
Protandim Protects Oligodendrocytes against an Oxidative Insult


Abstract
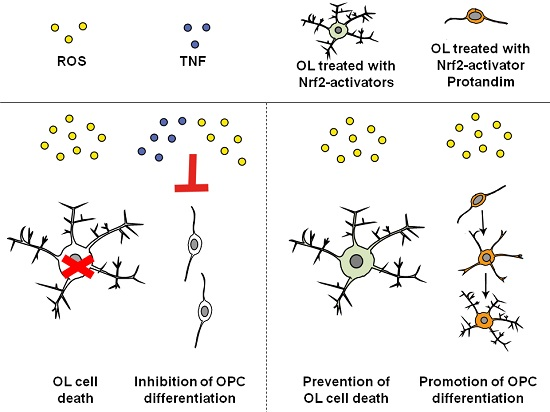
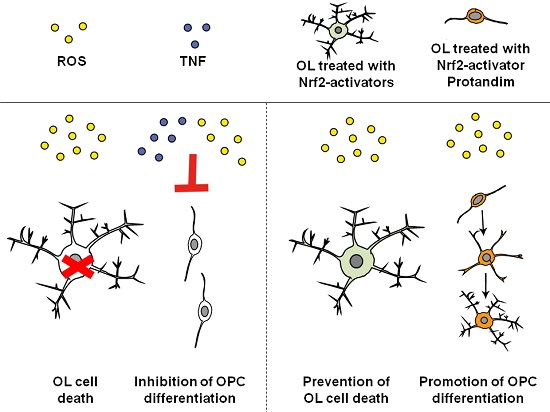
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Functional Analysis of Oligodendrocytes
2.3. Immunocytochemistry
2.4. Western Blotting
2.5. Antibody Characterization
2.6. Glutathione Assay
2.7. Statistical Analysis
3. Results
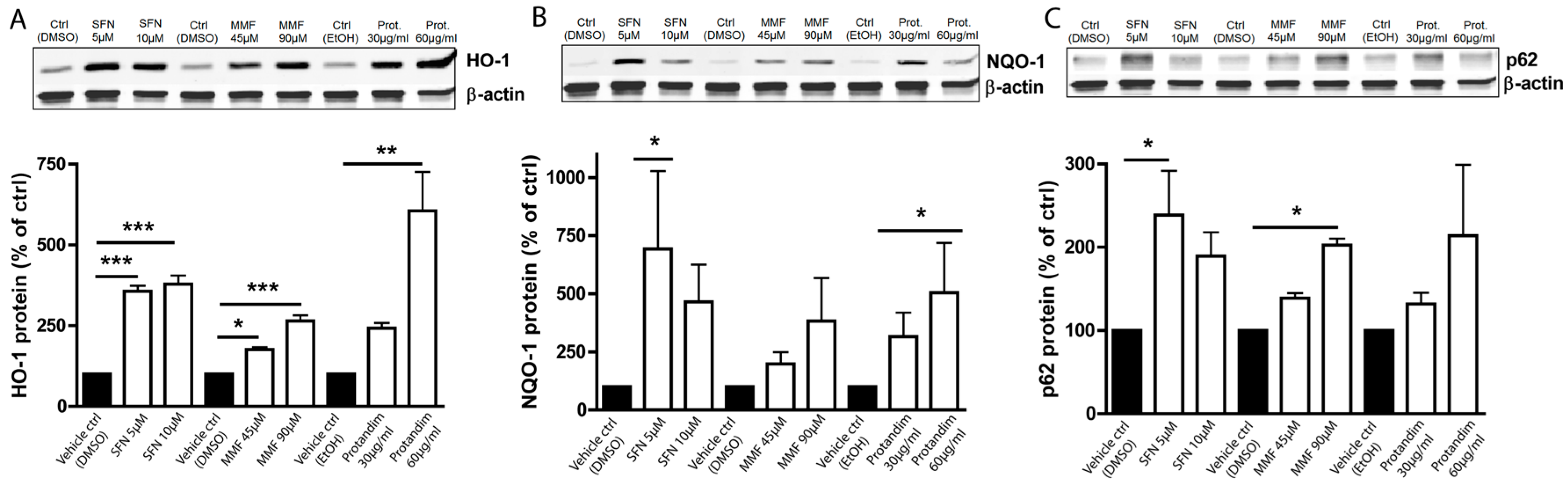
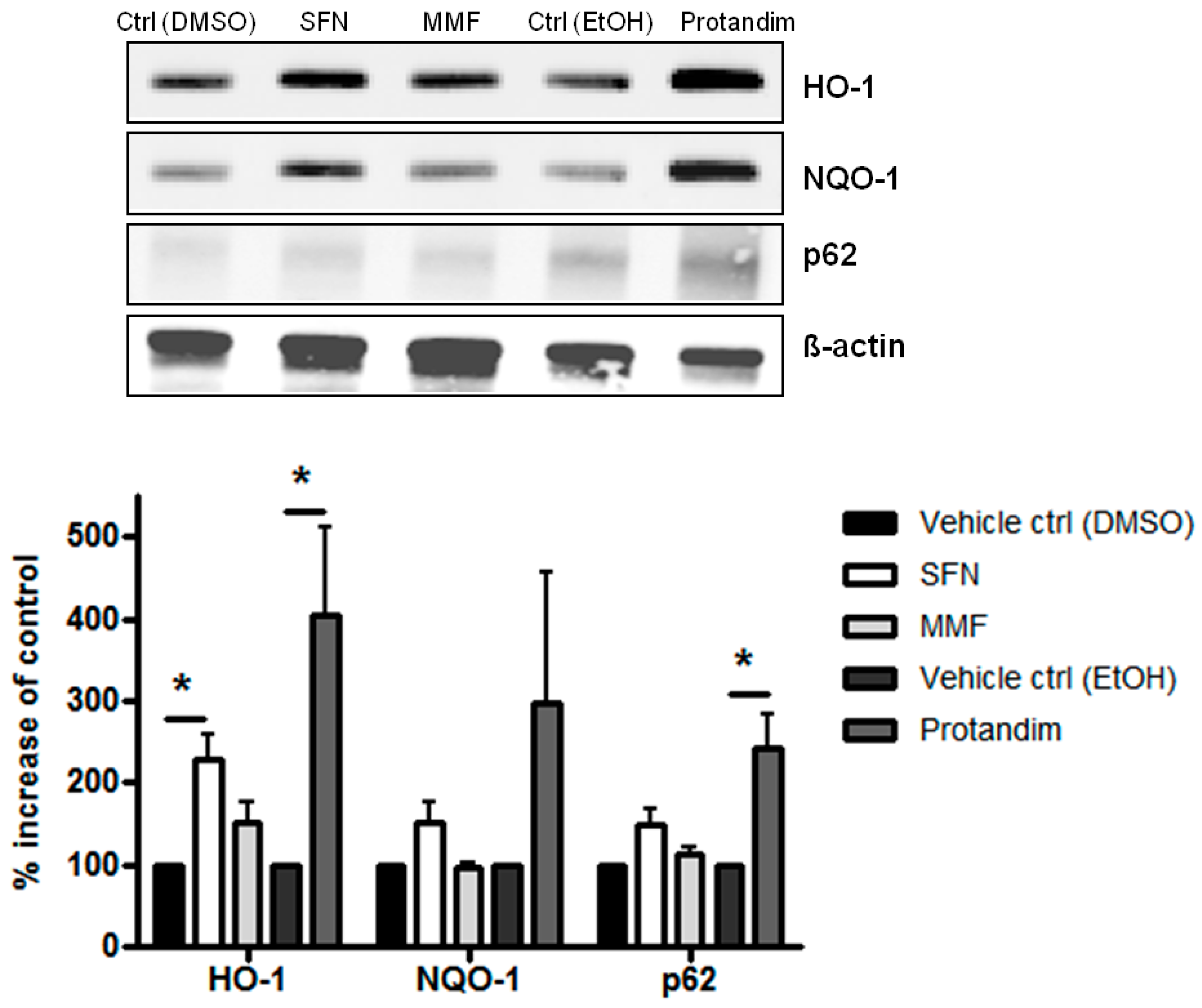
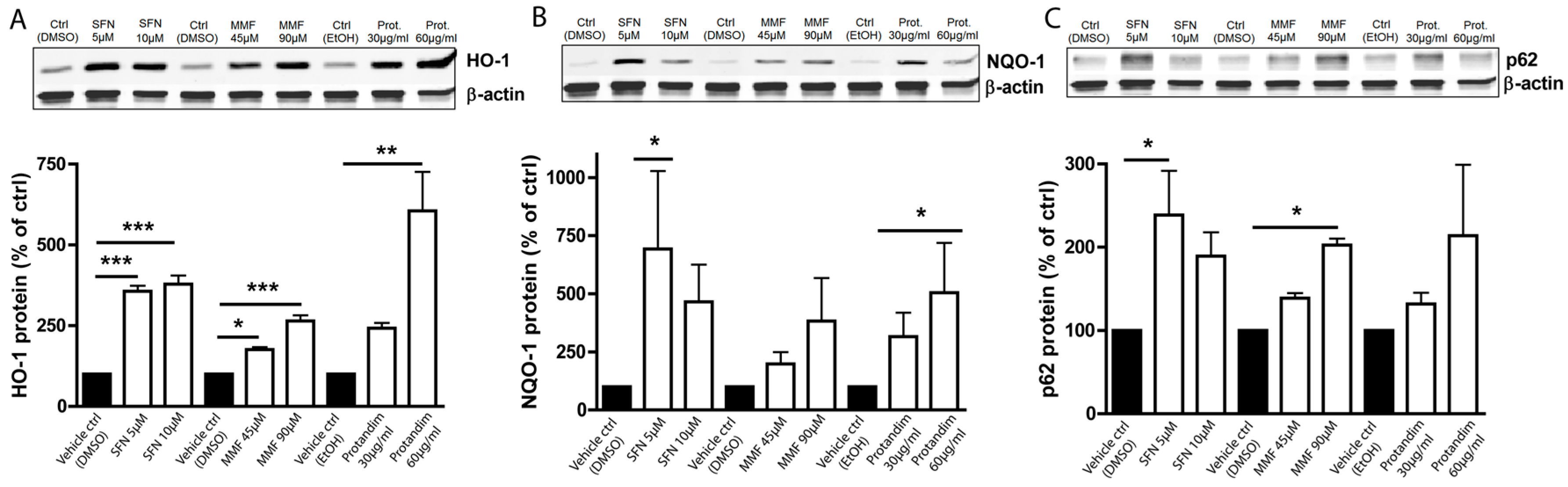
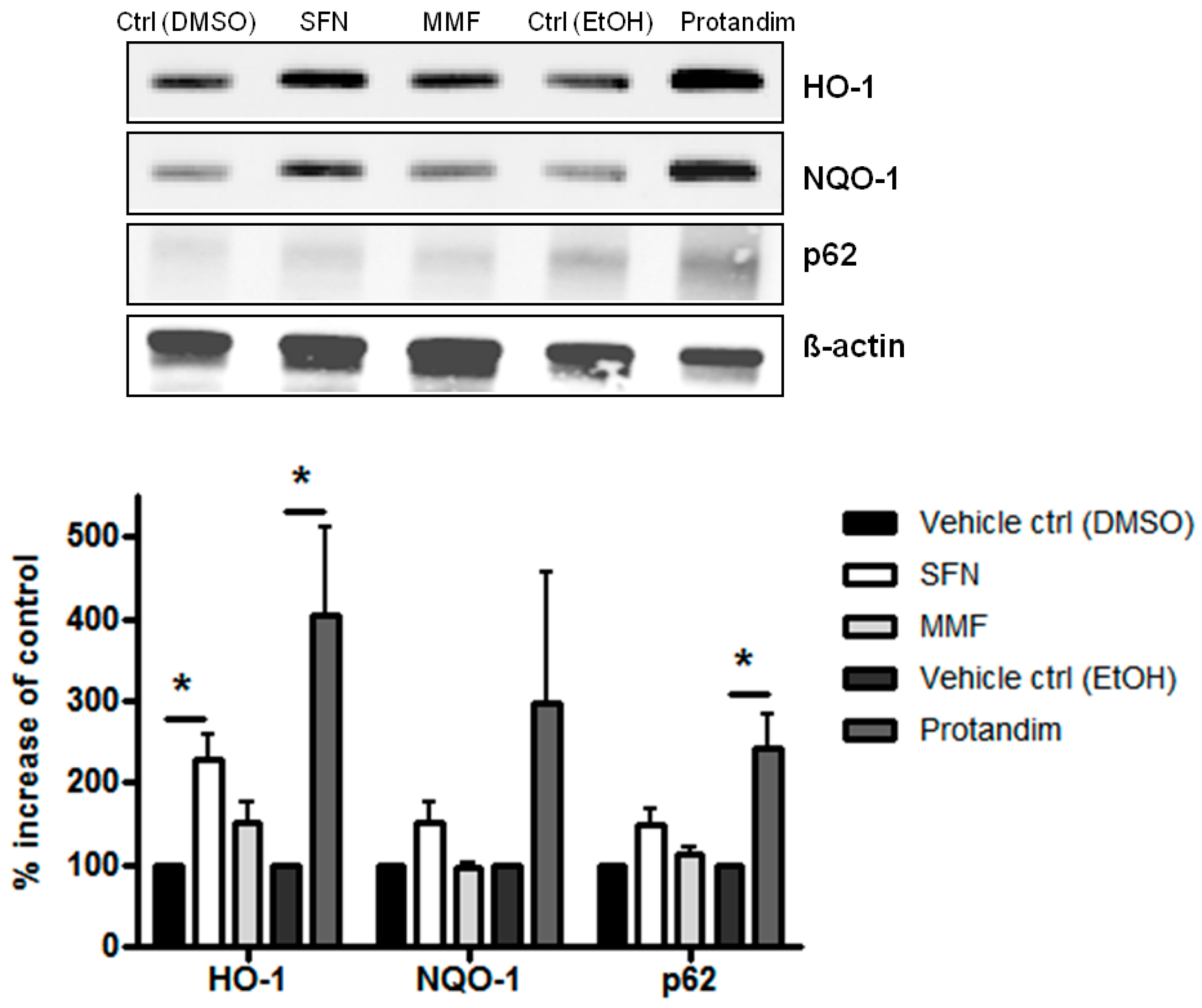
3.1. SFN, MMF and Protandim Increase Antioxidant Enzymes and p62 Expression in OLN-93 Cells
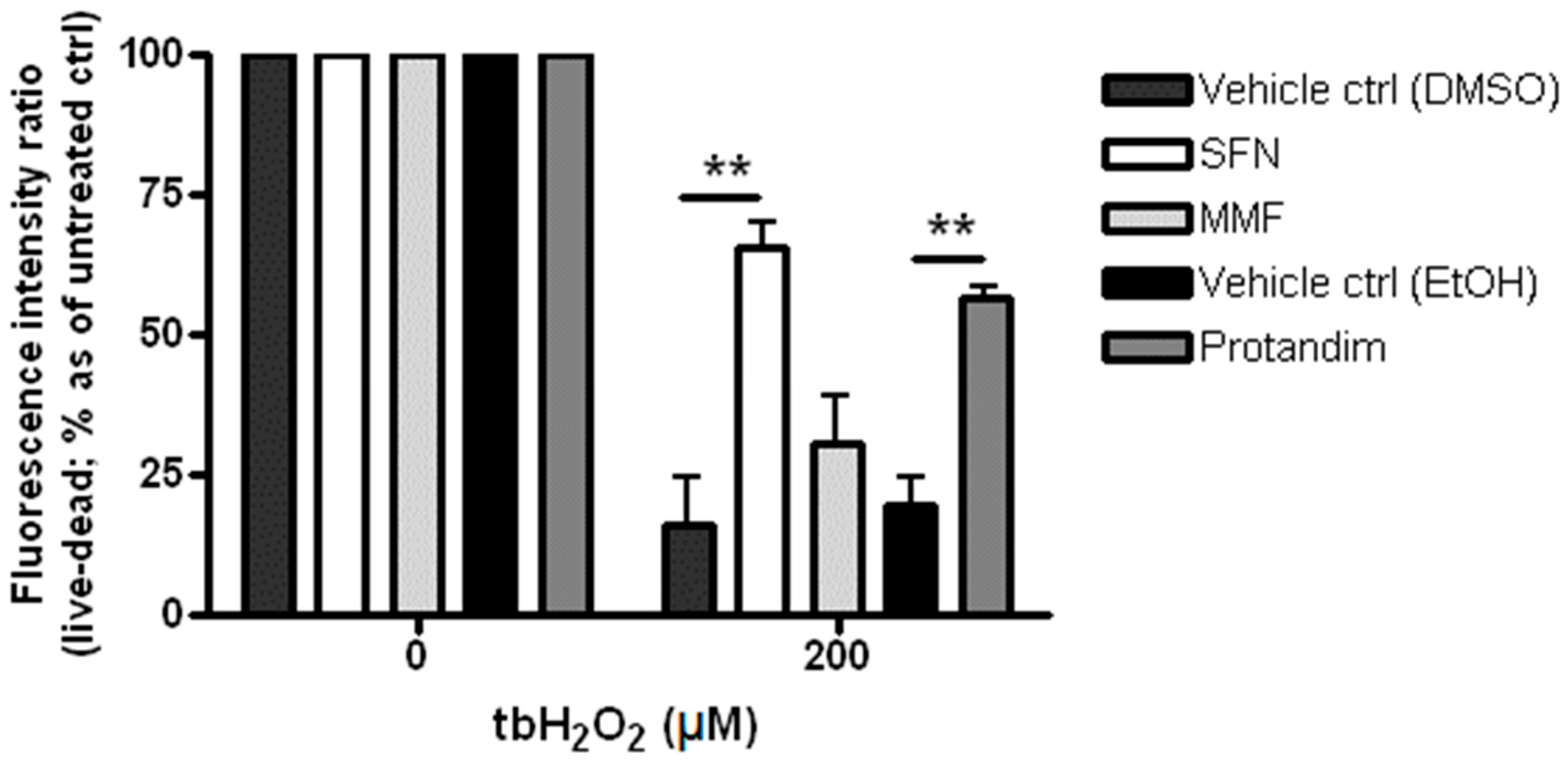
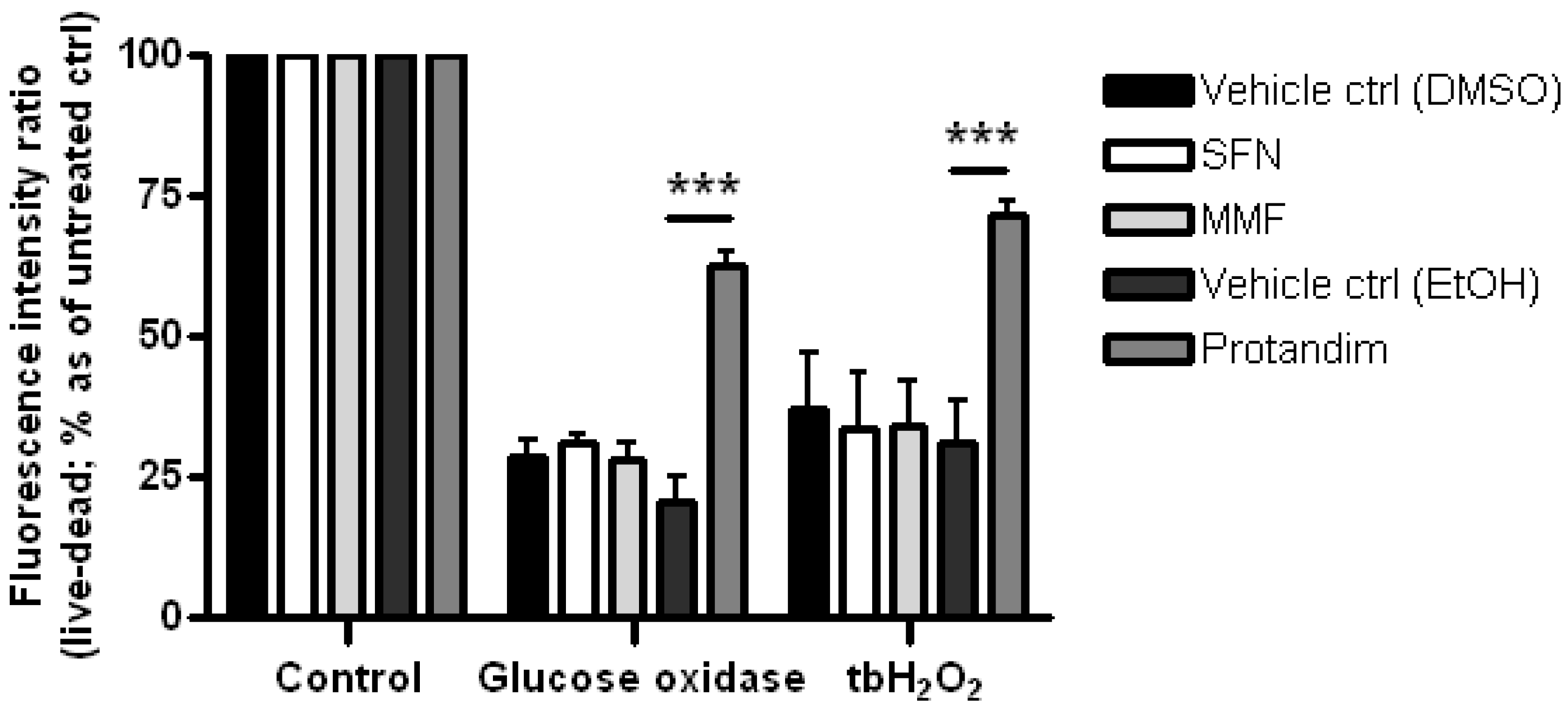
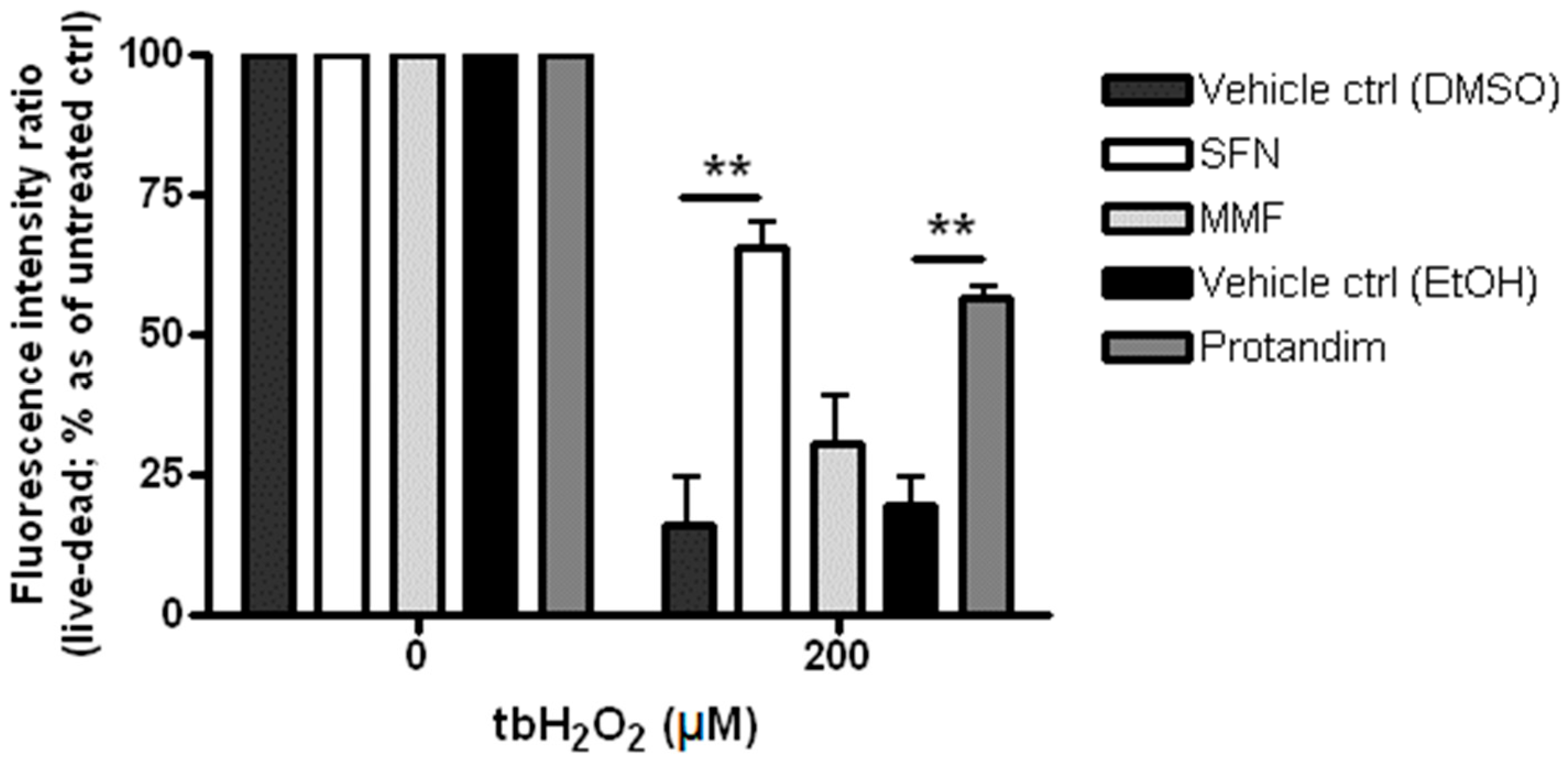
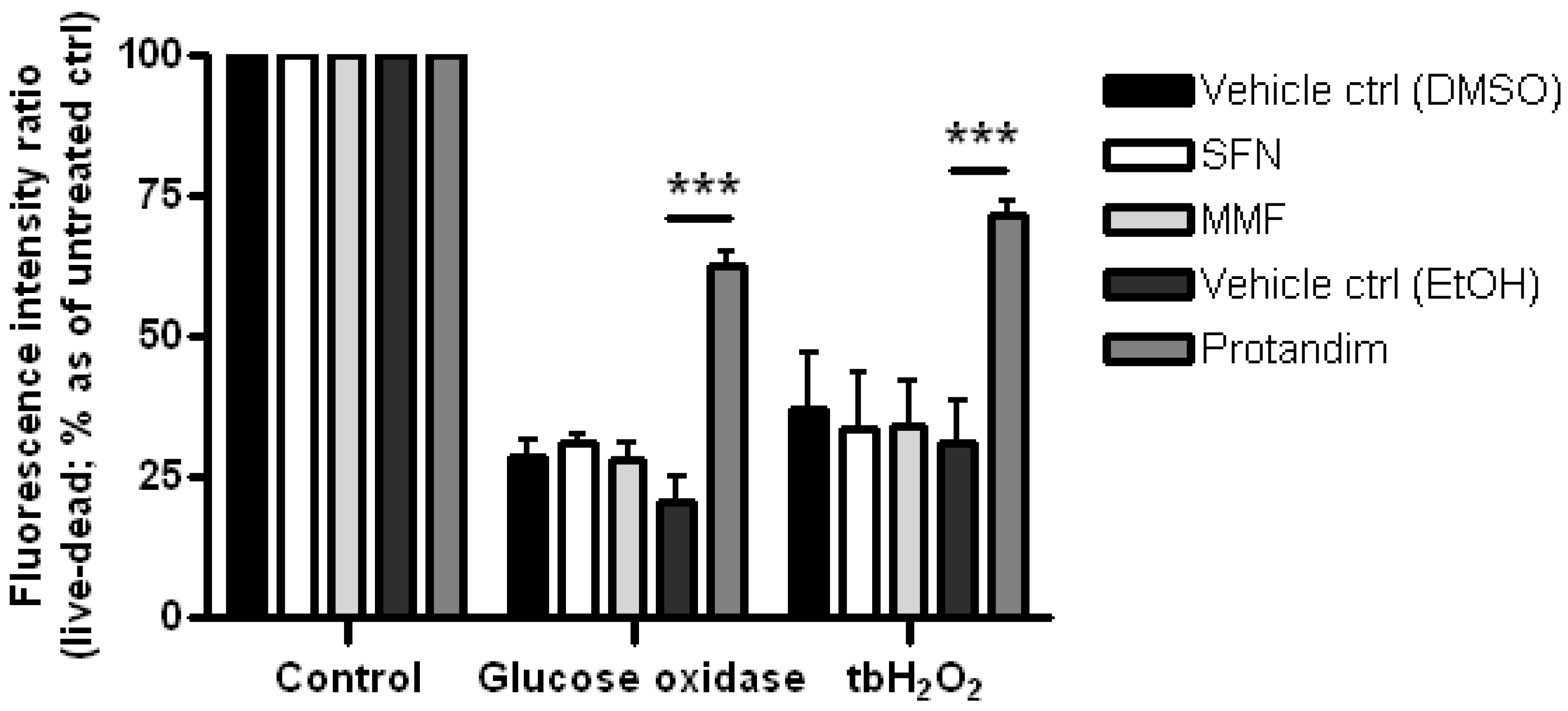
3.2. SFN and Protandim Prevent ROS-Induced Cell Death of OLN-93 Cells
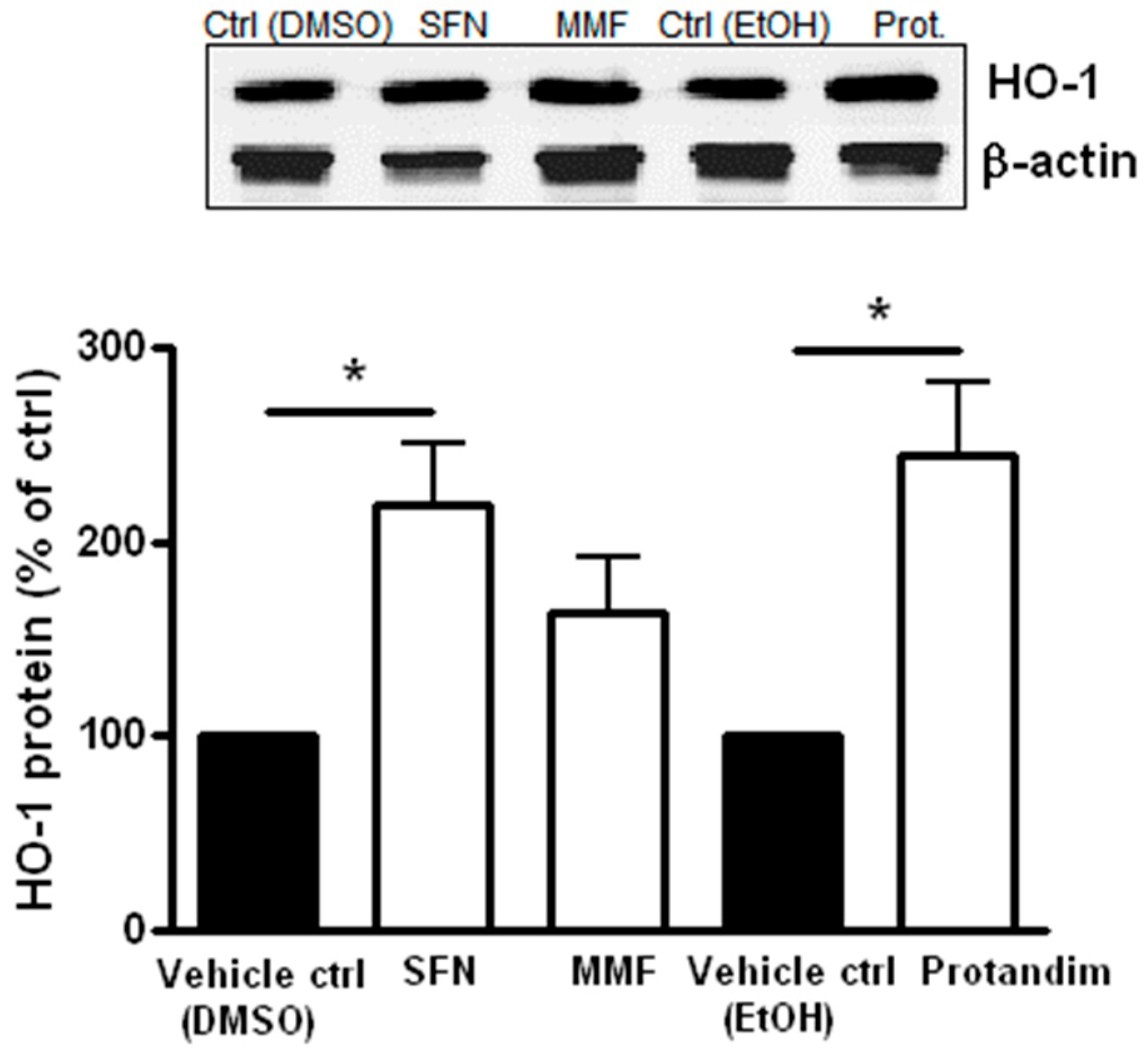
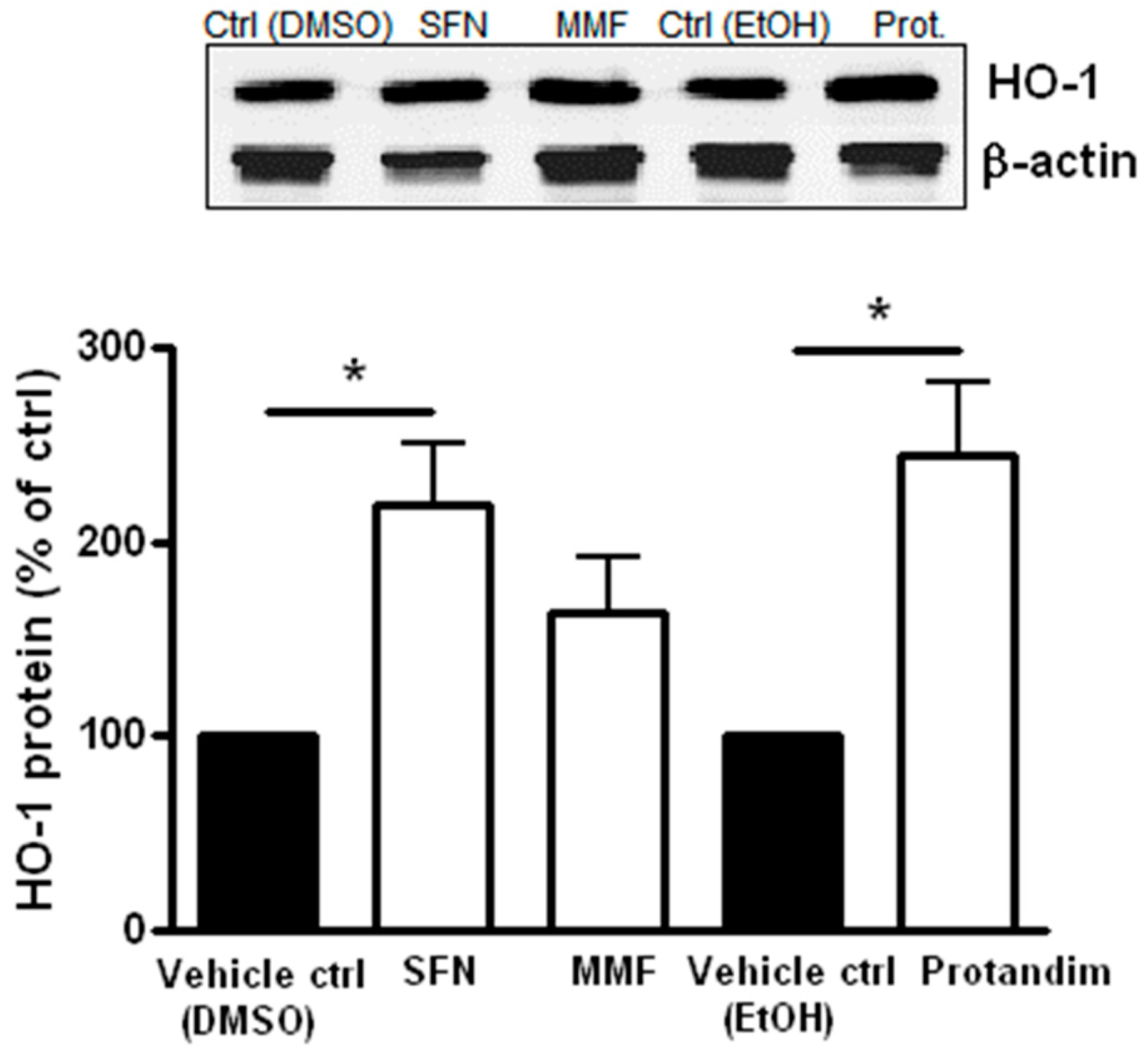
3.3. SFN and Protandim Increase Antioxidant Enzymes and p62 Expression in Mature Primary Rat OLs
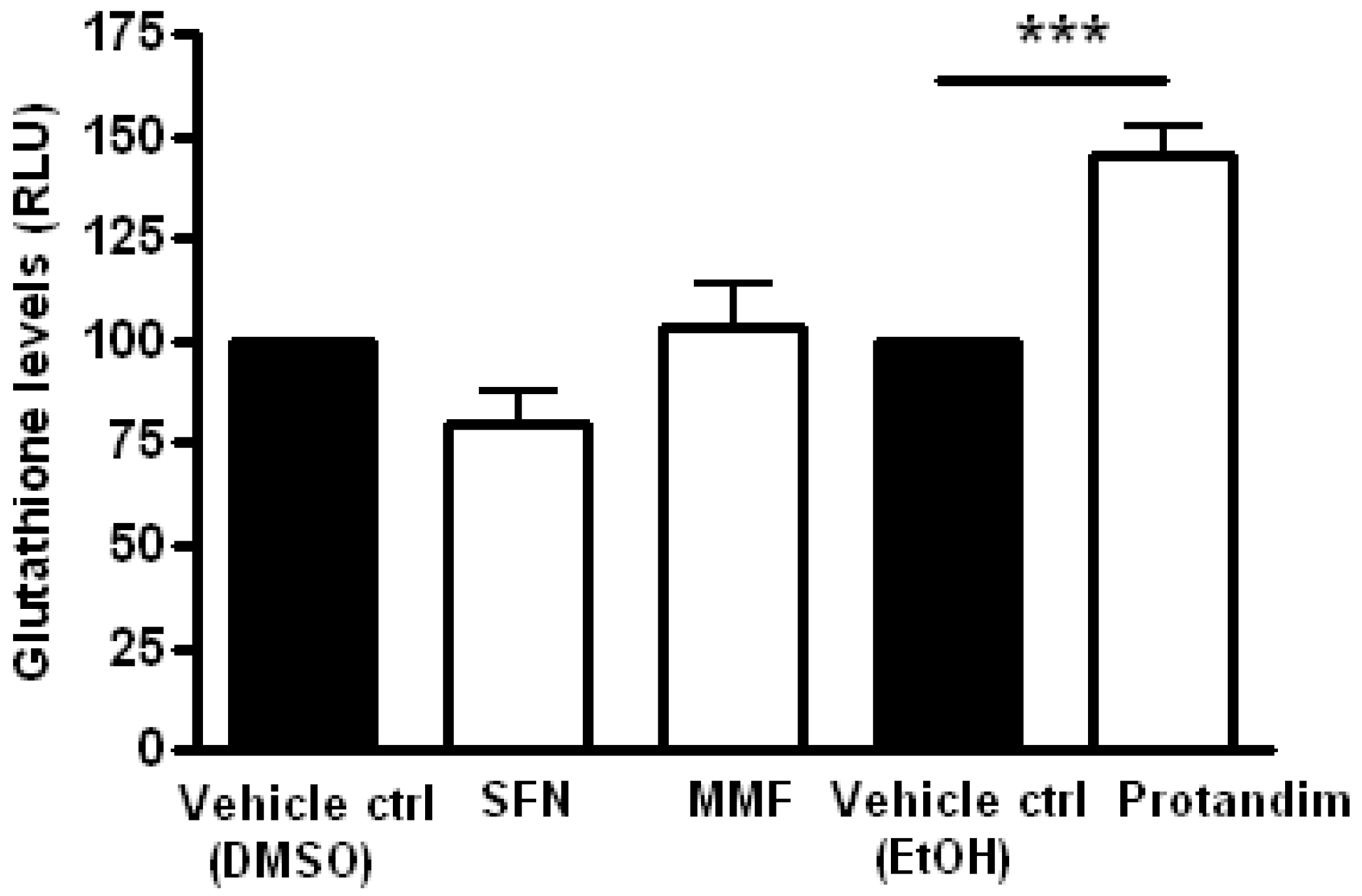
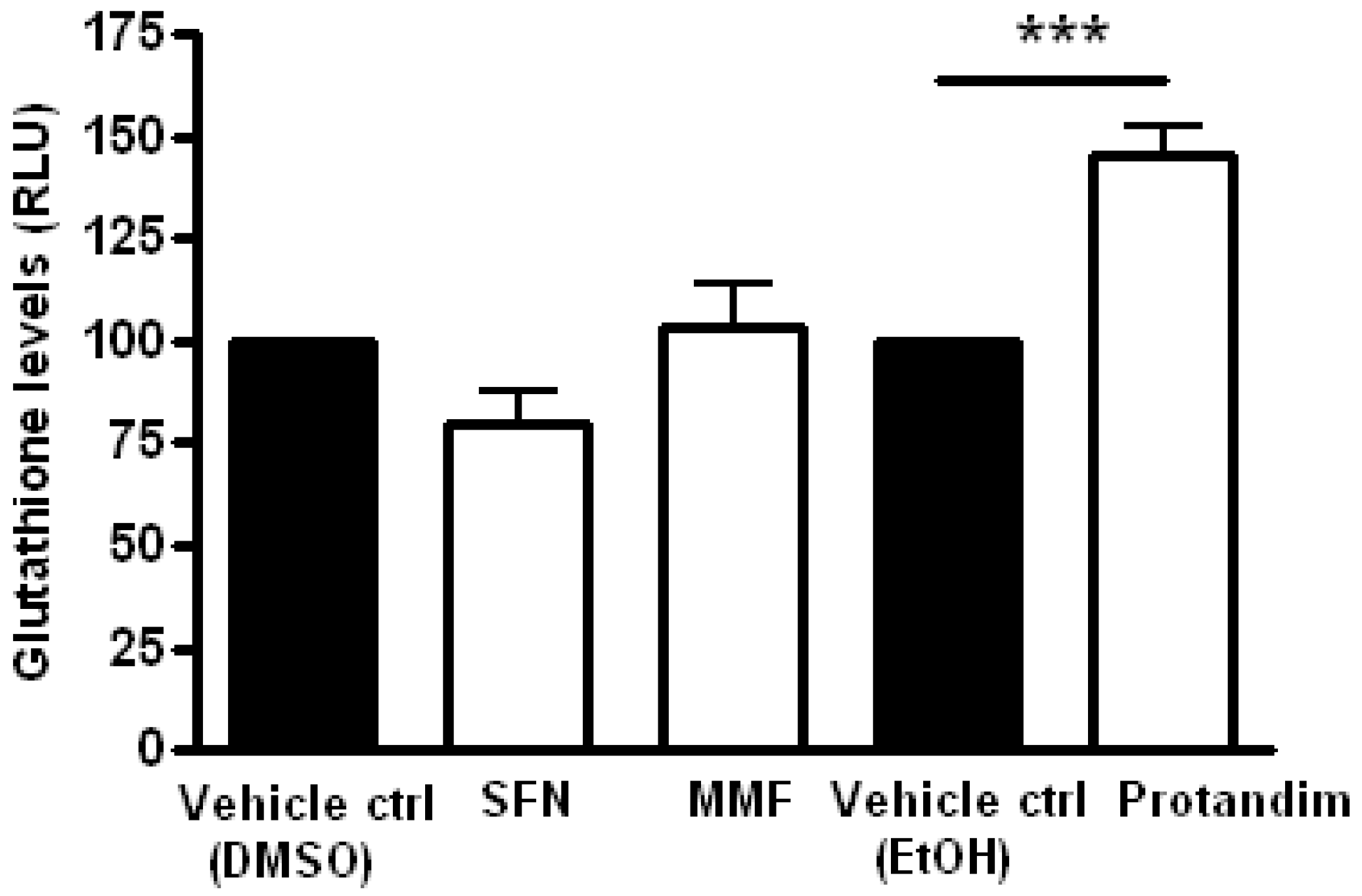
3.4. Protandim Increases Glutathione Levels in Mature Primary Rat OLs and Protects OLs from ROS-Induced Cell Death
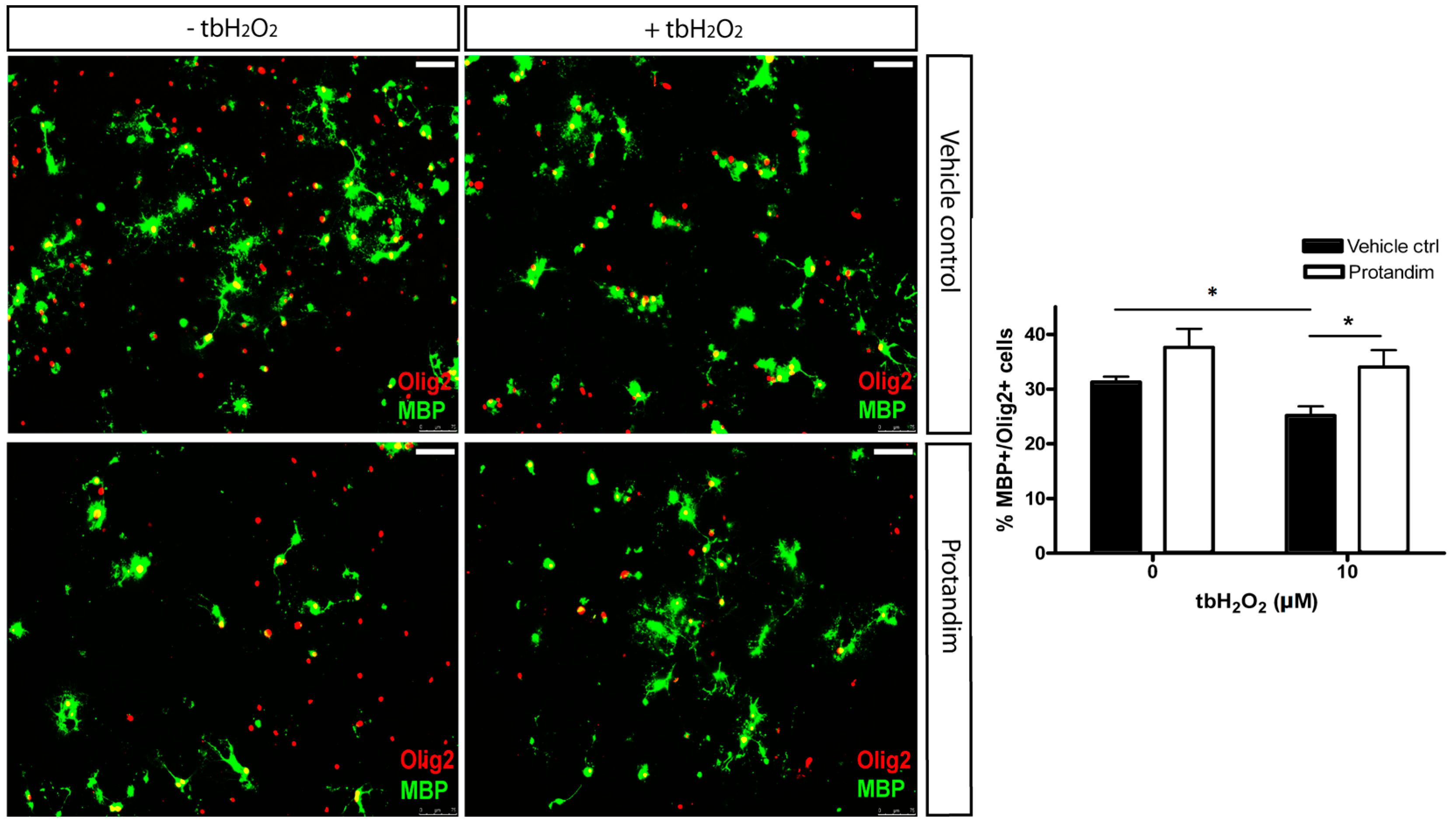
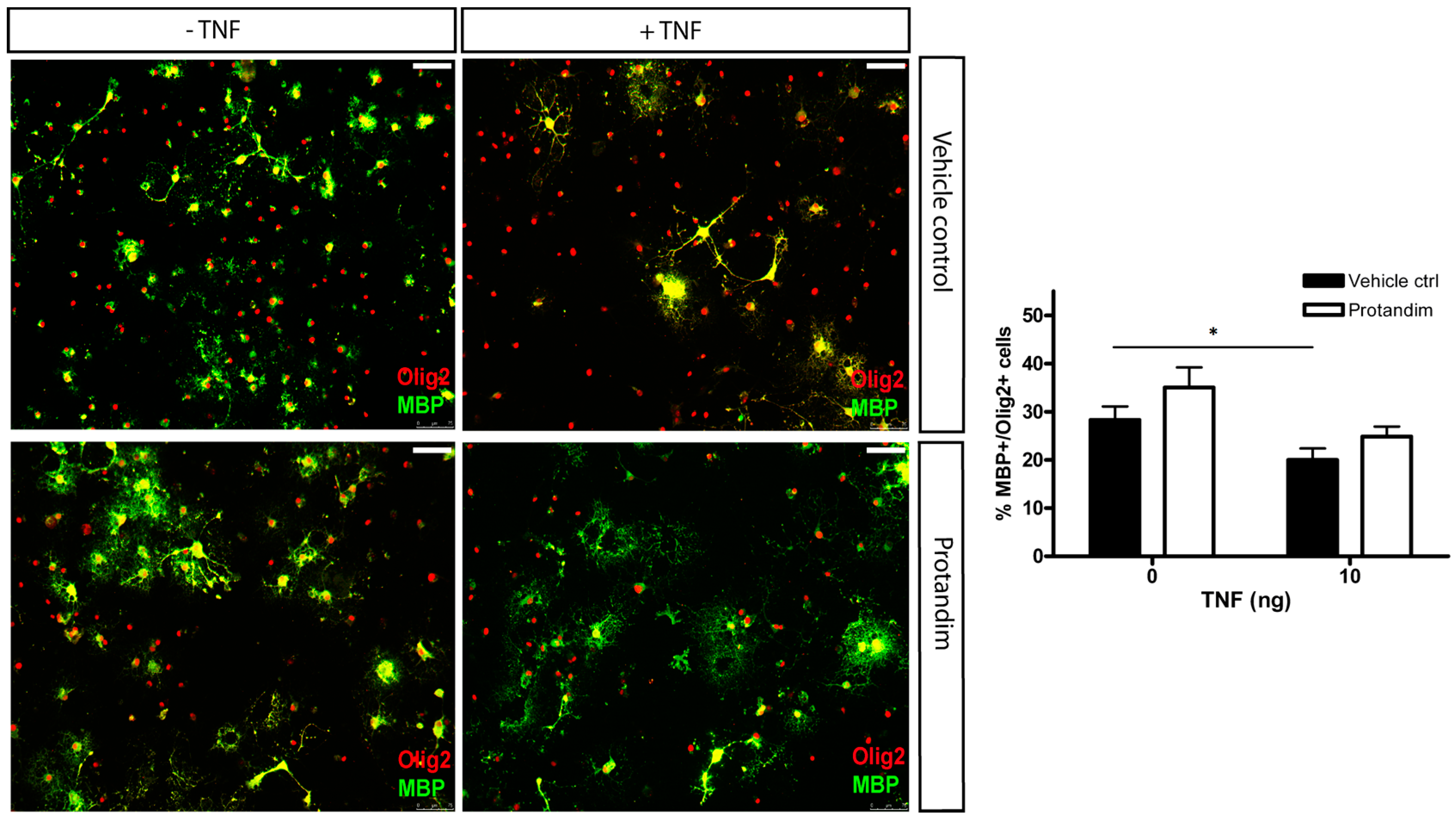
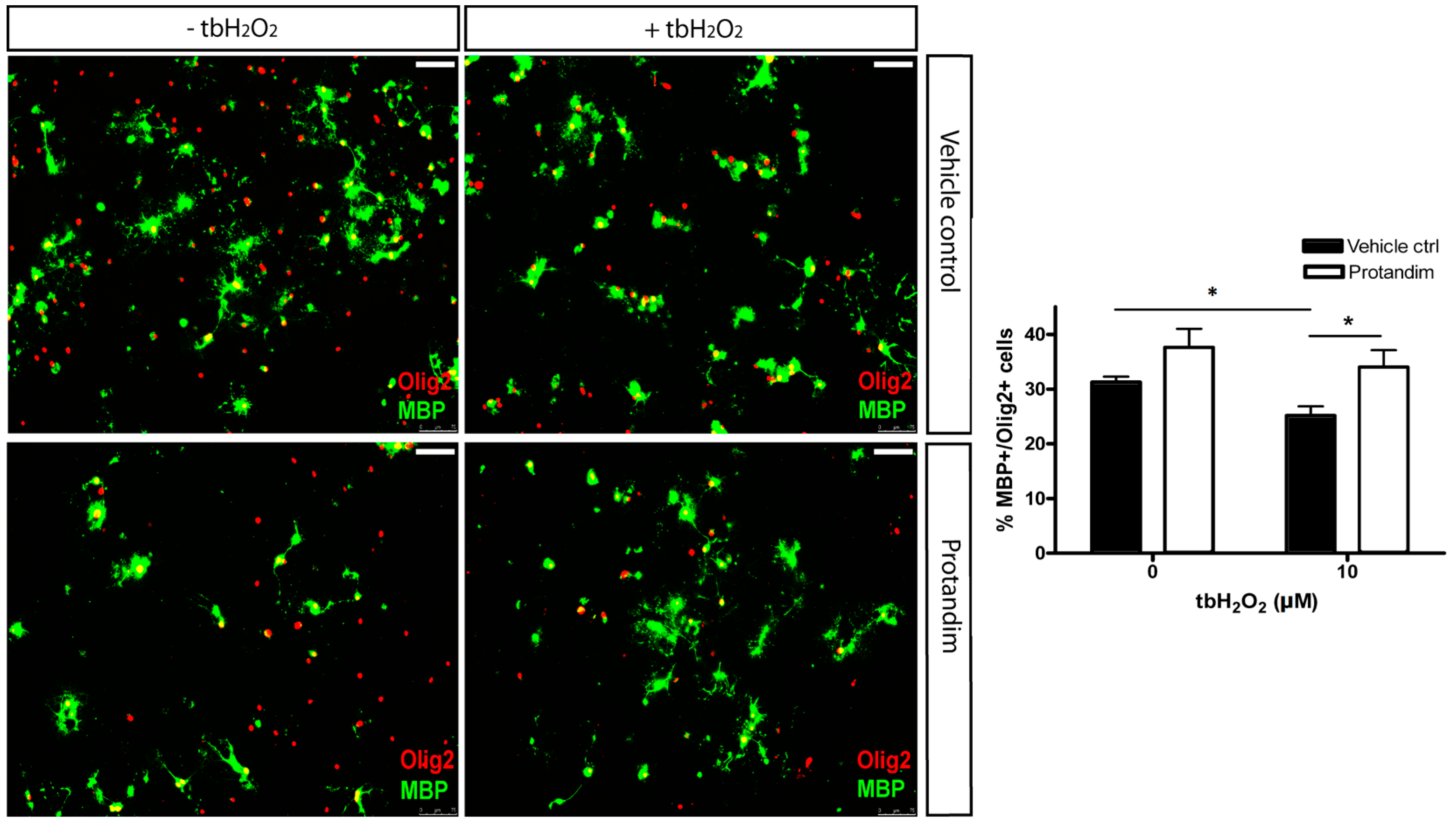
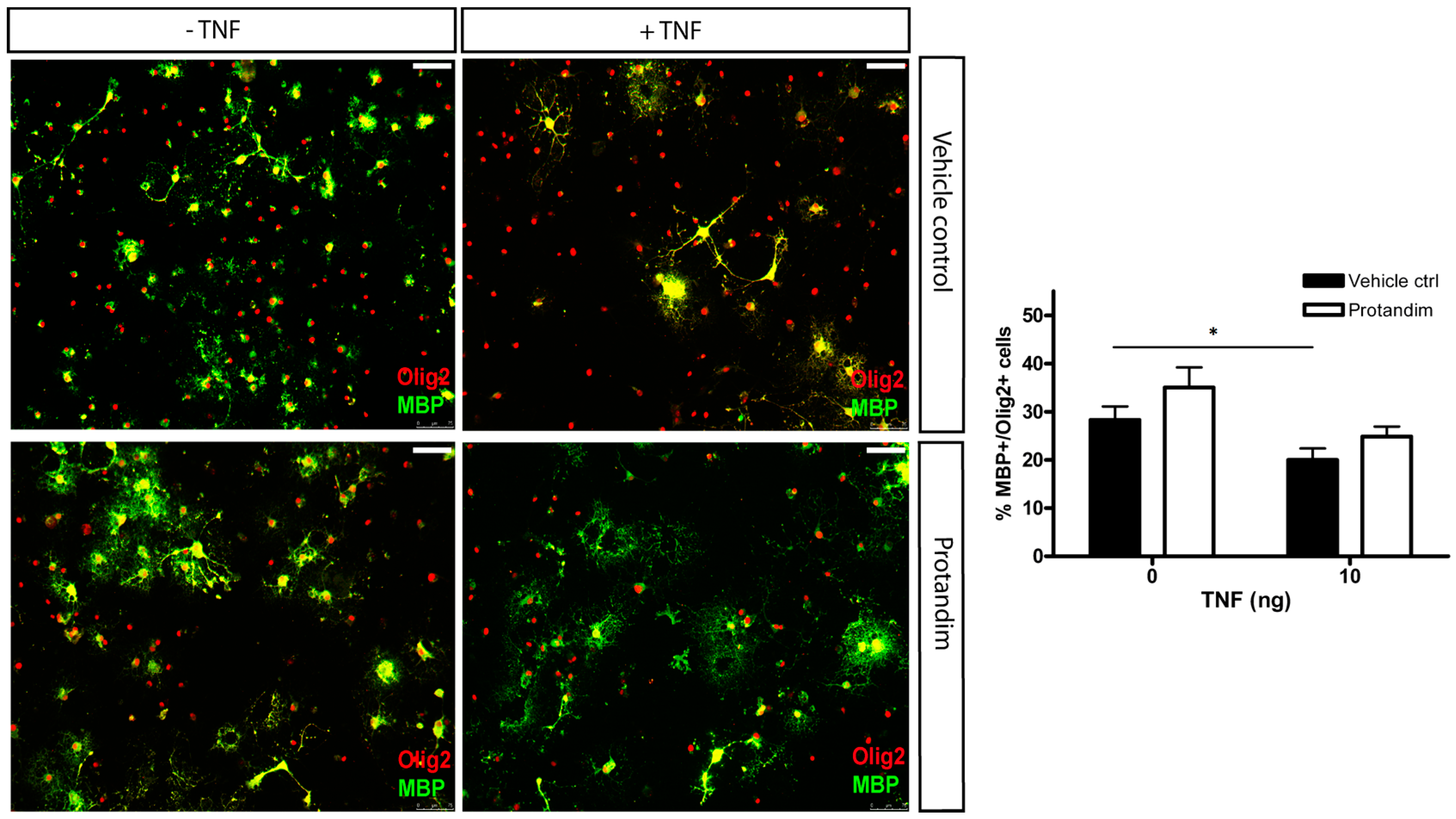
3.5. Protandim Promotes Differentiation of OPCs under Oxidative Conditions
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lassmann, H.; van Horssen, J. The molecular basis of neurodegeneration in multiple sclerosis. FEBS Lett. 2011, 585, 3715–3723. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Mahad, D.; van Horssen, J. Progressive multiple sclerosis: Pathology and pathogenesis. Nat. Rev. Neurol. 2012, 8, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Brück, W.; Schmied, M.; Suchanek, G.; Brück, Y.; Breitschopf, H.; Poser, S.; Piddlesden, S.; Lassmann, H. Oligodendrocytes in the early course of multiple sclerosis. Ann. Neurol. 1994, 35, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, K.; Suchanek, G.; Breitschopf, H.; Brück, W.; Budka, H.; Jellinger, K.; Lassmann, H. Patterns of oligodendroglia pathology in multiple sclerosis. Brain 1994, 117, 1311–1322. [Google Scholar] [CrossRef] [PubMed]
