

Rivastigmine Templates with Antioxidant Motifs—A Medicinal Chemist’s Toolbox Towards New Multipotent AD Drugs
 , , ,
, , ,  , and
, and
Abstract
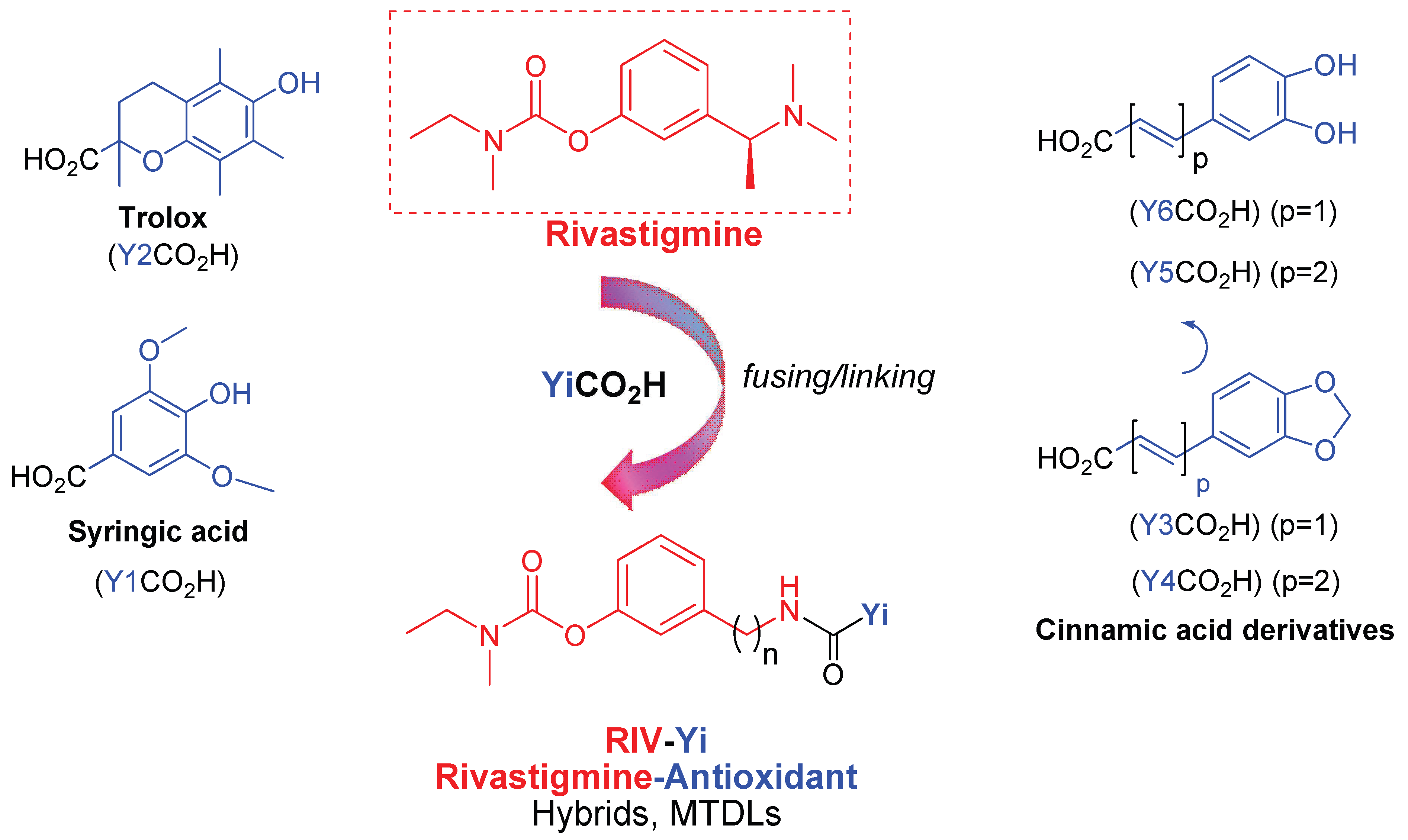
1. Introduction
2. Materials and Methods
2.1. Materials and Equipment
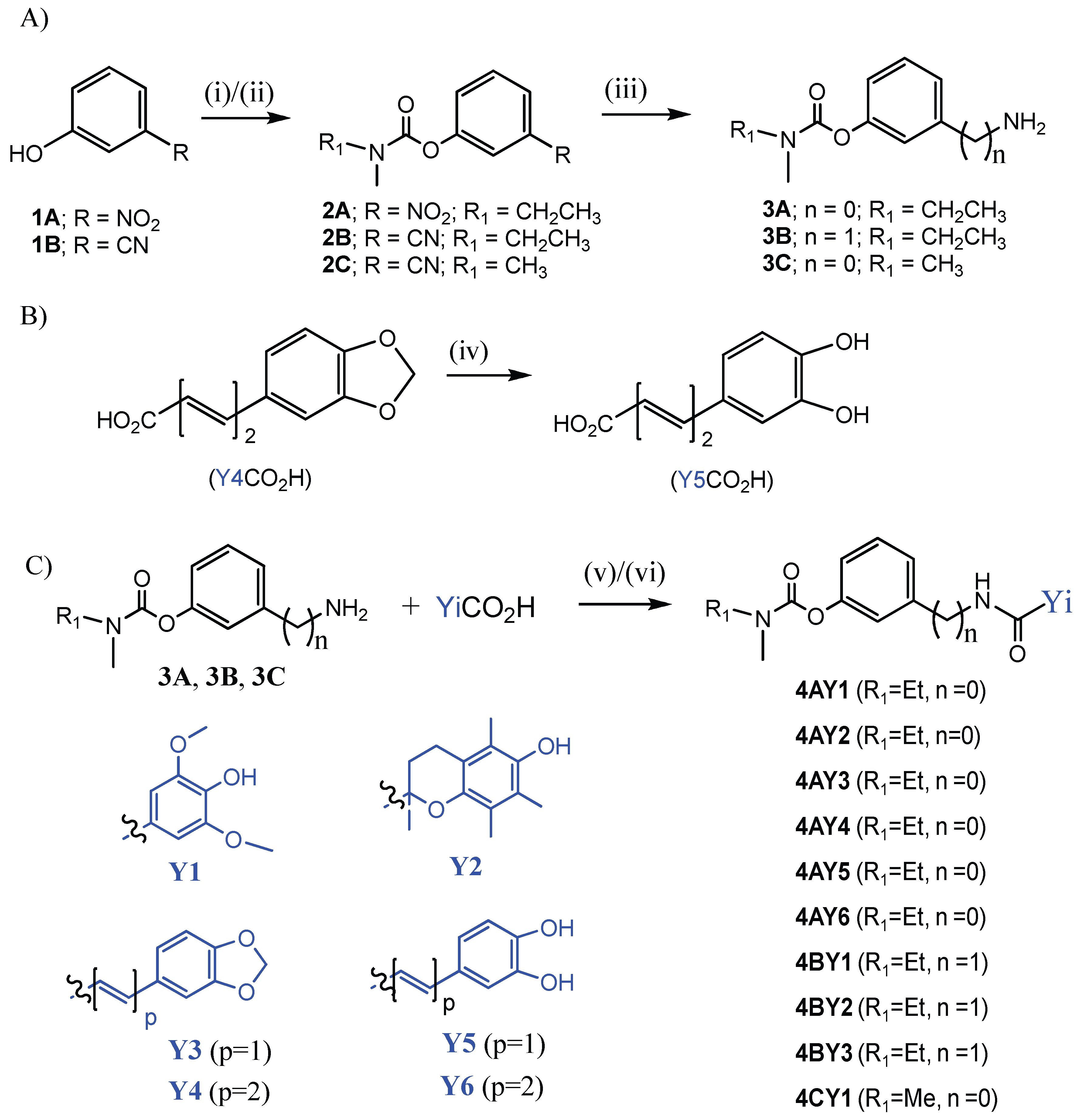
2.2. Synthesis of the Intermediate Compounds and Final RIV Hybrids
2.2.1. General Procedure for Synthesis of the Carbamates (2A, 2B, and 2C)
- 3-Nitrophenyl ethyl(methyl)carbamate (2A)
- 3-Cyanophenyl ethyl(methyl)carbamate (2B)
- 3-Nitrophenyl dimethylcarbamate (2C)
2.2.2. General Procedure for Synthesis of the Aminocarbamates (3A, 3B and 3C)
- 3-Aminophenyl ethyl(methyl)carbamate (3A)
- 3-(Aminomethyl)phenyl ethyl(methyl)carbamate (3B)
- 3-Aminophenyldimethylcarbamate (3C)
2.2.3. Synthesis of (2E,4E)-5-(3,4-Dihydroxyphenyl)penta-2,4-dienoic Acid (Y5CO2H)
2.2.4. Synthetic Procedures for the Target RIV Hybrids
General Procedure (Method A) for the Synthesis of the RIV Hybrids (4AY1, 4AY2, 4AY3, 4AY4, 4BY1, 4BY3, 4BY2, and 4CY1)
- 3-(4-Hydroxy-3,5-dimethoxybenzamido)phenyl ethyl(methyl)carbamate (4AY1)
- 3-(6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxamido)phenyl ethyl(methyl)carbamate (4AY2)
- (E)-3-(3-(Benzo[d][1,3]dioxol-5-yl)acrylamido)phenyl ethyl(methyl)carbamate (4AY3)
- 3-((2E,4E)-5-(Benzo[d][1,3]dioxol-5-yl)penta-2,4-dienamido)phenyl ethyl(methyl)carbamate (4AY4)
- 3-((4-Hydroxy-3,5-dimethoxybenzamido)methyl)phenyl ethyl(methyl)carbamate (4BY1)
- 3-((6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxamido)methyl)phenyl ethyl(methyl)carbamate (4BY2)
- (E)-3-((3-(Benzo[d][1,3]dioxol-5-yl)acrylamido)methyl)phenyl ethyl(methyl)carbamate (4BY3)
- 3-(4-Hydroxy-3,5-dimethoxybenzamido)phenyl dimethylcarbamate (4CY1)
General Procedure (Method B) for the Synthesis of the RIV Hybrids (4AY5, 4AY6)
- (E)-3-(3-(3,4-Dihydroxyphenyl)acrylamido)phenyl ethyl(methyl)carbamate (4AY5)
- 3-((2E,4E)-5-(3,4-Dihydroxyphenyl)penta-2,4-dienamido)phenyl ethyl(methyl)carbamate (4AY6)
2.3. Free-Radical-Scavenging Assays
2.4. Cholinesterase Inhibition
2.5. Inhibition of Aβ1–42 Self-Aggregation
2.6. Molecular Modeling Studies
2.6.1. Protein Structure Selection/Protein and Ligand Preparation
2.6.2. Molecular-Docking Calculations
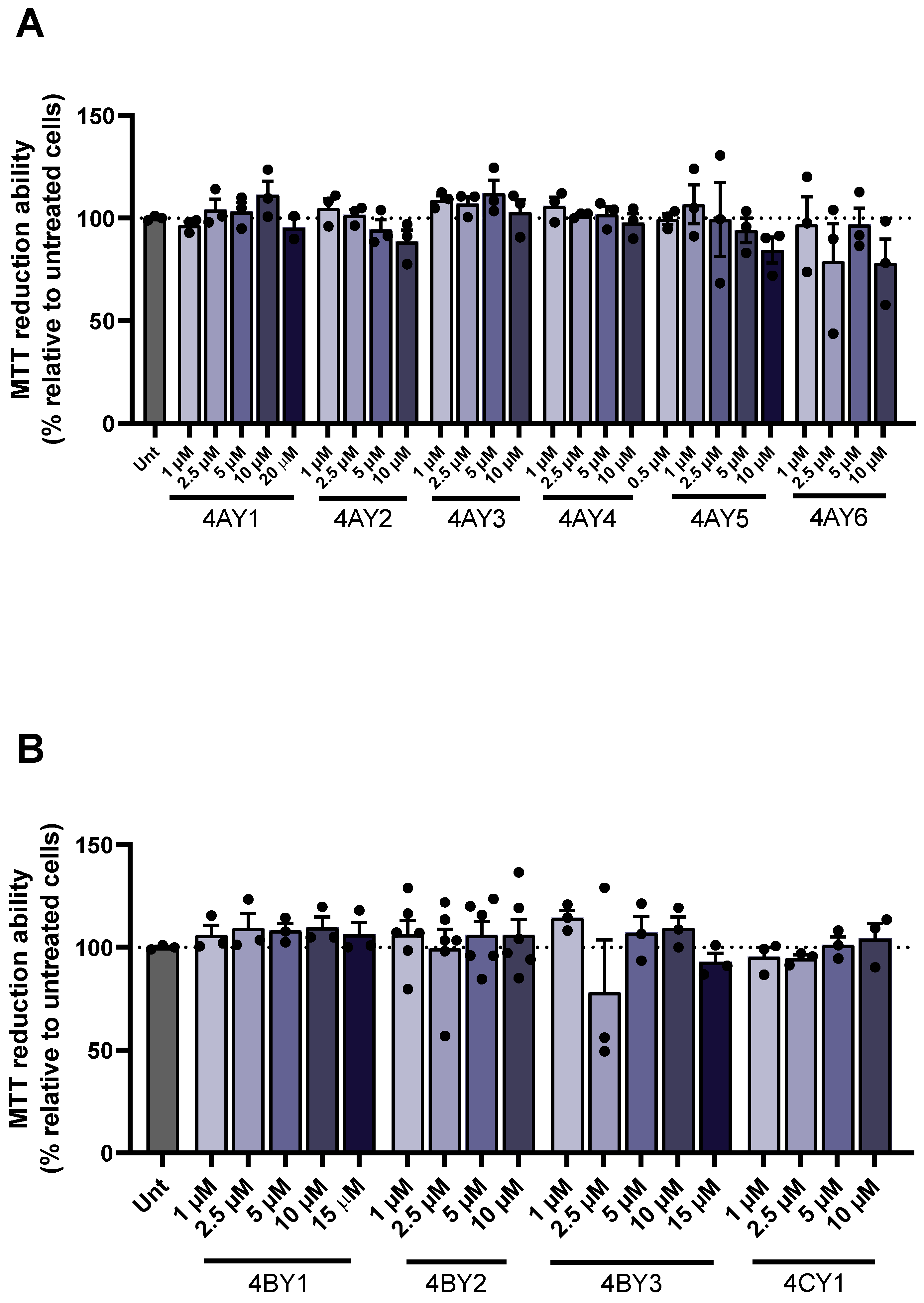
2.7. In Vitro Cell Assays
2.8. Pharmacokinetic and Physicochemical Properties
3. Results
3.1. Synthetic Pathway for the RIV-IND Hybrids
