Melatonin Therapy in Patients with Alzheimer’s Disease

Abstract
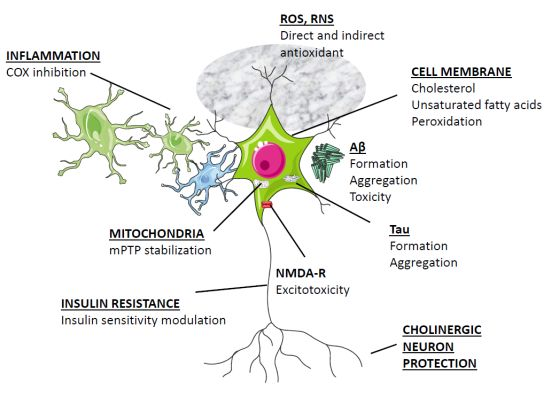
1. Introduction
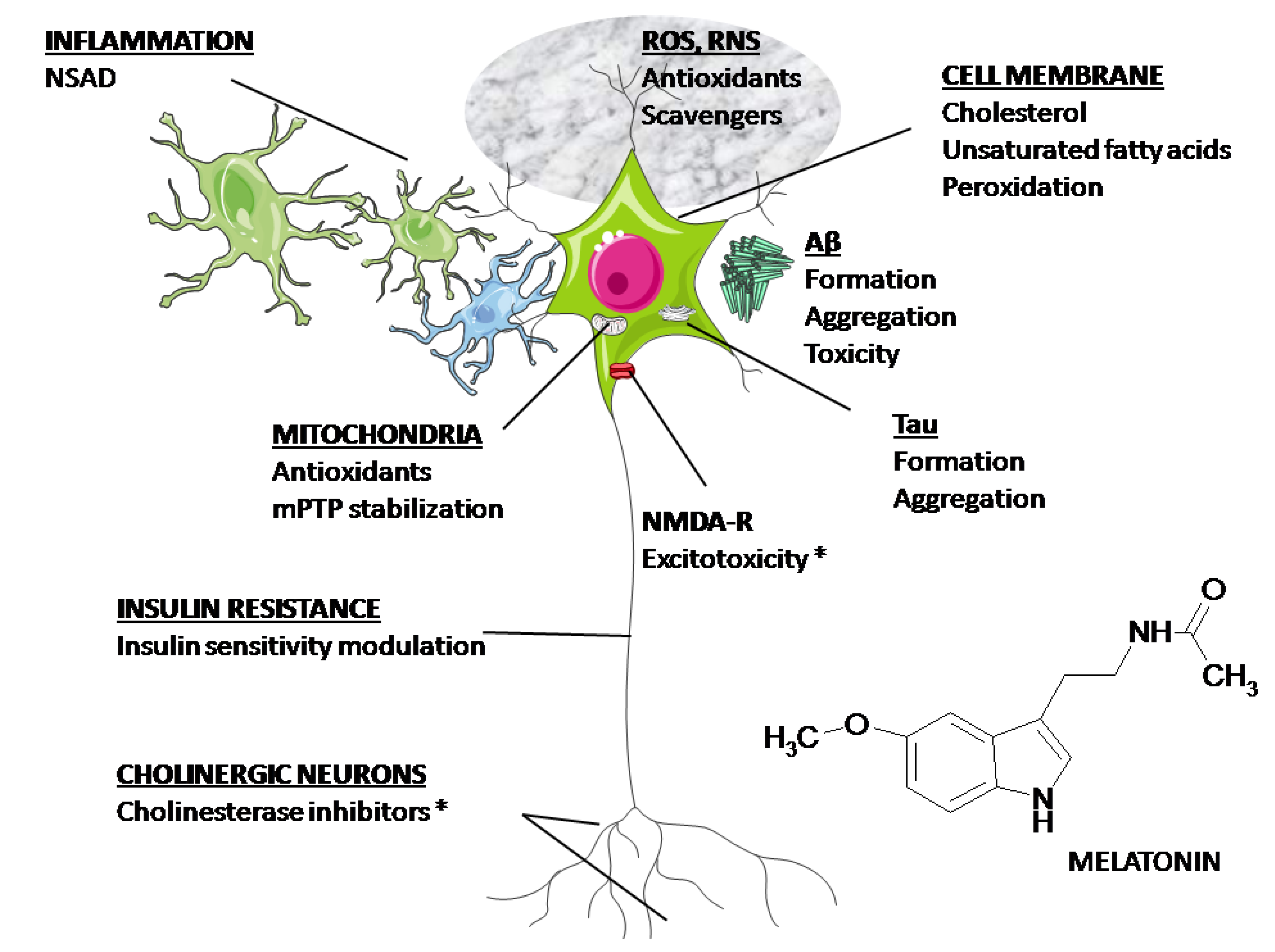
2. Basic Biology of Melatonin Relevant to Neurodegeneration
3. Overview of Melatonin Therapy for Alzheimer’s Disease—Theory and Mode of Action
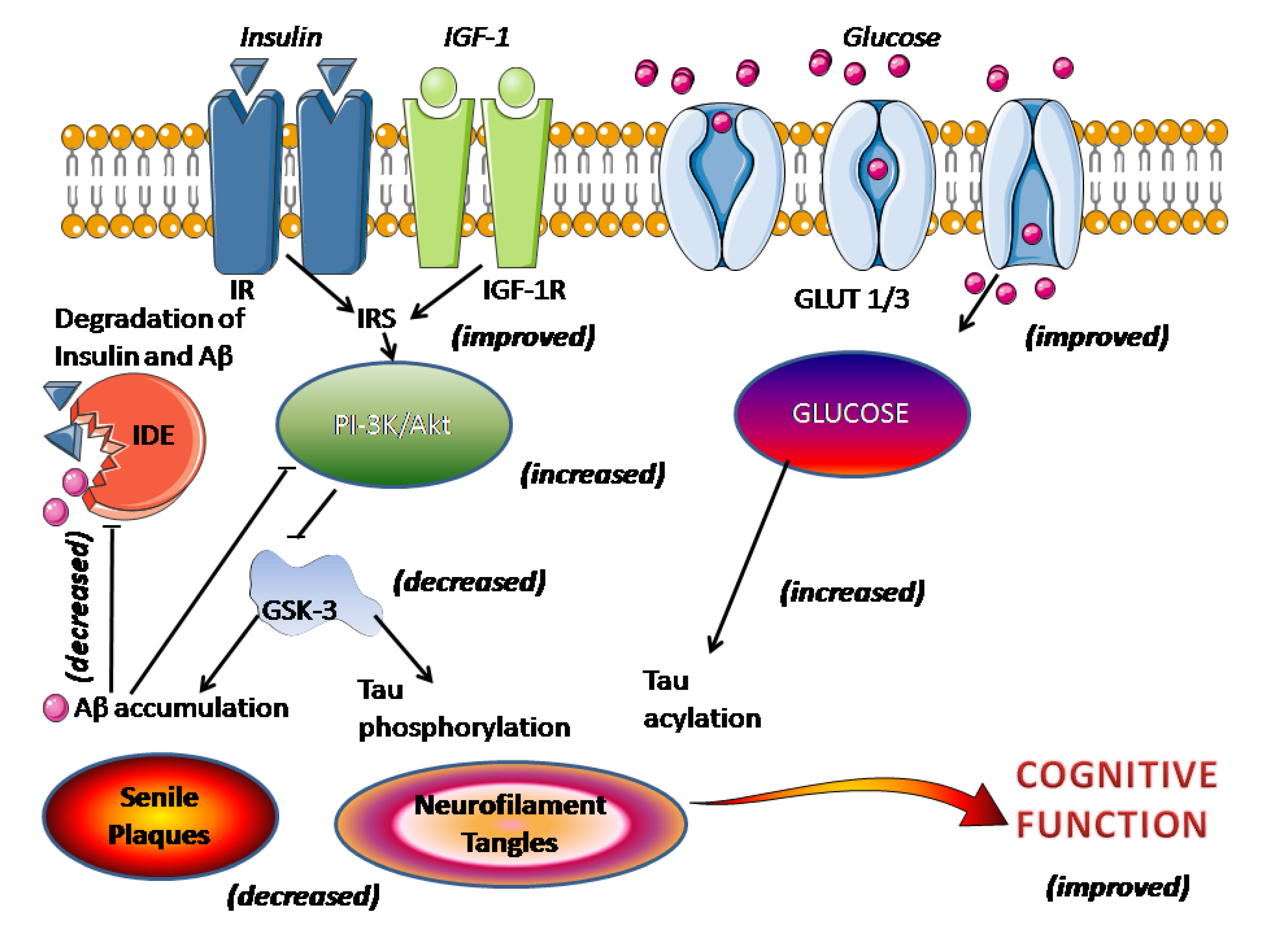
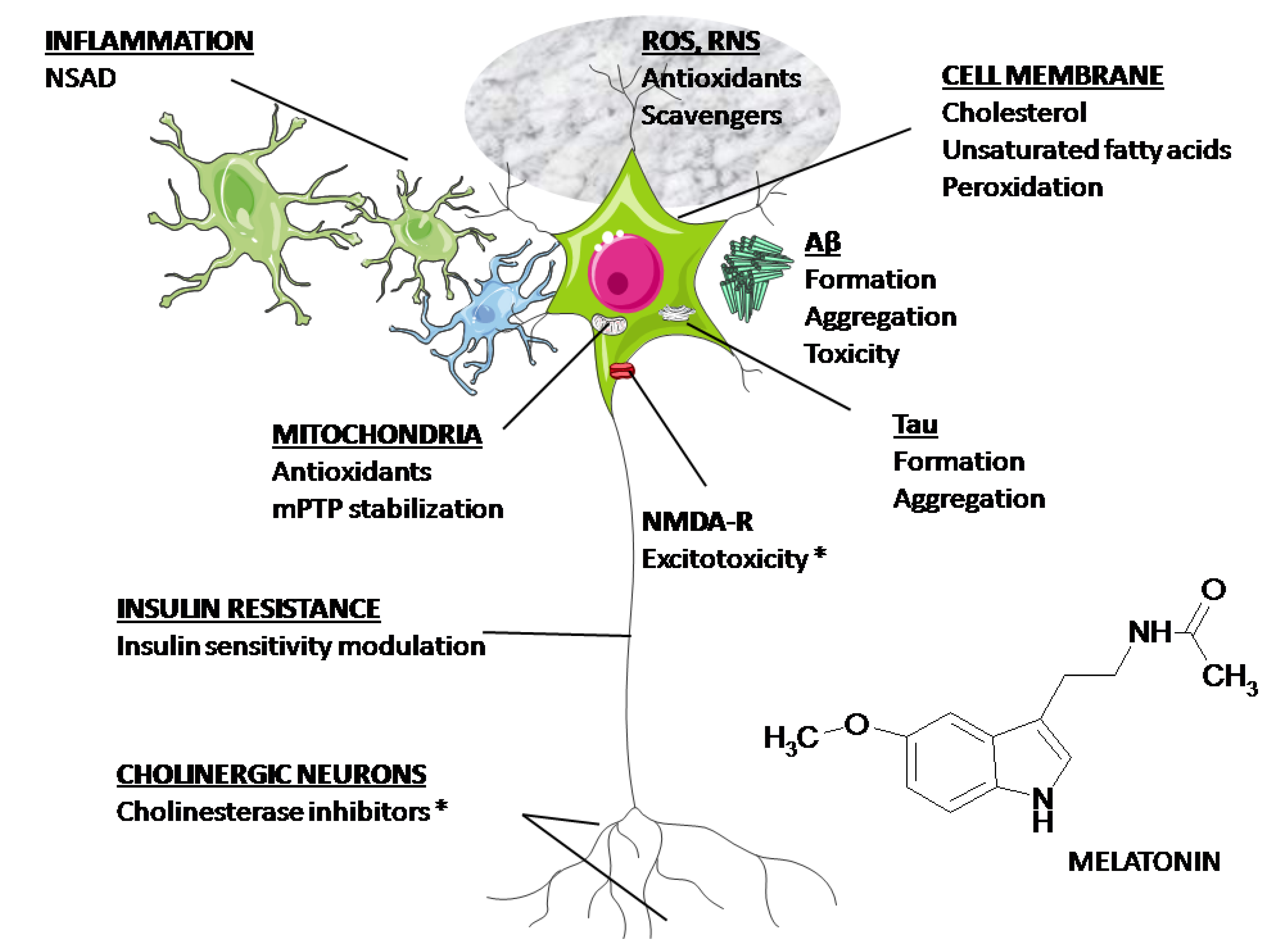
4. Clinical Aspects of Melatonin Application in AD

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Design | Subjects | Treatment | Time | Measured | Results | Reference |
|---|---|---|---|---|---|---|
| Open-label study | 10 AD patients | 3 mg melatonin p.o./daily at bed time | 3 weeks | Daily logs of sleep and wake quality completed by caretakers | 7 out of 10 dementia patients having sleep disorders treated with melatonin showed a significant decrease in sundowning and reduced variability of sleep onset time | [155] |
| Open-label study | 14 AD patients | 9 mg melatonin p.o./daily at bed time | 22 to 35 months | Daily logs of sleep and wake quality completed by caretakers. Neuro-psychological assessment | Sundowning was no longer detectable in 12 patients and persisted, although attenuated in 2 patients. A significant improvement of sleep quality was found. Lack of progression of the cognitive and behavioral signs of the disease during the time they received melatonin | [156] |
| Case report | Mono-zygotic twins with AD of 8 years duration | One of the patients was treated with melatonin 9 mg p.o./daily at bed time. | 36 months | Neuro-psychological assessment. Neuroimaging | Sleep and cognitive function severely impaired in the twin not receiving melatonin as compared to the melatonin-treated twin | [157] |
| Open-label study | 11 AD patients | 3 mg melatonin p.o./daily at bed time | 3 weeks | Daily logs of sleep and wake quality | Significant decrease in agitated behaviors in all three shifts; significant decrease in daytime sleepiness | [158] |
| Open-label, placebo-controlled trial | 14 AD patients | 6 mg melatonin p.o./daily at bed time or placebo | 4 weeks | Daily logs of sleep and wake quality completed by caretakers. Actigraphy | AD patients receiving melatonin showed a significantly reduced percentage of nighttime activity compared to a placebo group | [159] |
| Randomized double blind placebo-controlled cross over study | 25 AD patients | 6 mg of slow release melatonin p.o. or placebo at bed time | 7 weeks | Actigraphy | Melatonin had no effect on median total time asleep, number of awakenings or sleep efficiency | [160] |
| Open-label study | 45 AD patients | 6–9 mg melatonin p.o./daily at bed time | 4 months | Daily logs of sleep and wake quality completed by caretakers. Neuro-psychological assessment | Melatonin improved sleep and suppressed sundowning, an effect seen regardless of the concomitant medication employed | [161] |
| Randomized placebo-controlled clinical trial | 157 AD patients | 2.5-mg slow-release melatonin, or 10-mg melatonin or placebo at bed time | 2 months | Actigraphy. Caregiver ratings of sleep quality | Non significant trends for increased nocturnal total sleep time and decreased wake after sleep onset in the melatonin groups. Caregiver ratings of sleep quality showed a significant improvement in the 2.5-mg sustained-release melatonin group relative to placebo | [162] |
| Double-blind, placebo-controlled study | 20 AD patients | Placebo or 3 mg melatonin p.o./daily at bed time | 4 weeks | Actigraphy. Neuro-psychological assessment | Melatonin significantly prolonged the sleep time and decreased activity in the night. Cognitive function was improved by melatonin | [163] |
| Open-label study | 7 AD patients | 3 mg melatonin p.o./daily at bed time | 3 weeks | Actigraphy. Neuro-psychological assessment. | Complete remission of day-night rhythm disturbances or sundowning was seen in 4 patients, with partial remission in other 2 | [164] |
| Randomized placebo-controlled study | 17 AD patients | 3 mg melatonin p.o./daily at bed time (7 patients). Placebo (10 patients) | 2 weeks | Actigraphy. Neuro-psychological assessment. | In melatonin-treated group, actigraphic nocturnal activity and agitation showed significant reductions compared to baseline | [165] |
| Case report | 68-year-old man with AD who developed rapid eye movement (REM) sleep behavior disorder | 5–10 mg melatonin p.o./daily at bed time. | 20 months | Polysomno-graphy | Melatonin was effective to suppress REM sleep behavior disorder | [166] |
| Randomized placebo-controlled study | 50 AD patients | Morning light exposure (2500 lux, 1 h) and 5 mg melatonin (N = 16) or placebo (N = 17) in the evening | 10 weeks | Actigraphy | Light treatment alone did not improve nighttime sleep, daytime wake, or rest-activity rhythm. Light treatment plus melatonin increased daytime wake time and activity levels and strengthened the rest-activity rhythm | [167] |
| Randomized placebo-controlled study | 41 AD patients | Melatonin (8.5 mg immediate release and 1.5 mg sustained release) (N = 24) or placebo (N = 17) administered at 22:00 h | 10 days | Actigraphy | There were no significant effects of melatonin, compared with placebo, on sleep, circadian rhythms, or agitation | [168] |
5. Conclusions
| Design | Subjects | Treatment | Time | Measured | Results | Reference |
|---|---|---|---|---|---|---|
| Double-blind, placebo-controlled, crossover study | 10 patients with MCI | 6 mg melatonin p.o./daily at bed time | 10 days | Actigraphy. Neuro-psychological assessment | Melatonin enhanced the rest-activity rhythm and improved sleep quality. The ability to remember previously learned items improved along with a significant reduction in depressed mood | [206] |
| Double-blind, placebo-controlled pilot study | 26 patients with age-related MCI | 1 mg melatonin p.o. or placebo at bed time | 4 weeks | Sleep questionnaire and cognitive tests at baseline and at 4 weeks | Melatonin administration improved reported morning “restedness” and sleep latency after nocturnal awakening. It also improved scores on the California Verbal Learning Test-interference subtest. | [207] |
