




Transcriptomic Analysis Identifies Oxidative Stress-Related Hub Genes and Key Pathways in Sperm Maturation
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Considerations and Study Design
2.2. Participant Selection Criteria
2.3. Human Fibroblast Cultivation
2.4. Isolation and Characterization of Human Spermatogonial Stem Cells
2.5. Collection of Single Cells
2.6. Immunocytochemistry
2.7. Microarray Analysis
2.8. Collection of Single Cells by Per-Cell Basis
2.9. Top Genes Based on the Semantic Similarities of GO Terms
2.10. Construction of PPI Network
2.11. WGCNA
2.12. Models Set Up
2.13. Analysis of Human Metastatic and Cell–Cell Interactions
2.14. Development and Evaluation of Differentiation Models
2.15. Statistical Analysis
3. Results
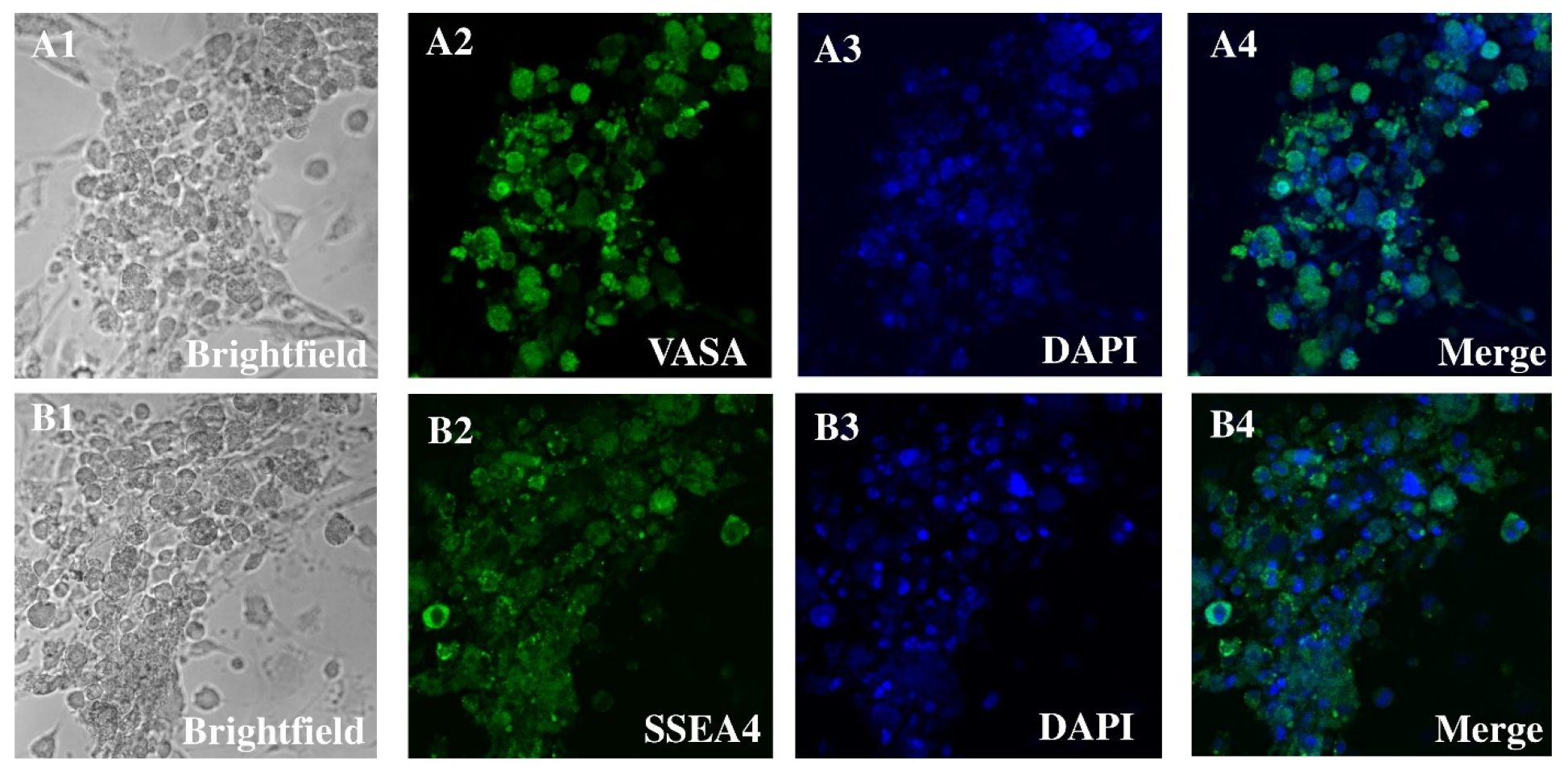
3.1. HSSCs Selection
3.2. Gene Expression in SSC Versus Fibroblast
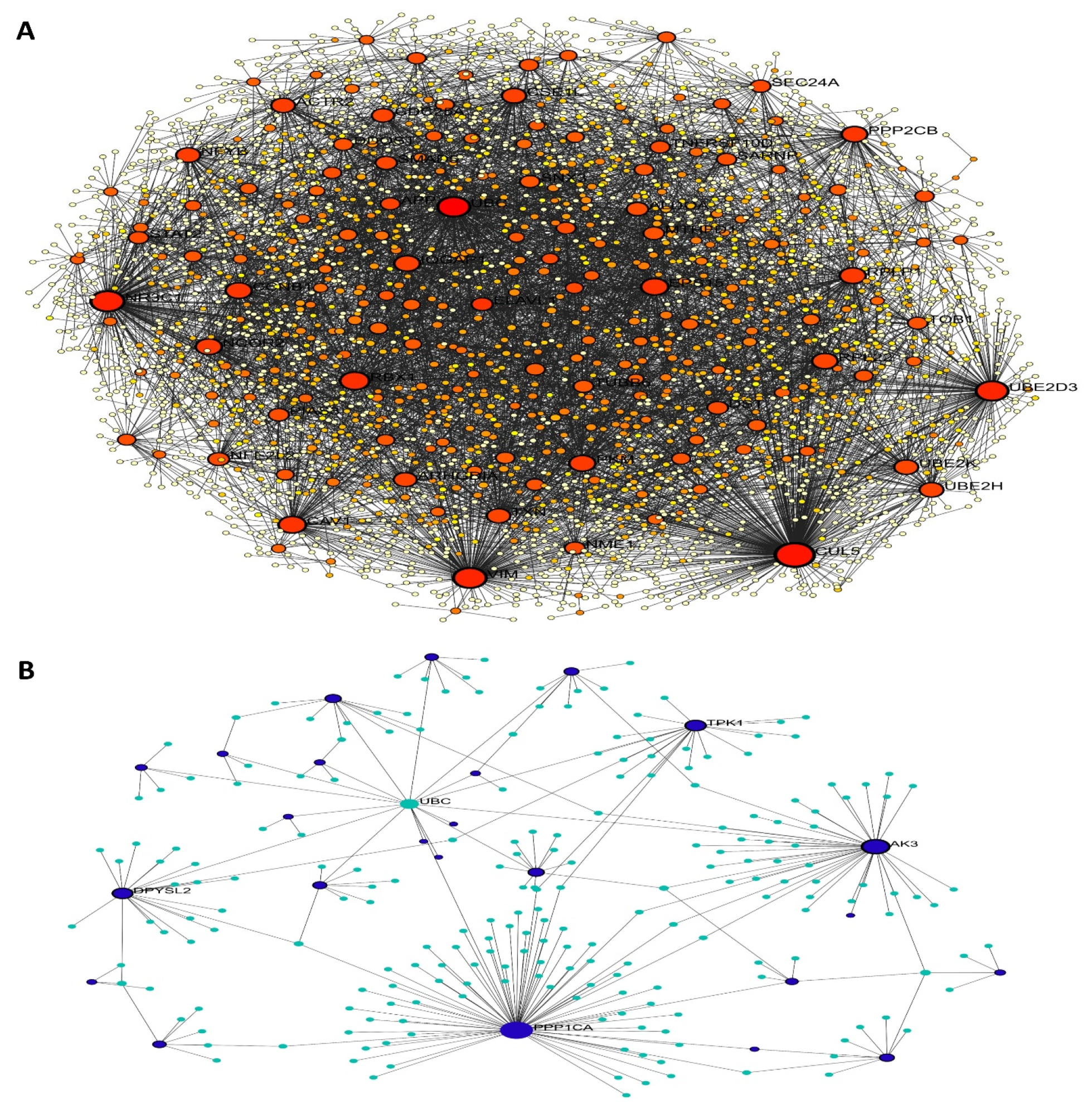
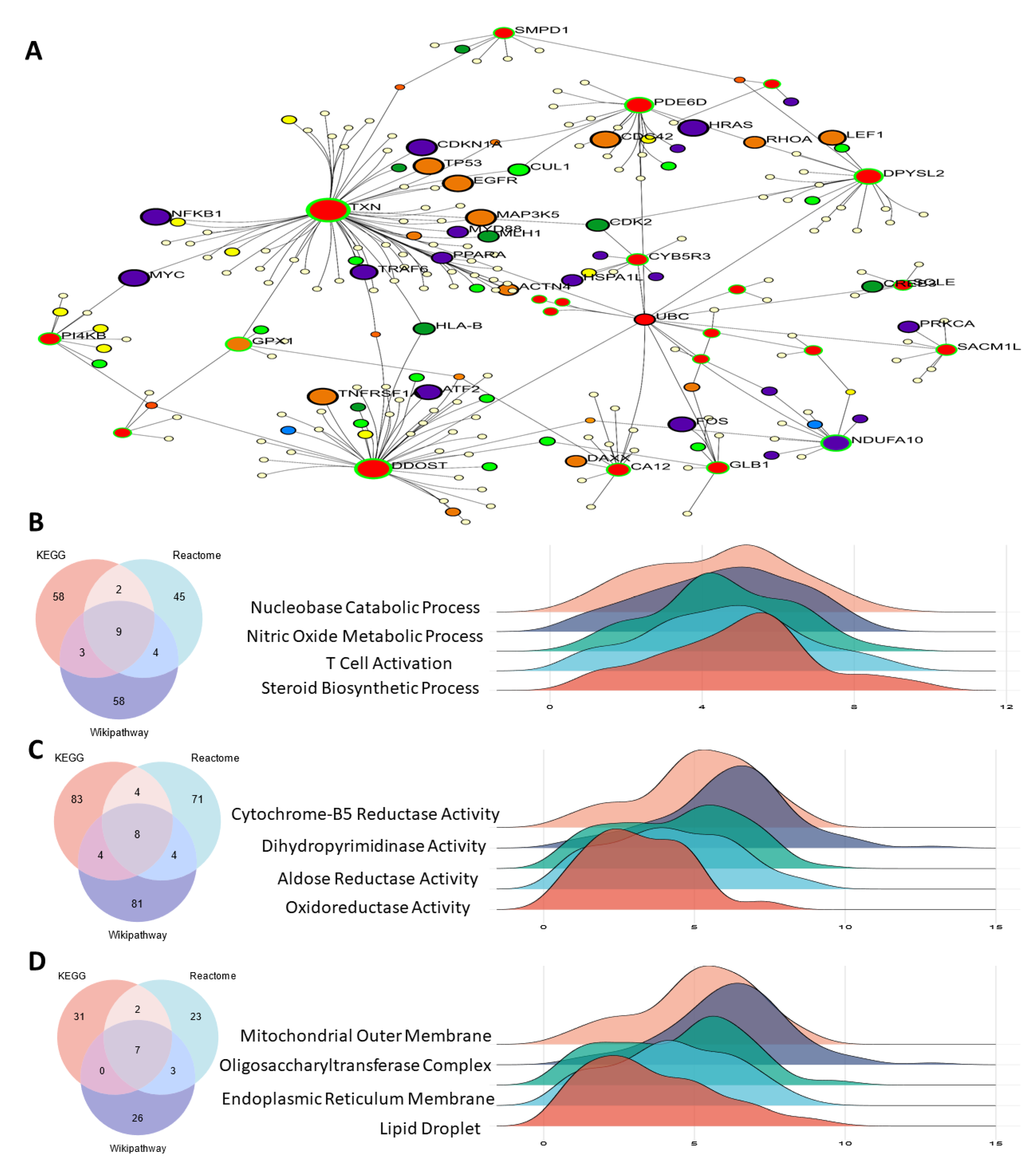
3.3. PPI Network of Oxidative Stress-Related Hub Genes
3.4. Functional Enrichment Analysis
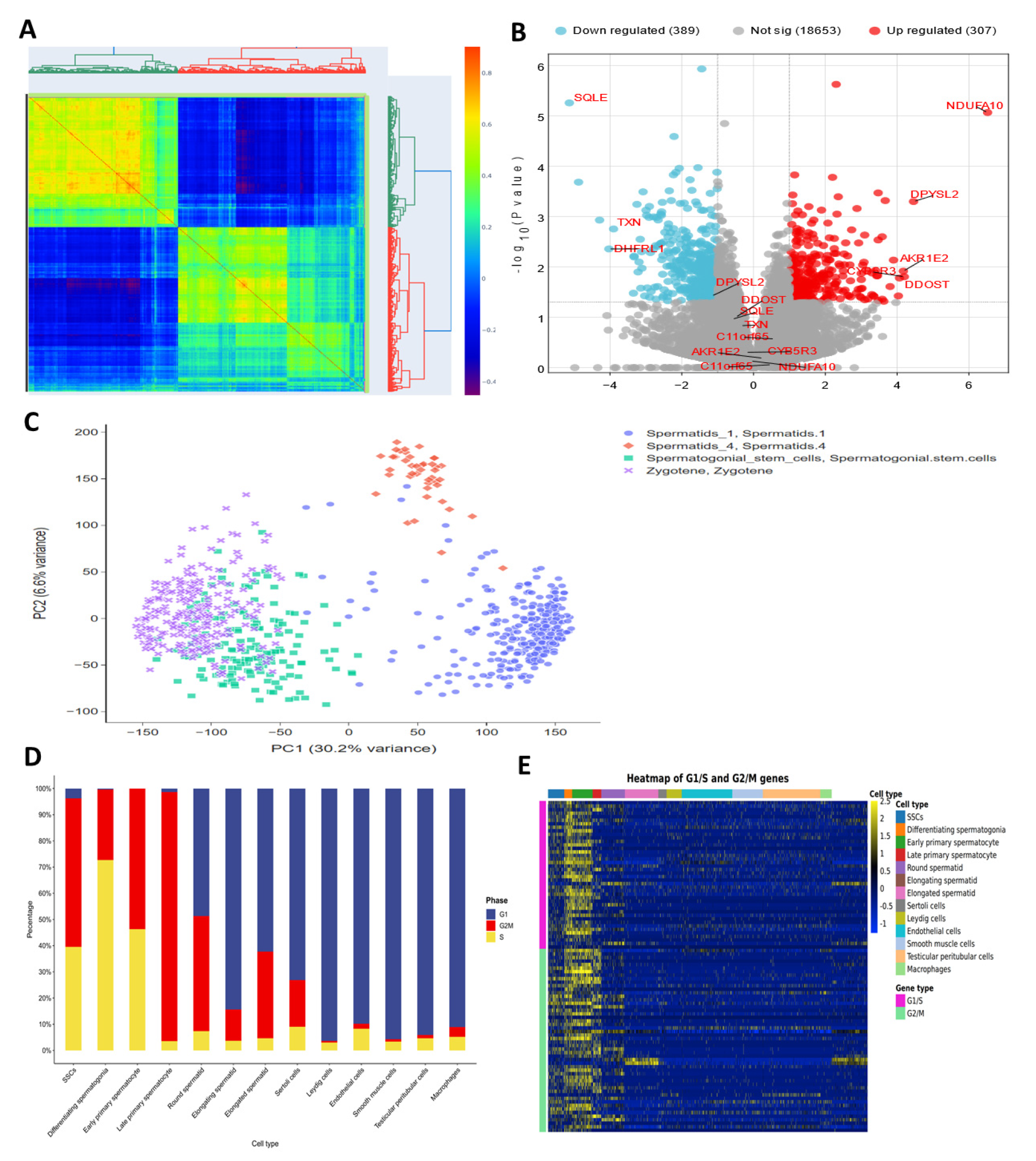
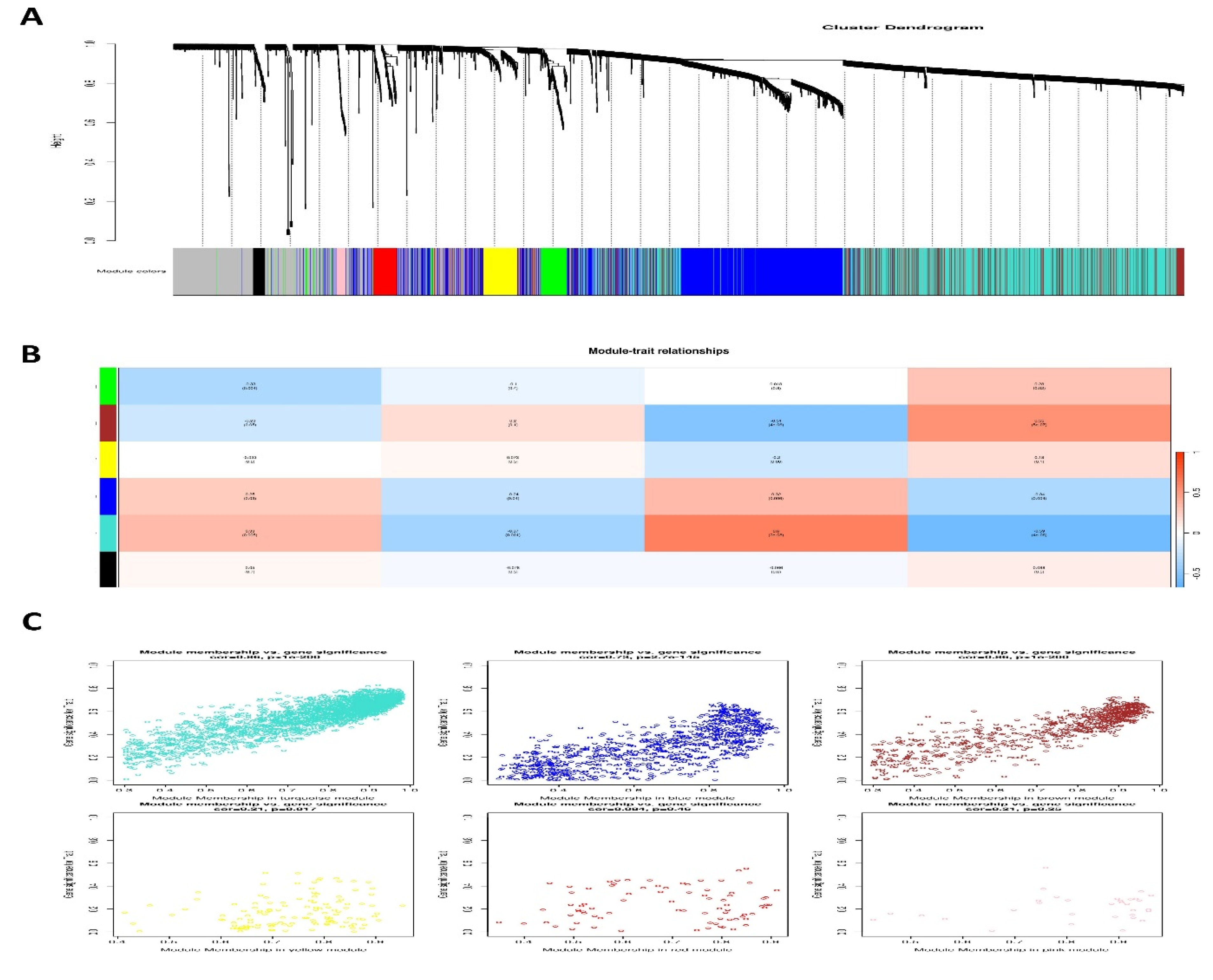
3.5. Weighted Gene Co-Expression Modules
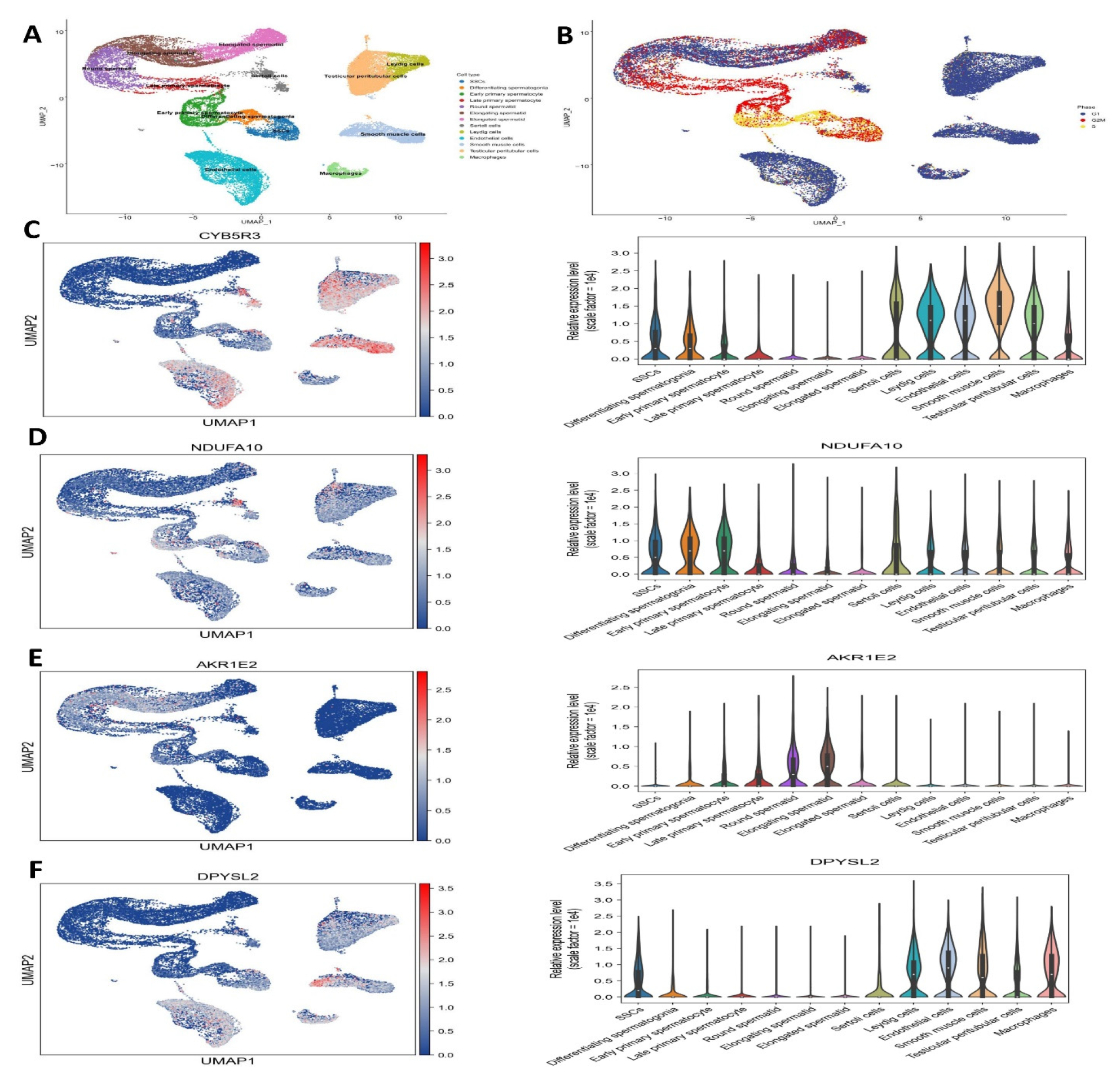
3.6. Single-Cell RNA Sequencing on Testicular Cells
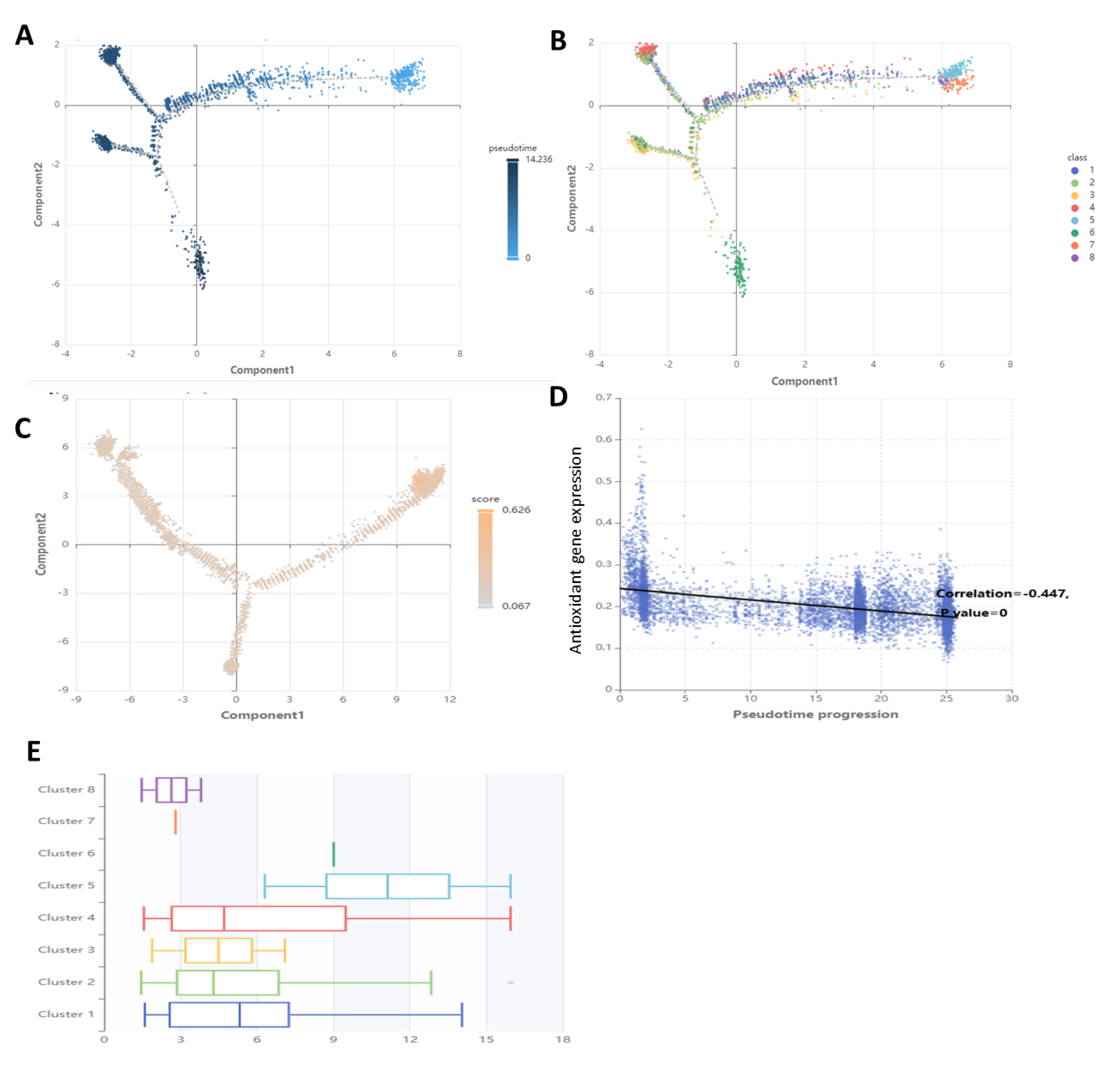
3.7. Performance of Antioxidant Gene Expression in SSCs Differentiation Models
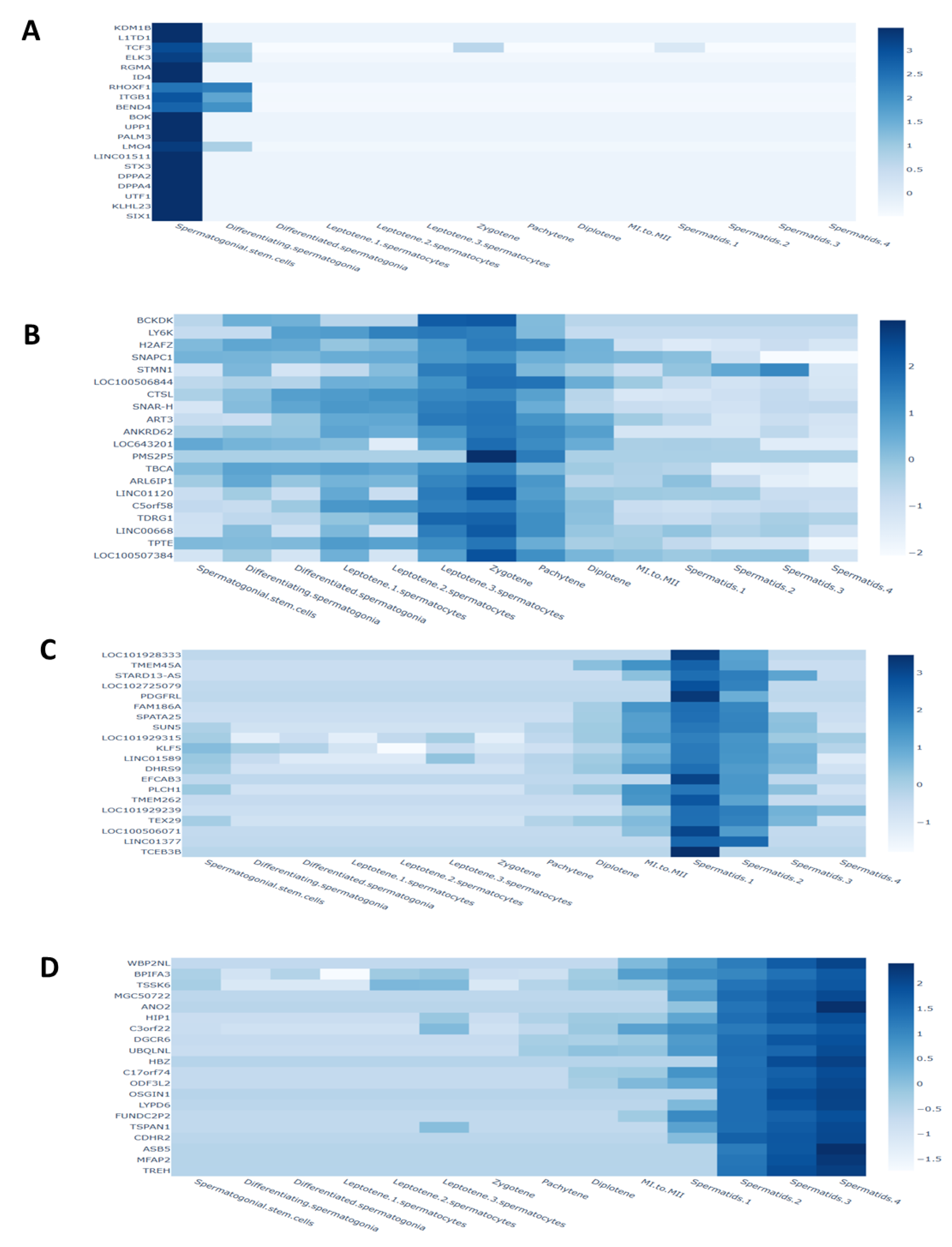
3.8. Analysis of Single-Cell RNA Sequencing for Antioxidant Gene Expression
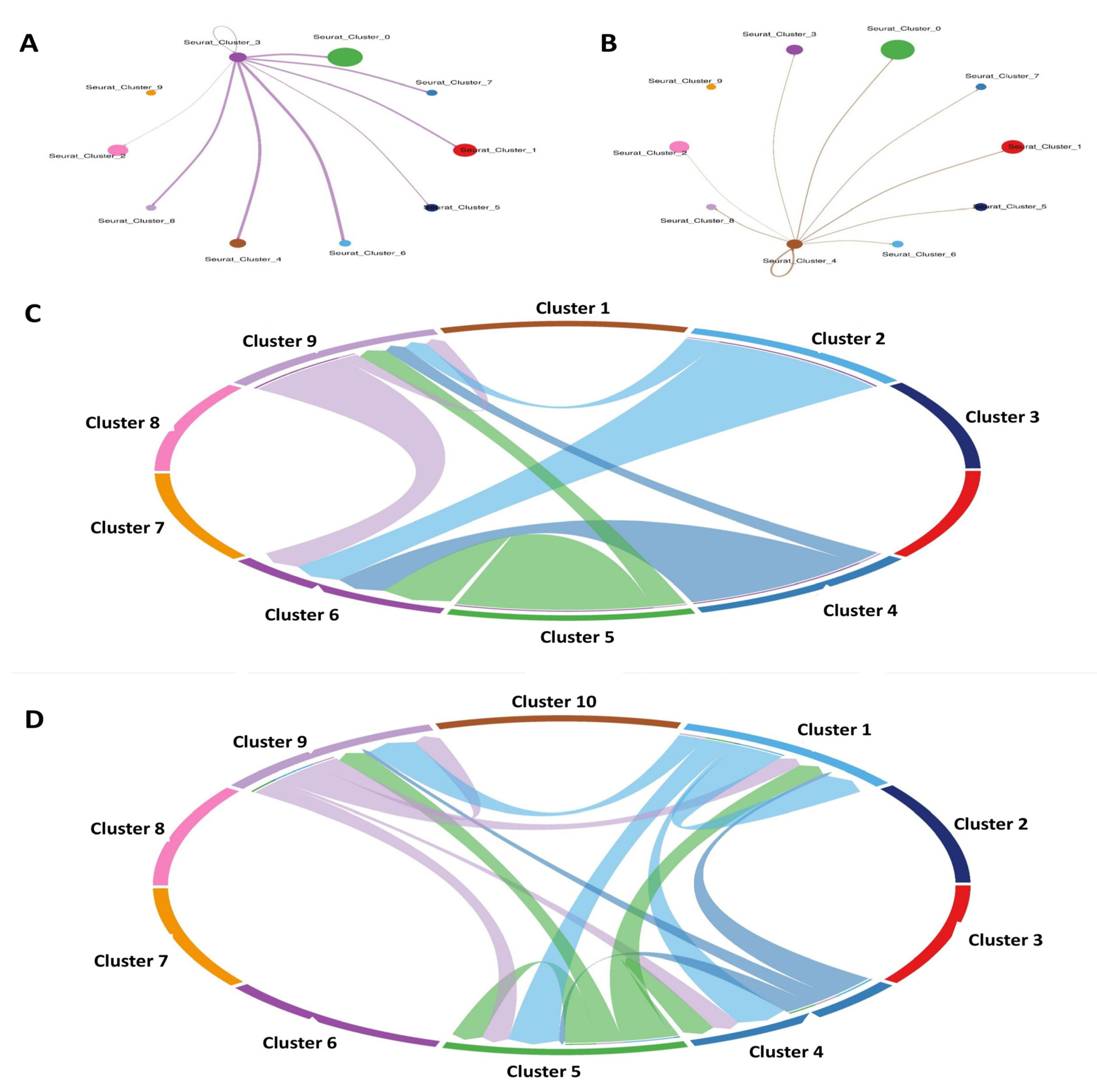
3.9. Scoring Cell–Cell Interactions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Azizi, H.; Karoii, D.H.; Skutella, T. Whole Exome Sequencing and In Silico Analysis of Human Sertoli in Patients with Non-Obstructive Azoospermia. Int. J. Mol. Sci. 2022, 23, 12570. [Google Scholar] [CrossRef]
- Amirian, M.; Azizi, H.; Hashemi Karoii, D.; Skutella, T. VASA protein and gene expression analysis of human non-obstructive azoospermia and normal by immunohistochemistry, immunocytochemistry, and bioinformatics analysis. Sci. Rep. 2022, 12, 17259. [Google Scholar] [CrossRef]