3.2. Free-Radical-Scavenging Evaluation
3.3. Inhibition of Cholinesterases
3.4. Inhibition of Self Aβ1–42 Aggregation
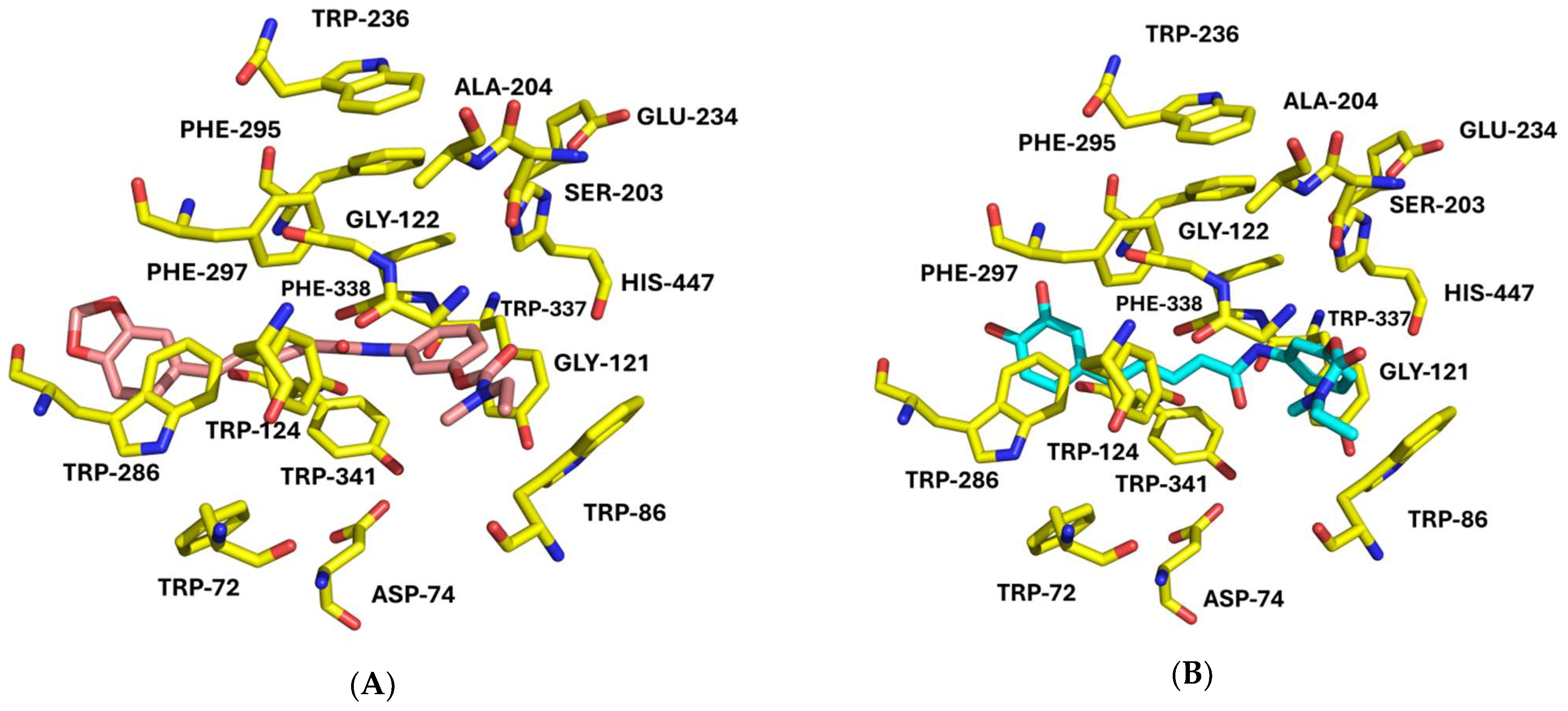
3.5. Molecular-Docking Studies
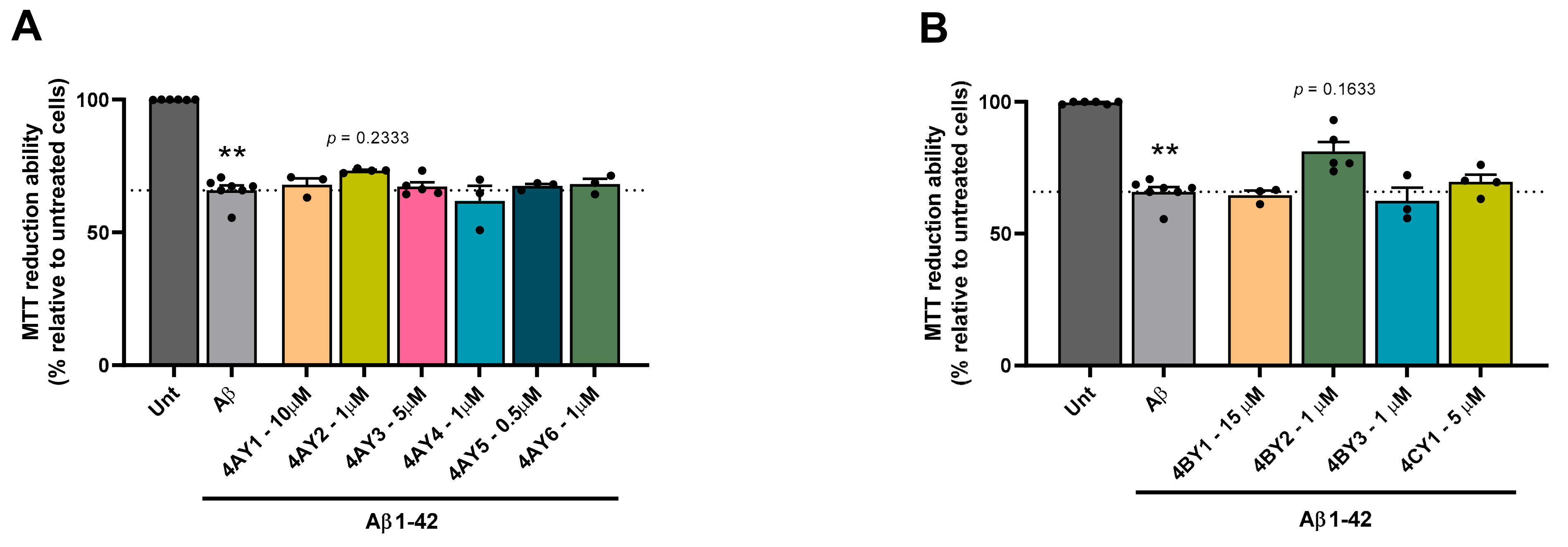
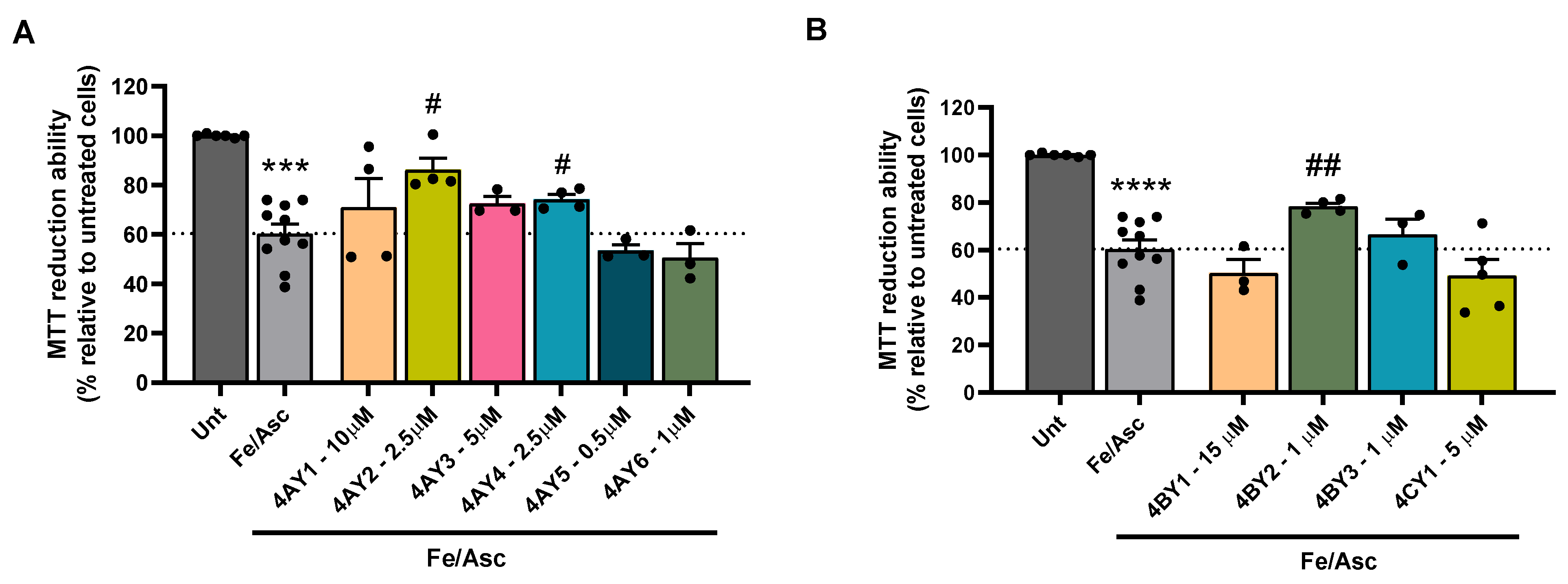
3.6. Assessment of Cell Viability and Protective Effects
3.7. Evaluation of Pharmacokinetic and Physicochemical Properties
4. Discussion
4.1. Synthesis of the RIV-IND Hybrids
4.2. Free-Radical-Scavenging Activity
4.3. Inhibition of Cholinesterase Activity
4.4. Inhibition of Self Aβ1–42 Aggregation
4.5. Molecular-Docking Studies
4.6. Relevance of Findings to Neurodegeneration and Therapeutic Potential
4.7. Prediction of Pharmacokinetic and Physicochemical Properties
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cummings, J.; Zhou, Y.; Lee, G.; Zhong, K.; Fonseca, J.; Cheng, F. Alzheimer’s disease drug development pipeline: 2024. Alzheimer’s Dement. 2024, 10, e12465. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, J.; Commas-Herrera, A.; D’Amico, F.; Farina, N.; Wong, J.; Orange, J.; Gaber, S.; Knapp, M.; Salcher-Konrad, M.; Stevens, M.; et al. The World Alzheimer Report 2019: Attitudes to Dementia; Alzheimer’s Disease International (ADI): London, UK, 2019. [Google Scholar]
- van der Kant, R.; Goldstein, L.S.B.; Ossenkoppele, R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosc. 2020, 21, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Scarpini, E.; Schelterns, P.; Feldman, H. Treatment of Alzheimer’s disease: Current status and new perspectives. Lancet Neurol. 2003, 2, 539–547. [Google Scholar] [CrossRef]
- Greenough, M.A.; Camakaris, J.; Bush, A.I. Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem. Int. 2013, 62, 540–555. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Nahed, P.; Kambar, M.E.Z.N.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s disease drug development pipeline: 2022. Alzheimer’s Dement. 2022, 8, e12295. [Google Scholar] [CrossRef]
- Zemek, F.; Drtinova, L.; Nepovimova, E.; Sepsova, V.; Korabecny, J.; Klimes, J.; Kuca, K. Outcomes of Alzheimer’s disease therapy with acetylcholinesterase inhibitors and memantine. Expert. Opin. Drug Saf. 2014, 13, 759–774. [Google Scholar] [CrossRef]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimer’s Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef]
