A Cystine-Rich Whey Supplement (Immunocal®) Delays Disease Onset and Prevents Spinal Cord Glutathione Depletion in the hSOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis


Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Supplement Content | % of Total Supplement |
|---|---|---|
| Whey proteins (serum albumin, α lactalbumin and lactoferrin) | 8.8–9.2 g | 88%–92% |
| Fat | ~0.05 g | <0.5% |
| Lactose | ~0.15 g | <1.5% |
| Minerals (Ca, Na) | ~0.30 g | <3.0% |
| Moisture | 0.5 g | ~5% |
2. Experimental Section
2.1. Mouse Model of ALS
| G93A SOD1 Transgenic | Males (n = 11) | Females (n = 12) |
|---|---|---|
| Onset (days ± SEM) | 90 ± 1.0 | 92.8 ± 1.4 |
| Survival (days ± SEM) | 123.3 ± 2.2 | 128.6 ± 3.1 |
| Group Number(s) | Treatment | Females | Males | Total N | Mean Onset (days) | SEM |
|---|---|---|---|---|---|---|
| 1 | NonTG | 11 | 13 | 24 | ||
| 2 | G93A SOD1 Control | 12 | 11 | 23 | 91 | 1 |
| 3 | ICAL (ad libitum) | 5 | 8 | 13 | 99 ** | 1 |
