic Fatty Liver Disease: Reality or Myth?" Antioxidants 7, no. 1: 12. https://doi.org/10.3390/antiox7010012
APA StyleHadi, H. E., Vettor, R., & Rossato, M. (2018). Vitamin E as a Treatment for Nonalcoholic Fatty Liver Disease: Reality or Myth? Antioxidants, 7(1), 12. https://doi.org/10.3390/antiox7010012