- Calhoun, A.; King, C.; Khoury, R.; Grossberg, G.T. An evaluation of memantine ER/donepezil for the treatment of Alzheimer’s disease. Expert. Opin. Pharmacother. 2018, 19, 1711–1717. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in early Alzheimer’s disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Pardo-Moreno, T.; Gonzalez-Acedo, A.; Rivas-Dominguez, A.; Garcia-Morales, V.; Garcia-Cozar, F.J.; Ramos-Rodriguez, J.J.; Melguizo-Rodriguez, L. Therapeutic approach to Alzheimer’s disease: Current treatments and new perspectives. Pharmaceutics 2022, 14, 1117. [Google Scholar] [CrossRef]
- Albertini, C.; Salerno, A.; Pinheiro, P.S.M.; Bolognesi, M.L. From combinations to multitarget-directed ligands: A continuum in Alzheimer’s disease polypharmacology. Med. Res. Rev. 2021, 41, 2606–2633. [Google Scholar] [CrossRef]
- Oset-Gasque, M.J.; Marco-Contelles, J. Alzheimer’s disease, the “One-Molecule, One-Target” paradigm, and the multitarget directed ligand approach. ACS Chem. Neurosci. 2018, 9, 401–403. [Google Scholar] [CrossRef]
- Santos, M.A.; Chand, K.; Chaves, S. Recent progress in repositioning Alzheimer’s disease drugs based on a multitarget strategy. Future Med. Chem. 2016, 8, 2113–2142. [Google Scholar] [CrossRef] [PubMed]
- Sampietro, A.; Perez-Areales, F.J.; Martinez, P.; Arce, E.M.; Galdeano, C.; Munoz-Torrero, D. Unveiling the multitarget anti-Alzheimer drug discovery landscape: A bibliometric analysis. Pharmaceuticals 2022, 15, 545. [Google Scholar] [CrossRef]
- Rossi, M.; Freschi, M.; Nascente, L.C.; Salerno, A.; Teixeira, S.M.V.; Nachon, F.; Chantegreil, F.; Soukup, O.; Prchal, L.; Malaguti, M.; et al. Sustainable drug discovery of multi-target-directed ligands for Alzheimer’s disease. J. Med. Chem. 2021, 64, 4972–4990. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Aguilera, Ó.M.; Hagenow, S.; Palomino-Antolin, A.; Farré-Alins, V.; Ismaili, L.; Joffrin, P.-L.; Jimeno, M.L.; Soukup, O.; Janočková, J.; Kalinowsky, L.; et al. Multitarget-directed ligands combining cholinesterase and monoamine oxidase inhibition with histamine H3R antagonism for neurodegenerative diseases. Angew. Chem. Int. Ed. Engl. 2017, 56, 12765–12769. [Google Scholar] [CrossRef]
- Hiremathad, A.; Keri, R.S.; Esteves, A.R.; Cardoso, S.M.; Chaves, S.; Santos, M.A. Novel Tacrine-hydroxyphenylbenzimidazole hybrids as potential multitarget drug candidates for Alzheimer’s disease. Eur. J. Med. Chem. 2018, 148, 255–267. [Google Scholar] [CrossRef]
