
Prenatal Bisphenol A Exposure Impairs Fetal Heart Development: Molecular and Structural Alterations with Sex-Specific Differences

 ,
,  ,
,  , ,
, ,  , and
, and 
Abstract

1. Introduction
2. Materials and Methods
2.1. Animals and Treatments
2.2. Western Blot Analysis
2.3. Gelatin Zymography for Matrix Metalloproteinase Activity Detection
2.4. Gene Expression Analysis by qPCR
2.5. Evaluation of Malondialdehyde (MDA) Levels
2.6. Detection of Protein Carbonylation
2.7. Histological Analysis
2.8. Immunofluorescence and Immunohistochemistry
2.9. Statistical Analysis
3. Results
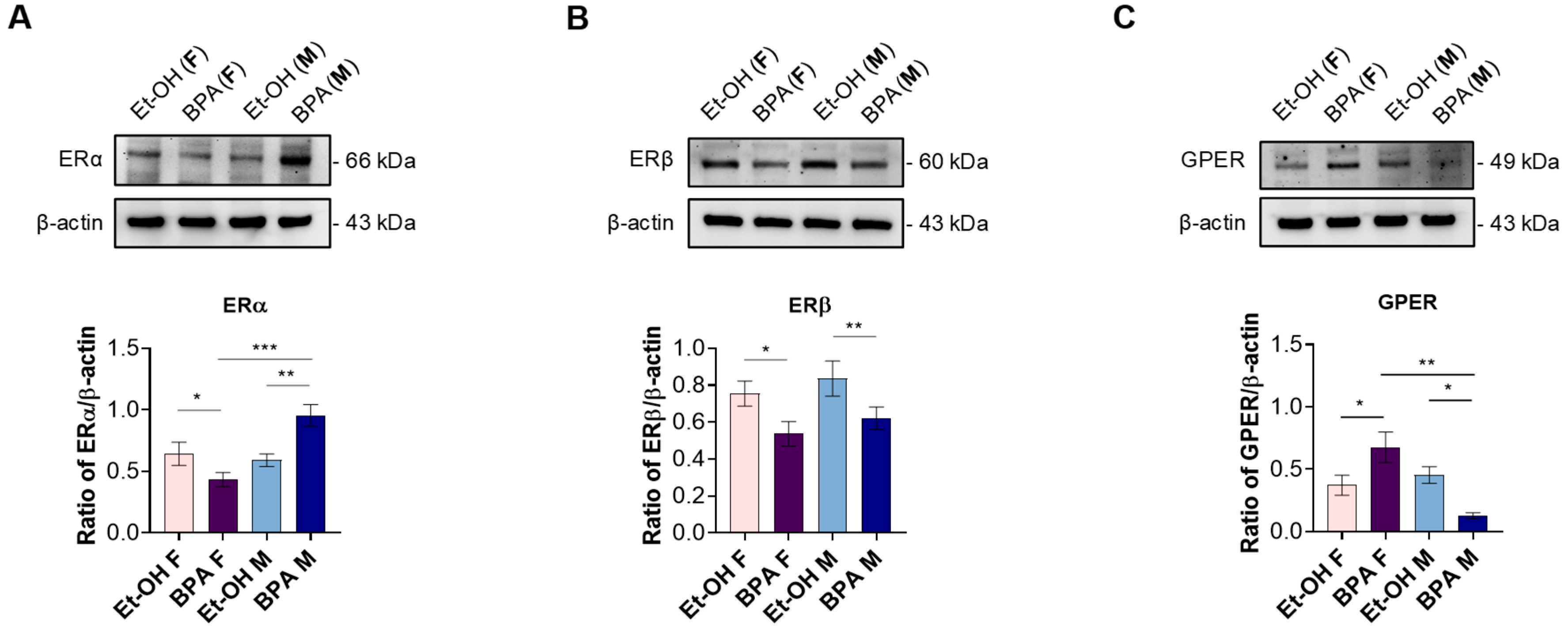
3.1. Prenatal BPA Exposure Alters Estrogen Receptor Expression in a Sex-Specific Manner in the Fetal Rat Heart
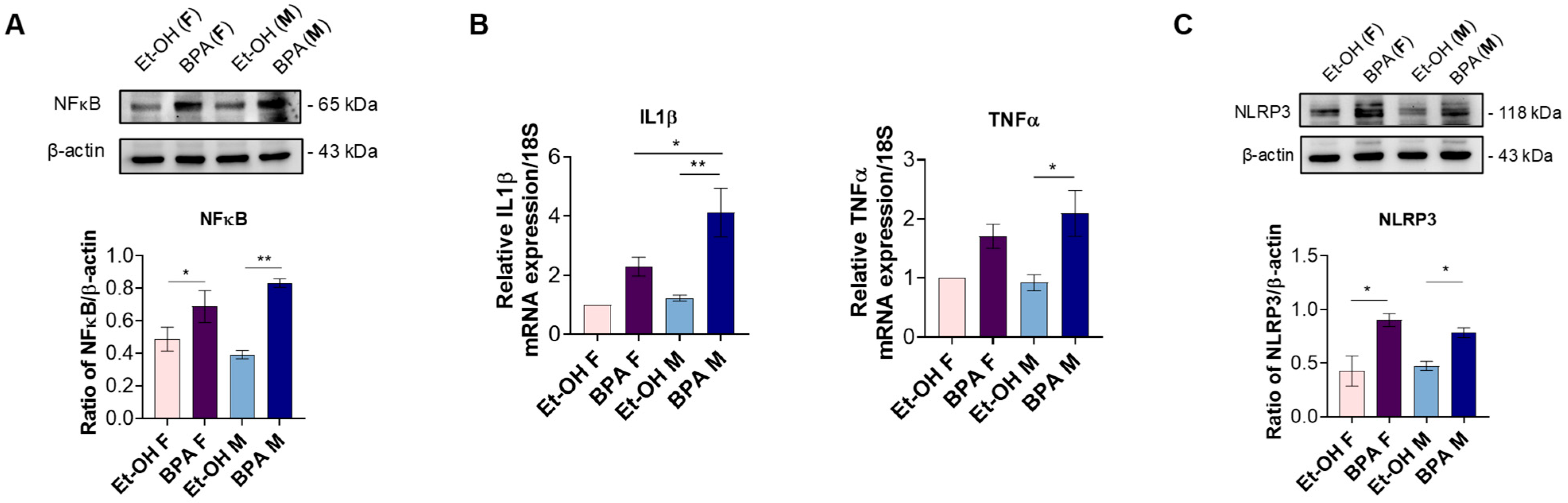
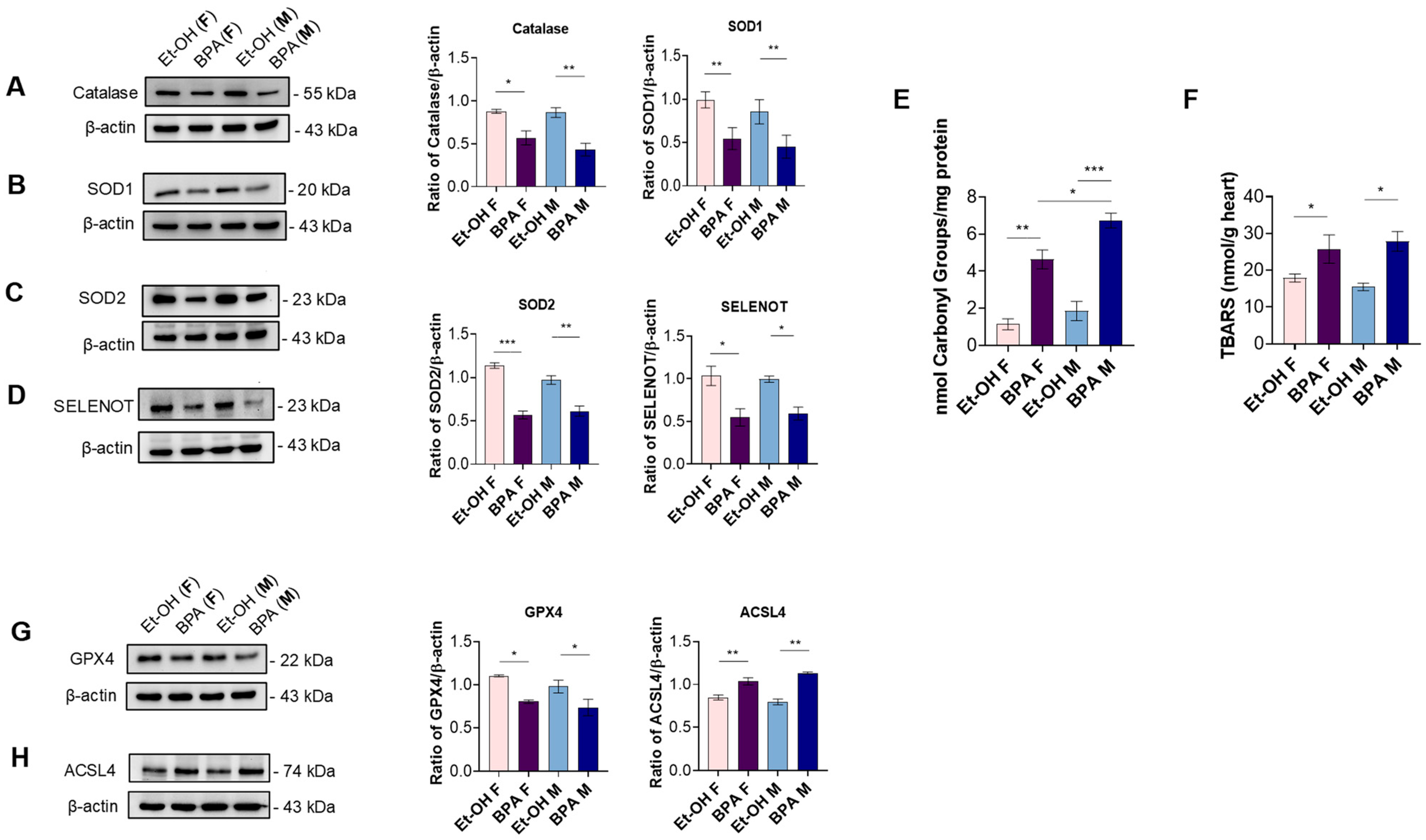
3.2. Prenatal BPA Exposure Triggers Inflammation, Oxidative Stress, and Ferroptosis in the Fetal Rat Heart
3.3. Prenatal BPA Exposure Affects Markers of Cardiac Distension, Remodeling, and Fibrosis in the Fetal Rat Heart
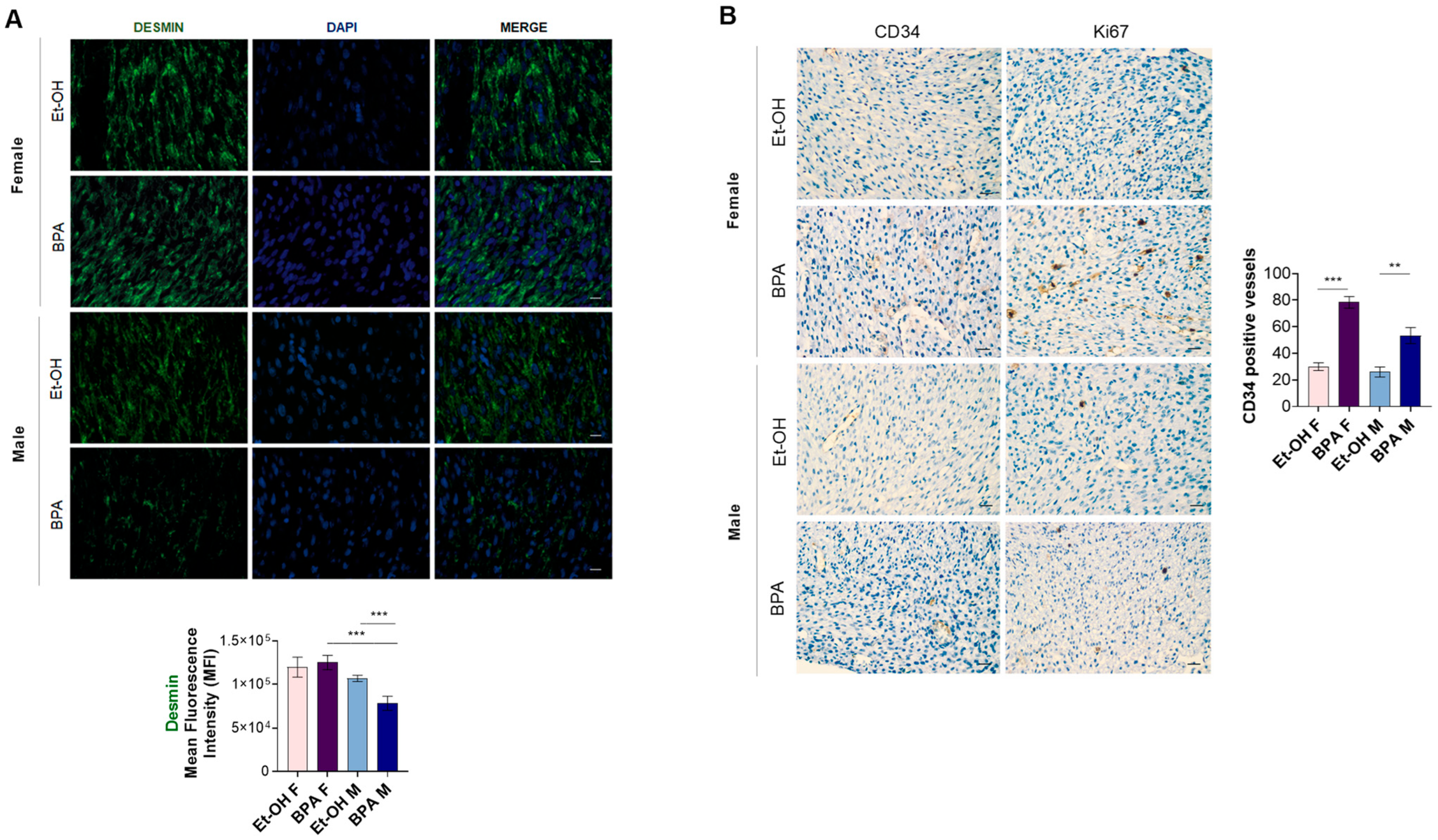
