

The Mitochondrial Permeability Transition Pore in Platelets: Mechanisms, Physiological Roles, and Therapeutic Perspectives


Abstract
1. Introduction
2. Biological Background: Platelet Mitochondria
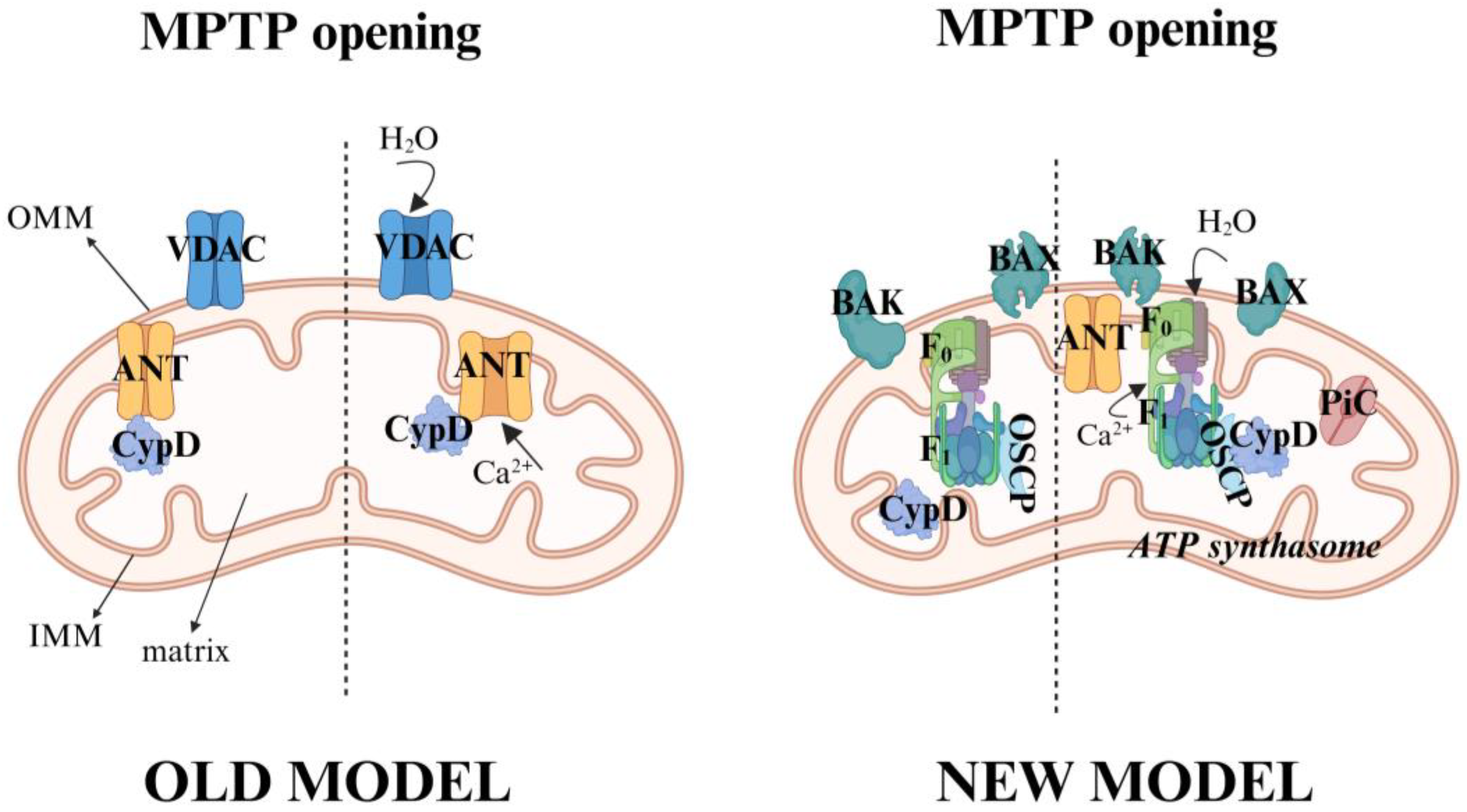
3. Mitochondrial Permeability Transition Pore (mPTP) in Platelets
3.1. Molecular Composition and Regulation
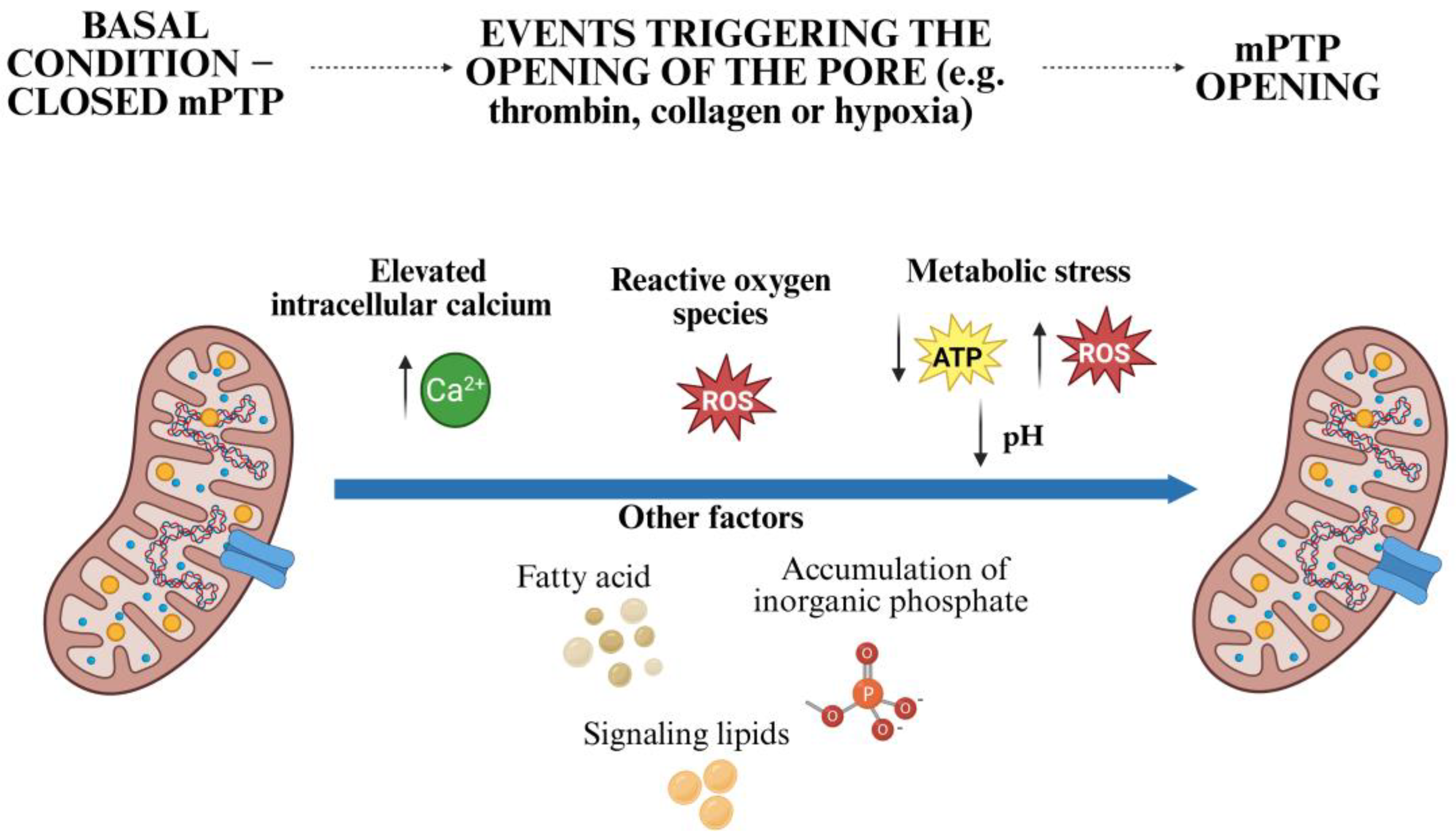
3.2. Triggers of mPTP Opening in Platelets
- (a)
- Elevated intracellular calcium: platelet activation by agonists such as thrombin, collagen, or adenosine diphosphate (ADP) leads to a rapid increase in cytosolic calcium, some of which is sequestered in the mitochondria via the MCU. Excessive mitochondrial calcium accumulation promotes opening of the mPTP [22,38].