- Lucchinetti, C.; Brück, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. A quantitative analysis of oligodendrocytes in multiple sclerosis lesions. A study of 113 cases. Brain 1999, 122, 2279–2295. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Tourtellotte, W.W.; Rudick, R.; Trapp, B.D. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N. Engl. J. Med. 2002, 346, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Schreibelt, G.; van Horssen, J.; van Rossum, S.; Dijkstra, C.D.; Drukarch, B.; de Vries, H.E. Therapeutic potential and biological role of endogenous antioxidant enzymes in multiple sclerosis pathology. Brain Res. Rev. 2007, 56, 322–330. [Google Scholar] [CrossRef] [PubMed]
- De Vries, H.E.; Witte, M.; Hondius, D.; Rozemuller, A.J.; Drukarch, B.; Hoozemans, J.; van Horssen, J. Nrf2-induced antioxidant protection: A promising target to counteract ROS-mediated damage in neurodegenerative disease? Free Radic. Biol. Med. 2008, 45, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Van Horssen, J.; Witte, M.E.; Schreibelt, G.; de Vries, H.E. Radical changes in multiple sclerosis pathogenesis. Biochim. Biophys. Acta. 2011, 1812, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Haider, L.; Fischer, M.T.; Frischer, J.M.; Bauer, J.; Hoftberger, R.; Botond, G.; Esterbauer, H.; Binder, C.J.; Witztum, J.L.; Lassmann, H. Oxidative damage in multiple sclerosis lesions. Brain 2011, 134, 1914–1924. [Google Scholar] [CrossRef] [PubMed]
- Van Horssen, J.; Schreibelt, G.; Drexhage, J.; Hazes, T.; Dijkstra, C.D.; van der Valk, P.; de Vries, H.E. Severe oxidative damage in multiple sclerosis lesions coincides with enhanced antioxidant enzyme expression. Free Radic. Biol. Med. 2008, 45, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.T.; Sharma, R.; Lim, J.L.; Haider, L.; Frischer, J.M.; Drexhage, J.; Mahad, D.; Bradl, M.; van Horssen, J.; Lassmann, H. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 2012, 135, 886–899. [Google Scholar] [CrossRef] [PubMed]
- Mitrovic, B.; Ignarro, L.J.; Montestruque, S.; Smoll, A.; Merrill, J.E. Nitric oxide as a potential pathological mechanism in demyelination: Its differential effects on primary glial cells in vitro. Neuroscience 1994, 61, 575–585. [Google Scholar] [CrossRef]