- Kawahara, T.; Suzuki, S.; Nakagawa, T.; Kamo, Y.; Kanouchi, M.; Fujita, M.; Hattori, M.; Suzuki, A.; Tanemura, K.; Yoshida, S.; et al. Age-Dependent Clonal Expansion of Non–Sperm-Forming Spermatogonial Stem Cells in Mouse Testes. Aging Cell 2025, 21, e70019. [Google Scholar] [CrossRef]
- Qamar, A.Y.; Hussain, T.; Rafique, M.K.; Bang, S.; Tanga, B.M.; Seong, G.; Fang, X.; Saadeldin, I.M.; Cho, J. The role of stem cells and their derived extracellular vesicles in restoring female and male fertility. Cells 2021, 10, 2460. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kan, Y.; Zhong, Y.; Jawad, M.; Wei, W.; Gu, K.; Gui, L.; Li, M. Generation of a normal long-term-cultured Chinese hook snout carp spermatogonial stem cell line capable of sperm production in vitro. Biology 2022, 11, 1069. [Google Scholar] [CrossRef] [PubMed]
- Diao, L.; Turek, P.J.; John, C.M.; Fang, F.; Reijo Pera, R.A. Roles of spermatogonial stem cells in spermatogenesis and fertility restoration. Front. Endocrinol. 2022, 13, 895528. [Google Scholar] [CrossRef] [PubMed]
- Jabari, A.; Gholami, K.; Khadivi, F.; Koruji, M.; Amidi, F.; Gilani, M.A.S.; Mahabadi, V.P.; Nikmahzar, A.; Salem, M.; Movassagh, S.A.; et al. In vitro complete differentiation of human spermatogonial stem cells to morphologic spermatozoa using a hybrid hydrogel of agarose and laminin. Int. J. Biol. Macromol. 2023, 235, 123801. [Google Scholar] [CrossRef]
- Karoii, H.; Azizi, H. OCT4 protein and gene expression analysis in the differentiation of spermatogonia stem cells into neurons by immunohistochemistry, immunocytochemistry, and bioinformatics analysis. Stem Cell Rev. Rep. 2023, 19, 1828–1844. [Google Scholar] [CrossRef]
- Zeitlinger, J.; Roy, S.; Ay, F.; Mathelier, A.; Medina-Rivera, A.; Mahony, S.; Sinha, S.; Ernst, J. Perspective on recent developments and challenges in regulatory and systems genomics. arXiv 2024, arXiv:2411.04363. [Google Scholar] [CrossRef]