- Chaves, S.; Várnagy, K.; Santos, M.A. Recent multi-target approaches on the development of anti-Alzheimer’s agents integrating metal chelation activity. Curr. Med. Chem. 2021, 28, 7247–7277. [Google Scholar] [CrossRef]
- Lane, R.M.; Potkin, S.G.; Enz, A. Targeting acetylcholinesterase and butyrylcholinesterase in dementia. Int. J. Neuropsychopharmacol. 2006, 9, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ma, C.; Li, Y.; Li, M.; Cui, T.; Zhao, X.; Li, Z.; Jia, H.; Wang, H.; Xiu, X.; et al. Design, synthesis and biological evaluation of carbamate derivatives incorporating multifunctional carrier scaffolds as pseudo-irreversible cholinesterase inhibitors for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2024, 265, 116071. [Google Scholar] [CrossRef]
- Vicente-Zurdo, D.; Rosales-Conrado, N.; León-González, M.E.; Brunetti, L.; Piemontese, L.; Pereira-Santos, A.R.; Cardoso, S.M.; Madrid, Y.; Chaves, S.; Santos, M.A. Novel rivastigmine derivatives as promising multi-target compounds for potential treatment of Alzheimer’s disease. Biomedicines 2022, 10, 1510. [Google Scholar] [CrossRef]
- Bon, L.; Bana’s, A.; Dias, I.; Melo-Marques, I.; Cardoso, S.M.; Chaves, S.; Santos, M.A. New multitarget rivastigmine–indole hybrids as potential drug candidates for Alzheimer’s disease. Pharmaceutics 2024, 16, 281. [Google Scholar] [CrossRef] [PubMed]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Kamaljeet; Singh, S.; Gupta, G.D.; Aran, K.R. Emerging role of antioxidants in Alzheimer’s disease: Insight into physiological, pathological mechanisms and management. Pharmaceut. Sci. Adv. 2024, 2, 100021. [Google Scholar] [CrossRef]
- Mezeiova, E.; Spilovska, K.; Nepovimova, E.; Gorecki, L.; Soukup, O.; Dolezal, R.; Malinak, D.; Janockova, J.; Jun, D.; Kuca, K.; et al. Profiling donepezil template into multipotent hybrids with antioxidant properties. J. Enz. Inhib. Med. Chem. 2018, 33, 583–606. [Google Scholar] [CrossRef]
- Cai, P.; Fang, S.-Q.; Yang, X.-L.; Wu, J.-J.; Liu, Q.-H.; Hong, H.; Wang, X.-B.; Kong, L.-Y. Rational design and multibiological profiling of novel donepezil−trolox hybrids against Alzheimer’s disease, with cholinergic, antioxidant, neuroprotective, and cognition enhancing properties. ACS Chem. Neurosci. 2017, 8, 2496–2511. [Google Scholar] [CrossRef]