- Husain, J.; Juurlink, B.H. Oligodendroglial precursor cell susceptibility to hypoxia is related to poor ability to cope with reactive oxygen species. Brain Res. 1995, 698, 86–94. [Google Scholar] [CrossRef]
- Thorburne, S.K.; Juurlink, B.H. Low glutathione and high iron govern the susceptibility of oligodendroglial precursors to oxidative stress. J. Neurochem. 1996, 67, 1014–1122. [Google Scholar] [CrossRef] [PubMed]
- Griot, C.; Vandevelde, M.; Richard, A.; Peterhans, E.; Stocker, R. Selective degeneration of oligodendrocytes mediated by reactive oxygen species. Free Radic. Res. Commun. 1990, 11, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Mitrovic, B.; Parkinson, J.; Merrill, J.E. An in vitro model of oligodendrocyte destruction by nitric oxide and its relevance to multiple sclerosis. Methods 1996, 10, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Oka, A.; Belliveau, M.J.; Rosenberg, P.A.; Volpe, J.J. Vulnerability of oligodendroglia to glutamate: Pharmacology, mechanisms, and prevention. J. Neurosci. 1993, 13, 1441–1453. [Google Scholar] [PubMed]
- Yonezawa, M.; Back, S.A.; Gan, X.; Rosenberg, P.A.; Volpe, J.J. Cystine deprivation induces oligodendroglial death: Rescue by free radical scavengers and by a diffusible glial factor. J. Neurochem. 1996, 67, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Back, S.A.; Luo, N.L.; Borenstein, N.S.; Levine, J.M.; Volpe, J.J.; Kinney, H.C. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J. Neurosci. 2001, 21, 1302–1312. [Google Scholar] [PubMed]
- Baud, O.; Greene, A.E.; Li, J.; Wang, H.; Volpe, J.J.; Rosenberg, P.A. Glutathione peroxidase-catalase cooperativity is required for resistance to hydrogen peroxide by mature rat oligodendrocytes. J. Neurosci. 2004, 24, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Fragoso, G.; Martínez-Bermúdez, A.K.; Liu, H.N.; Khorchid, A.; Chemtob, S.; Mushynski, W.E.; Almazan, G. Developmental differences in HO-induced oligodendrocyte cell death: Role of glutathione, mitogen activated protein kinases and caspase 3. J. Neurochem. 2004, 90, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Back, S.A.; Gan, X.; Li, Y.; Rosenberg, P.A.; Volpe, J.J. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J. Neurosci. 1998, 18, 6241–6253. [Google Scholar] [PubMed]
- Juurlink, B.H.; Thorburne, S.K.; Hertz, L. Peroxide-scavenging deficit underlies oligodendrocyte susceptibility to oxidative stress. Glia 1998, 22, 371–378. [Google Scholar] [CrossRef]