| 4 | ICAL (oral gavage) | 1 | 4 | 5 | 98 * | 3 |
| 5 | ICAL (oral gavage) + riluzole | 5 | 5 | 10 | 101 *** | 2 |
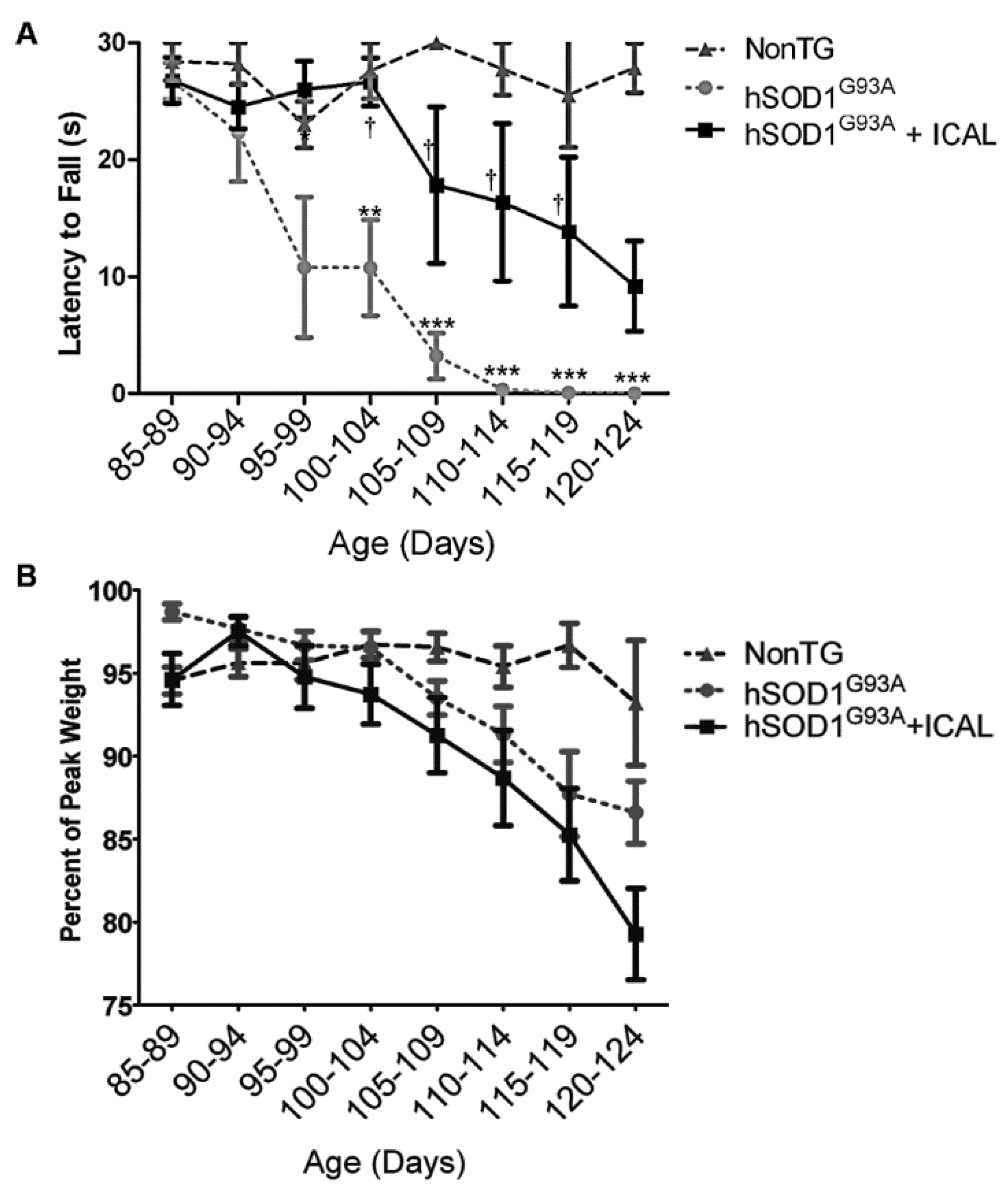
2.2. Clinical Tests
2.3. Tissue Processing
2.4. High Performance Liquid Chromatography with Electrochemical Detection
2.5. Isolation of Mitochondria from Lumbar Spinal Cord
2.6. Mitochondrial GSH Loading and Measurement of GSH Levels in Isolated Mitochondria
2.7. Statistical Analysis
3. Results
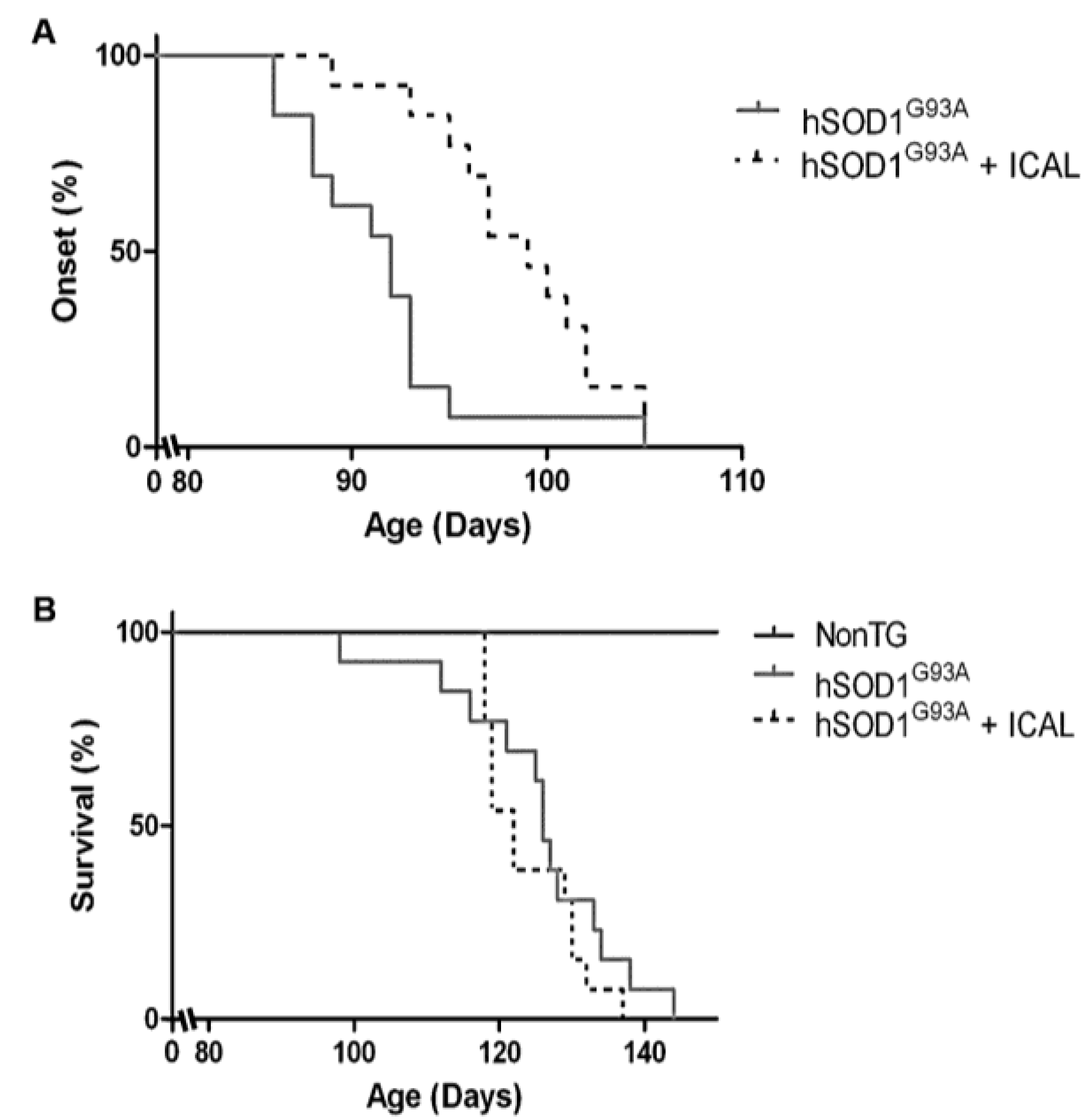
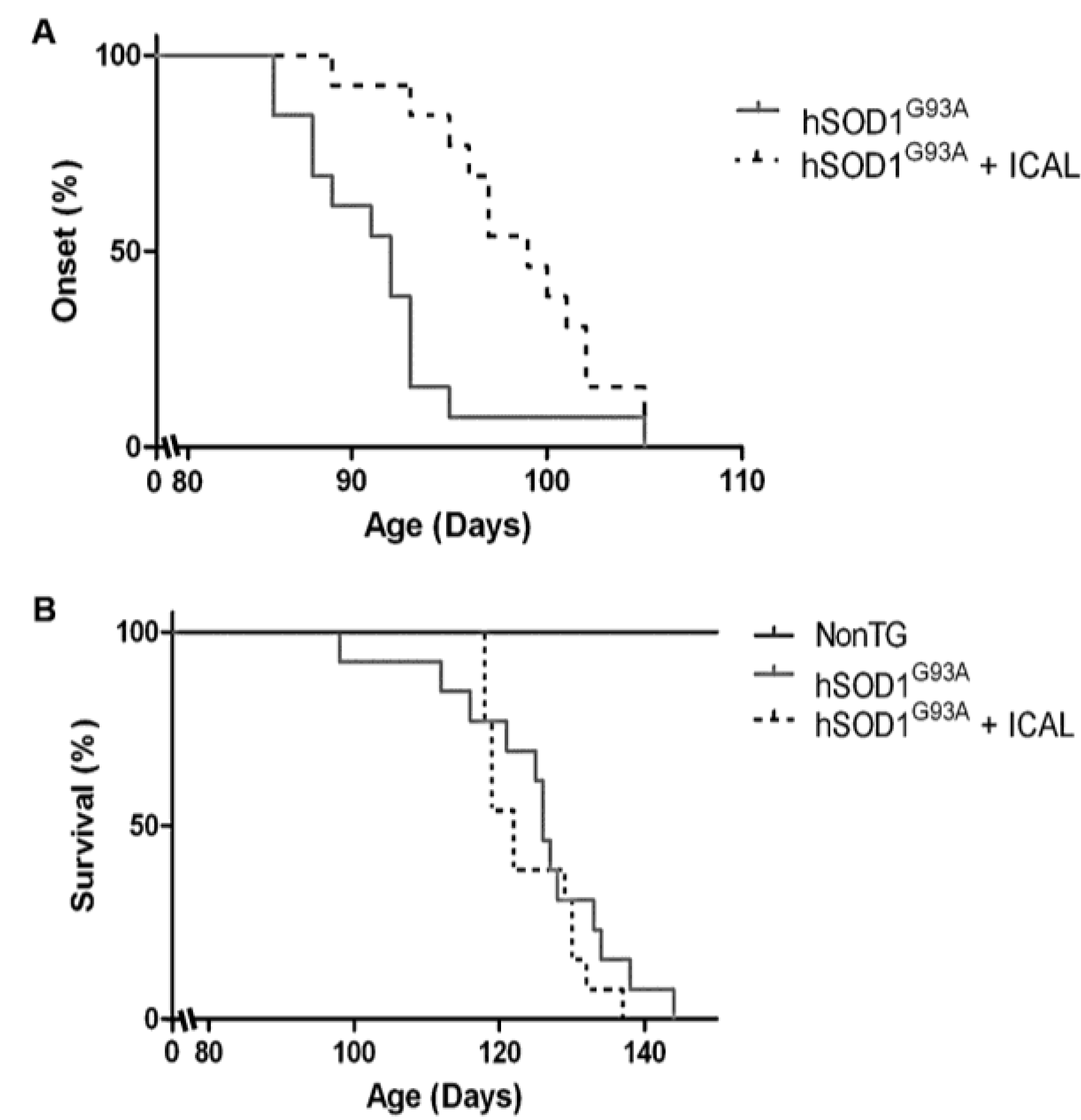
3.1. Immunocal® Delays Disease Onset, but Does Not Extend Lifespan in the hSOD1G93A Mouse Model of ALS
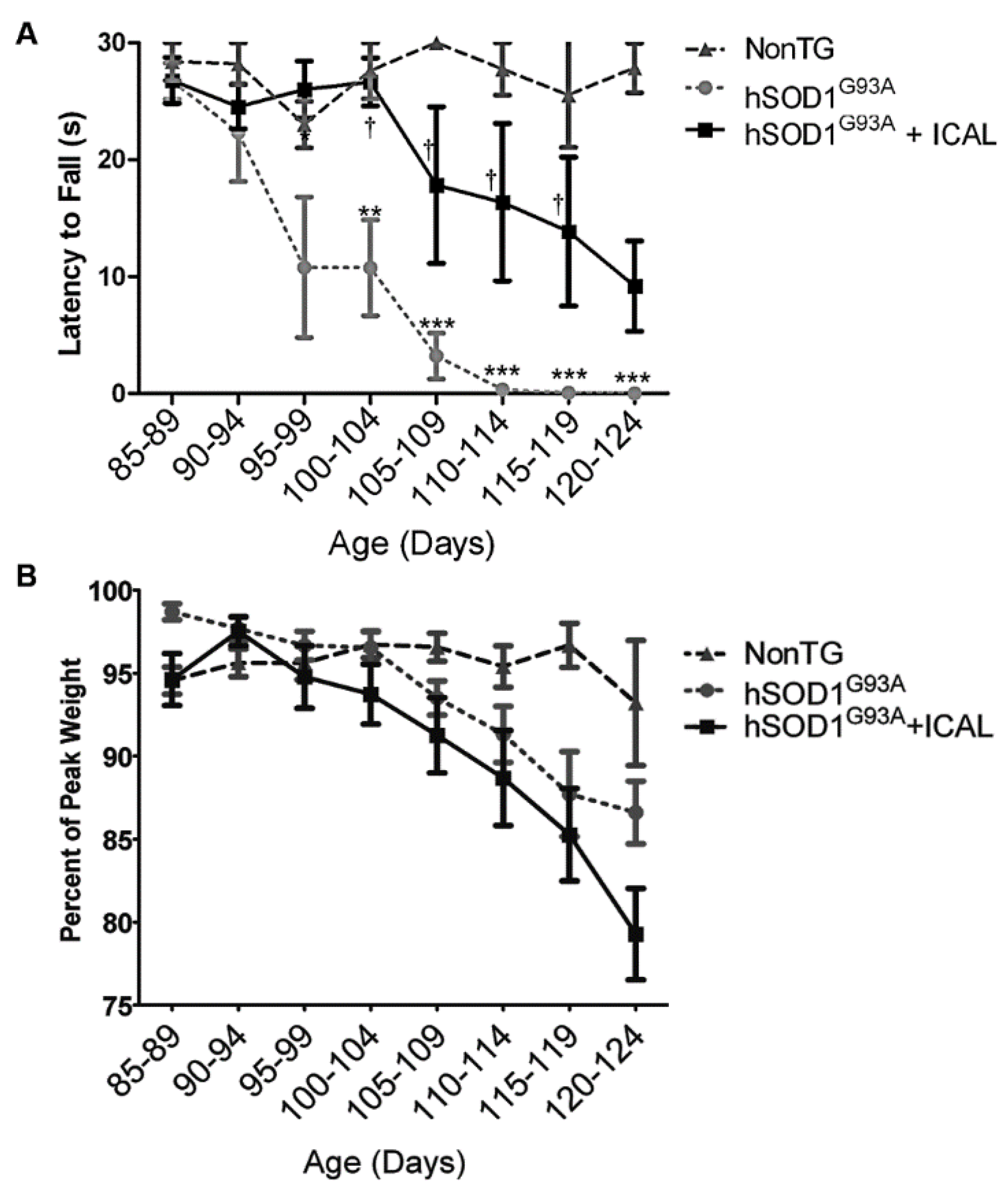
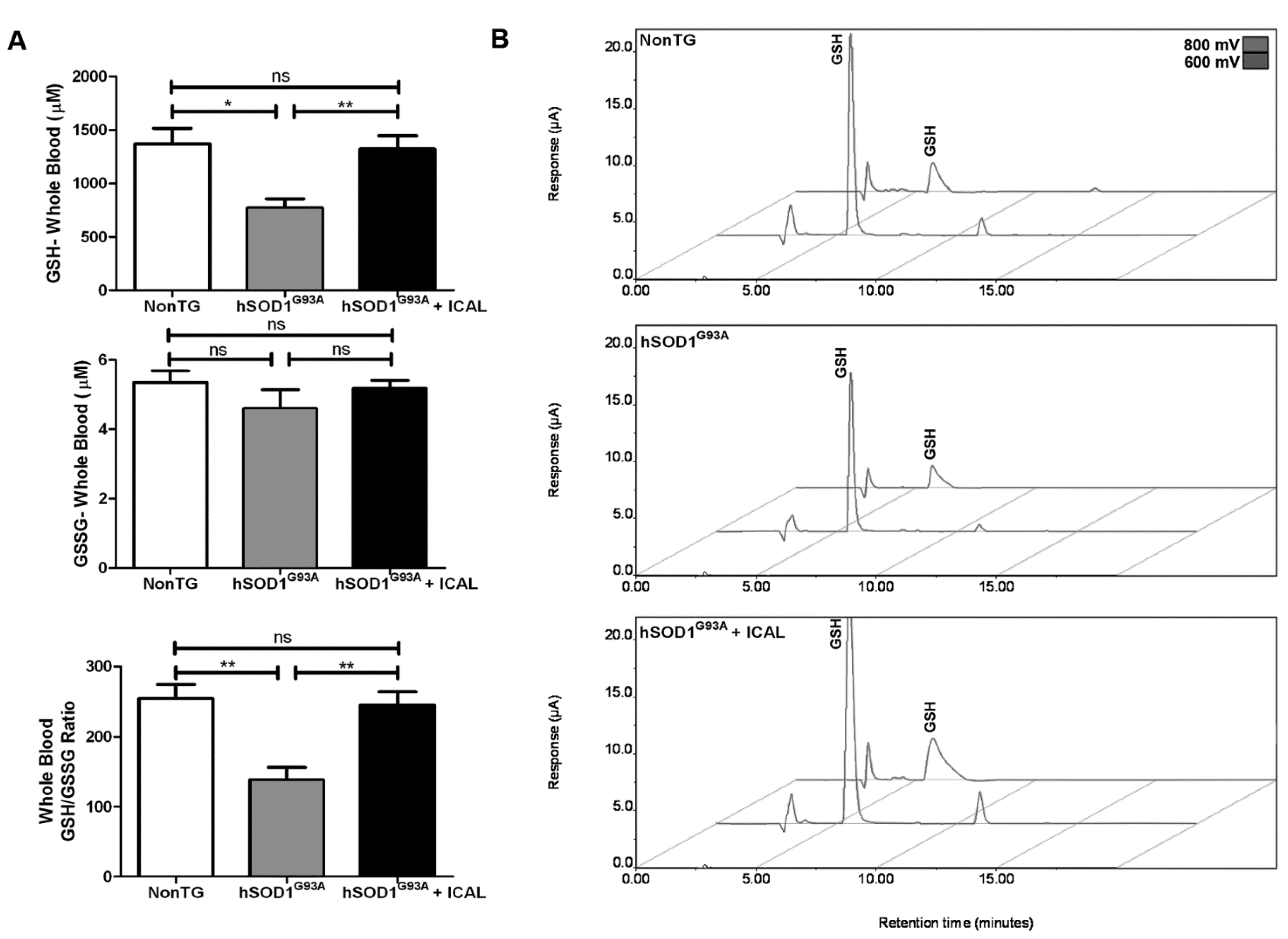
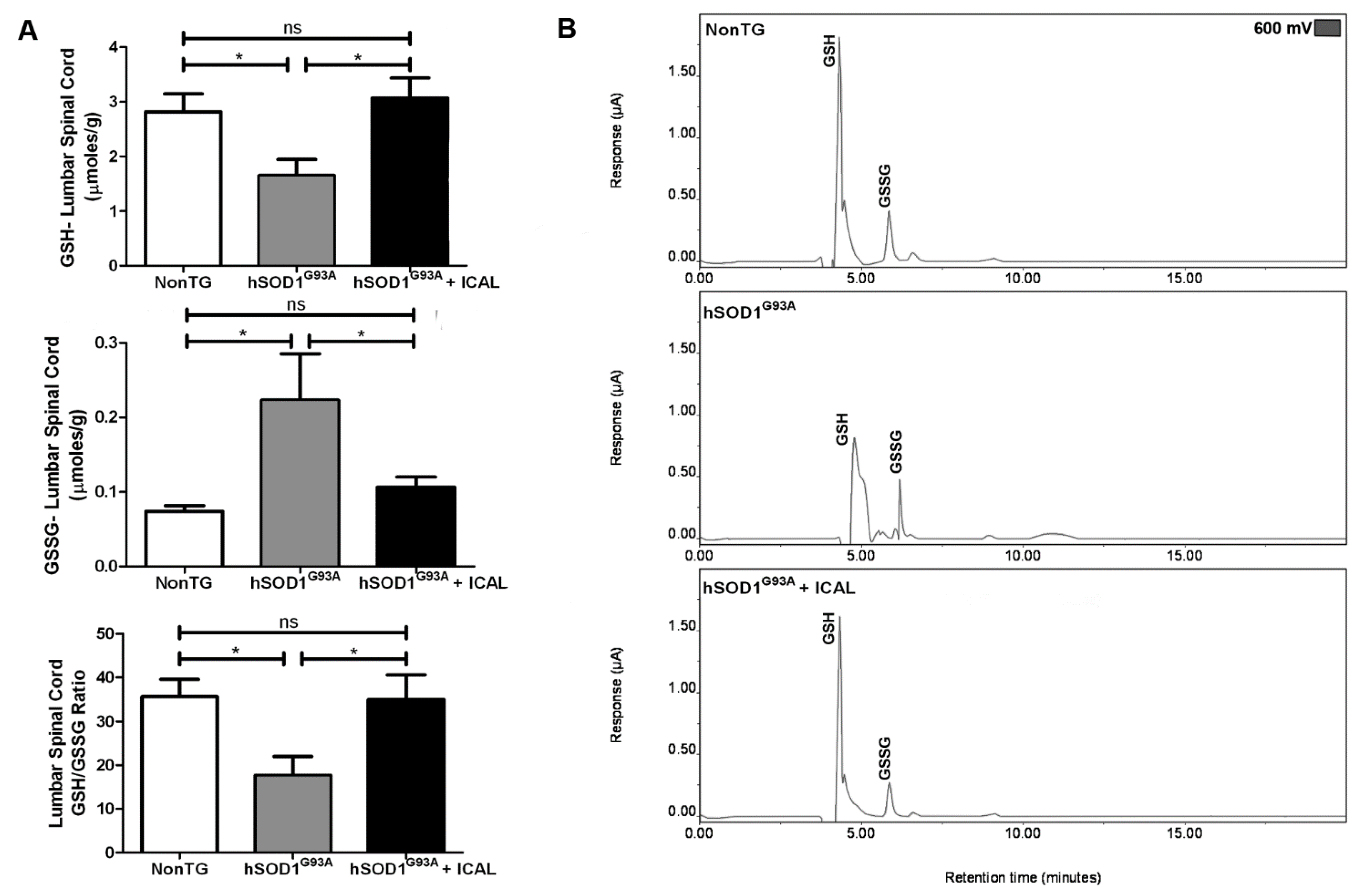
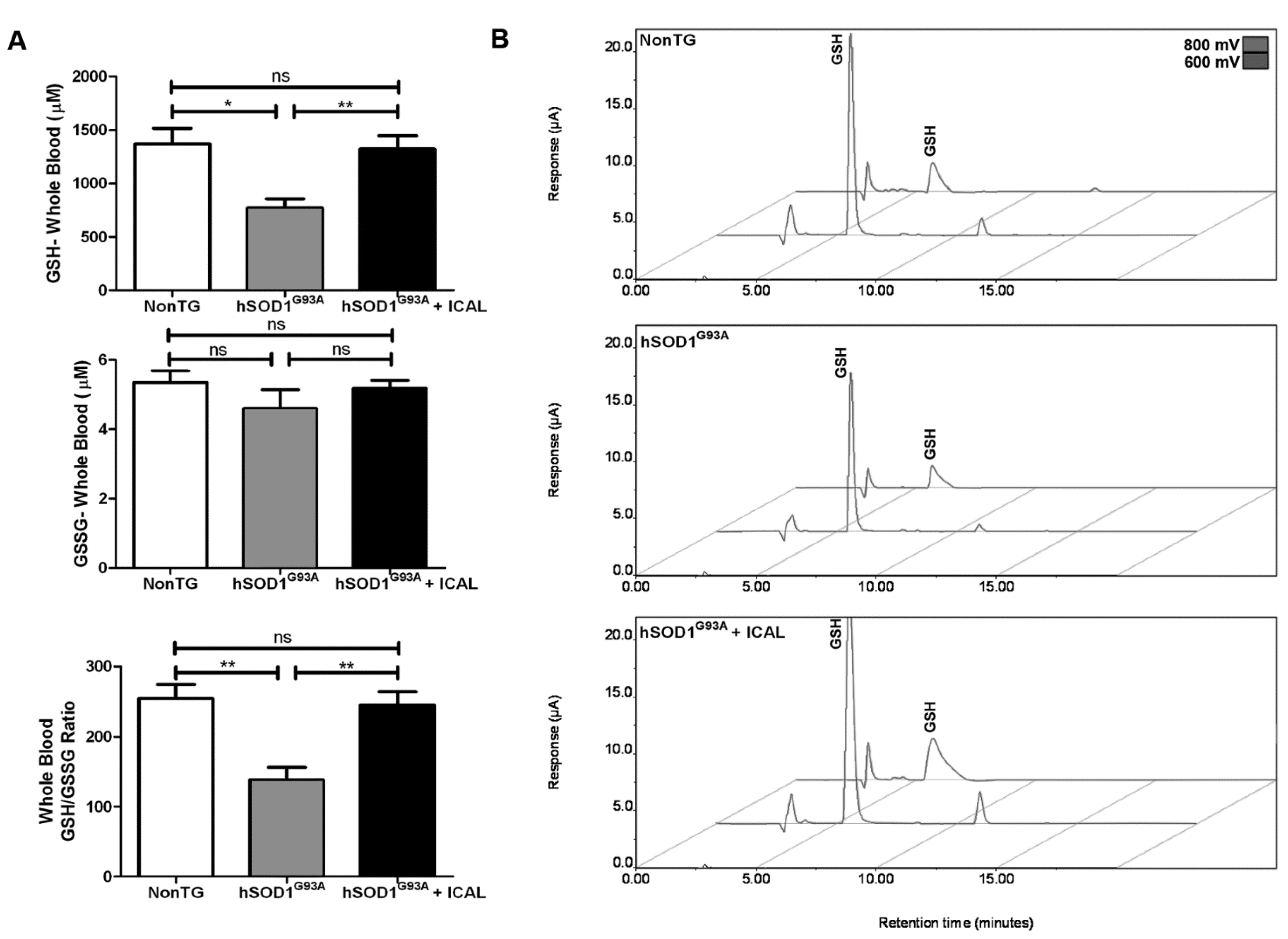
3.2. Immunocal® Prevents GSH Depletion in Whole Blood and Lumbar Spinal Cord Tissue of hSOD1G93A Mice
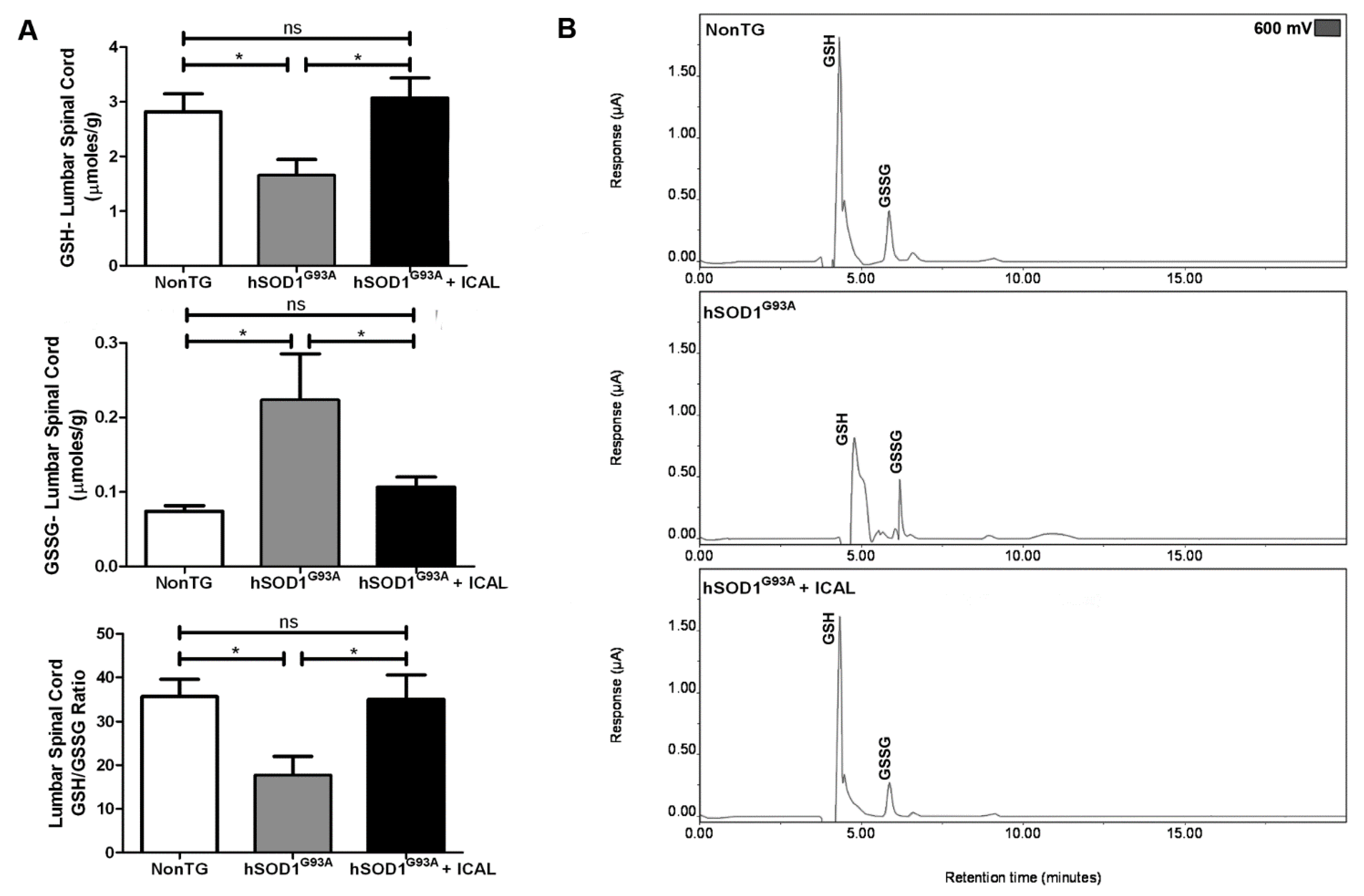
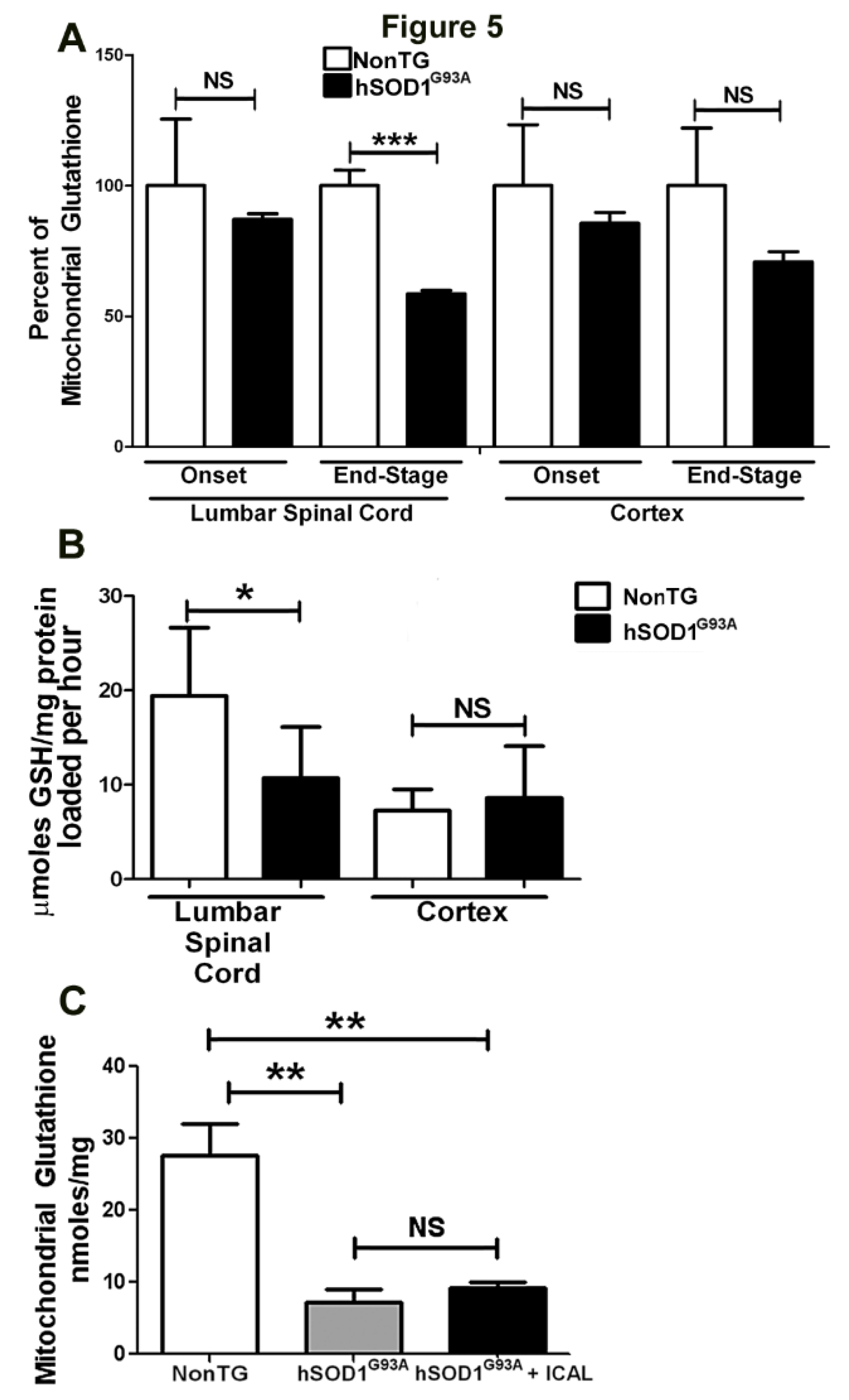