- Karoii, H.; Azizi, H. Functions and mechanism of noncoding RNA in regulation and differentiation of male mammalian reproduction. Cell Biochem. Funct. 2023, 41, 767–778. [Google Scholar] [CrossRef]
- Cheuquemán, C.; Maldonado, R. Non-coding RNAs and chromatin: Key epigenetic factors from spermatogenesis to transgenerational inheritance. Biol. Res. 2021, 54, 41. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Yeh, Y.H.; Maezawa, S.; Nakagawa, T.; Yoshida, S.; Namekawa, S.H. PRC1 directs PRC2-H3K27me3 deposition to shield adult spermatogonial stem cells from differentiation. Nucleic Acids Res. 2024, 52, 2306–2322. [Google Scholar] [CrossRef]
- Munakata, Y.; Hu, M.; Kitamura, Y.; Dani, R.G.; Bynder, A.L.; Fritz, A.S.; Schultz, R.M.; Namekawa, S.H. Chromatin remodeler CHD4 establishes chromatin states required for ovarian reserve formation, maintenance and male germ cell survival. Nucleic Acids Res. 2025, 53, gkaf008. [Google Scholar] [CrossRef] [PubMed]
- Hashemi Karoii, D.; Azizi, H.; Darvari, M.; Qorbanee, A.; Hawezy, D.J. Identification of novel cytoskeleton protein involved in spermatogenic cells and sertoli cells of non-obstructive azoospermia based on microarray and bioinformatics analysis. BMC Med. Genom. 2025, 18, 19. [Google Scholar] [CrossRef] [PubMed]
- Karoii, H.; Azizi, H.; Skutella, T. Altered G-Protein Transduction Protein Gene Expression in the Testis of Infertile Patients with Nonobstructive Azoospermia. DNA Cell Biol. 2023, 42, 617–637. [Google Scholar] [CrossRef]
- Karoii, H.; Azizi, H.; Skutella, T. Microarray and in silico analysis of DNA repair genes between human testis of patients with nonobstructive azoospermia and normal cells. Cell Biochem. Funct. 2022, 40, 865–879. [Google Scholar] [CrossRef]
- Karoii, H.; Azizi, H.; Skutella, T. Integrating Microarray Data and Single-Cell RNA-Seq Reveals Key Gene Involved in Spermatogonia Stem Cell Aging. Int. J. Mol. Sci. 2024, 25, 11653. [Google Scholar] [CrossRef]
- Azizi, H.; Karoii, D.H.; Skutella, T. Clinical management, differential diagnosis, follow-up and biomarkers of infertile men with nonobstructive azoospermia. Transl. Androl. Urol. 2024, 13, 359–362. [Google Scholar] [CrossRef]