3.4. Prenatal BPA Exposure Induces Histological Alterations in the Fetal Rat Heart
4. Discussion
Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACSL4 | acyl-CoA synthetase long-chain family member 4 |
| ANP | Atrial Natriuretic Peptide |
| BNP | Brain Natriuretic Peptide |
| BPA | Bisphenol A |
| BPA-GA | BPA-glucuronide |
| CAT | Catalase |
| Col1A1 | Collagen type I alpha 1 chain |
| Col3A1 | Collagen type III alpha 1 chain |
| CTGF | Connective tissue growth factor |
| CVD | Cardiovascular disease |
| ECM | Extracellular matrix |
| EDCs | Endocrine-disrupting chemicals |
| ER | Estrogen receptor |
| EtOH | Ethanol |
| GPR30/GPER | G protein-coupled estrogen receptor 30 |
| GPX4 | Glutathione peroxidase 4 |
| H.E. | Hematoxylin and eosin |
| hiPSC | Human-induced pluripotent stem cell |
| IL-1β | Interleukin-1β |
| MMPs | Matrix metalloproteinases |
| NF-κB | Nuclear Factor kappa B |
| NLRP3 | NOD-like receptor protein 3 |
| qPCR | Quantitative real-time PCR |
| SELENOT | Selenoprotein T |
| SERM | Selective estrogen receptor modulator |