- (b)
- (c)
- Persistent metabolic stress: pathological conditions such as ischemia reperfusion or sepsis lead to ATP depletion, intracellular acidosis, and further ROS production, which promotes the opening of the mPTP [32].
- (d)
- Other Factors: fatty acids, the accumulation of inorganic phosphate, and potentially certain signaling lipids can also sensitize the pore [25].
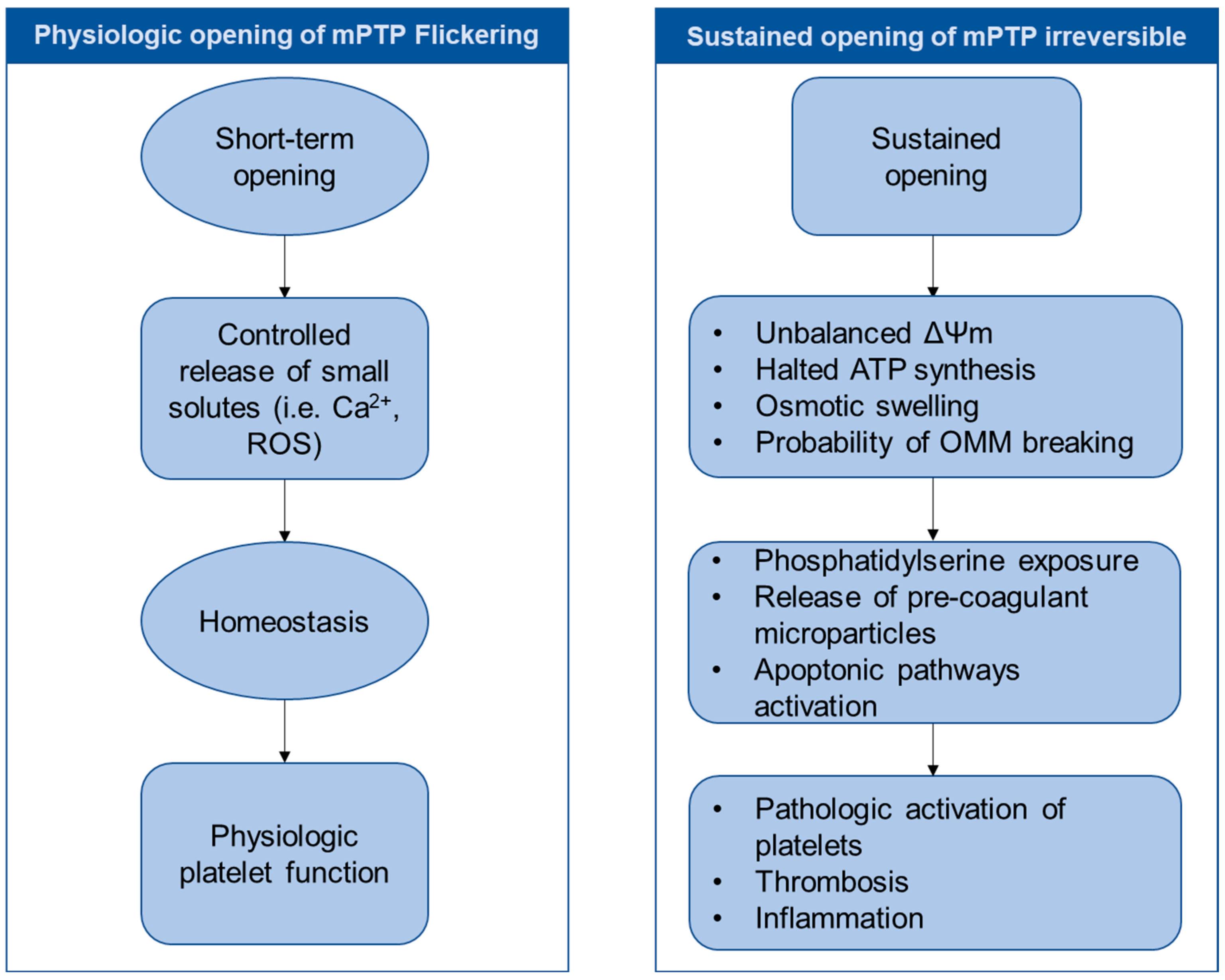
3.3. Unique Aspects of mPTP Behavior in Platelets
4. Mechanisms of mPTP Opening in Platelets
4.1. Mitochondrial Calcium Overload
4.2. Oxidative Stress and Reactive Oxygen Species (ROS)
4.3. Role of Cyclophilin D (CypD)
4.4. Energetic Stress and ATP Depletion
4.5. Involvement of the F1F0-ATP Synthase Complex
5. Physiological and Pathological Roles of mPTP in Platelets
5.1. Physiological Roles: Beyond Cell Death Signaling?
5.2. mPTP Opening and Platelet Clearance
- (a)
- Mitochondrial disfunction: structural changes in mitochondria, the collapse of ΔΨm, ATP depletion, and increased ROS production signal irreversible damage [7]. Swelling and fragmentation of the mitochondria are also evident.
- (b)
- (c)
- Microparticle shedding: platelets undergoing apoptosis-like changes can release microparticles that are enriched in procoagulant lipids and proteins and can contribute to thrombin generation [40].
5.3. Pathological Effects of Dysregulated mPTP Opening
- (a)
- Cardiovascular Diseases: In acute coronary syndromes (ACSs) and atherosclerosis, platelets exhibit increased mitochondrial ROS production, enhanced sensitivity to mPTP opening, and a procoagulant phenotype [6]. This increased mPTP activity promotes platelet hyperreactivity, contributes to thrombosis and can favor vascular occlusion. Targeting mPTP opening with pharmacological agents has shown promise in experimental models to reduce thrombus formation without impairing basal hemostasis [52].
- (b)
- Diabetes Mellitus: Platelets from patients with diabetes have mitochondrial dysfunction characterized by increased ROS, mitochondrial calcium overload, and a lower threshold for mPTP opening [53]. These mitochondrial abnormalities contribute to platelet hyperactivity, exacerbate endothelial damage, and may accelerate the development of vascular complications.
- (c)
- Sepsis: In sepsis, systemic inflammation and oxidative stress have a strong impact on platelet mitochondria. Enhanced mPTP opening can lead to platelet exhaustion, increased clearance, and thrombocytopenia and contributes to disseminated intravascular coagulation (DIC) through the release of procoagulant microparticles [54]. Paradoxically, the initial hyperactivation of platelets in sepsis may also be related to mitochondrial signaling before the onset of exhaustion.
- (d)
- Neurodegenerative Diseases: Emerging evidence links systemic mitochondrial dysfunction to neurodegenerative disorders such as Alzheimer’s and Parkinson’s disease. Platelet mitochondrial dysfunction, including dysregulated mPTP activity and altered redox state, may serve as a biomarker for systemic oxidative stress and mitochondrial impairment in these diseases [55].
- (e)
- Cancer and Inflammation: Platelets play a complex role in carcinogenesis (e.g., promoting metastasis) and in chronic inflammatory diseases. Research is ongoing but mitochondrial function, redox signaling, and potentially mPTP dysregulation likely influence platelet interactions with tumor and immune cells, contributing to these pathologies [56]. Targeting platelet mitochondria could offer new therapeutic opportunities in these contexts.