| Randomizeddouble blind, placebo-controlled study | 354 patients with age-related MCI | Prolonged release melatonin (Circadin, 2 mg) or placebo, 2 h before bedtime | 3 weeks | Leeds Sleep Evaluation and Pittsburgh Sleep QuestionnairesClinical Global Improvement scale score and quality of life. | Melatonin resulted in significant and clinically meaningful improvements in sleep quality, morning alertness, sleep onset latency and quality of life | [208] |
| Open-label, retrospective study | 60 MCI out-patients | 35 patients received daily 3–9 mg of a fast-release melatonin preparation p.o. at bedtime. Melatonin was given in addition to the standard medication | 9–24 months | Daily logs of sleep and wake quality. Initial and final neuro-psychological assessment. | Abnormally high Beck Depression Inventory scores decreased in melatonin-treated patients, concomitantly with an improvement in wakefulness and sleep quality. Patients treated with melatonin showed significantly better performance in neuropsychological assessment. | [174] |
| Long-term, double-blind, placebo-controlled, 2 × 2 factorial randomized study | 189 patients with age-related cognitive decay | Long-term daily treatment with whole-day bright (1000 lux) or dim (300 lux) light. Evening melatonin (2.5 mg) or placebo | 1 to 3.5 years | Standardized scales for cognitive and noncognitive symptoms, limitations of activities of daily living, and adverse effects assessed every 6 months. | Light attenuated cognitive deterioration and ameliorated depressive symptoms. Melatonin shortened sleep onset latency and increased sleep duration but adversely affected scores for depression. The combined treatment of bright light plus melatonin showed the best effects. | [209] |
| Prospective, randomized, double-blind, placebo-controlled, study | 22 patients with age-related cognitive decay | Patients received 2 months of melatonin (5 mg p.o./day) and 2 months of placebo | 2 months | Sleep disorders were evaluated with the Northside Hospital Sleep Medicine Institute test. Behavioral disorders were evaluated with the Yesavage Geriatric Depression Scale and Goldberg Anxiety Scale. | Melatonin treatment significantly improved sleep quality scores. Depression also improved significantly after melatonin administration. | [210] |
| Randomizeddouble-blind, placebo-controlled study | 25 MCI out-patients | 11 patients received an oily emulsion of docosa-hexaenoic acid-phospho-lipids containing melatonin (10 mg) and tryptophan (190 mg) | 12 weeks | Neuro-psychological assessment of orientation and cognitive functions, short-term and long-term memory, attentional abilities, executive functions, visuo-constructional and visuo-spatial abilities, language and mood. | Older adults with MCI had significant improvements in several measures of cognitive function when supplemented with the oily emulsion containing melatonin and tryptophan for 12 weeks, compared with the placebo. The antioxidant capacity of erythrocytes and membrane lipid composition improved after treatment. | [211,212] |
| Open-label, retrospective study | 96 MCI out-patients | 61 patients received daily 3–24 mg of a fast-release melatonin preparation p.o. at bedtime. Melatonin was given in addition to the standard medication | 15–60 months | Daily logs of sleep and wake quality. Initial and final neuro-psychological assessment. | Abnormally high Beck Depression Inventory scores decreased in melatonin-treated patients, concomitantly with an improvement in wakefulness and sleep quality. Patients treated with melatonin showed significantly better performance in neuropsychological assessment. Only 6 out of 61 patients treated with melatonin needed concomitant benzodiazepine treatment vs. 22 out of 35 MCI patients not receiving melatonin. | [175] |
Abbreviations
| Ach | acetylcholine |
| AChE | acetylcholinesterase |
| AD | Alzheimer’s disease |
| ADI | Alzheimer’s Disease International |
| AFMK | N1-acetyl-N2-formyl-5-methoxykynuramine |
| Akt | protein kinase identified in the AKT virus |
| AMK | N1-acetyl-5-methoxykynuramine |
| APP | amyloid precursor protein |
| Aβ | aggregated β-amyloid |
| Bcl-2 | B cell lymphoma proto-oncogene protein |
| ChAT | choline acetyltransferase |
| Cox | cyclooxygenase |
| CSF | cerebrospinal fluid |
| CYP1A1 | cytochrome P450 1A1 |
| CYP1A2 | cytochrome P450 1A2 |
| CYP2C19 | cytochrome P450 2C19 |
| CYP1B1 | cytochrome P450 1B1 |
| GABA | γ-aminobutyric acid |
| GPR50 | G-protein receptor 50 ortholog |
| GLUT-1 | glucose transporter-1 |
| GLUT-3 | glucose transporter-3 |
| GPx | glutathione peroxidase |
| GRd | glutathione reductase |
| GSH | reduced glutathione |
| GSK-3 | glycogen synthase kinase-3 |
| IDE | insulin-degrading enzyme |
| iNOS | inducible nitric oxide synthase |
| IGF-1 | Insulin-like growth factor 1 |
| MAP | microtubule-associated protein |
| MCI | mild cognitive impairment |
| mPTP | mitochondrial permeability transition pore |
| mRNA | messenger ribonucleic acid |
| MT1 | melatonin receptor 1 |
| MT2 | melatonin receptor 2 |
| NFκB | nuclear factor κB |
| NMDA | N-methyl-d-aspartate |
| nNOS | neuronal nitric oxide synthase |
| NO | nitric oxide |
| NSAD | non-steroidal anti-inflammatory drugs |
| PI3-K | phosphoinositide 3-kinase |
| PK | protein kinase |
| REM | rapid eye movement |
| RNS | reactive nitrogen species |
| ROR | retinoic acid receptor-related orphan receptor |
| ROS | reactive oxygen species |
| RZR | retinoid Z receptor |
| SCN | suprachiasmatic nuclei |
| SOD | superoxide dismutase |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Reiter, R.J.; Garcia, J.J.; Pie, J. Oxidative toxicity in models of neurodegeneration: Responses to melatonin. Restor. Neurol. Neurosci. 1998, 12, 135–142. [Google Scholar] [PubMed]
- ADI G8 Policy Briefing Reveals 135 Million People will Live with Dementia by 2050. Available online: http://www.alz.co.uk/news/g8-policy-brief-reveals-135-million-people-with-dementia-by-2050 (accessed on 1 April 2014).