- French, H.M.; Reid, M.; Mamontov, P.; Simmons, R.A.; Grinspan, J.B. Oxidative stress disrupts oligodendrocyte maturation. J. Neurosci. Res. 2009, 87, 3076–3087. [Google Scholar] [CrossRef] [PubMed]
- Selmaj, K.W.; Raine, C.S. Tumor necrosis factor mediates myelin and oligodendrocyte damage in vitro. Ann. Neurol. 1988, 23, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Baerwald, K.D.; Popko, B. Developing and mature oligodendrocytes respond differently to the immune cytokine interferon-gamma. J. Neurosci. Res. 1998, 52, 230–239. [Google Scholar] [CrossRef]
- Loui, J.C.; Magal, E.; Takayama, S.; Varon, S. CNTF protection of oligodendrocytes against natural and tumor necrosis factor-induced death. Science 1993, 259, 689–692. [Google Scholar]
- Ye, P.; D’Ercole, A.J. Insulin-like growth factor I protects oligodendrocytes from tumor necrosis factor-alpha-induced injury. Endocrinology 1999, 140, 3063–3072. [Google Scholar] [PubMed]
- Jana, M.; Pahan, K. Redox regulation of cytokine-mediated inhibition of myelin gene expression in human primary oligodendrocytes. Free Radic. Biol. Med. 2005, 39, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; de Marchi, E.; Patergnani, S.; Suski, J.M.; Celsi, F.; Bononi, A.; Giorgi, C.; Marchi, S.; Rimessi, A.; Duszyński, J.; et al. Tumor necrosis factor-alpha impairs oligodendroglial differentiation through a mitochondria-dependent process. Cell. Death Differ. 2014, 21, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.L.; Wilhelmus, M.M.; de Vries, H.E.; Drukarch, B.; Hoozemans, J.J.; van Horssen, J. Antioxidative defense mechanisms controlled by Nrf2: State-of-the-art and clinical perspectives in neurodegenerative diseases. Arch. Toxicol. 2014, 88, 1773–1786. [Google Scholar] [CrossRef] [PubMed]
- Kraft, A.D.; Johnson, D.A.; Johnson, J.A. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J. Neurosci. 2004, 24, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Moore, A.N.; Clifton, G.L.; Dash, P.K. Sulforaphane enhances aquaporin-4 expression and decreases cerebral edema following traumatic brain injury. J. Neurosci. Res. 2005, 82, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Moore, A.N.; Redell, J.B.; Dash, P.K. Enhancing expression of Nrf2-driven genes protects the blood brain barrier after brain injury. J. Neurosci. 