6. Targeting the mPTP in Platelets: Therapeutic Opportunities
6.1. Cyclophilin D Inhibitors: The Leading Strategy
- (a)
- Cyclosporin A (CsA): CsA inhibits mPTP opening by binding to CypD and preventing its interaction with putative pore components, thereby stabilizing the mitochondrial inner membrane and preserving mitochondrial integrity [62]. This preserves ΔΨm, limits ATP depletion and reduces the release of pro-apoptotic factors. CsA can also enhance the mitochondrial calcium-buffering capacity in several diseases [63].Preclinical studies support the cardioprotective effect of CsA and its analogue Sanglifehrin A in an ischemic myocardial infarction model [59,64,65,66,67,68]. In addition, CsA reduces the liver damage after ischemia–reperfusion injury [69], mitigates the stroke outcome, and improves mitochondrial function after transient middle cerebral artery occlusion [70]. However, a meta-analysis of randomized controlled trials showed that the administration of CsA does not protect the heart from reperfusion injury in clinical patients with myocardial infarction [71]. This non-effect may be related to its controversial activity on platelets.It has been reported that CsA can enhance platelet procoagulant activity, intracellular calcium mobilization, and platelet aggregation in response to ADP [72,73]. In contrast, others showed a protective effect of CsA on platelet apoptosis, reduced PS exposure, and the loss of ΔΨm induced by strong agonists such as collagen and thrombin [10,39,74].Recently, a potential link between cellular metabolism and mPTP regulation has been uncovered [43]. Platelets rely heavily on both aerobic glycolysis and mitochondrial oxidative phosphorylation for energy production, and perturbations in these metabolic pathways can affect mitochondrial dynamics and functions. In particular, specific mitochondrial and glycolytic enzymes may modulate mPTP opening either directly, through structural interactions, or indirectly by altering the redox balance, calcium handling, or nucleotide availability (ATP/ADP ratio).This suggests that metabolic reprogramming could be a novel avenue for the therapeutic modulation of mPTP activity. Agents targeting key metabolic enzymes or regulators such as hexokinase II, pyruvate dehydrogenase, or mitochondrial complex I may provide a dual benefit by preserving mitochondrial function and reducing platelet hyperactivation. This area remains poorly understood in the context of platelet physiology. However, its potential to influence the interactions between CypD and mPTP through metabolic control should be further investigated as a complementary strategy to direct mPTP inhibition.
- (b)
- Non-immunosuppressive CsA derivatives: Agents such as Debio-025 and NIM811, analogs of CsA, have a CypD-inhibiting effect without significant immunosuppression. They have shown promise in preclinical models by reducing the ischemia–reperfusion injury damage during hepatic surgery [69], as well as brain and heart damage post-cardiac arrest [75,76,77]. Their direct effect on platelet function is poorly studied [78]; some studies mention a reduction in platelet counts and the modulation of platelet-derived growth factor receptors after treatment with Debio-025 [79]. These compounds are currently being investigated for their ability to protect platelet mitochondria in cardiovascular and metabolic diseases.
- (c)
- Molecules unrelated to CsA: Recently, novel small-molecule CypD inhibitors (F759 and F83236) that are structurally unrelated to CsA have been developed. These agents promote the loss of mitochondrial membrane potential, reduce procoagulant platelet formation and the clotting time, and reduce fibrin formation when stimulated by dual-agonists (convulxin plus thrombin) without modifying P-selectin expression and integrin αIIbβ3 activation. In contrast to CsA, they did not enhance ADP-induced platelet aggregation [80].
- (d)
- Sirtuin 3 (SIRT3) activators: Enhancing endogenous protective mechanisms represents an additional strategy. SIRT3, a mitochondrial NAD+-dependent deacetylase, inhibits mPTP opening by deacetylating CypD at lysine 166 [51]. Compounds such as honokiol, a natural polyphenol from the Magnolia plant, activate SIRT3 and have shown protective effects in preclinical cardiac and neuronal models by improving mitochondrial function and reducing oxidative stress [81]. Although direct studies on platelet activation by honokiol are lacking, affecting SIRT3 activity could theoretically stabilize mitochondrial function in platelets, reduce oxidative stress, and prevent inappropriate platelet activation. Further research is needed to clarify the specific effects of SIRT3 agents such as honokiol on platelet function.
6.2. Compounds Binding to the Translocator Protein (TSPO) Located on the Outer Mitochondrial Membrane
6.3. Mitochondria-Targeted Antioxidants: Combating Oxidative Stress
- (a)
- (b)
- Natural antioxidants: Several natural compounds with antioxidant properties inhibit platelet activation, potentially via mechanisms involving mPTP modulation. Icariin and gallic acid, which are known to prevent the downstream activation of mPTP [60,61,90], decrease ROS production and platelet activation [91,92]. In addition, a selenium-containing protein from selenium-enriched Spirulina platensis counteracts oxidative damage by regulating mPTP opening [93]. Finally, Schisandrin B, a lignan from Schisandra chinensis, inhibits mPTP opening and preserves cardiomyocytes from anthracycline-induced cardiotoxicity by maintaining mitochondrial integrity and reducing oxidative stress [94,95].
6.4. Modulators of Mitochondrial Calcium Handling: Reducing the Trigger
- (a)
- MCU inhibitors: The direct pharmacological blockade of MCU (e.g., Ru360 or newer small molecules) can attenuate calcium-induced mPTP opening in isolated mitochondria and cells [96,97]. Despite these promising experimental approaches, the systemic effects of MCU inhibition need to be carefully evaluated due to the fundamental role of mitochondrial calcium in cellular metabolism. A major translational challenge is the limited membrane permeability of classical MCU inhibitors such as Ru360 [98], which significantly hampers their clinical applicability.Newer compounds such as Ru265 show improved cell permeability and efficacy in preclinical models, particular a reduction in ischemic brain injury at low doses. However, high doses have been associated with severe side effects such as fatal convulsions [99]. Although promising, the safety and therapeutic window of this drug in vivo remain to be well defined. These limitations need to be considered when evaluating the feasibility of MCU as a therapeutic target.
- (b)
- Indirect modulation via cytosolic calcium: An alternative strategy to prevent mPTP opening is the indirect modulation of mitochondrial calcium uptake by targeting cytosolic calcium entry mechanisms. In particular, store-operated calcium entry (SOCE), which is mediated by STIM1-Orai1, is crucial for maintaining platelet calcium influx after depletion of the intracellular calcium store. Excessive SOCE activity during pathological activation contributes to mitochondrial calcium overload, which promotes mPTP opening and downstream platelet dysfunction. The genetic or pharmacological inhibition of STIM1 or Orai1 impairs platelet calcium signaling, reduces aggregation, and protects against thrombotic events, supporting the therapeutic potential of targeting these channels to prevent calcium-induced mitochondrial dysfunction in platelets [100,101,102,103]. The pharmacological inhibition of SOCE channels such as BTP-2 or SKF-96365 attenuates the mitochondrial calcium increase, preserves ΔΨm, and reduces platelet activation [104,105].
6.5. Modulators of the F1F0-ATPase: Enhancing Mitochondrial Function
- (a)
- Inhibitors potentially acting via ATP synthase modulation: 1,2,3-triazole derivatives inhibit platelet aggregation induced by ADP or collagen [106], and 1,5-disubstituted-1,2,3-triazoles attenuate mPTP opening and reduce oxidative stress, thereby protecting cardiovascular cells from damage [107,108].new class of inhibitors of the F1F0-ATPase complex, the 1,3,8-Triazaspiro [4.5] decane derivatives, targeting the c subunit, reduced myocardial reperfusion injury [106], but their effects on platelet function, to our knowledge, are unknown. The quinoline-4-carboxamide ER-000444793, identified as an inhibitor of the mPTP complex [109], was mainly studied in vascular smooth muscle cells [110].
- (b)
6.6. Challenges and Future Directions: Moving Towards the Clinic
- What is the definitive molecular composition of platelet mPTP, and does it vary under different pathological conditions or between individuals?
- What are the precise molecular switches and post-translational modifications that distinguish transient, potentially physiological flickering, from persistent, pathological pore opening?
- How do mitochondrial dynamics (fusion/fission events) and interorganelle communication (e.g., with ER/DTS) overlap with mPTP regulation in platelets?