- Johnson, E.J.; Vishwanathan, R.; Johnson, M.A.; Hausman, D.B.; Davey, A.; Scott, T.M.; Green, R.C.; Miller, L.S.; Gearing, M.; Woodard, J.; et al. Relationship between serum and brain carotenoids, α-tocopherol, and retinol concentrations and cognitive performance in the oldest old from the Georgia Centenarian Study. J. Aging Res. 2013, 2013. [Google Scholar] [CrossRef]
- Bubenik, G.A.; Konturek, S.J. Melatonin and aging: Prospects for human treatment. J. Physiol Pharmacol. 2011, 62, 13–19. [Google Scholar] [PubMed]
- Claustrat, B.; Brun, J.; Chazot, G. The basic physiology and pathophysiology of melatonin. Sleep Med. Rev. 2005, 9, 11–24. [Google Scholar] [CrossRef]
- Hardeland, R.; Cardinali, D.P.; Srinivasan, V.; Spence, D.W.; Brown, G.M.; Pandi-Perumal, S.R. Melatonin—A pleiotropic, orchestrating regulator molecule. Prog. Neurobiol. 2011, 93, 350–384. [Google Scholar] [CrossRef]
- Venegas, C.; Garcia, J.A.; Escames, G.; Ortiz, F.; Lopez, A.; Doerrier, C.; Garcia-Corzo, L.; Lopez, L.C.; Reiter, R.J.; Acuña-Castroviejo, D. Extrapineal melatonin: Analysis of its subcellular distribution and daily fluctuations. J. Pineal Res. 2012, 52, 217–227. [Google Scholar] [CrossRef]
- Paredes, S.D.; Korkmaz, A.; Manchester, L.C.; Tan, D.X.; Reiter, R.J. Phytomelatonin: A review. J. Exp. Bot. 2009, 60, 57–69. [Google Scholar] [PubMed]
- Cardinali, D.P.; Lynch, H.J.; Wurtman, R.J. Binding of melatonin to human and rat plasma proteins. Endocrinology 1972, 91, 1213–1218. [Google Scholar] [CrossRef]
- Ma, X.; Idle, J.R.; Krausz, K.W.; Gonzalez, F.J. Metabolism of melatonin by human cytochromes p450. Drug Metab. Dispos. 2005, 33, 489–494. [Google Scholar] [CrossRef]
- Facciola, G.; Hidestrand, M.; von Bahr, C.; Tybring, G. Cytochrome P450 isoforms involved in melatonin metabolism in human liver microsomes. Eur. J. Clin. Pharmacol. 2001, 56, 881–888. [Google Scholar] [CrossRef]
- Skene, D.J.; Papagiannidou, E.; Hashemi, E.; Snelling, J.; Lewis, D.F.; Fernandez, M.; Ioannides, C. Contribution of CYP1A2 in the hepatic metabolism of melatonin: Studies with isolated microsomal preparations and liver slices. J. Pineal Res. 2001, 31, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Young, I.M.; Leone, R.M.; Francis, P.; Stovell, P.; Silman, R.E. Melatonin is metabolized to N-acetyl serotonin and 6-hydroxymelatonin in man. J. Clin. Endocrinol. Metab. 1985, 60, 114–119. [Google Scholar] [CrossRef]
- Hardeland, R.; Tan, D.X.; Reiter, R.J. Kynuramines, metabolites of melatonin and other indoles: The resurrection of an almost forgotten class of biogenic amines. J. Pineal Res. 2009, 47, 109–126. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchester, L.C.; Terron, M.P.; Flores, L.J.; Reiter, R.J. One molecule, many derivatives: A never-ending interaction of melatonin with reactive oxygen and nitrogen species? J. Pineal Res. 2007, 42, 28–42. [Google Scholar] [CrossRef]
- Waldhauser, F.; Waldhauser, M.; Lieberman, H.R.; Deng, M.H.; Lynch, H.J.; Wurtman, R.J. Bioavailability of oral melatonin in humans. Neuroendocrinology 1984, 39, 307–313. [Google Scholar] [CrossRef]
- Aldhous, M.; Franey, C.; Wright, J.; Arendt, J. Plasma concentrations of melatonin in man following oral absorption of different preparations. Br. J. Clin. Pharmacol. 1985, 19, 517–521. [Google Scholar] [CrossRef]
- Fourtillan, J.B.; Brisson, A.M.; Gobin, P.; Ingrand, I.; Decourt, J.P.; Girault, J. Bioavailability of melatonin in humans after day-time administration of D7 melatonin. Biopharm. Drug Dispos. 2000, 21, 15–22. [Google Scholar] [CrossRef]
- Dubocovich, M.L.; Delagrange, P.; Krause, D.N.; Sugden, D.; Cardinali, D.P.; Olcese, J. International Union of Basic and Clinical Pharmacology. LXXV. Nomenclature, classification, and pharmacology of G protein-coupled melatonin receptors. Pharmacol. Rev. 2010, 62, 343–380. [Google Scholar] [CrossRef]
- Levoye, A.; Dam, J.; Ayoub, M.A.; Guillaume, J.L.; Couturier, C.; Delagrange, P.; Jockers, R. The orphan GPR50 receptor specifically inhibits MT1 melatonin receptor function through heterodimerization. EMBO J. 2006, 25, 3012–3023. [Google Scholar] [CrossRef]
- Wiesenberg, I.; Missbach, M.; Kahlen, J.P.; Schrader, M.; Carlberg, C. Transcriptional activation of the nuclear receptor RZR alpha by the pineal gland hormone melatonin and identification of CGP 52608 as a synthetic ligand. Nucleic Acids Res. 1995, 23, 327–333. [Google Scholar] [CrossRef]
- Lardone, P.J.; Guerrero, J.M.; Fernandez-Santos, J.M.; Rubio, A.; Martin-Lacave, I.; Carrillo-Vico, A. Melatonin synthesized by T lymphocytes as a ligand of the retinoic acid-related orphan receptor. J. Pineal Res. 2011, 51, 454–462. [Google Scholar] [CrossRef]
- Galano, A.; Tan, D.X.; Reiter, R.J. Melatonin as a natural ally against oxidative stress: A physicochemical examination. J. Pineal Res. 2011, 51, 1–16. [Google Scholar] [CrossRef]
- Antolin, I.; Rodriguez, C.; Sainz, R.M.; Mayo, J.C.; Uria, H.; Kotler, M.L.; Rodriguez-Colunga, M.J.; Tolivia, D.; Menendez-Pelaez, A. Neurohormone melatonin prevents cell damage: Effect on gene expression for antioxidant enzymes. FASEB J. 1996, 10, 882–890. [Google Scholar] [PubMed]
- Pablos, M.I.; Reiter, R.J.; Ortiz, G.G.; Guerrero, J.M.; Agapito, M.T.; Chuang, J.I.; Sewerynek, E. Rhythms of glutathione peroxidase and glutathione reductase in brain of chick and their inhibition by light. Neurochem. Int. 1998, 32, 69–75. [Google Scholar] [CrossRef]
- Rodriguez, C.; Mayo, J.C.; Sainz, R.M.; Antolin, I.; Herrera, F.; Martin, V.; Reiter, R.J. Regulation of antioxidant enzymes: A significant role for melatonin. J. Pineal Res. 2004, 36, 1–9. [Google Scholar] [CrossRef]
- Jimenez-Ortega, V.; Cano, P.; Cardinali, D.P.; Esquifino, A.I. 24-Hour variation in gene expression of redox pathway enzymes in rat hypothalamus: Effect of melatonin treatment. Redox Rep. 2009, 14, 132–138. [Google Scholar] [CrossRef]
- Subramanian, P.; Mirunalini, S.; Pandi-Perumal, S.R.; Trakht, I.; Cardinali, D.P. Melatonin treatment improves the antioxidant status and decreases lipid content in brain and liver of rats. Eur. J. Pharmacol. 2007, 571, 116–119. [Google Scholar] [CrossRef]
- Kilanczyk, E.; Bryszewska, M. The effect of melatonin on antioxidant enzymes in human diabetic skin fibroblasts. Cell. Mol. Biol. Lett. 2003, 8, 333–336. [Google Scholar] [PubMed]
- Cardinali, D.P.; Ritta, M.N.; Fuentes, A.M.; Gimeno, M.F.; Gimeno, A.L. Prostaglandin E release by rat medial basal hypothalamus in vitro. Inhibition by melatonin at submicromolar concentrations. Eur. J. Pharmacol. 1980, 67, 151–153. [Google Scholar] [CrossRef]
- Deng, W.G.; Tang, S.T.; Tseng, H.P.; Wu, K.K. Melatonin suppresses macrophage cyclooxygenase-2 and inducible nitric oxide synthase expression by inhibiting p52 acetylation and binding. Blood 2006, 108, 518–524. [Google Scholar] [CrossRef]
- Costantino, G.; Cuzzocrea, S.; Mazzon, E.; Caputi, A.P. Protective effects of melatonin in zymosan-activated plasma-induced paw inflammation. Eur. J. Pharmacol. 1998, 363, 57–63. [Google Scholar] [CrossRef]
- Tan, D.; Reiter, R.J.; Chen, L.D.; Poeggeler, B.; Manchester, L.C.; Barlow-Walden, L.R. Both physiological and pharmacological levels of melatonin reduce DNA adduct formation induced by the carcinogen safrole. Carcinogenesis 1994, 15, 215–218. [Google Scholar] [CrossRef]
- Leon-Blanco, M.M.; Guerrero, J.M.; Reiter, R.J.; Pozo, D. RNA expression of human telomerase subunits TR and TERT is differentially affected by melatonin receptor agonists in the MCF-7 tumor cell line. Cancer Lett. 2004, 216, 73–80. [Google Scholar] [CrossRef]
- Urata, Y.; Honma, S.; Goto, S.; Todoroki, S.; Iida, T.; Cho, S.; Honma, K.; Kondo, T. Melatonin induces γ-glutamylcysteine synthetase mediated by activator protein-1 in human vascular endothelial cells. Free Radic. Biol. Med. 1999, 27, 838–847. [Google Scholar] [CrossRef]
- Poliandri, A.H.; Esquifino, A.I.; Cano, P.; Jimenez, V.; Lafuente, A.; Cardinali, D.P.; Duvilanski, B.H. In vivo protective effect of melatonin on cadmium-induced changes in redox balance and gene expression in rat hypothalamus and anterior pituitary. J. Pineal Res. 2006, 41, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Ortega, V.; Cano, P.; Scacchi, P.A.; Cardinali, D.P.; Esquifino, A.I. Cadmium-induced disruption in 24-h expression of clock and redox enzyme genes in rat medial basal hypothalamus. Prevention by melatonin. Front. Neurol. 2011, 2. [Google Scholar] [CrossRef]
- Shaikh, A.Y.; Xu, J.; Wu, Y.; He, L.; Hsu, C.Y. Melatonin protects bovine cerebral endothelial cells from hyperoxia-induced DNA damage and death. Neurosci. Lett. 1997, 229, 193–197. [Google Scholar] [CrossRef]