3.3. Immunocal® Does Not Prevent Depletion of Mitochondrial GSH in Lumbar Spinal Cord of hSOD1G93A Mice
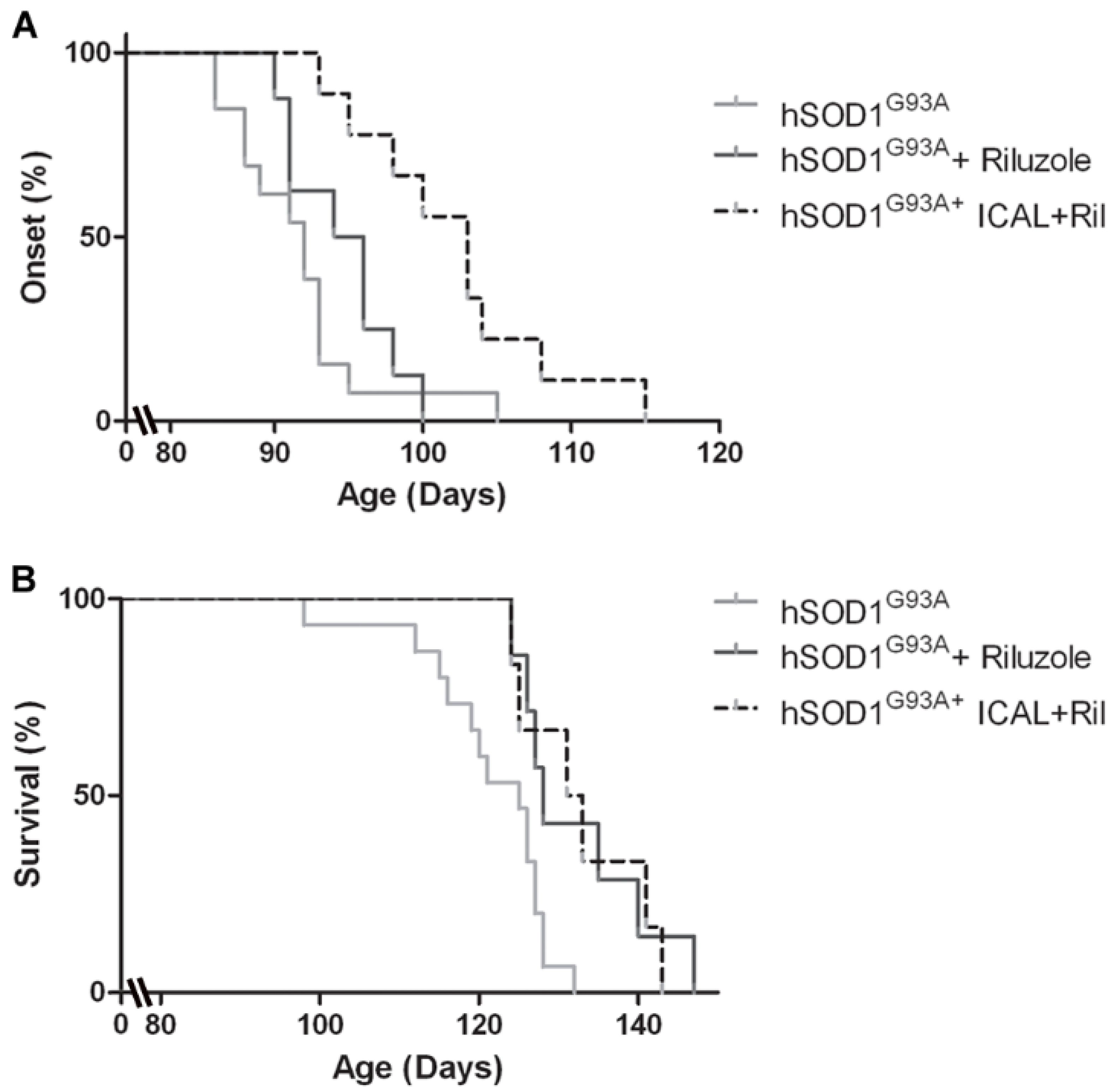
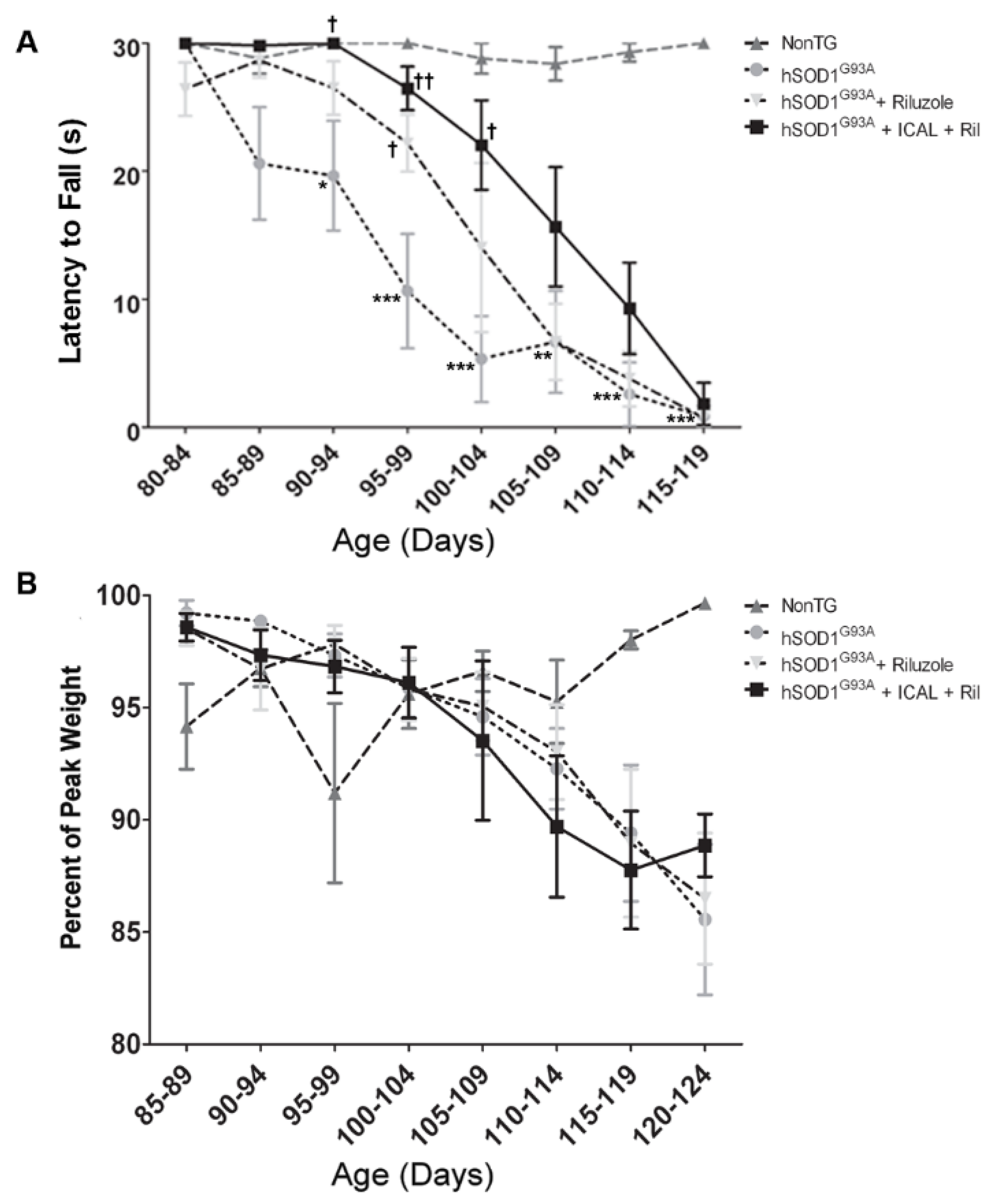
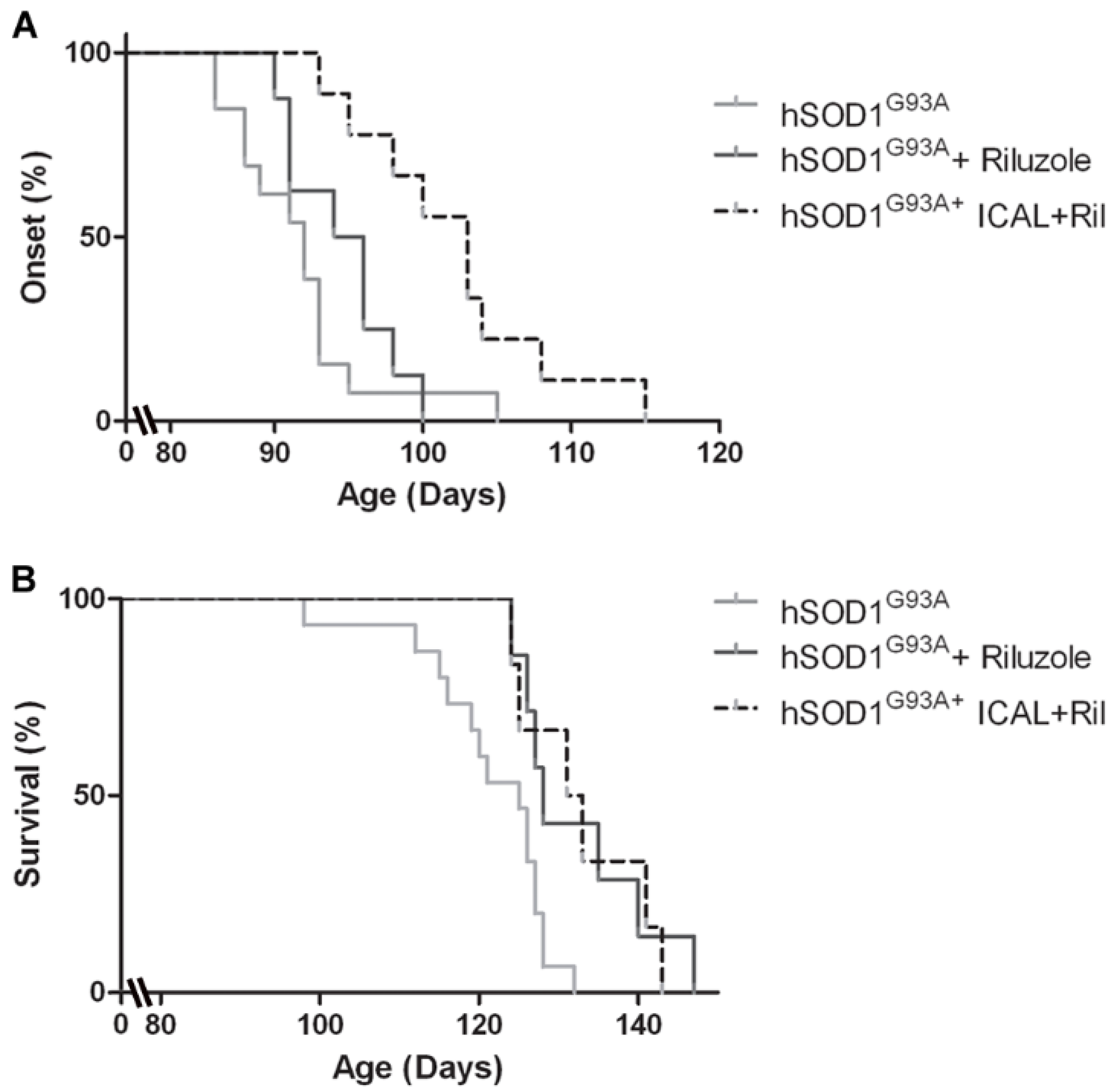
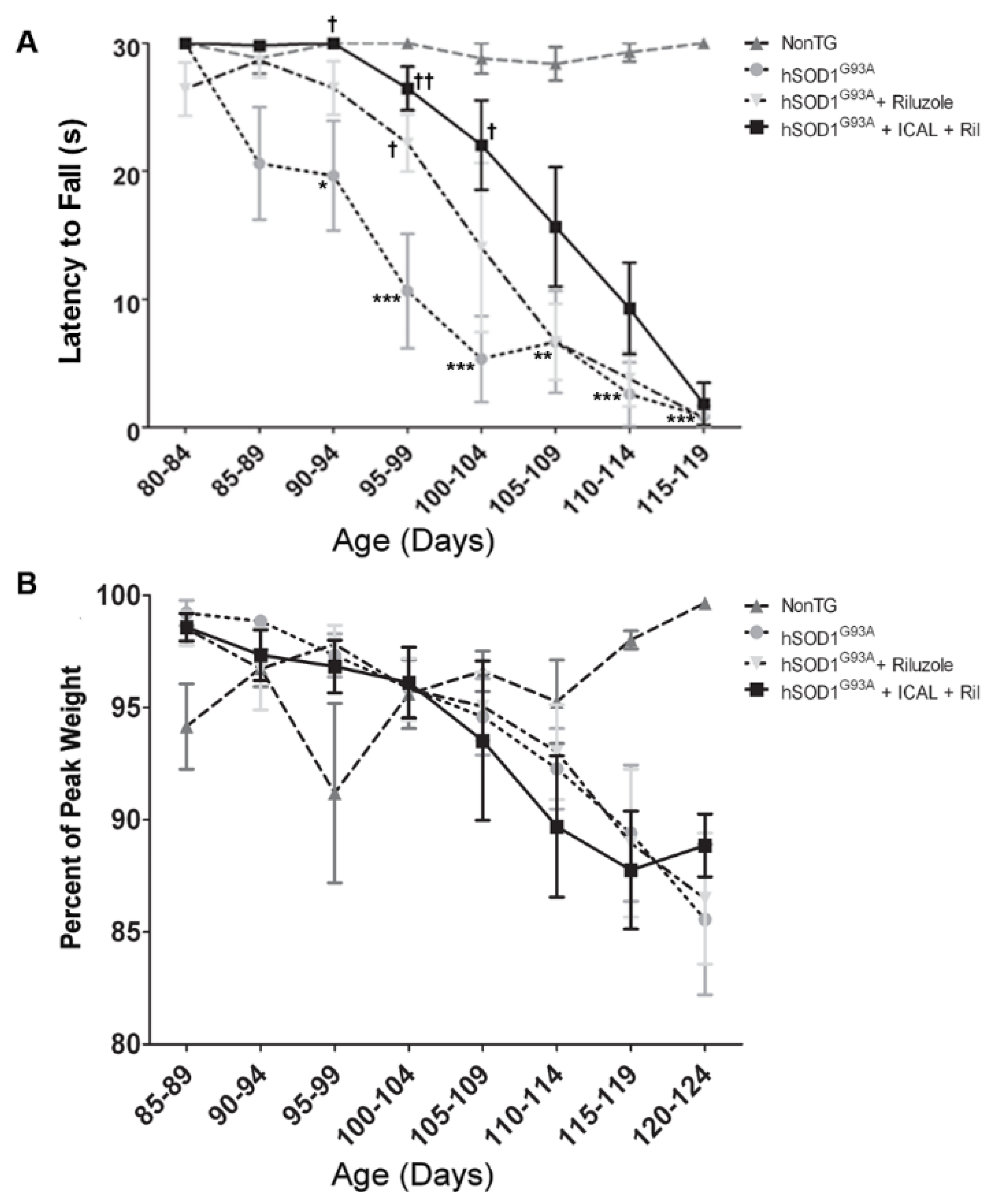
3.4. Combination Treatment with Immunocal® and Riluzole Shows Complementary Effects on Disease Onset and Survival in hSOD1G93A Mice
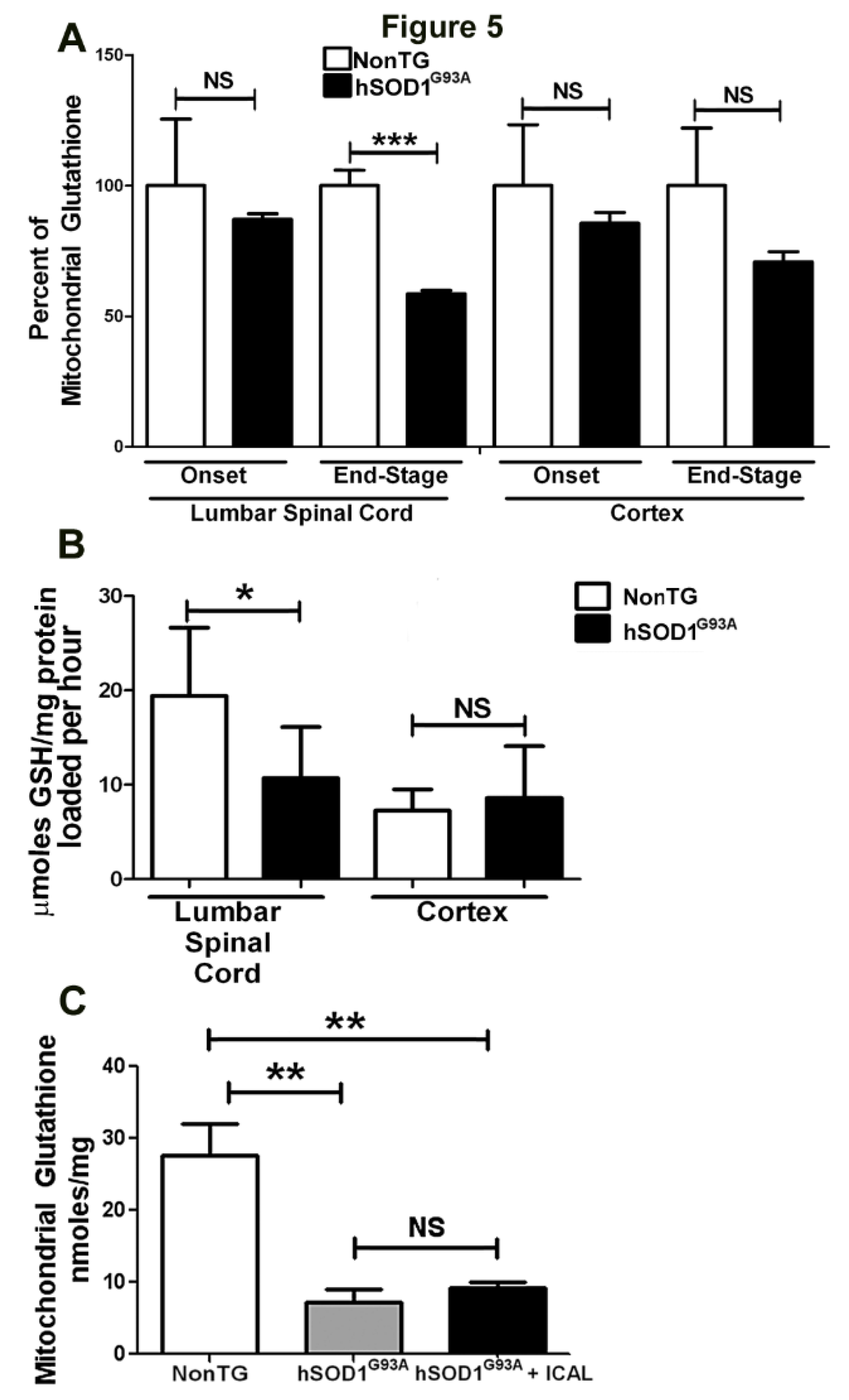
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tandan, R.; Bradley, W.G. Amyotrophic lateral sclerosis: Part 1. Clinical features, pathology, and ethical issues in management. Ann. Neurol. 1985, 18, 271–280. [Google Scholar] [CrossRef]
- Barber, S.C.; Shaw, P.J. Oxidative stress in ALS: Key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Cozzolino, M.; Carri, M.T. Mitochondrial dysfunction in ALS. Prog. Neurobiol. 2012, 97, 54–66. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Yim, M.B.; Kang, J.H.; Yim, H.S.; Kwak, H.S.; Chock, P.B.; Stadtman, E.R. A gain-of-function of an amyotrophic lateral sclerosis-associated Cu,Zn-superoxide dismutase mutant: An enhancement of free radical formation due to a decrease in Km for hydrogen peroxide. Proc. Natl. Acad. Sci. USA 1996, 93, 5709–5714. [Google Scholar] [CrossRef]
- Ferri, A.; Cozzolino, M.; Crosio, C.; Nencini, M.; Casciati, A.; Gralla, E.B.; Rotilio, G.; Valentine, J.S.; Carri, M.T. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc. Natl. Acad. Sci. USA 2006, 103, 13860–13865. [Google Scholar] [CrossRef] [Green Version]
- Andreassen, O.A.; Ferrante, R.J.; Klivenyi, P.; Klein, A.M.; Shinobu, L.A.; Epstein, C.J.; Beal, M.F. Partial deficiency of manganese superoxide dismutase exacerbates a transgenic mouse model of amyotrophic lateral sclerosis. Ann. Neurol. 2000, 47, 447–455. [Google Scholar] [CrossRef]
- Franco, R.; Cidlowski, J.A. Apoptosis and glutathione: Beyond an antioxidant. Cell Death Differ. 2009, 16, 1303–1314. [Google Scholar] [CrossRef]