- Davoodi Nik, B.; Hashemi Karoii, D.; Favaedi, R.; Ramazanali, F.; Jahangiri, M.; Movaghar, B.; Shahhoseini, M. Differential expression of ion channel coding genes in the endometrium of women experiencing recurrent implantation failures. Sci. Rep. 2024, 14, 19822. [Google Scholar] [CrossRef]
- Karoii, D.H.; Abroudi, A.S.; Darvar, M.; Djamali, M.; Azizi, H.; Skutella, T. Identification of novel long non-coding RNA involved in Sertoli cell of non-obstructive azoospermia based on microarray and bioinformatics analysis. Genomics 2025, 117, 111046. [Google Scholar] [CrossRef]
- Karoii, H.; Azizi, H. A review of protein-protein interaction and signaling pathway of Vimentin in cell regulation, morphology and cell differentiation in normal cells. J. Recept. Signal Transduct. 2022, 42, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Cicoli, M.; Conlon, J.P.; Maharana, A.; Parameswaran, S.; Quevedo, F.; Zavala, I. String cosmology: From the early universe to today. Phys. Rep. 2024, 1059, 1–155. [Google Scholar] [CrossRef]
- Franz, M.; Lopes, C.T.; Fong, D.; Kucera, M.; Cheung, M.; Siper, M.C.; Huck, G.; Dong, Y.; Sumer, O.; Bader, G.D. Cytoscape. js 2023 update: A graph theory library for visualization and analysis. Bioinformatics 2023, 39, btad031. [Google Scholar] [CrossRef] [PubMed]
- Hashemi Karoii, D.; Baghaei, H.; Abroudi, A.S.; Djamali, M.; Hasani Mahforoozmahalleh, Z.; Azizi, H.; Skutella, T. Alteration of the metabolite interconversion enzyme in sperm and Sertoli cell of non-obstructive azoospermia: A microarray data and in-silico analysis. Sci. Rep. 2024, 14, 25965. [Google Scholar] [CrossRef] [PubMed]
- Hashemi Karoii, D.; Bavandi, S.; Djamali, M.; Abroudi, A.S. Exploring the interaction between immune cells in the prostate cancer microenvironment combining weighted correlation gene network analysis and single-cell sequencing: An integrated bioinformatics analysis. Discov. Oncol. 2024, 15, 513. [Google Scholar] [CrossRef]