2007, 27, 10240–10248. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, L.O.; Kipp, M.; Lucius, R.; Pufe, T.; Wruck, C.J. Sulforaphane suppresses LPS-induced inflammation in primary rat microglia. Inflamm. Res. 2010, 59, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, K.; Kume, T.; Muto, C.; Takada-Takatori, Y.; Izumi, Y.; Sugimoto, H.; Akaike, A. Glutathione biosynthesis via activation of the nuclear factor E2-related factor 2 (Nrf2)--antioxidant-response element (ARE) pathway is essential for neuroprotective effects of sulforaphane and 6-(methylsulfinyl) hexyl isothiocyanate. J. Pharmacol. Sci. 2011, 115, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Cui, W.; Liu, J.; Li, R.; Liu, Q.; Xie, X.H.; Ge, X.L.; Zhang, J.; Song, X.J.; Wang, Y.; et al. Sulforaphane ameliorates the development of experimental autoimmune encephalomyelitis by antagonizing oxidative stress and Th17-related inflammation in mice. Exp. Neurol. 2013, 250, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.J.; Miller, D.H.; Phillips, J.T.; Hutchinson, M.; Havrdova, E.; Kita, M.; Yang, M.; Raghupathi, K.; Novas, M.; Sweetser, M.T.; et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N. Engl. J. Med. 2012, 367, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; Sheikh, S.I.; et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Scannevin, R.H.; Chollate, S.; Jung, M.Y.; Shackett, M.; Patel, H.; Bista, P.; Zeng, W.; Ryan, S.; Yamamoto, M.; Lukashev, M.; et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J. Pharmacol. Exp. Ther. 2012, 341, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Bogaard, H.J.; Natarajan, R.; Henderson, S.C.; Long, C.S.; Kraskauskas, D.; Smithson, L.; Ockaili, R.; McCord, J.M.; Voelkel, N.F. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation 2009, 120, 1951–1960. [Google Scholar] [CrossRef] [PubMed]
- Velmurugan, K.; Alam, J.; McCord, J.M.; Pugazhenthi, S. Synergistic induction of heme oxygenase-1 by the components of the antioxidant supplement Protandim. Free Radic. Biol. Med. 2009, 46, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Reuland, D.J.; Khademi, S.; Castle, C.J.; Irwin, D.C.; McCord, J.M.; Miller, B.F.; Hamilton, K.L. Upregulation of phase II enzymes through phytochemical activation of Nrf2 protects cardiomyocytes against oxidant stress. Free Radic. Biol. Med. 2013, 56, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Richter-Landsberg, C.; Heinrich, M. OLN-93: A new permanent oligodendroglia cell line derived from primary rat brain glial cultures. J. Neurosci. Res. 1996, 45, 161–173. [Google Scholar] [CrossRef]