- What are the most reliable and feasible methods to study mPTP dynamics (e.g., flickering vs. full opening) in circulating human platelets or in relevant in vivo models?
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACS | acute coronary syndromes |
| ADP | adenosine diphosphate |
| ANT | adenine nucleotide translocase |
| ATP | adenosine triphosphate |
| CsA | cyclosporin A |
| CypD | Cyclophilin D |
| DIC | disseminated intravascular coagulation |
| ER | endoplasmic reticulum |
| ETC | electron transport chain |
| GPVI | glycoprotein VI |
| ΔΨm | mitochondrial membrane potential |
| MCU | mitochondrial calcium uniporter |
| MnSOD | manganese superoxide dismutase |
| mPTP | mitochondrial permeability transition pore |
| mtDNA | mitochondrial DNA |
| mtROS | mitochondrial ROS |
| ROS | reactive oxygen species |
| SHRSP | spontaneously hypertensive stroke-prone rats |
| SIRT3 | sirtuin 3 |
| SOCE | store-operated calcium entry |
| TCA | tricarboxylic acid |
| TSPO | peripheral benzodiazepine receptor |
| VDAC | voltage-dependent anion channel |
References
- Hou, Y.; Carrim, N.; Wang, Y.; Gallant, R.C.; Marshall, A.; Ni, H. Platelets in hemostasis and thrombosis: Novel mechanisms of fibrinogen-independent platelet aggregation and fibronectin-mediated protein wave of hemostasis. J. Biomed. Res. 2015, 29, 437–444. [Google Scholar] [CrossRef]
- Trivigno, S.M.G.; Guidetti, G.F.; Barbieri, S.S.; Zara, M. Blood Platelets in Infection: The Multiple Roles of the Platelet Signalling Machinery. Int. J. Mol. Sci. 2023, 24, 7462. [Google Scholar] [CrossRef]
- Chaudhary, P.K.; Kim, S.; Kim, S. An Insight into Recent Advances on Platelet Function in Health and Disease. Int. J. Mol. Sci. 2022, 23, 6022. [Google Scholar] [CrossRef] [PubMed]
- Baaten, C.; Moenen, F.; Henskens, Y.M.C.; Swieringa, F.; Wetzels, R.J.H.; van Oerle, R.; Heijnen, H.F.G.; Ten Cate, H.; Holloway, G.P.; Beckers, E.A.M.; et al. Impaired mitochondrial activity explains platelet dysfunction in thrombocytopenic cancer patients undergoing chemotherapy. Haematologica 2018, 103, 1557–1567. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Gerle, C.; Halestrap, A.P.; Jonas, E.A.; Karch, J.; Mnatsakanyan, N.; Pavlov, E.; Sheu, S.S.; Soukas, A.A. Identity, structure, and function of the mitochondrial permeability transition pore: Controversies, consensus, recent advances, and future directions. Cell Death Differ. 2023, 30, 1869–1885. [Google Scholar] [CrossRef] [PubMed]
- Ajanel, A.; Campbell, R.A.; Denorme, F. Platelet mitochondria: The mighty few. Curr. Opin. Hematol. 2023, 30, 167–174. [Google Scholar] [CrossRef]
- Jobe, S.M.; Wilson, K.M.; Leo, L.; Raimondi, A.; Molkentin, J.D.; Lentz, S.R.; Di Paola, J. Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood 2008, 111, 1257–1265. [Google Scholar] [CrossRef]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef]
- Kinnally, K.W.; Peixoto, P.M.; Ryu, S.Y.; Dejean, L.M. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim. Biophys. Acta 2011, 1813, 616–622. [Google Scholar] [CrossRef]
- Leytin, V.; Allen, D.J.; Mutlu, A.; Gyulkhandanyan, A.V.; Mykhaylov, S.; Freedman, J. Mitochondrial control of platelet apoptosis: Effect of cyclosporin A, an inhibitor of the mitochondrial permeability transition pore. Lab. Investig. 2009, 89, 374–384. [Google Scholar] [CrossRef]
- Cao, Y.; Ma, W.; Liu, Z.; Pei, Y.; Zhu, Y.; Chen, F.; Zou, L.; Jiang, Y.; Liu, X.; Huang, J.; et al. Early predictive value of platelet function for clinical outcome in sepsis. J. Infect. 2022, 84, 628–636. [Google Scholar] [CrossRef]
- Liu, F.; Gamez, G.; Myers, D.R.; Clemmons, W.; Lam, W.A.; Jobe, S.M. Mitochondrially mediated integrin alphaIIbbeta3 protein inactivation limits thrombus growth. J. Biol. Chem. 2013, 288, 30672–30681. [Google Scholar] [CrossRef]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial dysfunction: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2024, 9, 124. [Google Scholar] [CrossRef]
- Hayashi, T.; Tanaka, S.; Hori, Y.; Hirayama, F.; Sato, E.F.; Inoue, M. Role of mitochondria in the maintenance of platelet function during in vitro storage. Transfus. Med. 2011, 21, 166–174. [Google Scholar] [CrossRef]
- Melchinger, H.; Jain, K.; Tyagi, T.; Hwa, J. Role of Platelet Mitochondria: Life in a Nucleus-Free Zone. Front. Cardiovasc. Med. 2019, 6, 153. [Google Scholar] [CrossRef]
- Bonora, M.; Patergnani, S.; Rimessi, A.; De Marchi, E.; Suski, J.M.; Bononi, A.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. ATP synthesis and storage. Purinergic Signal 2012, 8, 343–357. [Google Scholar] [CrossRef]
- Baccarelli, A.A.; Byun, H.M. Platelet mitochondrial DNA methylation: A potential new marker of cardiovascular disease. Clin. Epigenetics 2015, 7, 44. [Google Scholar] [CrossRef]
- Sarasija, S.; Norman, K.R. A gamma-Secretase Independent Role for Presenilin in Calcium Homeostasis Impacts Mitochondrial Function and Morphology in Caenorhabditis elegans. Genetics 2015, 201, 1453–1466. [Google Scholar] [CrossRef] [PubMed]
- Hekimi, S.; Wang, Y.; Noe, A. Mitochondrial ROS and the Effectors of the Intrinsic Apoptotic Pathway in Aging Cells: The Discerning Killers! Front. Genet. 2016, 7, 161. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.E.; Grant, A.R.; Simic, M.S.; Kohnz, R.A.; Nomura, D.K.; Durieux, J.; Riera, C.E.; Sanchez, M.; Kapernick, E.; Wolff, S.; et al. Lipid Biosynthesis Coordinates a Mitochondrial-to-Cytosolic Stress Response. Cell 2016, 166, 1539–1552.e16. [Google Scholar] [CrossRef] [PubMed]
- Richman, T.R.; Ermer, J.A.; Baker, J.; Siira, S.J.; Kile, B.T.; Linden, M.D.; Rackham, O.; Filipovska, A. Mitochondrial gene expression is required for platelet function and blood clotting. Cell Rep. 2023, 42, 113312. [Google Scholar] [CrossRef]
- Choo, H.J.; Saafir, T.B.; Mkumba, L.; Wagner, M.B.; Jobe, S.M. Mitochondrial calcium and reactive oxygen species regulate agonist-initiated platelet phosphatidylserine exposure. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2946–2955. [Google Scholar] [CrossRef] [PubMed]
- Davizon-Castillo, P.; McMahon, B.; Aguila, S.; Bark, D.; Ashworth, K.; Allawzi, A.; Campbell, R.A.; Montenont, E.; Nemkov, T.; D’Alessandro, A.; et al. TNF-alpha-driven inflammation and mitochondrial dysfunction define the platelet hyperreactivity of aging. Blood 2019, 134, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Diaz, F.; Moraes, C.T. Mitochondrial biogenesis and turnover. Cell Calcium 2008, 44, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell Cardiol. 2009, 46, 821–831. [Google Scholar] [CrossRef]