- Pablos, M.I.; Reiter, R.J.; Chuang, J.I.; Ortiz, G.G.; Guerrero, J.M.; Sewerynek, E.; Agapito, M.T.; Melchiorri, D.; Lawrence, R.; Deneke, S.M. Acutely administered melatonin reduces oxidative damage in lung and brain induced by hyperbaric oxygen. J. Appl. Physiol. 1997, 83, 354–358. [Google Scholar] [PubMed]
- Princ, F.G.; Juknat, A.A.; Maxit, A.G.; Cardalda, C.; Batlle, A. Melatonin’s antioxidant protection against δ-aminolevulinic acid-induced oxidative damage in rat cerebellum. J. Pineal Res. 1997, 23, 40–46. [Google Scholar] [CrossRef]
- Carneiro, R.C.; Reiter, R.J. δ-Aminolevulinic acid-induced lipid peroxidation in rat kidney and liver is attenuated by melatonin: An in vitro and in vivo study. J. Pineal Res. 1998, 24, 131–136. [Google Scholar] [CrossRef]
- Onuki, J.; Almeida, E.A.; Medeiros, M.H.; Di, M.P. Inhibition of 5-aminolevulinic acid-induced DNA damage by melatonin, N1-acetyl-N2-formyl-5-methoxykynuramine, quercetin or resveratrol. J. Pineal Res. 2005, 38, 107–115. [Google Scholar] [CrossRef]
- Erol, F.S.; Topsakal, C.; Ozveren, M.F.; Kaplan, M.; Ilhan, N.; Ozercan, I.H.; Yildiz, O.G. Protective effects of melatonin and vitamin E in brain damage due to gamma radiation: An experimental study. Neurosurg. Rev. 2004, 27, 65–69. [Google Scholar] [CrossRef]
- Shirazi, A.; Haddadi, G.H.; Asadi-Amoli, F.; Sakhaee, S.; Ghazi-Khansari, M.; Avand, A. Radioprotective effect of melatonin in reducing oxidative stress in rat lenses. Cell J. 2011, 13, 79–82. [Google Scholar] [PubMed]
- Taysi, S.; Memisogullari, R.; Koc, M.; Yazici, A.T.; Aslankurt, M.; Gumustekin, K.; Al, B.; Ozabacigil, F.; Yilmaz, A.; Tahsin, O.H. Melatonin reduces oxidative stress in the rat lens due to radiation-induced oxidative injury. Int. J. Radiat. Biol. 2008, 84, 803–808. [Google Scholar] [CrossRef]
- Lee, E.J.; Wu, T.S.; Lee, M.Y.; Chen, T.Y.; Tsai, Y.Y.; Chuang, J.I.; Chang, G.L. Delayed treatment with melatonin enhances electrophysiological recovery following transient focal cerebral ischemia in rats. J. Pineal Res. 2004, 36, 33–42. [Google Scholar] [CrossRef]
- Tai, S.H.; Hung, Y.C.; Lee, E.J.; Lee, A.C.; Chen, T.Y.; Shen, C.C.; Chen, H.Y.; Lee, M.Y.; Huang, S.Y.; Wu, T.S. Melatonin protects against transient focal cerebral ischemia in both reproductively active and estrogen-deficient female rats: The impact of circulating estrogen on its hormetic dose-response. J. Pineal Res. 2011, 50, 292–303. [Google Scholar] [CrossRef]
- Beni, S.M.; Kohen, R.; Reiter, R.J.; Tan, D.X.; Shohami, E. Melatonin-induced neuroprotection after closed head injury is associated with increased brain antioxidants and attenuated late-phase activation of NF-κB and AP-1. FASEB J. 2004, 18, 149–151. [Google Scholar] [PubMed]
- Tsai, M.C.; Chen, W.J.; Tsai, M.S.; Ching, C.H.; Chuang, J.I. Melatonin attenuates brain contusion-induced oxidative insult, inactivation of signal transducers and activators of transcription 1, and upregulation of suppressor of cytokine signaling-3 in rats. J. Pineal Res. 2011, 51, 233–245. [Google Scholar] [CrossRef]
- Kabadi, S.V.; Maher, T.J. Posttreatment with uridine and melatonin following traumatic brain injury reduces edema in various brain regions in rats. Ann. N. Y. Acad. Sci. 2010, 1199, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Manchester, L.C.; Tan, D.X. Neurotoxins: Free radical mechanisms and melatonin protection. Curr. Neuropharmacol. 2010, 8, 194–210. [Google Scholar] [CrossRef]
- Golombek, D.A.; Pevet, P.; Cardinali, D.P. Melatonin effects on behavior: Possible mediation by the central GABAergic system. Neurosci. Biobehav. Rev. 1996, 20, 403–412. [Google Scholar] [CrossRef]
- Caumo, W.; Levandovski, R.; Hidalgo, M.P. Preoperative anxiolytic effect of melatonin and clonidine on postoperative pain and morphine consumption in patients undergoing abdominal hysterectomy: A double-blind, randomized, placebo-controlled study. J. Pain 2009, 10, 100–108. [Google Scholar] [CrossRef]
- Louzada, P.R.; Paula Lima, A.C.; Mendonca-Silva, D.L.; Noel, F.; de Mello, F.G.; Ferreira, S.T. Taurine prevents the neurotoxicity of beta-amyloid and glutamate receptor agonists: Activation of GABA receptors and possible implications for Alzheimer’s disease and other neurological disorders. FASEB J. 2004, 18, 511–518. [Google Scholar] [CrossRef]
- Giusti, P.; Lipartiti, M.; Franceschini, D.; Schiavo, N.; Floreani, M.; Manev, H. Neuroprotection by melatonin from kainate-induced excitotoxicity in rats. FASEB J. 1996, 10, 891–896. [Google Scholar] [PubMed]
- Manev, H.; Uz, T.; Kharlamov, A.; Cagnoli, C.M.; Franceschini, D.; Giusti, P. In vivo protection against kainate-induced apoptosis by the pineal hormone melatonin: Effect of exogenous melatonin and circadian rhythm. Restor. Neurol. Neurosci. 1996, 9, 251–256. [Google Scholar] [PubMed]
- Cho, S.; Joh, T.H.; Baik, H.H.; Dibinis, C.; Volpe, B.T. Melatonin administration protects CA1 hippocampal neurons after transient forebrain ischemia in rats. Brain Res. 1997, 755, 335–338. [Google Scholar] [CrossRef]
- Kilic, E.; Ozdemir, Y.G.; Bolay, H.; Kelestimur, H.; Dalkara, T. Pinealectomy aggravates and melatonin administration attenuates brain damage in focal ischemia. J. Cereb. Blood Flow Metab. 1999, 19, 511–516. [Google Scholar] [PubMed]
- Furio, A.M.; Fontao, R.; Falco, N.; Ruiz, J.I.; Caccuri, R.L.; Cardinali, D.P. Neuroprotective effect of melatonin on glucocorticoid toxicity in the rat hippocampus. Open Physiol. J. 2008, 1, 23–27. [Google Scholar] [CrossRef]
- Dodd, S.; Maes, M.; Anderson, G.; Dean, O.M.; Moylan, S.; Berk, M. Putative neuroprotective agents in neuropsychiatric disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 42, 135–145. [Google Scholar] [CrossRef]
- Jiao, S.; Wu, M.M.; Hu, C.L.; Zhang, Z.H.; Mei, Y.A. Melatonin receptor agonist 2-iodomelatonin prevents apoptosis of cerebellar granule neurons via K+ current inhibition. J. Pineal Res. 2004, 36, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Koh, P.O. Melatonin prevents down-regulation of astrocytic phosphoprotein PEA-15 in ischemic brain injury. J. Pineal Res. 2011, 51, 381–386. [Google Scholar] [CrossRef]
- Radogna, F.; Diederich, M.; Ghibelli, L. Melatonin: A pleiotropic molecule regulating inflammation. Biochem. Pharmacol. 2010, 80, 1844–1852. [Google Scholar] [CrossRef]
- Peng, T.I.; Hsiao, C.W.; Reiter, R.J.; Tanaka, M.; Lai, Y.K.; Jou, M.J. mtDNA T8993G mutation-induced mitochondrial complex V inhibition augments cardiolipin-dependent alterations in mitochondrial dynamics during oxidative, Ca2+, and lipid insults in NARP cybrids: A potential therapeutic target for melatonin. J. Pineal Res. 2012, 52, 93–106. [Google Scholar] [CrossRef]
- Jou, M.J. Melatonin preserves the transient mitochondrial permeability transition for protection during mitochondrial Ca2+ stress in astrocyte. J. Pineal Res. 2011, 50, 427–435. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Sayeed, I.; Siemen, D.; Wolf, G.; Horn, T.F. Direct inhibition of the mitochondrial permeability transition pore: A possible mechanism responsible for anti-apoptotic effects of melatonin. FASEB J. 2004, 18, 869–871. [Google Scholar] [PubMed]
- Hardeland, R. Melatonin and the theories of aging: A critical appraisal of melatonin’s role in antiaging mechanisms. J. Pineal Res. 2013, 55, 325–356. [Google Scholar] [PubMed]
- Selkoe, D.J. Cell biology of protein misfolding: The examples of Alzheimer’s and Parkinson’s diseases. Nat. Cell Biol. 2004, 6, 1054–1061. [Google Scholar] [CrossRef]
- Lahiri, D.K.; Ghosh, C. Interactions between melatonin, reactive oxygen species, and nitric oxide. Ann. N. Y. Acad. Sci. 1999, 893, 325–330. [Google Scholar] [CrossRef]
- Matsubara, E.; Bryant-Thomas, T.; Pacheco, Q.J.; Henry, T.L.; Poeggeler, B.; Herbert, D.; Cruz-Sanchez, F.; Chyan, Y.J.; Smith, M.A.; Perry, G.; et al. Melatonin increases survival and inhibits oxidative and amyloid pathology in a transgenic model of Alzheimer’s disease. J. Neurochem. 2003, 85, 1101–1108. [Google Scholar] [CrossRef]
- Lahiri, D.K.; Chen, D.; Ge, Y.W.; Bondy, S.C.; Sharman, E.H. Dietary supplementation with melatonin reduces levels of amyloid β-peptides in the murine cerebral cortex. J. Pineal Res. 2004, 36, 224–231. [Google Scholar] [CrossRef]
- Song, W.; Lahiri, D.K. Melatonin alters the metabolism of the beta-amyloid precursor protein in the neuroendocrine cell line PC12. J. Mol. Neurosci. 1997, 9, 75–92. [Google Scholar] [CrossRef]
- Zhang, Y.C.; Wang, Z.F.; Wang, Q.; Wang, Y.P.; Wang, J.Z. Melatonin attenuates β-amyloid-induced inhibition of neurofilament expression. Acta Pharmacol. Sin. 2004, 25, 447–451. [Google Scholar] [PubMed]