- Karoii, D.H.; Azizi, H.; Amirian, M. Signaling pathways and protein–protein interaction of vimentin in invasive and migration cells: A review. Cell Reprogram. 2022, 24, 165–174. [Google Scholar] [CrossRef]
- Karoii, D.H.; Azizi, H.; Skutella, T. Whole transcriptome analysis to identify non-coding RNA regulators and hub genes in sperm of non-obstructive azoospermia by microarray, single-cell RNA sequencing, weighted gene co-expression network analysis, and mRNA-miRNA-lncRNA interaction analysis. BMC Genom. 2024, 25, 583. [Google Scholar] [CrossRef]
- Niazi Tabar, A.; Azizi, H.; Hashemi Karoii, D.; Skutella, T. Testicular Localization and Potential Function of Vimentin Positive Cells during Spermatogonial Differentiation Stages. Animals 2022, 12, 268. [Google Scholar] [CrossRef]
- Shams, A.A.; Vesal, S.; Karoii, D.H.; Vesali, S.; Alizadeh, A.; Shahhoseini, M. Paternal trans fatty acid and vitamin E diet affect the expression pattern of androgen signaling pathway genes in the testis of rat offspring. Theriogenology 2025, 231, 1–10. [Google Scholar] [CrossRef]
- Gao, X.M.; Zhou, X.H.; Jia, M.W.; Wang, X.Z.; Liu, D. Identification of key genes in sepsis by WGCNA. Prev. Med. 2023, 172, 107540. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, X.; Liu, S.; Zhao, C.; Miao, Y.; Jin, L.; Wang, D.; Zhou, L. Cyp17a1 is required for female sex determination and male fertility by regulating sex steroid biosynthesis in fish. Endocrinology 2021, 162, bqab205. [Google Scholar] [CrossRef]
- Staicu, F.D.; Martínez-Soto, J.C.; Canovas, S.; Matás, C. Nitric oxide-targeted protein phosphorylation during human sperm capacitation. Sci. Rep. 2021, 11, 20979. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Matsui, Y. Metabolic control of germline formation and differentiation in mammals. Sexual Dev. 2022, 16, 388–403. [Google Scholar] [CrossRef] [PubMed]
- Gualtieri, R.; Kalthur, G.; Barbato, V.; Longobardi, S.; Di Rella, F.; Adiga, S.K.; Talevi, R. Sperm oxidative stress during in vitro manipulation and its effects on sperm function and embryo development. Antioxidants 2021, 10, 1025. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Jasso, D.E.; López-Guzmán, S.F.; Bermúdez-Cruz, R.M.; Oviedo, N. Novel aspects of cAMP-response element modulator (CREM) role in spermatogenesis and male fertility. Int. J. Mol. Sci. 2023, 24, 12558. [Google Scholar] [CrossRef]