- Cao, Y.; Zhang, L.; Sun, S.; Yi, Z.; Jiang, X.; Jia, D. Neuroprotective effects of syringic acid against OGD/R-induced injury in cultured hippocampal neuronal cells. Int. J. Mol. Med. 2016, 38, 567–573. [Google Scholar] [CrossRef]
- Quintanova, C.; Keri, R.S.; Marques, S.M.; Fernandes, M.G.; Cardoso, S.M.; Serralheiro, M.L.; Santos, M.A. Design, Synthesis and bioevaluation of tacrine-cinnamate based hybrids as potential bifunctional anti-Alzheimer drug candidates. MedChemComm 2015, 6, 1969–1977. [Google Scholar] [CrossRef]
- Chavarria, D.; Cagide, F.; Pinto, M.; Gomes, L.R.; Low, J.N.; Borges, F. Development of piperic acid-based monoamine oxidase inhibitors: Synthesis, structural characterization and biological evaluation. J. Mol. Struct. 2019, 1182, 298–307. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Perrin, D.D. Purification of Laboratory Chemicals, 4th ed.; Butterworth-Heinemann: Oxford, UK, 1999. [Google Scholar]
- Bartolini, M.; Bertucci, C.; Bolognesi, M.L.; Cavalli, A.; Melchiorre, C.; Andrisano, V. Insight into the kinetic of amyloid beta (1-42) peptide self-aggregation: Elucidation of inhibitors’ mechanism of action. ChemBioChem 2007, 8, 2152–2161. [Google Scholar] [CrossRef] [PubMed]
- Chao, X.; He, X.; Yang, Y.; Zhou, X.; Jin, M.; Liu, S.; Cheng, Z.; Liu, P.; Wang, Y.; Yu, J.; et al. Design, synthesis and pharmacological evaluation of novel tacrine-caffeic acid hybrids as multi-targeted compounds against Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2012, 22, 6498–6502. [Google Scholar] [CrossRef]
- Crystallographic Structure Factors and Electron Density, Resolution and R-Value, RCSB PDB-101. 2025. Available online: https://pdb101.rcsb.org/learn/guide-to-understanding-pdb-data/crystallographic-data (accessed on 5 February 2025).
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef]
- Brus, B.; Košak, U.; Turk, S.; Pišlar, A.; Coquelle, N.; Kos, J.; Stojan, J.; Colletier, J.-P.; Gobec, S. Discovery, biological evaluation, and crystal structure of a novel nanomolar selective butyrylcholinesterase inhibitor. J. Med. Chem. 2014, 57, 8167–8179. [Google Scholar] [CrossRef]
- Košak, U.; Knez, D.; Coquelle, N.; Brus, B.; Pišlar, A.; Nachon, F.; Brazzolotto, X.; Kos, J.; Colletier, J.; Gobe, S. N-propargylpiperidines with naphthalene-2-carboxamide or naphthalene-2-sulfonamide moieties: Potential multifunctional anti-Alzheimer’s agents. Bioorg. Med. Chem. 2017, 25, 633–645. [Google Scholar] [CrossRef]