- Maier, O.; van der Heide, T.; van Dam, A.M.; Baron, W.; de Vries, H.E.; Hoekstra, D. Alteration of the extracellular matrix interferes with raft association of neurofascin in oligodendrocytes. Potential significance for multiple sclerosis? Mol. Cell Neurosci. 2005, 28, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Nijland, P.G.; Witte, M.E.; van het Hof, B.; van der Pol, S.; Bauer, J.; Lassmann, H.; van der Valk, P.; de Vries, H.E.; van Horssen, J. Astroglial PGC-1α increases mitochondrial antioxidant capacity and suppresses inflammation: Implications for multiple sclerosis. Acta. Neuropathol. Commun. 2014, 2, 170. [Google Scholar] [CrossRef] [PubMed]
- Witte, M.E.; Nijland, P.G.; Drexhage, J.A.; Gerritsen, W.; Geerts, D.; van het Hof, B.; Reijerkerk, A.; de Vries, H.E.; van der Valk, P.; van Horssen, J. Reduced expression of PGC-1α partly underlies mitochondrial changes and correlates with neuronal loss in multiple sclerosis cortex. Acta. Neuropathol. 2013, 125, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Relvas, J.B.; Setzu, A.; Baron, W.; Buttery, P.C.; LaFlamme, S.E.; Franklin, R.J.; ffrench-Constant, C. Expression of dominant-negative and chimeric subunits reveals an essential role for beta1 integrin during myelination. Curr. Biol. 2011, 11, 1039–1043. [Google Scholar] [CrossRef]
- Inui, T.; Chano, T.; Takikita-Suzuki, M.; Nishikawa, M.; Yamamoto, G.; Okabe, H. Association of p62/SQSTM1 excess and oral carcinogenesis. PLoS ONE 2013, 8, e74398. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shi, S.; Gu, Z.; Du, Y.; Liu, M.; Yan, S.; Gao, J.; Li, J.; Shao, Y.; Zhong, W.; et al. Impaired autophagic function in rat islets with aging. Age (Dordr.) 2013, 35, 1531–1544. [Google Scholar] [CrossRef] [PubMed]
- Cheishvili, D.; Dietrich, P.; Maayan, C.; Even, A.; Weil, M.; Dragatsis, I.; Razin, A. IKAP deficiency in an FD mouse model and in oligodendrocyte precursor cells results in downregulation of genes involved in oligodendrocyte differentiation and myelin formation. PLoS ONE 2014, 9, e94612. [Google Scholar] [CrossRef] [PubMed]