- Olivieri, G.; Hess, C.; Savaskan, E.; Ly, C.; Meier, F.; Baysang, G.; Brockhaus, M.; Muller-Spahn, F. Melatonin protects SHSY5Y neuroblastoma cells from cobalt-induced oxidative stress, neurotoxicity and increased β-amyloid secretion. J. Pineal Res. 2001, 31, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.; Kulhanek, D.; Nowlin, J.; Jones, R.; Pratico, D.; Rokach, J.; Stackman, R. Chronic melatonin therapy fails to alter amyloid burden or oxidative damage in old Tg2576 mice: Implications for clinical trials. Brain Res. 2005, 1037, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef]
- Donnelly, P.S.; Caragounis, A.; Du, T.; Laughton, K.M.; Volitakis, I.; Cherny, R.A.; Sharples, R.A.; Hill, A.F.; Li, Q.X.; Masters, C.L.; et al. Selective intracellular release of copper and zinc ions from bis (thiosemicarbazonato) complexes reduces levels of Alzheimer disease amyloid-β peptide. J. Biol. Chem. 2008, 283, 4568–4577. [Google Scholar] [CrossRef]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guenette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef]
- Poeggeler, B.; Miravalle, L.; Zagorski, M.G.; Wisniewski, T.; Chyan, Y.J.; Zhang, Y.; Shao, H.; Bryant-Thomas, T.; Vidal, R.; Frangione, B.; et al. Melatonin reverses the profibrillogenic activity of apolipoprotein E4 on the Alzheimer amyloid Aβ peptide. Biochemistry 2001, 40, 14995–15001. [Google Scholar] [CrossRef]
- Pappolla, M.; Bozner, P.; Soto, C.; Shao, H.; Robakis, N.K.; Zagorski, M.; Frangione, B.; Ghiso, J. Inhibition of Alzheimer β-fibrillogenesis by melatonin. J. Biol. Chem. 1998, 273, 7185–7188. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Chang, Y.; Cheng, Y.; Zhang, B.L.; Qu, Z.W.; Qin, C.; Zhang, J.T. Melatonin alleviates behavioral deficits associated with apoptosis and cholinergic system dysfunction in the APP 695 transgenic mouse model of Alzheimer’s disease. J. Pineal Res. 2004, 37, 129–136. [Google Scholar] [CrossRef]
- Zatta, P.; Tognon, G.; Carampin, P. Melatonin prevents free radical formation due to the interaction between β-amyloid peptides and metal ions [AlIII, ZnII, CuII, MnII, FeII]. J. Pineal Res. 2003, 35, 98–103. [Google Scholar] [CrossRef]
- Furio, A.M.; Cutrera, R.A.; Castillo Thea, V.; Perez, L.S.; Riccio, P.; Caccuri, R.L.; Brusco, L.L.; Cardinali, D.P. Effect of melatonin on changes in locomotor activity rhythm of Syrian hamsters injected with β amyloid peptide 25–35 in the suprachiasmatic nuclei. Cell. Mol. Neurobiol. 2002, 22, 699–709. [Google Scholar] [CrossRef]
- Shen, Y.X.; Xu, S.Y.; Wei, W.; Sun, X.X.; Yang, J.; Liu, L.H.; Dong, C. Melatonin reduces memory changes and neural oxidative damage in mice treated with d-galactose. J. Pineal Res. 2002, 32, 173–178. [Google Scholar] [CrossRef]
- Rosales-Corral, S.; Tan, D.X.; Reiter, R.J.; Valdivia-Velazquez, M.; Martinez-Barboza, G.; Acosta-Martinez, J.P.; Ortiz, G.G. Orally administered melatonin reduces oxidative stress and proinflammatory cytokines induced by amyloid-β peptide in rat brain: A comparative, in vivo study versus vitamin C and, E. J. Pineal Res. 2003, 35, 80–84. [Google Scholar] [CrossRef]
- Olcese, J.M.; Cao, C.; Mori, T.; Mamcarz, M.B.; Maxwell, A.; Runfeldt, M.J.; Wang, L.; Zhang, C.; Lin, X.; Zhang, G.; et al. Protection against cognitive deficits and markers of neurodegeneration by long-term oral administration of melatonin in a transgenic model of Alzheimer disease. J. Pineal Res. 2009, 47, 82–96. [Google Scholar] [CrossRef]
- Dragicevic, N.; Copes, N.; O’Neal-Moffitt, G.; Jin, J.; Buzzeo, R.; Mamcarz, M.; Tan, J.; Cao, C.; Olcese, J.M.; Arendash, G.W.; et al. Melatonin treatment restores mitochondrial function in Alzheimer’s mice: A mitochondrial protective role of melatonin membrane receptor signaling. J. Pineal Res. 2011, 51, 75–86. [Google Scholar] [CrossRef]
- Brion, J.P.; Anderton, B.H.; Authelet, M.; Dayanandan, R.; Leroy, K.; Lovestone, S.; Octave, J.N.; Pradier, L.; Touchet, N.; Tremp, G. Neurofibrillary tangles and tau phosphorylation. Biochem. Soc. Symp. 2001, 81–88. [Google Scholar]
- Billingsley, M.L.; Kincaid, R.L. Regulated phosphorylation and dephosphorylation of tau protein: Effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem. J. 1997, 323, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Khatoon, S.; Grundke-Iqbal, I.; Iqbal, K. Brain levels of microtubule-associated protein tau are elevated in Alzheimer’s disease: A radioimmuno-slot-blot assay for nanograms of the protein. J. Neurochem. 1992, 59, 750–753. [Google Scholar] [CrossRef]
- Iqbal, K.; Alonso, A.C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 2005, 1739, 198–210. [Google Scholar] [CrossRef]
- Deng, Y.Q.; Xu, G.G.; Duan, P.; Zhang, Q.; Wang, J.Z. Effects of melatonin on wortmannin-induced tau hyperphosphorylation. Acta Pharmacol. Sin. 2005, 26, 519–526. [Google Scholar] [CrossRef]
- Li, S.P.; Deng, Y.Q.; Wang, X.C.; Wang, Y.P.; Wang, J.Z. Melatonin protects SH-SY5Y neuroblastoma cells from calyculin A-induced neurofilament impairment and neurotoxicity. J. Pineal Res. 2004, 36, 186–191. [Google Scholar] [CrossRef]
- Li, X.C.; Wang, Z.F.; Zhang, J.X.; Wang, Q.; Wang, J.Z. Effect of melatonin on calyculin A-induced tau hyperphosphorylation. Eur. J. Pharmacol. 2005, 510, 25–30. [Google Scholar] [CrossRef]
- Xiong, Y.F.; Chen, Q.; Chen, J.; Zhou, J.; Wang, H.X. Melatonin reduces the impairment of axonal transport and axonopathy induced by calyculin A. J. Pineal Res. 2011, 50, 319–327. [Google Scholar] [CrossRef]
- Benitez-King, G.; Tunez, I.; Bellon, A.; Ortiz, G.G.; Anton-Tay, F. Melatonin prevents cytoskeletal alterations and oxidative stress induced by okadaic acid in N1E-115 cells. Exp. Neurol. 2003, 182, 151–159. [Google Scholar] [CrossRef]
- Montilla-Lopez, P.; Munoz-Agueda, M.C.; Feijoo, L.M.; Munoz-Castaneda, J.R.; Bujalance-Arenas, I.; Tunez-Finana, I. Comparison of melatonin versus vitamin C on oxidative stress and antioxidant enzyme activity in Alzheimer’s disease induced by okadaic acid in neuroblastoma cells. Eur. J. Pharmacol. 2002, 451, 237–243. [Google Scholar] [CrossRef]
- Montilla, P.; Feijoo, M.; Munoz, M.C.; Munoz-Castaneda, J.R.; Bujalance, I.; Tunez, I. Effect of melatonin on the oxidative stress in N1E-115 cells is not mediated by MT1 receptors. J. Physiol. Biochem. 2003, 59, 263–268. [Google Scholar] [CrossRef]
- Wang, Y.P.; Li, X.T.; Liu, S.J.; Zhou, X.W.; Wang, X.C.; Wang, J.Z. Melatonin ameliorated okadaic-acid induced Alzheimer-like lesions. Acta Pharmacol. Sin. 2004, 25, 276–280. [Google Scholar] [PubMed]
- Liu, S.J.; Wang, J.Z. Alzheimer-like tau phosphorylation induced by wortmannin in vivo and its attenuation by melatonin. Acta Pharmacol. Sin. 2002, 23, 183–187. [Google Scholar] [PubMed]
- Wang, X.C.; Zhang, J.; Yu, X.; Han, L.; Zhou, Z.T.; Zhang, Y.; Wang, J.Z. Prevention of isoproterenol-induced tau hyperphosphorylation by melatonin in the rat. Sheng Li Xue Bao 2005, 57, 7–12. [Google Scholar] [PubMed]
- Zhu, L.Q.; Wang, S.H.; Ling, Z.Q.; Wang, D.L.; Wang, J.Z. Effect of inhibiting melatonin biosynthesis on spatial memory retention and tau phosphorylation in rat. J. Pineal Res. 2004, 37, 71–77. [Google Scholar] [CrossRef]
- Gomez-Ramos, A.; Diaz-Nido, J.; Smith, M.A.; Perry, G.; Avila, J. Effect of the lipid peroxidation product acrolein on tau phosphorylation in neural cells. J. Neurosci. Res. 2003, 71, 863–870. [Google Scholar] [CrossRef]
- Lovell, M.A.; Xiong, S.; Xie, C.; Davies, P.; Markesbery, W.R. Induction of hyperphosphorylated tau in primary rat cortical neuron cultures mediated by oxidative stress and glycogen synthase kinase-3. J. Alzheimers Dis. 2004, 6, 659–671. [Google Scholar] [PubMed]
- Kenyon, C.J. The genetics of ageing. Nature 2010, 464, 504–512. [Google Scholar] [CrossRef]
- Schuster, C.; Williams, L.M.; Morris, A.; Morgan, P.J.; Barrett, P. The human MT1 melatonin receptor stimulates cAMP production in the human neuroblastoma cell line SH-SY5Y cells via a calcium-calmodulin signal transduction pathway. J. Neuroendocrinol. 2005, 17, 170–178. [Google Scholar] [CrossRef]
- Peschke, E.; Muhlbauer, E.; Musshoff, U.; Csernus, V.J.; Chankiewitz, E.; Peschke, D. Receptor (MT1) mediated influence of melatonin on cAMP concentration and insulin secretion of rat insulinoma cells INS-1. J. Pineal Res. 2002, 33, 63–71. [Google Scholar] [CrossRef]
- Witt-Enderby, P.A.; MacKenzie, R.S.; McKeon, R.M.; Carroll, E.A.; Bordt, S.L.; Melan, M.A. Melatonin induction of filamentous structures in non-neuronal cells that is dependent on expression of the human MT1 melatonin receptor. Cell Motil. Cytoskelet. 2000, 46, 28–42. [Google Scholar] [CrossRef]
- Rivera-Bermudez, M.A.; Gerdin, M.J.; Earnest, D.J.; Dubocovich, M.L. Regulation of basal rhythmicity in protein kinase C activity by melatonin in immortalized rat suprachiasmatic nucleus cells. Neurosci. Lett. 2003, 346, 37–40. [Google Scholar] [CrossRef]