- Viayna, E.; Coquelle, N.; Cieslikiewicz-Bouet, M.; Cisternas, P.; Oliva, C.A.; Sánchez-López, E.; Ettcheto, M.; Bartolini, M.; De Simone, A.; Ricchini, M.; et al. Discovery of a potent dual inhibitor of acetylcholinesterase and butyrylcholinesterase with antioxidant activity that alleviates Alzheimer-like pathology in old APP/PS1 mice. J. Med. Chem. 2021, 64, 812–839. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE) 2024.0601; Chemical Computing Group ULC: Montreal, QC, Canada, 2025; Available online: https://www.chemcomp.com/en/index.htm (accessed on 1 June 2024).
- Fadlan, A.; Nusantoro, Y.R. The effect of energy minimization on the molecular docking of acetone-based oxindole derivatives. J. Kim. Dan. Pendidik. Kim. 2021, 6, 69. [Google Scholar] [CrossRef]
- Maestro, Version 9.3; Schrödinger Inc.: Portland, OR, USA, 2012.
- Che, X.; Liu, Q.; Zhang, L. An accurate and universal protein-small molecule batch docking solution using Autodock Vina. Results Eng. 2023, 19, 101335. [Google Scholar] [CrossRef]
- Hoffmann, R. An extended Hückel theory. I. Hydrocarbons. J. Chem. Phys. 1963, 39, 1397–1412. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Gehlhaar, D.K.; Verkhivker, G.M.; Rejto, P.A.; Sherman, C.J.; Fogel, D.B.; Fogel, L.J.; Freer, S.T. Molecular recognition of the inhibitor AG-1343 by HIV-1 protease: Conformationally flexible docking by evolutionary programming. Chem. Biol. 1995, 2, 317–324. [Google Scholar] [CrossRef]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided. Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef]
- Mooij, W.T.M.; Verdonk, M.L. General and targeted statistical potentials for protein-ligand interactions. Proteins Struct. Funct. Genet. 2005, 61, 272–287. [Google Scholar] [CrossRef]
- McNutt, A.T.; Francoeur, P.; Aggarwal, R.; Masuda, T.; Meli, R.; Ragoza, M.; Sunseri, J.; Koes, D.R. GNINA 1.0: Molecular docking with deep learning. J. Cheminform. 2021, 13, 43. [Google Scholar] [CrossRef]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Neudert, G.; Klebe, G. fconv: Format conversion, manipulation and feature computation of molecular data. Bioinformatics 2011, 27, 1021–1022. [Google Scholar] [CrossRef]
- Bouysset, C.; Fiorucci, S. ProLIF: A library to encode molecular interactions as fingerprints. J. Cheminform. 2021, 13, 72. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- QikProp, Version 2.5; Schrödinger, LLC.: New York, NY, USA, 2005.