- Thiessen, A.; Schmidth, M.M.; Dringen, R. Fumaric acid dialkyl esters deprive cultured rat oligodendroglial cells of glutathione and upregulate the expression of heme oxygenase 1. Neurosci. Lett. 2010, 475, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Hybertson, B.M.; Goa, B.; Bose, K.B.; McCord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Aspects Med. 2011, 32, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P. Redefining oxidative stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Cidlowski, J.A. Apoptosis and glutathione: beyond an antioxidant. Cell Death Differ. 2009, 16, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.; Sah, S.; Nath, S.K. Interaction between glutathione and apoptosis in systemic lupus erythematosus. Autoimmun. Rev. 2013, 12, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Kaidery, N.A.; Banerjee, R.; Yang, L.; Smirnova, N.A.; Hushpulian, D.M.; Liby, K.T.; Williams, C.R.; Yamamoto, M.; Kensler, T.W.; Ratan, R.R.; et al. Targeting Nrf2-mediated gene transcription by extremely potent synthetic triterpenoids attenuate dopaminergic neurotoxicity in the MPTP mouse model of Parkinson’s disease. Antioxid. Redox Signal. 2013, 18, 139–157. [Google Scholar] [CrossRef] [PubMed]
- Schilling, S.; Goelz, S.; Linker, R.; Luehder, F.; Gold, R. Fumaric acid esters are effective in chronic experimental autoimmune encephalomyelitis and suppress macrophage infiltration. Clin. Exp. Immunol. 2006, 145, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Licht-Mayer, S.; Wimmer, I.; Traffehn, S.; Metz, I.; Brück, W.; Bauer, J.; Bradl, M.; Lassmann, H. Cell type-specific Nrf2 expression in multiple sclerosis lesions. Acta. Neuropathol. 2015, 130, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Heo, S.R.; Han, A.M.; Kwon, Y.K.; Joung, I. p62 protects SH-SY5Y neuroblastoma cells against H2O2-induced injury through the PDK1/Akt pathway. Neurosci. Lett. 2009, 450, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Voelkel, N.F.; Bogaard, H.J.; Al Husseini, A.; Farkas, L.; Gomez-Arroyo, J.; Natarajan, R. Antioxidants for the treatment of patients with severe angioproliferative pulmonary hypertension? Antioxid. Redox Signal. 2013, 18, 1810–1817. [Google Scholar] [CrossRef] [PubMed]