- Benitez-King, G.; Rios, A.; Martinez, A.; Anton-Tay, F. In vitro inhibition of Ca2+/calmodulin-dependent kinase II activity by melatonin. Biochim. Biophys. Acta 1996, 1290, 191–196. [Google Scholar] [CrossRef]
- Chan, A.S.; Lai, F.P.; Lo, R.K.; Voyno-Yasenetskaya, T.A.; Stanbridge, E.J.; Wong, Y.H. Melatonin MT1 and MT2 receptors stimulate c-Jun N-terminal kinase via pertussis toxin-sensitive and -insensitive G proteins. Cell Signal. 2002, 14, 249–257. [Google Scholar] [CrossRef]
- Arends, Y.M.; Duyckaerts, C.; Rozemuller, J.M.; Eikelenboom, P.; Hauw, J.J. Microglia, amyloid and dementia in alzheimer disease. A correlative study. Neurobiol. Aging 2000, 21, 39–47. [Google Scholar] [PubMed]
- Combadiere, C.; Feumi, C.; Raoul, W.; Keller, N.; Rodero, M.; Pezard, A.; Lavalette, S.; Houssier, M.; Jonet, L.; Picard, E.; et al. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J. Clin. Investig. 2007, 117, 2920–2928. [Google Scholar] [CrossRef]
- Streit, W.J.; Mrak, R.E.; Griffin, W.S. Microglia and neuroinflammation: A pathological perspective. J. Neuroinflamm. 2004, 1. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, G.; Liu, L.; Xu, S. Suppressive effects of melatonin on amyloid-β-induced glial activation in rat hippocampus. Arch. Med. Res. 2007, 38, 284–290. [Google Scholar] [CrossRef]
- Stuchbury, G.; Munch, G. Alzheimer’s associated inflammation, potential drug targets and future therapies. J. Neural Transm. 2005, 112, 429–453. [Google Scholar] [CrossRef]
- Park, S.Y.; Jin, M.L.; Kim, Y.H.; Kim, Y.; Lee, S.J. Anti-inflammatory effects of aromatic-turmerone through blocking of NF-κB, JNK, and p38 MAPK signaling pathways in amyloid beta-stimulated microglia. Int. Immunopharmacol. 2012, 14, 13–20. [Google Scholar] [CrossRef]
- Lau, W.W.; Ng, J.K.; Lee, M.M.; Chan, A.S.; Wong, Y.H. Interleukin-6 autocrine signaling mediates melatonin MT1/2 receptor-induced STAT3 Tyr705 phosphorylation. J. Pineal Res. 2012, 52, 477–489. [Google Scholar] [CrossRef]
- Mohan, N.; Sadeghi, K.; Reiter, R.J.; Meltz, M.L. The neurohormone melatonin inhibits cytokine, mitogen and ionizing radiation induced NF-κB. Biochem. Mol. Biol. Int. 1995, 37, 1063–1070. [Google Scholar] [PubMed]
- Chuang, J.I.; Mohan, N.; Meltz, M.L.; Reiter, R.J. Effect of melatonin on NF-κB DNA-binding activity in the rat spleen. Cell Biol. Int. 1996, 20, 687–692. [Google Scholar] [CrossRef]
- Struble, R.G.; Cork, L.C.; Whitehouse, P.J.; Price, D.L. Cholinergic innervation in neuritic plaques. Science 1982, 216, 413–415. [Google Scholar] [CrossRef] [PubMed]
- Samuel, W.; Masliah, E.; Hill, L.R.; Butters, N.; Terry, R. Hippocampal connectivity and Alzheimer’s dementia: Effects of synapse loss and tangle frequency in a two-component model. Neurology 1994, 44, 2081–2088. [Google Scholar] [CrossRef]
- Terry, A.V., Jr.; Buccafusco, J.J. The cholinergic hypothesis of age and Alzheimer’s disease-related cognitive deficits: Recent challenges and their implications for novel drug development. J. Pharmacol. Exp. Ther. 2003, 306, 821–827. [Google Scholar] [CrossRef]
- Rinne, J.O.; Laine, M.; Hiltunen, J.; Erkinjuntti, T. Semantic decision making in early probable AD: A PET activation study. Brain Res. Cogn. Brain Res. 2003, 18, 89–96. [Google Scholar] [CrossRef]
- Spencer, J.P.; Middleton, L.J.; Davies, C.H. Investigation into the efficacy of the acetylcholinesterase inhibitor, donepezil, and novel procognitive agents to induce γ oscillations in rat hippocampal slices. Neuropharmacology 2010, 59, 437–443. [Google Scholar] [CrossRef]
- Guermonprez, L.; Ducrocq, C.; Gaudry-Talarmain, Y.M. Inhibition of acetylcholine synthesis and tyrosine nitration induced by peroxynitrite are differentially prevented by antioxidants. Mol. Pharmacol. 2001, 60, 838–846. [Google Scholar] [PubMed]
- Feng, Z.; Cheng, Y.; Zhang, J.T. Long-term effects of melatonin or 17 β-estradiol on improving spatial memory performance in cognitively impaired, ovariectomized adult rats. J. Pineal Res. 2004, 37, 198–206. [Google Scholar] [CrossRef]
- Tang, F.; Nag, S.; Shiu, S.Y.; Pang, S.F. The effects of melatonin and Ginkgo biloba extract on memory loss and choline acetyltransferase activities in the brain of rats infused intracerebroventricularly with β-amyloid 1-40. Life Sci. 2002, 71, 2625–2631. [Google Scholar] [CrossRef]
- Agrawal, R.; Tyagi, E.; Shukla, R.; Nath, C. A study of brain insulin receptors, AChE activity and oxidative stress in rat model of ICV STZ induced dementia. Neuropharmacology 2009, 56, 779–787. [Google Scholar] [CrossRef]
- Fernandez-Bachiller, M.I.; Perez, C.; Campillo, N.E.; Paez, J.A.; Gonzalez-Munoz, G.C.; Usan, P.; Garcia-Palomero, E.; Lopez, M.G.; Villarroya, M.; Garcia, A.G.; et al. Tacrine-melatonin hybrids as multifunctional agents for Alzheimer’s disease, with cholinergic, antioxidant, and neuroprotective properties. ChemMedChem 2009, 4, 828–841. [Google Scholar] [CrossRef]
- Spuch, C.; Antequera, D.; Isabel Fernandez-Bachiller, M.; Isabel Rodriguez-Franco, M.; Carro, E. A new tacrine-melatonin hybrid reduces amyloid burden and behavioral deficits in a mouse model of Alzheimer’s disease. Neurotox. Res. 2010, 17, 421–431. [Google Scholar] [CrossRef]
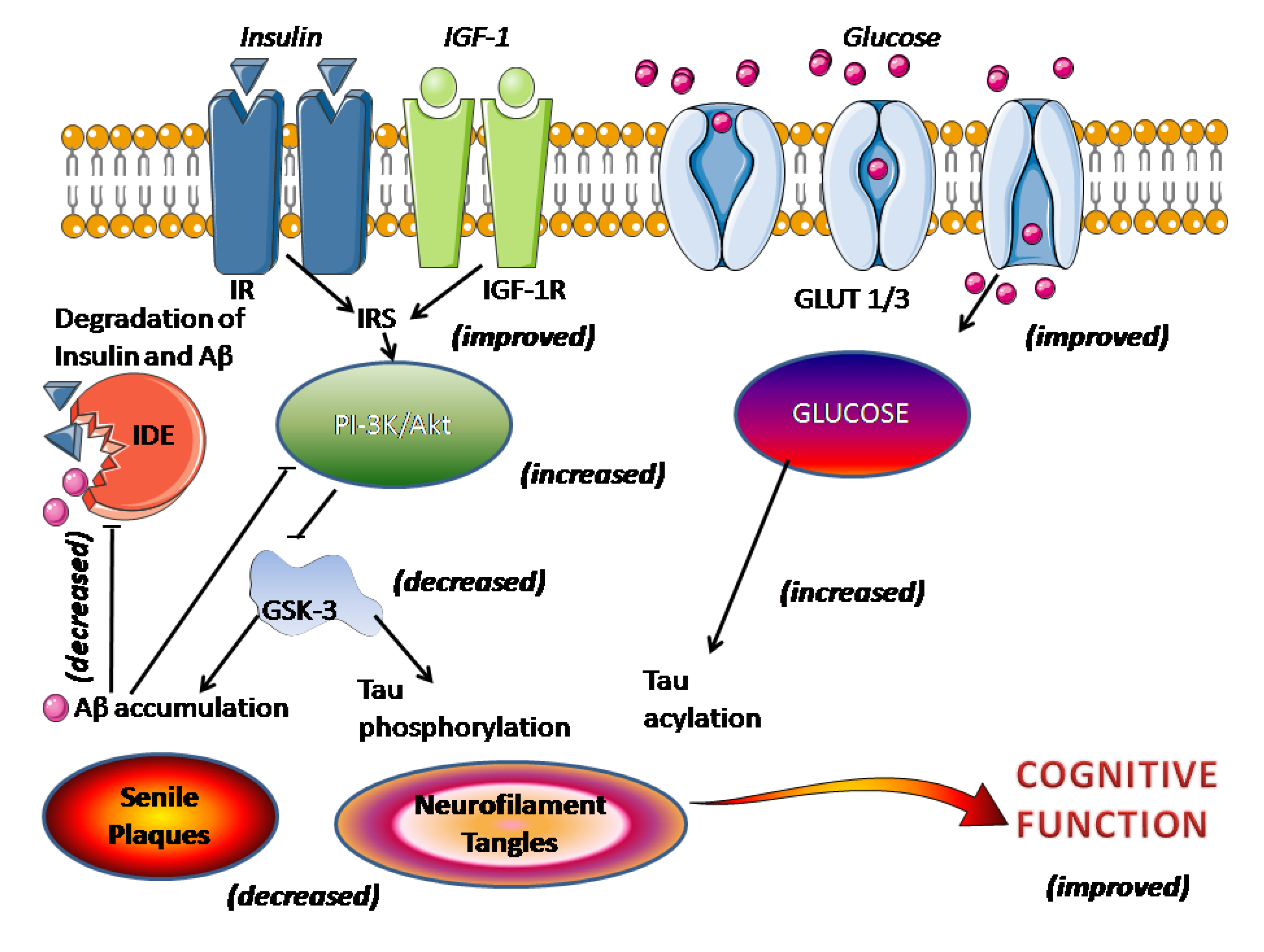
- O’Neill, C.; Kiely, A.P.; Coakley, M.F.; Manning, S.; Long-Smith, C.M. Insulin and IGF-1 signalling: Longevity, protein homoeostasis and Alzheimer’s disease. Biochem. Soc. Trans. 2012, 40, 721–727. [Google Scholar] [CrossRef]
- Hildreth, K.L.; van Pelt, R.E.; Schwartz, R.S. Obesity, insulin resistance, and Alzheimer’s disease. Obesity 2012, 20, 1549–1557. [Google Scholar] [CrossRef]
- De la Monte, S.M. Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 35–66. [Google Scholar] [CrossRef]
- Thambisetty, M.; Jeffrey, M.E.; Yang, A.; Dolan, H.; Marano, C.; Zonderman, A.B.; Troncoso, J.C.; Zhou, Y.; Wong, D.F.; Ferrucci, L.; et al. Glucose intolerance, insulin resistance, and pathological features of Alzheimer disease in the Baltimore Longitudinal Study of Aging. JAMA Neurol. 2013, 70, 1167–1172. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Cano, P.; Jimenez-Ortega, V.; Esquifino, A.I. Melatonin and the metabolic syndrome: Physiopathologic and therapeutical implications. Neuroendocrinology 2011, 93, 133–142. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Bernasconi, P.A.; Reynoso, R.; Toso, C.F.; Scacchi, P. Melatonin may curtail the metabolic syndrome: Studies on initial and fully established fructose-induced metabolic syndrome in rats. Int. J. Mol. Sci. 2013, 14, 2502–2514. [Google Scholar] [CrossRef]
- Magri, F.; Locatelli, M.; Balza, G.; Molla, G.; Cuzzoni, G.; Fioravanti, M.; Solerte, S.B.; Ferrari, E. Changes in endocrine circadian rhythms as markers of physiological and pathological brain aging. Chronobiol. Int. 1997, 14, 385–396. [Google Scholar] [CrossRef]
- Zhou, J.N.; Liu, R.Y.; Kamphorst, W.; Hofman, M.A.; Swaab, D.F. Early neuropathological Alzheimer’s changes in aged individuals are accompanied by decreased cerebrospinal fluid melatonin levels. J. Pineal Res. 2003, 35, 125–130. [Google Scholar] [CrossRef]
- Skene, D.J.; Vivien-Roels, B.; Sparks, D.L.; Hunsaker, J.C.; Pevet, P.; Ravid, D.; Swaab, D.F. Daily variation in the concentration of melatonin and 5-methoxytryptophol in the human pineal gland: Effect of age and Alzheimer’s disease. Brain Res. 1990, 528, 170–174. [Google Scholar] [CrossRef]
- Ohashi, Y.; Okamoto, N.; Uchida, K.; Iyo, M.; Mori, N.; Morita, Y. Daily rhythm of serum melatonin levels and effect of light exposure in patients with dementia of the Alzheimer’s type. Biol. Psychiatry 1999, 45, 1646–1652. [Google Scholar] [CrossRef]
- Liu, R.Y.; Zhou, J.N.; van Heerikhuize, J.; Hofman, M.A.; Swaab, D.F. Decreased melatonin levels in postmortem cerebrospinal fluid in relation to aging, Alzheimer’s disease, and apolipoprotein E-epsilon4/4 genotype. J. Clin. Endocrinol. Metab. 1999, 84, 323–327. [Google Scholar] [PubMed]
- Mishima, K.; Tozawa, T.; Satoh, K.; Matsumoto, Y.; Hishikawa, Y.; Okawa, M. Melatonin secretion rhythm disorders in patients with senile dementia of Alzheimer’s type with disturbed sleep-waking. Biol. Psychiatry 1999, 45, 417–421. [Google Scholar] [CrossRef]
- Skene, D.J.; Swaab, D.F. Melatonin rhythmicity: Effect of age and Alzheimer’s disease. Exp. Gerontol. 2003, 38, 199–206. [Google Scholar] [CrossRef]
- Wu, Y.H.; Feenstra, M.G.; Zhou, J.N.; Liu, R.Y.; Torano, J.S.; van Kan, H.J.; Fischer, D.F.; Ravid, R.; Swaab, D.F. Molecular changes underlying reduced pineal melatonin levels in Alzheimer disease: Alterations in preclinical and clinical stages. J. Clin. Endocrinol. Metab. 2003, 88, 5898–5906. [Google Scholar] [CrossRef]