- Griffith, O.W.; Meister, A. Origin and turnover of mitochondrial glutathione. Proc. Natl. Acad. Sci. USA 1985, 82, 4668–4672. [Google Scholar] [CrossRef]
- Lash, L.H. Mitochondrial glutathione transport: Physiological, pathological and toxicological implications. Chem. Biol. Interact. 2006, 163, 54–67. [Google Scholar] [CrossRef]
- Babu, G.N.; Kumar, A.; Chandra, R.; Puri, S.K.; Singh, R.L.; Kalita, J.; Misra, U.K. Oxidant-antioxidant imbalance in the erythrocytes of sporadic amyotrophic lateral sclerosis patients correlates with the progression of disease. Neurochem. Int. 2008, 52, 1284–1289. [Google Scholar] [CrossRef]
- Chi, L.; Ke, Y.; Luo, C.; Gozal, D.; Liu, R. Depletion of reduced glutathione enhances motor neuron degeneration in vitro and in vivo. Neuroscience 2007, 144, 991–1003. [Google Scholar] [CrossRef]
- Pesaresi, M.G.; Amori, I.; Giorgi, C.; Ferri, A.; Fiorenzo, P.; Gabanella, F.; Salvatore, A.M.; Giorgio, M.; Pelicci, P.G.; Pinton, P.; et al. Mitochondrial redox signalling by p66Shc mediates ALS-like disease through Rac1 inactivation. Hum. Mol. Genet. 2011, 20, 4196–4208. [Google Scholar] [CrossRef]