- Kokoszka, J.E.; Waymire, K.G.; Levy, S.E.; Sligh, J.E.; Cai, J.; Jones, D.P.; MacGregor, G.R.; Wallace, D.C. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 2004, 427, 461–465. [Google Scholar] [CrossRef]
- Varanyuwatana, P.; Halestrap, A.P. The roles of phosphate and the phosphate carrier in the mitochondrial permeability transition pore. Mitochondrion 2012, 12, 120–125. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Sheiko, T.; Craigen, W.J.; Molkentin, J.D. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 2007, 9, 550–555. [Google Scholar] [CrossRef]
- Szabo, I.; Zoratti, M. The mitochondrial permeability transition pore may comprise VDAC molecules. I. Binary structure and voltage dependence of the pore. FEBS Lett. 1993, 330, 201–205. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Sileikyte, J.; Petronilli, V.; Zulian, A.; Dabbeni-Sala, F.; Tognon, G.; Nikolov, P.; Bernardi, P.; Ricchelli, F. Regulation of the inner membrane mitochondrial permeability transition by the outer membrane translocator protein (peripheral benzodiazepine receptor). J. Biol. Chem. 2011, 286, 1046–1053. [Google Scholar] [CrossRef]
- Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Gatliff, J.; Campanella, M. The 18 kDa translocator protein (TSPO): A new perspective in mitochondrial biology. Curr. Mol. Med. 2012, 12, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabo, I.; et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [CrossRef] [PubMed]
- Carraro, M.; Bernardi, P. The mitochondrial permeability transition pore in Ca(2+) homeostasis. Cell Calcium 2023, 111, 102719. [Google Scholar] [CrossRef]
- Gonzalez-Correa, J.A.; De La Cruz, J.P.; Lucena, M.I.; Sanchez de la Cuesta, F. Effect of cyclosporin A on platelet aggregation and thromboxane/prostacyclin balance in a model of extrahepatic cholestasis in the rat. Thromb. Res. 1996, 81, 367–381. [Google Scholar] [CrossRef]
- Murphy, E. Cyclophilin D regulation of the mitochondrial permeability transition pore. Curr. Opin. Physiol. 2022, 25, 100486. [Google Scholar] [CrossRef]
- Obydennyy, S.I.; Sveshnikova, A.N.; Ataullakhanov, F.I.; Panteleev, M.A. Dynamics of calcium spiking, mitochondrial collapse and phosphatidylserine exposure in platelet subpopulations during activation. J. Thromb. Haemost. 2016, 14, 1867–1881. [Google Scholar] [CrossRef]
- Remenyi, G.; Szasz, R.; Friese, P.; Dale, G.L. Role of mitochondrial permeability transition pore in coated-platelet formation. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 467–471. [Google Scholar] [CrossRef]
- Jackson, S.P.; Schoenwaelder, S.M. Procoagulant platelets: Are they necrotic? Blood 2010, 116, 2011–2018. [Google Scholar] [CrossRef]
- Di Buduo, C.A.; Balduini, A.; Moccia, F. Pathophysiological Significance of Store-Operated Calcium Entry in Megakaryocyte Function: Opening New Paths for Understanding the Role of Calcium in Thrombopoiesis. Int. J. Mol. Sci. 2016, 17, 2055. [Google Scholar] [CrossRef]
- Ebbeling, L.; Robertson, C.; McNicol, A.; Gerrard, J.M. Rapid ultrastructural changes in the dense tubular system following platelet activation. Blood 1992, 80, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Ghatge, M.; Nayak, M.K.; Flora, G.D.; Kumskova, M.; Jain, A.; Patel, R.B.; Lin, Z.; Usachev, Y.M.; Chauhan, A.K. Mitochondrial calcium uniporter b deletion inhibits platelet function and reduces susceptibility to arterial thrombosis. J. Thromb. Haemost. 2023, 21, 2163–2174. [Google Scholar] [CrossRef] [PubMed]
- Kholmukhamedov, A.; Janecke, R.; Choo, H.J.; Jobe, S.M. The mitochondrial calcium uniporter regulates procoagulant platelet formation. J. Thromb. Haemost. 2018, 16, 2315–2321. [Google Scholar] [CrossRef] [PubMed]
- Polina, I.; Mishra, J.; Cypress, M.W.; Landherr, M.; Valkov, N.; Chaput, I.; Nieto, B.; Mende, U.; Zhang, P.; Jhun, B.S.; et al. Mitochondrial Ca (2+) uniporter (MCU) variants form plasma-membrane channels. bioRxiv 2023. [Google Scholar] [CrossRef]
- Amadio, P.; Sandrini, L.; Zara, M.; Barbieri, S.S.; Ieraci, A. NADPH-oxidases as potential pharmacological targets for thrombosis and depression comorbidity. Redox Biol. 2024, 70, 103060. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef]
- Kulkarni, P.P.; Ekhlak, M.; Sonkar, V.K.; Dash, D. Mitochondrial ATP generation in stimulated platelets is essential for granule secretion but dispensable for aggregation and procoagulant activity. Haematologica 2022, 107, 1209–1213. [Google Scholar] [CrossRef]
- Hafner, A.V.; Dai, J.; Gomes, A.P.; Xiao, C.Y.; Palmeira, C.M.; Rosenzweig, A.; Sinclair, D.A. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging 2010, 2, 914–923. [Google Scholar] [CrossRef]
- Naryzhnaya, N.V.; Maslov, L.N.; Oeltgen, P.R. Pharmacology of mitochondrial permeability transition pore inhibitors. Drug Dev. Res. 2019, 80, 1013–1030. [Google Scholar] [CrossRef]
- Sivitz, W.I.; Yorek, M.A. Mitochondrial dysfunction in diabetes: From molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox Signal 2010, 12, 537–577. [Google Scholar] [CrossRef] [PubMed]
- Grundler, K.; Angstwurm, M.; Hilge, R.; Baumann, P.; Annecke, T.; Crispin, A.; Sohn, H.Y.; Massberg, S.; Kraemer, B.F. Platelet mitochondrial membrane depolarization reflects disease severity in patients with sepsis and correlates with clinical outcome. Crit. Care 2014, 18, R31. [Google Scholar] [CrossRef] [PubMed]
- Johri, A.; Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, Y.; Guo, Y.; Shi, X.; Chen, X.; Feng, W.; Wu, L.L.; Zhang, J.; Yu, S.; Wang, Y.; et al. An Overview: The Diversified Role of Mitochondria in Cancer Metabolism. Int. J. Biol. Sci. 2023, 19, 897–915. [Google Scholar] [CrossRef]
- Briston, T.; Selwood, D.L.; Szabadkai, G.; Duchen, M.R. Mitochondrial Permeability Transition: A Molecular Lesion with Multiple Drug Targets. Trends Pharmacol. Sci. 2019, 40, 50–70. [Google Scholar] [CrossRef]
- Li, Y.; Sun, J.; Wu, R.; Bai, J.; Hou, Y.; Zeng, Y.; Zhang, Y.; Wang, X.; Wang, Z.; Meng, X. Mitochondrial MPTP: A Novel Target of Ethnomedicine for Stroke Treatment by Apoptosis Inhibition. Front. Pharmacol. 2020, 11, 352. [Google Scholar] [CrossRef]
- Mendoza, A.; Patel, P.; Robichaux, D.; Ramirez, D.; Karch, J. Inhibition of the mPTP and Lipid Peroxidation Is Additively Protective Against I/R Injury. Circ. Res. 2024, 134, 1292–1305. [Google Scholar] [CrossRef]