- Salazar, P.B.; Dupuy, F.G.; Fiori, M.C.; Stanfield, S.M.; McCord, J.; Altenberg, G.A.; Minahk, C.J. Nanodisc-associated acetylcholinesterase as a novel model system of physiological relevant membrane-bound cholinesterases. Inhibition by phenolic compounds. Biochem. Bioph. Acta—Biomembr. 2024, 1866, 184389. [Google Scholar] [CrossRef]
- Bosak, A.; Gazic, I.; Vinkovic, V.; Kovarik, Z. Stereoselective inhibition of human, mouse, and horse cholinesterases by bambuterol enantiomers. Chem. Biol. Interact. 2008, 175, 192–195. [Google Scholar] [CrossRef]
- Xu, Q.; Zhang, L.; Xia, G.; Zhan, D.; Zhu, J.; Zang, H. Synthesis and biological activity of trolox amide derivatives. Braz. J. Pharm. Sci. 2022, 58, e18887. [Google Scholar] [CrossRef]
- Chen, J.; Yang, J.; Ma, L.; Li, J.; Shahzad, N.; Kim, C.K. Structure-antioxidant activity relationship of methoxy, phenolic hydroxyl, and carboxylic acid groups of phenolic acids. Sci. Rep. 2020, 10, 2611. [Google Scholar] [CrossRef] [PubMed]
- Chavarria, D.; Silva, T.; Martins, D.; Bravo, J.; Summavielle, T.; Garrido, J.; Borges, F. Exploring cinnamic acid scaffold: Development of promising neuroprotective lipophilic antioxidants. MedChemComm 2015, 6, 1043–1053. [Google Scholar] [CrossRef]
- Kumar, J.; Shankar, G.; Kumar, S.; Thomas, J.; Singh, N.; Srikrishna, S.; Satija, J.; Krishnamurthy, S.; Modi, G.; Mishra, S.K. Extraction, isolation, synthesis, and biological evaluation of novel piperic acid derivatives for the treatment of Alzheimer’s disease. Mol. Divers. 2023, 28, 1439–1458. [Google Scholar] [CrossRef]
- Lee, S.Y.; Hwang, I.Y.; Jeong, C.S. Protective effects of cinnamic acid derivatives on gastric lesion. Nat. Prod. Sci. 2017, 23, 299–305. [Google Scholar] [CrossRef]
- Forero-Doria, O.; Guzmán, L.; Venturini, W.; Zapata-Gomez, F.; Duarte, Y.; Camargo-Ayala, L.; Echeverría, C.; Echeverría, J. O-Alkyl derivatives of ferulic and syringic acid as lipophilic antioxidants: Effect of the length of the alkyl chain on the improvement of the thermo-oxidative stability of sunflower oil. RSC Adv. 2024, 14, 22513–22524. [Google Scholar] [CrossRef]
- Pagano, K.; Tomaselli, S.; Molinari, H.; Ragona, L. Natural compounds as inhibitors of Aβ peptide aggregation: Chemical requirements and molecular mechanisms. Front. Neurosci. 2020, 14, 619667. [Google Scholar] [CrossRef] [PubMed]
- Ballard, C.G. Advances in the treatment of Alzheimer’s disease: Benefits of dual cholinesterase inhibition. Eur. Neurol. 2002, 47, 64–70. [Google Scholar] [CrossRef]
- Harel, M.; Sussman, J.L.; Krejci, E.; Bon, S.; Chanal, P.; Massoulie, J.; Silman, I. Conversion of acetylcholinesterase to butyrylcholinesterase: Modelling and mutagenesis. Proc. Natl. Acad. Sci. USA. 1992, 89, 10827–10831. [Google Scholar] [CrossRef]
- Chatonnet, A.; Lockridge, O. Comparison of butyrylcholinesterase and acetylcholinesterase. Biochem. J. 1989, 260, 625–634. [Google Scholar] [CrossRef]
- Rosenberry, T.L.; Brazzolotto, X.; Macdonald, I.R.; Wandhammer, M.; Trovaslet-Leroy, M.; Darvesh, S.; Nachon, F. Comparison of the binding of reversible inhibitors to human butyrylcholinesterase and acetylcholinesterase: A crystallographic, kinetic and calorimetric study. Molecules 2017, 22, 2098. [Google Scholar] [CrossRef]
- Dighe, S.N.; Deora, G.S.; Mora, E.; Nachon, F.; Chan, S.; Parat, M.O.; Brazzolotto, X.; Ross, B.P. Discovery and structure-activity relationships of a highly selective butyrylcholinesterase inhibitor by structure-based virtual screening. J. Med. Chem. 2016, 59, 7683–7689. [Google Scholar] [CrossRef]
- Sliwoski, G.R.; Meiler, J.; Lowe, E.W. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef]
- Silva, D.F.; Selfridge, J.E.; Lu, J.; Lezi, E.; Cardoso, S.M.; Swerdlow, R.H. Mitochondrial abnormalities in Alzheimer’s disease: Possible targets for therapeutic intervention. Adv. Pharmacol. 2012, 64, 83–126. [Google Scholar] [CrossRef] [PubMed]
- Candeias, E.; Pereira-Santos, A.R.; Empadinhas, N.; Cardoso, S.M.; Esteves, A.R.F. The gut-brain axis in Alzheimer’s and Parkinson’s diseases: The catalytic role of mitochondria. J. Alzheimer’s Dis. 2024, 100, 413–429. [Google Scholar] [CrossRef]