- Olsson, B.; Johansson, M.; Gabrielsson, J.; Bolme, P. Pharmacokinetics and bioavailability of reduced and oxidized N-acetylcysteine. Eur. J. Clin. Pharmacol. 1988, 34, 77–82. [Google Scholar] [CrossRef]
- Zeevalk, G.D.; Manzino, L.; Sonsalla, P.K.; Bernard, L.P. Characterization of intracellular elevation of glutathione (GSH) with glutathione monoethyl ester and GSH in brain and neuronal cultures: Relevance to Parkinson’s disease. Exp. Neurol. 2007, 203, 512–520. [Google Scholar] [CrossRef]
- Bounous, G.; Gold, P. The biological activity of undenatured dietary whey proteins: Role of glutathione. Clin. Investig. Med. 1991, 14, 296–309. [Google Scholar]
- Bounous, G.; Baruchel, S.; Falutz, J.; Gold, P. Whey proteins as a food supplement in HIV-seropositive individuals. Clin. Investig. Med. 1993, 16, 204–209. [Google Scholar]
- De Rosa, S.C.; Zaretsky, M.D.; Dubs, J.G.; Roederer, M.; Anderson, M.; Green, A.; Mitra, D.; Watanabe, N.; Nakamura, H.; Tjioe, I.; et al. N-acetylcysteine replenishes glutathione in HIV infection. Eur. J. Clin. Investig. 2000, 30, 915–929. [Google Scholar] [CrossRef]
- PDR Staff. Physician’s Desk Reference, 67th ed.; PDR Network: Montvale, NJ, USA, 2013. [Google Scholar]
- Baruchel, S.; Bounous, G.; Gold, P. Place for an antioxidant therapy in human immunodeficiency virus (HIV) infection. J. Nutr. 1994, 112, 1747–1755. [Google Scholar]
- Grey, V.; Mohammed, S.R.; Smountas, A.A.; Bahlool, R.; Lands, L.C. Improved glutathione status in young adult patients with cystic fibrosis supplemented with whey protein. J. Cyst. Fibros. 2003, 2, 195–198. [Google Scholar] [CrossRef]
- Leitner, M.; Menzies, S.; Lutz, C. Working with ALS Mice: Guidelines for Preclinical Testing & Colony Management. Available online: http://jaxmice.jax.org/literature/factsheet/working_with_ALS_mice.pdf (accesssed on 04 June 2014).