- Hu, S.S.; Wang, T.Y.; Ni, L.; Hu, F.X.; Yue, B.W.; Zheng, Y.; Wang, T.L.; Kumar, A.; Wang, Y.Y.; Wang, J.E.; et al. Icariin Ameliorates D-galactose-induced Cell Injury in Neuron-like PC12 Cells by Inhibiting MPTP Opening. Curr. Med. Sci. 2024, 44, 748–758. [Google Scholar] [CrossRef]
- Zhou, Z.; Li, W.; Ni, L.; Wang, T.; Huang, Y.; Yu, Y.; Hu, M.; Liu, Y.; Wang, J.; Huang, X.; et al. Icariin improves oxidative stress injury during ischemic stroke via inhibiting mPTP opening. Mol. Med. 2024, 30, 77. [Google Scholar] [CrossRef]
- Gutierrez-Aguilar, M.; Baines, C.P. Structural mechanisms of cyclophilin D-dependent control of the mitochondrial permeability transition pore. Biochim. Biophys. Acta 2015, 1850, 2041–2047. [Google Scholar] [CrossRef]
- Mishra, J.; Davani, A.J.; Natarajan, G.K.; Kwok, W.M.; Stowe, D.F.; Camara, A.K.S. Cyclosporin A Increases Mitochondrial Buffering of Calcium: An Additional Mechanism in Delaying Mitochondrial Permeability Transition Pore Opening. Cells 2019, 8, 1052. [Google Scholar] [CrossRef]
- Hefler, J.; Marfil-Garza, B.A.; Campbell, S.; Freed, D.H.; Shapiro, A.M.J. Preclinical systematic review & meta-analysis of cyclosporine for the treatment of myocardial ischemia-reperfusion injury. Ann. Transl. Med. 2022, 10, 954. [Google Scholar] [CrossRef] [PubMed]
- Alanova, P.; Alan, L.; Neckar, J.; Ostadal, B.; Kolar, F. Cardioprotective Effect of Chronic Hypoxia Involves Inhibition of Mitochondrial Permeability Transition Pore Opening. Physiol. Res. 2024, 73, 881–884. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, G.; Matoba, T.; Nakano, Y.; Nagaoka, K.; Ishikita, A.; Nakano, K.; Funamoto, D.; Sunagawa, K.; Egashira, K. Nanoparticle-Mediated Targeting of Cyclosporine A Enhances Cardioprotection Against Ischemia-Reperfusion Injury Through Inhibition of Mitochondrial Permeability Transition Pore Opening. Sci. Rep. 2016, 6, 20467. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.R.; Yu, L.N. Cardioprotective effects of cyclosporine A in an in vivo model of myocardial ischemia and reperfusion. Acta Anaesthesiol. Scand. 2007, 51, 909–913. [Google Scholar] [CrossRef]
- Clarke, S.J.; McStay, G.P.; Halestrap, A.P. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J. Biol. Chem. 2002, 277, 34793–34799. [Google Scholar] [CrossRef]
- Hefler, J.; Pawlick, R.; Marfil-Garza, B.A.; Thiesen, A.; Cuesta-Gomez, N.; Hatami, S.; Freed, D.H.; Karvellas, C.; Bigam, D.L.; Shapiro, A.M.J. Protective effects of cyclosporine and its analog NIM-811 in a murine model of hepatic ischemia-reperfusion injury. Liver Res. 2024, 8, 46–53. [Google Scholar] [CrossRef]
- Ong, E.; Clottes, P.; Leon, C.; Guedouari, H.; Gallo-Bona, N.; Lo Grasso, M.; Motter, L.; Bolbos, R.; Ovize, M.; Nighogossian, N.; et al. Mitochondria dysfunction, a potential cytoprotection target against ischemia-reperfusion injury in a mouse stroke model. Neurotherapeutics 2025, 22, e00549. [Google Scholar] [CrossRef]
- Song, K.; Wang, S.; Qi, D. Effects of Cyclosporine on Reperfusion Injury in Patients: A Meta-Analysis of Randomized Controlled Trials. Oxid. Med. Cell Longev. 2015, 2015, 287058. [Google Scholar] [CrossRef] [PubMed]
- Tomasiak, M.; Rusak, T.; Gacko, M.; Stelmach, H. Cyclosporine enhances platelet procoagulant activity. Nephrol. Dial. Transplant. 2007, 22, 1750–1756. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.C.; Judge, H.M.; Allen, B.R.; Heptinstall, S. Platelet aggregation and intracellular calcium mobilisation responses are enhanced by cyclosporin A but not by pimecrolimus (SDZ ASM 981). Platelets 2002, 13, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.K.; Argaiz, E.R.; Mercado, J.E.; Maul, H.O.; Garza, J.; Enriquez, A.B.; Abdel-Monem, H.; Prakasam, A.; Andreeff, M.; Thiagarajan, P. Platelet senescence and phosphatidylserine exposure. Transfusion 2010, 50, 2167–2175. [Google Scholar] [CrossRef]
- Jahandiez, V.; Cour, M.; Bochaton, T.; Abrial, M.; Loufouat, J.; Gharib, A.; Varennes, A.; Ovize, M.; Argaud, L. Fast therapeutic hypothermia prevents post-cardiac arrest syndrome through cyclophilin D-mediated mitochondrial permeability transition inhibition. Basic. Res. Cardiol. 2017, 112, 35. [Google Scholar] [CrossRef]
- Cour, M.; Abrial, M.; Jahandiez, V.; Loufouat, J.; Belaidi, E.; Gharib, A.; Varennes, A.; Monneret, G.; Thibault, H.; Ovize, M.; et al. Ubiquitous protective effects of cyclosporine A in preventing cardiac arrest-induced multiple organ failure. J. Appl. Physiol. (1985) 2014, 117, 930–936. [Google Scholar] [CrossRef]
- Gomez, L.; Thibault, H.; Gharib, A.; Dumont, J.M.; Vuagniaux, G.; Scalfaro, P.; Derumeaux, G.; Ovize, M. Inhibition of mitochondrial permeability transition improves functional recovery and reduces mortality following acute myocardial infarction in mice. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1654–H1661. [Google Scholar] [CrossRef]
- Hansson, M.J.; Mattiasson, G.; Mansson, R.; Karlsson, J.; Keep, M.F.; Waldmeier, P.; Ruegg, U.T.; Dumont, J.M.; Besseghir, K.; Elmer, E. The nonimmunosuppressive cyclosporin analogs NIM811 and UNIL025 display nanomolar potencies on permeability transition in brain-derived mitochondria. J. Bioenerg. Biomembr. 2004, 36, 407–413. [Google Scholar] [CrossRef]
- Waldmeier, P.C.; Feldtrauer, J.J.; Qian, T.; Lemasters, J.J. Inhibition of the mitochondrial permeability transition by the nonimmunosuppressive cyclosporin derivative NIM811. Mol. Pharmacol. 2002, 62, 22–29. [Google Scholar] [CrossRef]
- Van Bael, J.; Vandenbulcke, A.; Ahmed-Belkacem, A.; Guichou, J.F.; Pawlotsky, J.M.; Samyn, J.; Barendrecht, A.D.; Maas, C.; De Meyer, S.F.; Vanhoorelbeke, K.; et al. Small-Molecule Cyclophilin Inhibitors Potently Reduce Platelet Procoagulant Activity. Int. J. Mol. Sci. 2023, 24, 7163. [Google Scholar] [CrossRef]
- Pillai, V.B.; Kanwal, A.; Fang, Y.H.; Sharp, W.W.; Samant, S.; Arbiser, J.; Gupta, M.P. Honokiol, an activator of Sirtuin-3 (SIRT3) preserves mitochondria and protects the heart from doxorubicin-induced cardiomyopathy in mice. Oncotarget 2017, 8, 34082–34098. [Google Scholar] [CrossRef]
- Skals, M.; Bjaelde, R.G.; Reinholdt, J.; Poulsen, K.; Vad, B.S.; Otzen, D.E.; Leipziger, J.; Praetorius, H.A. Bacterial RTX toxins allow acute ATP release from human erythrocytes directly through the toxin pore. J. Biol. Chem. 2014, 289, 19098–19109. [Google Scholar] [CrossRef]
- Bordet, T.; Buisson, B.; Michaud, M.; Drouot, C.; Galéa, P.; Delaage, P.; Akentieva, N.P.; Evers, A.S.; Covey, D.F.; Ostuni, M.A. Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J. Pharmacol. Exp. Ther. 2007, 322, 709–720. [Google Scholar] [CrossRef]