- Ardekani, O.S.; Letafati, A.; Dehkordi, S.E.; Farahani, A.V.; Bahari, M.; Mahdavi, B.; Ariamand, N.; Taghvaei, M.; Kohkalani, M.; Pirkooh, A.A.; et al. From infection to infertility: A review of the role of human papillomavirus-induced oxidative stress on reproductive health and infertility. Eur. J. Med. Res. 2025, 30, 339. [Google Scholar] [CrossRef]
- Yi, Z.Y.; Liang, Q.X.; Zhou, Q.; Yang, L.; Meng, Q.R.; Li, J.; Lin, Y.H.; Cao, Y.P.; Zhang, C.H. Maternal total sleep deprivation causes oxidative stress and mitochondrial dysfunction in oocytes associated with fertility decline in mice. PLoS ONE 2024, 19, e0306152. [Google Scholar] [CrossRef]
- Darwish, S.F.; Moustafa, Y.M.; Abdel Mageed, S.S.; Hassan, G.S.; Mangoura, S.A.; Aly, S.H.; Mansour, M.A.; Raouf, A.A.; Sallam, A.A.M.; Fawzi, S.F.; et al. Insecticides and testicular health: Mechanisms of injury and protective natural products. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2025, 2, 1–31. [Google Scholar] [CrossRef]
- Abdulle, A.E.; Diercks, G.F.; Feelisch, M.; Mulder, D.J.; Goor, H.V. The Role of Oxidative Stress in the Development of Systemic Sclerosis Related Vasculopathy. Front. Physiol 2018, 9, 1177. [Google Scholar] [CrossRef]
- Divvela, S.S.K.; Gallorini, M.; Gellisch, M.; Patel, G.D.; Saso, L.; Brand-Saberi, B. Navigating redox imbalance: The role of oxidative stress in embryonic development and long-term health outcomes. Front. Cell Dev. Biol. 2025, 13, 1521336. [Google Scholar] [CrossRef]
- Schreiber, M.; Ghanem, N.; Rahimi, M.; Habermann, H.; Tholen, E.; Hoelker, M.; Held-Hoelker, E. Developmental stage specific effect of Mito-TEMPO on the expression outline of antioxidant genes, ROS balance and cryo-resilience of bovine IVP embryos. Theriogenology 2025, 236, 105–113. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhang, S.; Nayak, R.; Věchtová, P.; Schumacher, F.; Linhartová, P.; Gazo, I.; Linhartová, Z.; Waghmare, S.G.; Kleuser, B.; et al. Fertilization by short-term stored sperm alters DNA methylation patterns at single-base resolution in common carp (Cyprinus carpio) embryos. Rev. Fish Biol. Fish. 2024, 34, 1167–1187. [Google Scholar] [CrossRef]
- Kaltsas, A.; Dimitriadis, F.; Zachariou, A.; Sofikitis, N.; Chrisofos, M. Phosphodiesterase Type 5 Inhibitors in Male Reproduction: Molecular Mechanisms and Clinical Implications for Fertility Management. Cells 2025, 14, 120. [Google Scholar] [CrossRef]
- Xie, C.T.; Zhang, H.L.; Li, Y.; Li, Q.; Wen, Y.X.; Liu, J.Y.; Han, F. Single-cell RNA-seq and pathological phenotype reveal the functional atlas and precise roles of Sox30 in testicular cell development and differentiation. Cell Death Dis. 2025, 16, 110. [Google Scholar] [CrossRef] [PubMed]
- Dou, W.; Sun, B.; Miao, Y.; Huang, D.; Xiao, J. Single-cell transcriptome sequencing reveals Wolbachia-mediated modification in early stages of Drosophila spermatogenesis. Proc. R. Soc. B 2023, 290, 20221963. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, Z.; Zhu, M.; Yu, W.; Mao, W.; Mao, D.; Wang, F.; Wan, Y. Histone lactylation regulates early embryonic development through m6A methyltransferase METTL3 in goats. Int. J. Biol. Macromol. 2025, 309, 142858. [Google Scholar] [CrossRef] [PubMed]
- Talibova, G.; Bilmez, Y.; Ozturk, S. DNA double-strand break repair in male germ cells during spermatogenesis and its association with male infertility development. DNA Repair 2022, 118, 103386. [Google Scholar] [CrossRef]
- Lee, S.H.; Rinaudo, P.F. Metabolic Regulation of preimplantation Embryo Development in vivo and in vitro: Molecular Mechanisms and insights. Biochem. Biophys. Res. Commun. 2024, 726, 150256. [Google Scholar] [CrossRef]
- Newman, H.; Catt, S.; Vining, B.; Vollenhoven, B.; Horta, F. DNA repair and response to sperm DNA damage in oocytes and embryos, and the potential consequences in ART: A systematic review. Mol. Hum. Reprod. 2022, 28, gaab071. [Google Scholar] [CrossRef]