- Joe, B.; Vijaykumar, M.; Lokesh, B.R. Biological properties of curcumin-cellular and molecular mechanisms of action. Crit. Rev. Food Sci. Nutr. 2004, 44, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.; Weinreb, O.; Amit, T.; Youdim, M.B. Cell signaling pathways in the neuroprotective actions of the green tea polyphenol (−)-epigallocatechin-3-gallate: Implications for neurodegenerative diseases. J. Neurochem. 2004, 88, 1555–1569. [Google Scholar] [CrossRef] [PubMed]
- Kishore, K.; Singh, M. Effect of bacosides, alcoholic extract of Bacopa monniera Linn. (brahmi), on experimental amnesia in mice. Indian J. Exp. Biol. 2005, 43, 640–645. [Google Scholar] [PubMed]
- Xie, L.; Li, X.K.; Takahara, S. Curcumin has bright prospects for the treatment of multiple sclerosis. Int. Immunopharmacol. 2011, 11, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Li, X.K.; Funeshima-Fuji, N.; Kimura, H.; Matsumoto, Y.; Isaka, Y.; Takahara, S. Amelioration of experimental autoimmune encephalomyelitis by curcumin treatment through inhibition of IL-17 production. Int. Immunopharmacol. 2009, 9, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Kanakasabai, S.; Casalini, E.; Walline, C.C.; Mo, C.; Chearwae, W.; Bright, J.J. Differential regulation of CD4(+) T helper cell responses by curcumin in experimental autoimmune encephalomyelitis. J. Nutr. Biochem. 2012, 23, 1498–1507. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, J.L.; Van der Pol, S.M.A.; Baron, W.; McCord, J.M.; De Vries, H.E.; Van Horssen, J. Protandim Protects Oligodendrocytes against an Oxidative Insult. Antioxidants 2016, 5, 30. https://doi.org/10.3390/antiox5030030
Lim JL, Van der Pol SMA, Baron W, McCord JM, De Vries HE, Van Horssen J. Protandim Protects Oligodendrocytes against an Oxidative Insult. Antioxidants. 2016; 5(3):30. https://doi.org/10.3390/antiox5030030
Chicago/Turabian StyleLim, Jamie L., Susanne M. A. Van der Pol, Wia Baron, Joe M. McCord, Helga E. De Vries, and Jack Van Horssen. 2016. "Protandim Protects Oligodendrocytes against an Oxidative Insult" Antioxidants 5, no. 3: 30. https://doi.org/10.3390/antiox5030030
APA StyleLim, J. L., Van der Pol, S. M. A., Baron, W., McCord, J. M., De Vries, H. E., & Van Horssen, J. (2016). Protandim Protects Oligodendrocytes against an Oxidative Insult. Antioxidants, 5(3), 30. https://doi.org/10.3390/antiox5030030