- Schaller, S.; Paradis, S.; Ngoh, G.A.; Assaly, R.; Buisson, B.; Drouot, C.; Ostuni, M.A.; Lacapere, J.J.; Bassissi, F.; Bordet, T.; et al. TRO40303, a new cardioprotective compound, inhibits mitochondrial permeability transition. J. Pharmacol. Exp. Ther. 2010, 333, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Le Lamer, S.; Paradis, S.; Rahmouni, H.; Chaimbault, C.; Michaud, M.; Culcasi, M.; Afxantidis, J.; Latreille, M.; Berna, P.; Berdeaux, A.; et al. Translation of TRO40303 from myocardial infarction models to demonstration of safety and tolerance in a randomized Phase I trial. J. Transl. Med. 2014, 12, 38. [Google Scholar] [CrossRef] [PubMed]
- Atar, D.; Arheden, H.; Berdeaux, A.; Bonnet, J.L.; Carlsson, M.; Clemmensen, P.; Cuvier, V.; Danchin, N.; Dubois-Rande, J.L.; Engblom, H.; et al. Effect of intravenous TRO40303 as an adjunct to primary percutaneous coronary intervention for acute ST-elevation myocardial infarction: MITOCARE study results. Eur. Heart J. 2015, 36, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Sonkar, V.K.; Kumar, R.; Jensen, M.; Wagner, B.A.; Sharathkumar, A.A.; Miller, F.J., Jr.; Fasano, M.; Lentz, S.R.; Buettner, G.R.; Dayal, S. Nox2 NADPH oxidase is dispensable for platelet activation or arterial thrombosis in mice. Blood Adv. 2019, 3, 1272–1284. [Google Scholar] [CrossRef]
- Firsov, A.M.; Rybalkina, I.G.; Kotova, E.A.; Rokitskaya, T.I.; Tashlitsky, V.N.; Korshunova, G.A.; Rybalkin, S.D.; Antonenko, Y.N. A conjugate of decyltriphenylphosphonium with plastoquinone can carry cyclic adenosine monophosphate, but not cyclic guanosine monophosphate, across artificial and natural membranes. Biochim. Biophys. Acta Biomembr. 2018, 1860, 329–334. [Google Scholar] [CrossRef]
- Mendez, D.; Arauna, D.; Fuentes, F.; Araya-Maturana, R.; Palomo, I.; Alarcon, M.; Sebastian, D.; Zorzano, A.; Fuentes, E. Mitoquinone (MitoQ) Inhibits Platelet Activation Steps by Reducing ROS Levels. Int. J. Mol. Sci. 2020, 21, 6192. [Google Scholar] [CrossRef]
- Teixeira, J.; Oliveira, C.; Cagide, F.; Amorim, R.; Garrido, J.; Borges, F.; Oliveira, P.J. Discovery of a new mitochondria permeability transition pore (mPTP) inhibitor based on gallic acid. J. Enzyme Inhib. Med. Chem. 2018, 33, 567–576. [Google Scholar] [CrossRef]
- Chang, S.S.; Lee, V.S.; Tseng, Y.L.; Chang, K.C.; Chen, K.B.; Chen, Y.L.; Li, C.Y. Gallic Acid Attenuates Platelet Activation and Platelet-Leukocyte Aggregation: Involving Pathways of Akt and GSK3beta. Evid. Based Complement. Alternat Med. 2012, 2012, 683872. [Google Scholar] [CrossRef]
- Li, X.; Yang, H.F.; Chen, Y.; Pei, L.J.; Jiang, R. Effect of the icariin on endothelial microparticles, endothelial progenitor cells, platelets, and erectile function in spontaneously hypertensive rats. Andrology 2022, 10, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zhang, L.; Hui, X.; Sun, X.; Yang, J.; Wang, J.; Wu, H.; Wang, X.; Zheng, Z.; Che, F.; et al. Selenium-containing protein from selenium-enriched Spirulina platensis antagonizes oxygen glucose deprivation-induced neurotoxicity by inhibiting ROS-mediated oxidative damage through regulating MPTP opening. Pharm. Biol. 2021, 59, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Tang, H.; Ai, W.; Zeng, Q.; Yang, H.; Zhu, F.; Wei, Y.; Feng, R.; Wen, L.; Pu, P.; et al. Schisandrin B Antagonizes Cardiotoxicity Induced by Pirarubicin by Inhibiting Mitochondrial Permeability Transition Pore (mPTP) Opening and Decreasing Cardiomyocyte Apoptosis. Front. Pharmacol. 2021, 12, 733805. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Hu, W.; Xu, H. Schisandrin B Alleviates LPS Induced Mitochondrial Damage in C28I2 Cells. J. Membr. Biol. 2024, 257, 107–114. [Google Scholar] [CrossRef]
- Kwong, J.Q.; Lu, X.; Correll, R.N.; Schwanekamp, J.A.; Vagnozzi, R.J.; Sargent, M.A.; York, A.J.; Zhang, J.; Bers, D.M.; Molkentin, J.D. The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell Rep. 2015, 12, 15–22. [Google Scholar] [CrossRef]
- Woods, J.J.; Nemani, N.; Shanmughapriya, S.; Kumar, A.; Zhang, M.; Nathan, S.R.; Thomas, M.; Carvalho, E.; Ramachandran, K.; Srikantan, S.; et al. A Selective and Cell-Permeable Mitochondrial Calcium Uniporter (MCU) Inhibitor Preserves Mitochondrial Bioenergetics after Hypoxia/Reoxygenation Injury. ACS Cent. Sci. 2019, 5, 153–166. [Google Scholar] [CrossRef]
- Matlib, M.A.; Zhou, Z.; Knight, S.; Ahmed, S.; Choi, K.M.; Krause-Bauer, J.; Phillips, R.; Altschuld, R.; Katsube, Y.; Sperelakis, N.; et al. Oxygen-bridged dinuclear ruthenium amine complex specifically inhibits Ca2+ uptake into mitochondria in vitro and in situ in single cardiac myocytes. J. Biol. Chem. 1998, 273, 10223–10231. [Google Scholar] [CrossRef]
- Novorolsky, R.J.; Nichols, M.; Kim, J.S.; Pavlov, E.V.; Woods, J.J.; Wilson, J.J.; Robertson, G.S. The cell-permeable mitochondrial calcium uniporter inhibitor Ru265 preserves cortical neuron respiration after lethal oxygen glucose deprivation and reduces hypoxic/ischemic brain injury. J. Cereb. Blood Flow. Metab. 2020, 40, 1172–1181. [Google Scholar] [CrossRef]
- Ambily, A.; Kaiser, W.J.; Pierro, C.; Chamberlain, E.V.; Li, Z.; Jones, C.I.; Kassouf, N.; Gibbins, J.M.; Authi, K.S. The role of plasma membrane STIM1 and Ca(2+)entry in platelet aggregation. STIM1 binds to novel proteins in human platelets. Cell Signal 2014, 26, 502–511. [Google Scholar] [CrossRef]
- Lang, F.; Munzer, P.; Gawaz, M.; Borst, O. Regulation of STIM1/Orai1-dependent Ca2+ signalling in platelets. Thromb. Haemost. 2013, 110, 925–930. [Google Scholar] [CrossRef]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef]
- Braun, A.; Varga-Szabo, D.; Kleinschnitz, C.; Pleines, I.; Bender, M.; Austinat, M.; Bosl, M.; Stoll, G.; Nieswandt, B. Orai1 (CRACM1) is the platelet SOC channel and essential for pathological thrombus formation. Blood 2009, 113, 2056–2063. [Google Scholar] [CrossRef]
- Harper, M.T.; Poole, A.W. Store-operated calcium entry and non-capacitative calcium entry have distinct roles in thrombin-induced calcium signalling in human platelets. Cell Calcium 2011, 50, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.M.; Munzer, P.; Borst, O.; Kraemer, B.F.; Schmid, E.; Urban, B.; Lindemann, S.; Ruth, P.; Gawaz, M.; Lang, F. Ion channels in the regulation of platelet migration. Biochem. Biophys. Res. Commun. 2011, 415, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Moura, L.A.; de Almeida, A.C.; da Silva, A.V.; de Souza, V.R.; Ferreira, V.F.; Menezes, M.V.; Kaiser, C.R.; Ferreira, S.B.; Fuly, A.L. Synthesis, Anticlotting and Antiplatelet Effects of 1,2,3-Triazoles Derivatives. Med. Chem. 2016, 12, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Algieri, V.; Algieri, C.; Maiuolo, L.; De Nino, A.; Pagliarani, A.; Tallarida, M.A.; Trombetti, F.; Nesci, S. 1,5-Disubstituted-1,2,3-triazoles as inhibitors of the mitochondrial Ca(2+) -activated F(1) F(O) -ATP(hydrol)ase and the permeability transition pore. Ann. N. Y. Acad. Sci. 2021, 1485, 43–55. [Google Scholar] [CrossRef]