- Savaskan, E.; Ayoub, M.A.; Ravid, R.; Angeloni, D.; Fraschini, F.; Meier, F.; Eckert, A.; Muller-Spahn, F.; Jockers, R. Reduced hippocampal MT2 melatonin receptor expression in Alzheimer’s disease. J. Pineal Res. 2005, 38, 10–16. [Google Scholar] [CrossRef]
- Savaskan, E.; Olivieri, G.; Meier, F.; Brydon, L.; Jockers, R.; Ravid, R.; Wirz-Justice, A.; Muller-Spahn, F. Increased melatonin 1a-receptor immunoreactivity in the hippocampus of Alzheimer’s disease patients. J. Pineal Res. 2002, 32, 59–62. [Google Scholar] [CrossRef]
- Weldemichael, D.A.; Grossberg, G.T. Circadian rhythm disturbances in patients with Alzheimer’s disease: A review. Int. J. Alzheimers Dis. 2010, 2010. [Google Scholar] [CrossRef]
- Klaffke, S.; Staedt, J. Sundowning and circadian rhythm disorders in dementia. Acta Neurol. Belg. 2006, 106, 168–175. [Google Scholar] [PubMed]
- Cardinali, D.P.; Furio, A.M.; Brusco, L.I. Clinical aspects of melatonin intervention in Alzheimer’s disease progression. Curr. Neuropharmacol. 2010, 8, 218–227. [Google Scholar] [CrossRef]
- De Jonghe, A.; Korevaar, J.C.; van Munster, B.C.; de Rooij, S.E. Effectiveness of melatonin treatment on circadian rhythm disturbances in dementia. Are there implications for delirium? A systematic review. Int. J. Geriatr. Psychiatry 2010, 25, 1201–1208. [Google Scholar] [CrossRef]
- Pappolla, M.A.; Chyan, Y.J.; Poeggeler, B.; Frangione, B.; Wilson, G.; Ghiso, J.; Reiter, R.J. An assessment of the antioxidant and the antiamyloidogenic properties of melatonin: Implications for Alzheimer’s disease. J. Neural Transm. 2000, 107, 203–231. [Google Scholar] [CrossRef]
- Wu, Y.H.; Swaab, D.F. The human pineal gland and melatonin in aging and Alzheimer’s disease. J. Pineal Res. 2005, 38, 145–152. [Google Scholar] [CrossRef]
- Wu, Y.H.; Zhou, J.N.; van Heerikhuize, J.; Jockers, R.; Swaab, D.F. Decreased MT1 melatonin receptor expression in the suprachiasmatic nucleus in aging and Alzheimer’s disease. Neurobiol. Aging 2007, 28, 1239–1247. [Google Scholar] [CrossRef]
- Fainstein, I.; Bonetto, A.; Brusco, L.I.; Cardinali, D.P. Effects of melatonin in elderly patients with sleep disturbance. A pilot study. Curr. Ther. Res. 1997, 58, 990–1000. [Google Scholar] [CrossRef]
- Brusco, L.I.; Marquez, M.; Cardinali, D.P. Melatonin treatment stabilizes chronobiologic and cognitive symptoms in Alzheimer’s disease. Neuro Endocrinol. Lett. 1998, 19, 111–115. [Google Scholar]
- Brusco, L.I.; Marquez, M.; Cardinali, D.P. Monozygotic twins with Alzheimer’s disease treated with melatonin: Case report. J. Pineal Res. 1998, 25, 260–263. [Google Scholar] [CrossRef]
- Cohen-Mansfield, J.; Garfinkel, D.; Lipson, S. Melatonin for treatment of sundowning in elderly persons with dementia—A preliminary study. Arch. Gerontol. Geriatr. 2000, 31, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Mishima, K.; Okawa, M.; Hozumi, S.; Hishikawa, Y. Supplementary administration of artificial bright light and melatonin as potent treatment for disorganized circadian rest-activity and dysfunctional autonomic and neuroendocrine systems in institutionalized demented elderly persons. Chronobiol. Int. 2000, 17, 419–432. [Google Scholar] [CrossRef]
- Serfaty, M.; Kennell-Webb, S.; Warner, J.; Blizard, R.; Raven, P. Double blind randomised placebo controlled trial of low dose melatonin for sleep disorders in dementia. Int. J. Geriatr. Psychiatry 2002, 17, 1120–1127. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Brusco, L.I.; Liberczuk, C.; Furio, A.M. The use of melatonin in Alzheimer’s disease. Neuro Endocrinol. Lett. 2002, 23, 20–23. [Google Scholar] [PubMed]
- Singer, C.; Tractenberg, R.E.; Kaye, J.; Schafer, K.; Gamst, A.; Grundman, M.; Thomas, R.; Thal, L.J. A multicenter, placebo-controlled trial of melatonin for sleep disturbance in Alzheimer’s disease. Sleep 2003, 26, 893–901. [Google Scholar] [PubMed]
- Asayama, K.; Yamadera, H.; Ito, T.; Suzuki, H.; Kudo, Y.; Endo, S. Double blind study of melatonin effects on the sleep-wake rhythm, cognitive and non-cognitive functions in Alzheimer type dementia. J. Nippon Med. Sch. 2003, 70, 334–341. [Google Scholar] [CrossRef]
- Mahlberg, R.; Kunz, D.; Sutej, I.; Kuhl, K.P.; Hellweg, R. Melatonin treatment of day-night rhythm disturbances and sundowning in Alzheimer disease: An open-label pilot study using actigraphy. J. Clin. Psychopharmacol. 2004, 24, 456–459. [Google Scholar] [PubMed]
- Mahlberg, R.; Walther, S. Actigraphy in agitated patients with dementia. Monitoring treatment outcomes. Z. Gerontol. Geriatr. 2007, 40, 178–184. [Google Scholar] [CrossRef]
- Anderson, K.N.; Jamieson, S.; Graham, A.J.; Shneerson, J.M. REM sleep behaviour disorder treated with melatonin in a patient with Alzheimer’s disease. Clin. Neurol. Neurosurg. 2008, 110, 492–495. [Google Scholar] [CrossRef]
- Dowling, G.A.; Burr, R.L.; van Someren, E.J.; Hubbard, E.M.; Luxenberg, J.S.; Mastick, J.; Cooper, B.A. Melatonin and bright-light treatment for rest-activity disruption in institutionalized patients with Alzheimer’s disease. J. Am. Geriatr. Soc. 2008, 56, 239–246. [Google Scholar] [CrossRef]
- Gehrman, P.R.; Connor, D.J.; Martin, J.L.; Shochat, T.; Corey-Bloom, J.; Ancoli-Israel, S. Melatonin fails to improve sleep or agitation in double-blind randomized placebo-controlled trial of institutionalized patients with Alzheimer disease. Am. J. Geriatr. Psychiatry 2009, 17, 166–169. [Google Scholar] [CrossRef]
- Farias, S.T.; Mungas, D.; Reed, B.R.; Harvey, D.; DeCarli, C. Progression of mild cognitive impairment to dementia in clinic- vs community-based cohorts. Arch. Neurol. 2009, 66, 1151–1157. [Google Scholar] [PubMed]
- Davies, L.; Wolska, B.; Hilbich, C.; Multhaup, G.; Martins, R.; Simms, G.; Beyreuther, K.; Masters, C.L. A4 amyloid protein deposition and the diagnosis of Alzheimer’s disease: Prevalence in aged brains determined by immunocytochemistry compared with conventional neuropathologic techniques. Neurology 1988, 38, 1688–1693. [Google Scholar] [CrossRef]
- Price, J.L.; Morris, J.C. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann. Neurol. 1999, 45, 358–368. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 1995, 16, 271–278. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Evolution of neuronal changes in the course of Alzheimer’s disease. J. Neural Transm. Suppl. 1998, 53, 127–140. [Google Scholar] [CrossRef]
- Furio, A.M.; Brusco, L.I.; Cardinali, D.P. Possible therapeutic value of melatonin in mild cognitive impairment. A retrospective study. J. Pineal Res. 2007, 43, 404–409. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Vigo, D.E.; Olivar, N.; Vidal, M.F.; Furio, A.M.; Brusco, L.I. Therapeutic application of melatonin in mild cognitive impairment. Am. J. Neurodegener Dis. 2012, 1, 280–291. [Google Scholar] [PubMed]
- Wu, Y.H.; Swaab, D.F. Disturbance and strategies for reactivation of the circadian rhythm system in aging and Alzheimer’s disease. Sleep Med. 2007, 8, 623–636. [Google Scholar] [CrossRef]
- Pandi-Perumal, S.R.; BaHammam, A.S.; Brown, G.M.; Spence, D.W.; Bharti, V.K.; Kaur, C.; Hardeland, R.; Cardinali, D.P. Melatonin antioxidative defense: Therapeutical implications for aging and neurodegenerative processes. Neurotox. Res. 2013, 23, 267–300. [Google Scholar] [CrossRef]
- Rosales-Corral, S.A.; Acuña-Castroviejo, D.; Coto-Montes, A.; Boga, J.A.; Manchester, L.C.; Fuentes-Broto, L.; Korkmaz, A.; Ma, S.; Tan, D.X.; Reiter, R.J. Alzheimer’s disease: Pathological mechanisms and the beneficial role of melatonin. J. Pineal Res. 2012, 52, 167–202. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchester, L.C.; Sanchez-Barcelo, E.; Mediavilla, M.D.; Reiter, R.J. Significance of high levels of endogenous melatonin in mammalian cerebrospinal fluid and in the central nervous system. Curr. Neuropharmacol. 2010, 8, 162–167. [Google Scholar] [CrossRef]
- Legros, C.; Chesneau, D.; Boutin, J.A.; Barc, C.; Malpaux, B. Melatonin from cerebrospinal fluid, but not from blood, reaches sheep cerebral tissues under physiological conditions. J. Neuroendocrinol. 2014. [Google Scholar] [CrossRef]
- Monti, J.M.; Alvarino, F.; Cardinali, D.P.; Savio, I.; Pintos, A. Polysomnographic study of the effect of melatonin on sleep in elderly patients with chronic primary insomnia. Arch. Gerontol. Geriatr. 1999, 28, 85–98. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Srinivasan, V.; Brzezinski, A.; Brown, G.M. Melatonin and its analogs in insomnia and depression. J. Pineal Res. 2012, 52, 365–375. [Google Scholar] [CrossRef]
- Rajaratnam, S.M.; Polymeropoulos, M.H.; Fisher, D.M.; Roth, T.; Scott, C.; Birznieks, G.; Klerman, E.B. Melatonin agonist tasimelteon (VEC-162) for transient insomnia after sleep-time shift: Two randomised controlled multicentre trials. Lancet 2009, 373, 482–491. [Google Scholar] [CrossRef]