- Weydt, P.; Hong, S.Y.; Kliot, M.; Moller, T. Assessing disease onset and progression in the SOD1 mouse model of ALS. Neuroreport 2003, 14, 1051–1054. [Google Scholar] [PubMed]
- Ludolph, A.C.; Bendotti, C.; Blaugrund, E.; Chio, A.; Greensmith, L.; Loeffler, J.P.; Mead, R.; Niessen, H.G.; Petri, S.; Pradat, P.F.; et al. Guidelines for preclinical animal research in ALS/MND: A consensus meeting. Amyotroph. Lateral Scler. 2010, 11, 38–45. [Google Scholar] [CrossRef]
- Jarrett, S.G.; Milder, J.B.; Liang, L.P.; Patel, M. The ketogenic diet increases mitochondrial glutathione levels. J. Neurochem. 2008, 106, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.P.; Patel, M. Mitochondrial oxidative stress and increased seizure susceptibility in Sod2−/+ mice. Free Radic. Biol. Med. 2004, 36, 542–554. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Kirchhof, D.; Manning, E.; Joseph, J.W.; Linseman, D.A. Mitochondrial glutathione transport is a key determinant of neuronal susceptibility to oxidative and nitrosative stress. J. Biol. Chem. 2013, 288, 5091–5101. [Google Scholar] [CrossRef]
- Miller, R.G.; Mitchell, J.D.; Lyon, M.; Moore, D.H. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst. Rev. 2012. [Google Scholar] [CrossRef]
- Gurney, M.E.; Cutting, F.B.; Zhai, P.; Doble, A.; Taylor, C.P.; Andrus, P.K.; Hall, E.D. Benefit of Vitamin E, riluzole, and gabapentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann. Neurol. 1996, 39, 147–157. [Google Scholar] [CrossRef]
- Pearce, R.K.; Owen, A.; Daniel, S.; Jenner, P.; Marsden, C.D. Alterations in the distribution of glutathione in the substantia nigra in Parkinson’s disease. J. Neural Transm. 1997, 104, 661–677. [Google Scholar] [CrossRef]
- Calabrese, V.; Sultana, R.; Scapagnini, G.; Guagliano, E.; Sapienza, M.; Bella, R.; Kanski, J.; Pennisi, G.; Mancuso, C.; Stella, A.M.; et al. Nitrosative stress, cellular stress response, and thiol homeostasis in patients with Alzheimerʼs disease. Antioxid. Redox Signal. 2006, 8, 1975–1986. [Google Scholar] [CrossRef]
- Iguchi, Y.; Katsuno, M.; Takagi, S.; Ishigaki, S.; Niwa, J.; Hasegawa, M.; Tanaka, F.; Sobue, G. Oxidative stress induced by glutathione depletion reproduces pathological modifications of TDP-43 linked to TDP-43 proteinopathies. Neurobiol. Dis. 2012, 45, 862–870. [Google Scholar] [CrossRef]
- Vargas, M.R.; Johnson, D.A.; Johnson, J.A. Decreased glutathione accelerates neurological deficit and mitochondrial pathology in familial ALS-linked hSOD1(G93A) mice model. Neurobiol. Dis. 2011, 43, 543–551. [Google Scholar] [CrossRef]
- Lands, L.C.; Grey, V.L.; Smountas, A.A. Effect of supplementation with a cysteine donor on muscular performance. J. Appl. Physiol. 1999, 87, 1381–1385. [Google Scholar] [PubMed]
- Winter, A.W.; Ross, E.K.; Linseman, D.A.; University of Denver, Denver, CO, USA. Unpublished work. 2014.
- Cova, E.; Bongioanni, P.; Cereda, C.; Metelli, M.R.; Salvaneschi, L.; Bernuzzi, S.; Guareschi, S.; Rossi, B.; Ceroni, M. Time course of oxidant markers and antioxidant defenses in subgroups of amyotrophic lateral sclerosis patients. Neurochem. Int. 2010, 56, 687–693. [Google Scholar] [CrossRef]
- Muyderman, H.; Hutson, P.G.; Matusica, D.; Rogers, M.L.; Rush, R.A. The human G93A-superoxide dismutase-1 mutation, mitochondrial glutathione and apoptotic cell death. Neurochem. Res. 2009, 34, 1847–1856. [Google Scholar] [CrossRef]
- Liu, R.; Li, B.; Flanagan, S.W.; Oberley, L.W.; Gozal, D.; Qiu, M. Increased mitochondrial antioxidative activity or decreased oxygen free radical propagation prevent mutant SOD1-mediated motor neuron cell death and increase amyotrophic lateral sclerosis-like transgenic mouse survival. J. Neurochem. 2002, 80, 488–500. [Google Scholar] [CrossRef]
- Ferri, A.; Nencini, M.; Cozzolino, M.; Carrara, P.; Moreno, S.; Carri, M.T. Inflammatory cytokines increase mitochondrial damage in motoneuronal cells expressing mutant SOD1. Neurobiol. Dis. 2008, 32, 454–460. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Marquardt, K.; Lash, L.H.; Linseman, D.A. Bcl-2 is a novel interacting partner for the 2-oxoglutarate carrier and a key regulator of mitochondrial glutathione. Free Radic. Biol. Med. 2012, 52, 410–419. [Google Scholar] [CrossRef]
- Mu, X.; He, J.; Anderson, D.W.; Trojanowski, J.Q.; Springer, J.E. Altered expression of Bcl-2 and bax mRNA in amyotrophic lateral sclerosis spinal cord motor neurons. Ann. Neurol. 1996, 40, 379–386. [Google Scholar] [CrossRef]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H., Jr. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 2004, 43, 19–30. [Google Scholar] [CrossRef]
- Xu, F.; Putt, D.A.; Matherly, L.H.; Lash, L.H. Modulation of expression of rat mitochondrial 2-oxoglutarate carrier in NRK-52E cells alters mitochondrial transport and accumulation of glutathione and susceptibility to chemically induced apoptosis. J. Pharmacol. Exp. Ther. 2006, 316, 1175–1186. [Google Scholar] [CrossRef]
- Papadimitriou, D.; Le Verche, V.; Jacquier, A.; Ikiz, B.; Przeborski, S.; Re, D.B. Inflammation in ALS and SMA: Sorting out the good from the evil. Neurobiol. Dis. 2010, 37, 493–502. [Google Scholar] [CrossRef]
- Philips, T.; Robberecht, W. Neuroinflammation in amyotrophic lateral scelerosis: Role of glial activation in motor neuron disease. Lancet Neurol. 2011, 10, 253–263. [Google Scholar] [CrossRef]
- Lee, M.; Cho, T.; Jantaratnotai, N.; Wang, Y.T.; McGeer, E.; McGeer, P.L. Depletion of GSH in glial cells incudces neurotoxicity: Relevance to aging and degenerative neurological diseases. FASEB J. 2010, 24, 2533–2545. [Google Scholar] [CrossRef]
- Lee, M.; Kwon, B.M.; Suk, K.; McGeer, E.; McGeer, P.L. Effects of obovatol on GSH depleted glia-mediated neurotoxicity and oxidative damage. J. Neuroimmune Pharmacol. 2012, 7, 173–186. [Google Scholar] [CrossRef]
- Moss, D.W.; Bates, T.E. Activation of murine microglial cells lines by lipopolysaccharide and interferon-γ causes NO-mediated decreases in mitochondrial and cellular function. Eur. J. Neurosci. 2001, 13, 529–538. [Google Scholar] [CrossRef]
- Barger, S.W.; Goodwin, M.E.; Porter, M.M.; Beggs, M.L. Glutamate release from activated microglia requires the oxidative burst and lipid peroxidation. J. Neurochem. 2007, 101, 1205–1213. [Google Scholar] [CrossRef]
- Seib, T.M.; Patel, S.A.; Bridges, R.J. Regulation of the system xC− cystine/glutamate exchanger by intracellular glutathione levels in rat astrocytes primary cultures. Glia 2011, 59, 1387–1401. [Google Scholar] [CrossRef]
- Gabryel, B.; Malecki, A. Ebselen attenuates oxidative stress in ischemic astrocytes depleted of glutathione. Comarpison with glutathione precursors. Pharmacol. Rep. 2006, 58, 381–392. [Google Scholar] [PubMed]
- Scott, S.; Kranz, J.E.; Cole, J.; Lincecum, J.M.; Thompson, K.; Kelly, N.; Bostrom, A.; Theodoss, J.; Al-Nakhala, B.M.; Vieira, F.G.; et al. Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph. Lateral Scler. 2008, 9, 4–15. [Google Scholar] [CrossRef]
- Wang, H.; O’Reilly, E.J.; Weisskopf, M.G.; Logroscino, G.; McCullough, M.L.; Schatzkin, A.; Kolonel, L.N.; Ascherio, A. Vitamin E intake and risk of amyotrophic lateral sclerosis: A pooled analysis of data from 5 prospective cohort studies. Am. J. Epidemiol. 2011, 173, 595–602. [Google Scholar] [CrossRef]
- Michal Freedman, D.; Kuncl, R.W.; Weinstein, S.J.; Malila, N.; Virtamo, J.; Albanes, D. Vitamin E serum levels and controlled supplementation and risk of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 246–251. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ross, E.K.; Winter, A.N.; Wilkins, H.M.; Sumner, W.A.; Duval, N.; Patterson, D.; Linseman, D.A. A Cystine-Rich Whey Supplement (Immunocal®) Delays Disease Onset and Prevents Spinal Cord Glutathione Depletion in the hSOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. Antioxidants 2014, 3, 843-865. https://doi.org/10.3390/antiox3040843
Ross EK, Winter AN, Wilkins HM, Sumner WA, Duval N, Patterson D, Linseman DA. A Cystine-Rich Whey Supplement (Immunocal®) Delays Disease Onset and Prevents Spinal Cord Glutathione Depletion in the hSOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. Antioxidants. 2014; 3(4):843-865. https://doi.org/10.3390/antiox3040843
Chicago/Turabian StyleRoss, Erika K., Aimee N. Winter, Heather M. Wilkins, Whitney A. Sumner, Nathan Duval, David Patterson, and Daniel A. Linseman. 2014. "A Cystine-Rich Whey Supplement (Immunocal®) Delays Disease Onset and Prevents Spinal Cord Glutathione Depletion in the hSOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis" Antioxidants 3, no. 4: 843-865. https://doi.org/10.3390/antiox3040843
APA StyleRoss, E. K., Winter, A. N., Wilkins, H. M., Sumner, W. A., Duval, N., Patterson, D., & Linseman, D. A. (2014). A Cystine-Rich Whey Supplement (Immunocal®) Delays Disease Onset and Prevents Spinal Cord Glutathione Depletion in the hSOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. Antioxidants, 3(4), 843-865. https://doi.org/10.3390/antiox3040843