- Algieri, C.; Bernardini, C.; Marchi, S.; Forte, M.; Tallarida, M.A.; Bianchi, F.; La Mantia, D.; Algieri, V.; Stanzione, R.; Cotugno, M.; et al. 1,5-disubstituted-1,2,3-triazoles counteract mitochondrial dysfunction acting on F(1)F(O)-ATPase in models of cardiovascular diseases. Pharmacol. Res. 2023, 187, 106561. [Google Scholar] [CrossRef]
- Briston, T.; Lewis, S.; Koglin, M.; Mistry, K.; Shen, Y.; Hartopp, N.; Katsumata, R.; Fukumoto, H.; Duchen, M.R.; Szabadkai, G.; et al. Identification of ER-000444793, a Cyclophilin D-independent inhibitor of mitochondrial permeability transition, using a high-throughput screen in cryopreserved mitochondria. Sci. Rep. 2016, 6, 37798. [Google Scholar] [CrossRef]
- Koval, O.M.; Nguyen, E.K.; Mittauer, D.J.; Ait-Aissa, K.; Chinchankar, W.C.; Grumbach, I.M. Regulation of Smooth Muscle Cell Proliferation by Mitochondrial Ca2+ in Type 2 Diabetes. Int. J. Mol. Sci. 2023, 24, 12897. [Google Scholar] [CrossRef]
- Nesci, S.; Algieri, C.; Tallarida, M.A.; Stanzione, R.; Marchi, S.; Pietrangelo, D.; Trombetti, F.; D’Ambrosio, L.; Forte, M.; Cotugno, M.; et al. Molecular mechanisms of naringenin modulation of mitochondrial permeability transition acting on F(1)F(O)-ATPase and counteracting saline load-induced injury in SHRSP cerebral endothelial cells. Eur. J. Cell Biol. 2024, 103, 151398. [Google Scholar] [CrossRef]
- Huang, M.; Deng, M.; Nie, W.; Zou, D.; Wu, H.; Xu, D. Naringenin Inhibits Platelet Activation and Arterial Thrombosis Through Inhibition of Phosphoinositide 3-Kinase and Cyclic Nucleotide Signaling. Front. Pharmacol. 2021, 12, 722257. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Del Zar, M.M.; Vacas, M.I. The effects of melatonin in human platelets. Acta Physiol. Pharmacol. Ther. Latinoam. 1993, 43, 1–13. [Google Scholar]
- Bohm, A.; Lauko, V.; Dostalova, K.; Balanova, I.; Varga, I.; Bezak, B.; Jajcay, N.; Moravcik, R.; Lazurova, L.; Slezak, P.; et al. In-vitro antiplatelet effect of melatonin in healthy individuals and patients with type 2 diabetes mellitus. J. Endocrinol. Investig. 2023, 46, 2493–2500. [Google Scholar] [CrossRef]
- Algieri, C.; Bernardini, C.; Cugliari, A.; Granata, S.; Trombetti, F.; Glogowski, P.A.; Fabbri, M.; Morciano, G.; Pedriali, G.; Pinton, P.; et al. Melatonin rescues cell respiration impaired by hypoxia/reoxygenation in aortic endothelial cells and affects the mitochondrial bioenergetics targeting the F(1)F(O)-ATPase. Redox Biol. 2025, 82, 103605. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Strategy | Primary Target | Mechanism of Action | Examples | Advantages | Limitations/Challenges |
|---|---|---|---|---|---|
| Cyclophilin D (CypD) Inhibitors | CypD | Prevents mPTP opening by binding CypD | Cyclosporin A (CsA), Sanglifehrin A | Mitochondrial protection; reduced ischemic damage | Immunosuppression, limited long-term use; ineffective in clinical trials; potential pro-thrombotic effects |
| Non-immunosuppressive inhibition of CypD | Debio-025, NIM811 | Avoids immunosuppression; reduces ischemia–reperfusion damage | Platelet effects poorly characterized; limited clinical validation | ||
| Novel small-molecule CypD inhibitors (CsA-unrelated) | F759, F83236 | Reduce platelet procoagulant activity without affecting integrin signaling | Still experimental; lack of human data | ||
| SIRT3 | Promotes CypD deacetylation and inhibits mPTP | Honokiol | Neuroprotective and cardioprotective; reduces oxidative stress | No direct data on platelets; potential systemic off-target effects | |
| TSPO Ligands | Translocator Protein (TSPO) | Stabilizes mitochondrial membranes | TRO40303, TRO19622 | Effective in reducing infarct sizes in animal models | Failed efficacy in Phase II trials; unclear mitochondrial specificity |
| Mitochondria-Targeted Antioxidants | Mitochondrial ROS | Scavenge ROS within mitochondria | MitoTEMPO, SkQ1, MitoQ | Improves mitochondrial function; prevents platelet dysfunction under oxidative stress | Long-term safety in humans unclear; dose optimization needed |
| Natural antioxidants modulating mPTP and reducing ROS | Icariin, Gallic Acid, Schisandrin B, Spirulina protein | Natural origin; multiple antioxidant and antiplatelet effects | Variability in bioavailability and formulation; limited clinical data | ||
| Modulation of Mitochondrial Calcium Handling | MCU (Mitochondrial Calcium Uniporter) | Inhibits mitochondrial calcium uptake to prevent mPTP | Ru360, other MCU blockers | Reduces calcium-triggered mitochondrial dysfunction | May impair essential metabolic processes regulated by calcium |
| SOCE (STIM1-Orai1) | Limits cytosolic calcium entry and mitochondrial overload | BTP-2, SKF-96365 | Reduces platelet activation and thrombotic risk | Risk of systemic immunosuppression; lacks platelet-specific targeting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lonobile, C.; Di Nubila, A.; Simone, R.; Hushi, M.; Barbieri, S.S. The Mitochondrial Permeability Transition Pore in Platelets: Mechanisms, Physiological Roles, and Therapeutic Perspectives. Antioxidants 2025, 14, 923. https://doi.org/10.3390/antiox14080923
Lonobile C, Di Nubila A, Simone R, Hushi M, Barbieri SS. The Mitochondrial Permeability Transition Pore in Platelets: Mechanisms, Physiological Roles, and Therapeutic Perspectives. Antioxidants. 2025; 14(8):923. https://doi.org/10.3390/antiox14080923
Chicago/Turabian StyleLonobile, Chiara, Alessia Di Nubila, Rosa Simone, Matilda Hushi, and Silvia Stella Barbieri. 2025. "The Mitochondrial Permeability Transition Pore in Platelets: Mechanisms, Physiological Roles, and Therapeutic Perspectives" Antioxidants 14, no. 8: 923. https://doi.org/10.3390/antiox14080923
APA StyleLonobile, C., Di Nubila, A., Simone, R., Hushi, M., & Barbieri, S. S. (2025). The Mitochondrial Permeability Transition Pore in Platelets: Mechanisms, Physiological Roles, and Therapeutic Perspectives. Antioxidants, 14(8), 923. https://doi.org/10.3390/antiox14080923