- Mulchahey, J.J.; Goldwater, D.R.; Zemlan, F.P. A single blind, placebo controlled, across groups dose escalation study of the safety, tolerability, pharmacokinetics and pharmacodynamics of the melatonin analog β-methyl-6-chloromelatonin. Life Sci. 2004, 75, 1843–1856. [Google Scholar] [CrossRef]
- McKenna, J.T.; Christie, M.A.; Jeffrey, B.A.; McCoy, J.G.; Lee, E.; Connolly, N.P.; Ward, C.P.; Strecker, R.E. Chronic ramelteon treatment in a mouse model of Alzheimer’s disease. Arch. Ital. Biol. 2012, 150, 5–14. [Google Scholar] [PubMed]
- Ursing, C.; Hartter, S.; von Bahr, C.; Tybring, G.; Bertilsson, L.; Rojdmark, S. Does hepatic metabolism of melatonin affect the endogenous serum melatonin level in man? J. Endocrinol. Investig. 2002, 25, 459–462. [Google Scholar] [CrossRef]
- Hartter, S.; Grozinger, M.; Weigmann, H.; Roschke, J.; Hiemke, C. Increased bioavailability of oral melatonin after fluvoxamine coadministration. Clin. Pharmacol. Ther. 2000, 67, 1–6. [Google Scholar] [CrossRef]
- Hartter, S.; Nordmark, A.; Rose, D.M.; Bertilsson, L.; Tybring, G.; Laine, K. Effects of caffeine intake on the pharmacokinetics of melatonin, a probe drug for CYP1A2 activity. Br. J. Clin. Pharmacol. 2003, 56, 679–682. [Google Scholar] [CrossRef]
- Souetre, E.; Salvati, E.; Belugou, J.L.; de Galeani, B.; Krebs, B.; Ortonne, J.P.; Darcourt, G. 5-Methoxypsoralen increases the plasma melatonin levels in humans. J. Investig. Dermatol. 1987, 89, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Garde, E.; Micic, S.; Knudsen, K.; Angelo, H.R.; Wulf, H.C. 8-Methoxypsoralen increases daytime plasma melatonin levels in humans through inhibition of metabolism. Photochem. Photobiol. 1994, 60, 475–480. [Google Scholar] [CrossRef]
- Mauviard, F.; Raynaud, F.; Geoffriau, M.; Claustrat, B.; Pevet, P. 5-Methoxypsoralen inhibits 6-hydroxylation of melatonin in the rat. Biol. Signals 1995, 4, 32–41. [Google Scholar] [CrossRef]
- Weishaupt, J.H.; Bartels, C.; Polking, E.; Dietrich, J.; Rohde, G.; Poeggeler, B.; Mertens, N.; Sperling, S.; Bohn, M.; Huther, G.; et al. Reduced oxidative damage in ALS by high-dose enteral melatonin treatment. J. Pineal Res. 2006, 41, 313–323. [Google Scholar] [CrossRef]
- Chahbouni, M.; Escames, G.; Venegas, C.; Sevilla, B.; Garcia, J.A.; Lopez, L.C.; Munoz-Hoyos, A.; Molina-Carballo, A.; Acuña-Castroviejo, D. Melatonin treatment normalizes plasma pro-inflammatory cytokines and nitrosative/oxidative stress in patients suffering from Duchenne muscular dystrophy. J. Pineal Res. 2010, 48, 282–289. [Google Scholar] [CrossRef]
- Voordouw, B.C.; Euser, R.; Verdonk, R.E.; Alberda, B.T.; de Jong, F.H.; Drogendijk, A.C.; Fauser, B.C.; Cohen, M. Melatonin and melatonin-progestin combinations alter pituitary-ovarian function in women and can inhibit ovulation. J. Clin. Endocrinol. Metab. 1992, 74, 108–117. [Google Scholar] [PubMed]
- Nickkholgh, A.; Schneider, H.; Sobirey, M.; Venetz, W.P.; Hinz, U.; Pelzl le , H.; Gotthardt, D.N.; Cekauskas, A.; Manikas, M.; Mikalauskas, S.; et al. The use of high-dose melatonin in liver resection is safe: First clinical experience. J. Pineal Res. 2011, 50, 381–388. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Pandi-Perumal, S.R.; Niles, L.P. Melatonin and Its Receptors: Biological Function in Circadian Sleep-Wake Regulation. In Neurochemistry of Sleep and Wakefulness; Monti, J.M., Pandi-Perumal, S.R., Sinton, C.M., Eds.; Cambridge University Press: Cambridge, UK, 2008; pp. 283–314. [Google Scholar]
- Dagan, Y.; Zisapel, N.; Nof, D.; Laudon, M.; Atsmon, J. Rapid reversal of tolerance to benzodiazepine hypnotics by treatment with oral melatonin: A case report. Eur. Neuropsychopharmacol. 1997, 7, 157–160. [Google Scholar] [CrossRef]
- Garfinkel, D.; Zisapel, N.; Wainstein, J.; Laudon, M. Facilitation of benzodiazepine discontinuation by melatonin: A new clinical approach. Arch. Intern. Med. 1999, 159, 2456–2460. [Google Scholar] [CrossRef]
- Siegrist, C.; Benedetti, C.; Orlando, A.; Beltran, J.M.; Tuchscherr, L.; Noseda, C.M.; Brusco, L.I.; Cardinali, D.P. Lack of changes in serum prolactin, FSH, TSH, and estradiol after melatonin treatment in doses that improve sleep and reduce benzodiazepine consumption in sleep-disturbed, middle-aged, and elderly patients. J. Pineal Res. 2001, 30, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Kunz, D.; Bineau, S.; Maman, K.; Milea, D.; Toumi, M. Benzodiazepine discontinuation with prolonged-release melatonin: Hints from a German longitudinal prescription database. Expert Opin. Pharmacother. 2012, 13, 9–16. [Google Scholar] [CrossRef]
- Clay, E.; Falissard, B.; Moore, N.; Toumi, M. Contribution of prolonged-release melatonin and anti-benzodiazepine campaigns to the reduction of benzodiazepine and Z-drugs consumption in nine European countries. Eur. J. Clin. Pharmacol. 2013, 69, 1–10. [Google Scholar] [PubMed]
- Leger, D.; Laudon, M.; Zisapel, N. Nocturnal 6-sulfatoxymelatonin excretion in insomnia and its relation to the response to melatonin replacement therapy. Am. J. Med. 2004, 116, 91–95. [Google Scholar] [CrossRef]
- Zhdanova, I.V.; Wurtman, R.J.; Regan, M.M.; Taylor, J.A.; Shi, J.P.; Leclair, O.U. Melatonin treatment for age-related insomnia. J. Clin. Endocrinol. Metab. 2001, 86, 4727–4730. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.J.; Nutt, D.J.; Alford, C.; Argyropoulos, S.V.; Baldwin, D.S.; Bateson, A.N.; Britton, T.C.; Crowe, C.; Dijk, D.J.; Espie, C.A.; et al. British Association for Psychopharmacology consensus statement on evidence-based treatment of insomnia, parasomnias and circadian rhythm disorders. J. Psychopharmacol. 2010, 24, 1577–1601. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Furio, A.M.; Brusco, L.I. The use of chronobiotics in the resynchronization of the sleep/wake cycle. Therapeutical application in the early phases of Alzheimer’s disease. Recent Pat. Endocr. Metab. Immune Drug Discov. 2011, 5, 80–90. [Google Scholar] [CrossRef]
- Jean-Louis, G.; von Gizycki, H.; Zizi, F. Melatonin effects on sleep, mood, and cognition in elderly with mild cognitive impairment. J. Pineal Res. 1998, 25, 177–183. [Google Scholar] [CrossRef]
- Peck, J.S.; LeGoff, D.B.; Ahmed, I.; Goebert, D. Cognitive effects of exogenous melatonin administration in elderly persons: A pilot study. Am. J. Geriatr. Psychiatry 2004, 12, 432–436. [Google Scholar] [PubMed]
- Wade, A.G.; Ford, I.; Crawford, G.; McMahon, A.D.; Nir, T.; Laudon, M.; Zisapel, N. Efficacy of prolonged release melatonin in insomnia patients aged 55–80 years: Quality of sleep and next-day alertness outcomes. Curr. Med. Res. Opin. 2007, 23, 2597–2605. [Google Scholar] [CrossRef]
- Riemersma-van der Lek, R.F.; Swaab, D.F.; Twisk, J.; Hol, E.M.; Hoogendijk, W.J.; van Someren, E.J. Effect of bright light and melatonin on cognitive and noncognitive function in elderly residents of group care facilities: A randomized controlled trial. JAMA 2008, 299, 2642–2655. [Google Scholar] [CrossRef]
- Garzon, C.; Guerrero, J.M.; Aramburu, O.; Guzman, T. Effect of melatonin administration on sleep, behavioral disorders and hypnotic drug discontinuation in the elderly: A randomized, double-blind, placebo-controlled study. Aging Clin. Exp. Res. 2009, 21, 38–42. [Google Scholar] [CrossRef]
- Cazzola, R.; Rondanelli, M.; Faliva, M.; Cestaro, B. Effects of DHA-phospholipids, melatonin and tryptophan supplementation on erythrocyte membrane physico-chemical properties in elderly patients suffering from mild cognitive impairment. Exp. Gerontol. 2012, 47, 974–978. [Google Scholar] [CrossRef]
- Rondanelli, M.; Opizzi, A.; Faliva, M.; Mozzoni, M.; Antoniello, N.; Cazzola, R.; Savare, R.; Cerutti, R.; Grossi, E.; Cestaro, B. Effects of a diet integration with an oily emulsion of DHA-phospholipids containing melatonin and tryptophan in elderly patients suffering from mild cognitive impairment. Nutr. Neurosci. 2012, 15, 46–54. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cardinali, D.P.; Vigo, D.E.; Olivar, N.; Vidal, M.F.; Brusco, L.I. Melatonin Therapy in Patients with Alzheimer’s Disease. Antioxidants 2014, 3, 245-277. https://doi.org/10.3390/antiox3020245
Cardinali DP, Vigo DE, Olivar N, Vidal MF, Brusco LI. Melatonin Therapy in Patients with Alzheimer’s Disease. Antioxidants. 2014; 3(2):245-277. https://doi.org/10.3390/antiox3020245
Chicago/Turabian StyleCardinali, Daniel P., Daniel E. Vigo, Natividad Olivar, María F. Vidal, and Luis I. Brusco. 2014. "Melatonin Therapy in Patients with Alzheimer’s Disease" Antioxidants 3, no. 2: 245-277. https://doi.org/10.3390/antiox3020245
APA StyleCardinali, D. P., Vigo, D. E., Olivar, N., Vidal, M. F., & Brusco, L. I. (2014). Melatonin Therapy in Patients with Alzheimer’s Disease. Antioxidants, 3(2), 245-277. https://doi.org/10.3390/antiox3020245