- Yang, Q.; Xie, Y.; Pan, B.; Cheng, Y.; Zhu, Y.; Fei, X.; Li, X.; Yu, J.; Chen, Z.; Li, J.; et al. The Expression and Epigenetic Characteristics of the HSF2 Gene in Cattle-Yak and the Correlation with Its Male Sterility. Animals 2024, 14, 1410. [Google Scholar] [CrossRef]
- Venditti, M.; Ben Rhouma, M.; Romano, M.Z.; Messaoudi, I.; Reiter, R.J.; Minucci, S. Altered expression of DAAM1 and PREP induced by cadmium toxicity is counteracted by melatonin in the rat testis. Genes 2021, 12, 1016. [Google Scholar] [CrossRef]
- Avena, M.V.; Funes, A.K.; Monclus, M.Á.; Boarelli, P.V.; Barbisan, L.F.; Bernal-López, M.R.; Gómez-Huelgas, R.; Lancellotti, T.E.S.; Fornés, M.W. Cholesterol and SREBP2 Dynamics During Spermatogenesis Stages in Rabbits: Effects of High-Fat Diet and Protective Role of Extra Virgin Olive Oil. Int. J. Mol. Sci. 2025, 26, 4062. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shakeri Abroudi, A.; Azizi, H.; Qadir, V.A.; Djamali, M.; Alsaffar, M.F.; Skutella, T. Transcriptomic Analysis Identifies Oxidative Stress-Related Hub Genes and Key Pathways in Sperm Maturation. Antioxidants 2025, 14, 936. https://doi.org/10.3390/antiox14080936
Shakeri Abroudi A, Azizi H, Qadir VA, Djamali M, Alsaffar MF, Skutella T. Transcriptomic Analysis Identifies Oxidative Stress-Related Hub Genes and Key Pathways in Sperm Maturation. Antioxidants. 2025; 14(8):936. https://doi.org/10.3390/antiox14080936
Chicago/Turabian StyleShakeri Abroudi, Ali, Hossein Azizi, Vyan A. Qadir, Melika Djamali, Marwa Fadhil Alsaffar, and Thomas Skutella. 2025. "Transcriptomic Analysis Identifies Oxidative Stress-Related Hub Genes and Key Pathways in Sperm Maturation" Antioxidants 14, no. 8: 936. https://doi.org/10.3390/antiox14080936
APA StyleShakeri Abroudi, A., Azizi, H., Qadir, V. A., Djamali, M., Alsaffar, M. F., & Skutella, T. (2025). Transcriptomic Analysis Identifies Oxidative Stress-Related Hub Genes and Key Pathways in Sperm Maturation. Antioxidants, 14(8), 936. https://doi.org/10.3390/antiox14080936