- Silva, D.F.; Candeias, E.; Esteves, A.R.; Magalhães, J.D.; Ferreira, I.L.; Nunes-Costa, D.; Rego, A.C.; Empadinhas, N.; Cardoso, S.M. Microbial BMAA elicits mitochondrial dysfunction, innate immunity activation, and Alzheimer’s disease features in cortical neurons. J. Neuroinflammation 2020, 17, 332. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.F.; Esteves, A.R.; Arduino, D.M.; Oliveira, C.R.; Cardoso, S.M. Amyloid-β-induced mitochondrial dysfunction impairs the autophagic lysosomal pathway in a tubulin dependent pathway. J. Alzheimer’s Dis. 2011, 26, 565–581. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | n | % AA a | AA a EC50 (μM) | AChE Inhib b IC50 (μM) | BChE Inhib b IC50 (μM) | SI c | % Aβ42 Self-Agg Inhib d | |
| 4AY1 | Et | 0 | <50 | - | >200 | 31 ± 2 | - | 20 ± 2 | |
| 4AY2 | Et | 0 | 95 ± 3 | 18.1 ± 0.4 | 69 ± 7 | 36 ± 3 | 1.9 | 45 ± 4 | |
| 4AY3 | Et | 0 | <50 | - | 50.5 ± 0.1 | 3.1 ± 0.4 | 16.3 | 45 ± 4 | |
| 4AY4 | Et | 0 | <50 | - | 14.51 ± 0.04 | 15.2 ± 0.4 | 0.9 | 23 ± 3 e | |
| 4AY5 | Et | 0 | 96 ± 2 | 15.7 ± 0.4 | 110 ± 5 | 7.2 ± 0.3 | 15.3 | 82 ± 5 | |
| 4AY6 | Et | 0 | 93.1 ± 0.8 | 28.0 ± 0.4 | 5 ± 1 | 5.7 ± 0.6 | 0.9 | 75 ± 7 | |
| 4BY1 | Et | 1 | <50 | - | >200 | 75.0 ± 0.6 | - | 58 ± 6 | |
| 4BY2 | Et | 1 | 93.9 ± 0.4 | 20.0 ± 0.8 | 32 ± 2 | 3.7 ± 0.1 | 8.6 | 47 ± 3 | |
| 4BY3 | Et | 1 | <50 | - | 91 ± 6 | 0.9 ± 0.2 | 101 | 29 ± 3 | |
| 4CY1 | Me | 0 | <50 | - | >200 | 68 ± 7 | - | 31 ± 3 e | |
| Trolox | - | - | - | 13.8 ± 0.2 | - | - | - | - | |
| Rivastigmine f | - | - | - | - | 32 ± 1 | 0.39 ± 0.09 | 82.3 | - | |
| Curcumin | - | - | - | - | - | - | - | 77 ± 1 | |
| One-way ANOVA | - | - | CL = 20 µM e | CL = 40 µM | |||||
| Compounds | 4AY(2,5,6), 4BY2 | 4AY(2,5,6), 4BY2, Trolox | 4AY(2–6), 4BY(2,3), rivastigmine | 4AY(1–6), 4BY(1–3), 4CY1, rivastigmine | 4AY4, 4CY1 | 4AY(1–3,5,6), 4BY(1–3), curcumin | |||
| Numerator | 4.134 | 91.01 | 2528.7 | 1468.4 | 120.12 | 1901.6 | |||
| Denominator | 4.108 | 0.274 | 33.22 | 10.40 | 15.01 | 25.79 | |||
| F value | F(3,8) = 1.006 | F(4,10) = 331.6 | F(7,8) = 76.13 | F(10,11) = 141.2 | F(1,6) = 8.002 | F(8,27) = 73.74 | |||
| p-value | 0.439 | <0.001 | <0.001 | <0.001 | 0.030 | <0.001 | |||
| Compound | MW a | PSA b | clog Po/w c | log K (HSA) d | log BB e | Caco-2 Permeab. f | MDCK Permeab. g | % Oral Absorption h |
|---|---|---|---|---|---|---|---|---|
| 4AY1 | 374.393 | 99.757 | 3.079 | 0.043 | −0.783 | 1437 | 732 | 91 |
| 4AY2 | 426.511 | 88.438/ 92.26 | 4.479/ 4.565 | 0.759/ 0.784 | −0.66/ −0.699 | 1449/ 1420 | 739/ 723 | 86/ 87 |
| 4AY3 | 368.388 | 91.183 | 3.444 | 0.143 | −0.615 | 1733 | 896 | 100 |
| 4AY4 | 394.426 | 91.452 | 4.123 | 0.327 | −0.797 | 1699 | 877 | 100 |
| 4AY5 | 356.377 | 115.137 | 2.512 | 0.032 | −1.953 | 175 | 75 | 72 |
| 4AY6 | 382.415 | 115.078 | 3.143 | 0.187 | −2.183 | 170 | 73 | 73 |
| 4BY1 | 388.419 | 102.759 | 3.19 | 0.129 | −1.153 | 793 | 385 | 83 |
| 4BY2 | 440.538 | 79.002/ 94.477 | 3.882/ 3.979 | 0.338/ 0.507 | −0.189/ −0.895 | 2193/ 513 | 1987/ 476 | 94/ 81 |
| 4BY3 | 382.415 | 94.46 | 3.627 | 0.252 | −0.949 | 1016 | 503 | 91 |
| 4CY1 | 360.366 | 100.414 | 2.697 | −0.039 | −0.845 | 1122 | 561 | 88 |
| Rivastigmine | 250.34 | 38.483 | 2.448 | −0.133 | 0.475 | 1381 | 776 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dias, I.; Emmanuel, M.; Vogt, P.; Guerreiro-Oliveira, C.; Melo-Marques, I.; Cardoso, S.M.; Guedes, R.C.; Chaves, S.; Santos, M.A. Rivastigmine Templates with Antioxidant Motifs—A Medicinal Chemist’s Toolbox Towards New Multipotent AD Drugs. Antioxidants 2025, 14, 921. https://doi.org/10.3390/antiox14080921
Dias I, Emmanuel M, Vogt P, Guerreiro-Oliveira C, Melo-Marques I, Cardoso SM, Guedes RC, Chaves S, Santos MA. Rivastigmine Templates with Antioxidant Motifs—A Medicinal Chemist’s Toolbox Towards New Multipotent AD Drugs. Antioxidants. 2025; 14(8):921. https://doi.org/10.3390/antiox14080921
Chicago/Turabian StyleDias, Inês, Marlène Emmanuel, Paul Vogt, Catarina Guerreiro-Oliveira, Inês Melo-Marques, Sandra M. Cardoso, Rita C. Guedes, Sílvia Chaves, and M. Amélia Santos. 2025. "Rivastigmine Templates with Antioxidant Motifs—A Medicinal Chemist’s Toolbox Towards New Multipotent AD Drugs" Antioxidants 14, no. 8: 921. https://doi.org/10.3390/antiox14080921
APA StyleDias, I., Emmanuel, M., Vogt, P., Guerreiro-Oliveira, C., Melo-Marques, I., Cardoso, S. M., Guedes, R. C., Chaves, S., & Santos, M. A. (2025). Rivastigmine Templates with Antioxidant Motifs—A Medicinal Chemist’s Toolbox Towards New Multipotent AD Drugs. Antioxidants, 14(8), 921. https://doi.org/10.3390/antiox14080921