| SOD | Superoxide dismutase |
| TGF-β | Transforming growth factor-β |
| TNF-α | Tumor necrosis factor-α |
References
- Hanson, M.A.; Gluckman, P.D. Early Developmental Conditioning of Later Health and Disease: Physiology or Pathophysiology? Physiol. Rev. 2014, 94, 1027–1076. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J.P. The Origins of the Developmental Origins Theory. J. Intern. Med. 2007, 261, 412–417. [Google Scholar] [CrossRef]
- Batra, V.; Norman, E.; Morgan, H.L.; Watkins, A.J. Parental Programming of Offspring Health: The Intricate Interplay between Diet, Environment, Reproduction and Development. Biomolecules 2022, 12, 1289. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, V.; Cardoso, R.C.; Puttabyatappa, M. Developmental Programming, a Pathway to Disease. Endocrinology 2016, 157, 1328–1340. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, M.A.; Fleming, T.P.; Watkins, A.J. Periconceptional Environment and the Developmental Origins of Disease. J. Endocrinol. 2019, 242, T33–T49. [Google Scholar] [CrossRef]
- Fleming, T.P.; Watkins, A.J.; Velazquez, M.A.; Mathers, J.C.; Prentice, A.M.; Stephenson, J.; Barker, M.; Saffery, R.; Yajnik, C.S.; Eckert, J.J.; et al. Origins of Lifetime Health around the Time of Conception: Causes and Consequences. Lancet 2018, 391, 1842–1852. [Google Scholar] [CrossRef]
- Alexander, B.T.; Dasinger, J.H.; Intapad, S. Fetal Programming and Cardiovascular Pathology. In Comprehensive Physiology; Wiley: Hoboken, NJ, USA, 2015; pp. 997–1025. [Google Scholar]
- Barker, D.J. Fetal Origins of Cardiovascular Disease. Ann. Med. 1999, 31 (Suppl. 1), 3–6. [Google Scholar] [CrossRef]
- Barker, D.J.P. Fetal Origins of Coronary Heart Disease. BMJ 1995, 311, 171–174. [Google Scholar] [CrossRef]
- Meyer, K.; Zhang, L. Fetal Programming of Cardiac Function and Disease. Reprod. Sci. 2007, 14, 209–216. [Google Scholar] [CrossRef]
- Wagner, N.; Wagner, K.-D. Transcriptional Regulation of Cardiac Development and Disease. Int. J. Mol. Sci. 2022, 23, 2945. [Google Scholar] [CrossRef]
- Ward, E.J.; Bert, S.; Fanti, S.; Malone, K.M.; Maughan, R.T.; Gkantsinikoudi, C.; Prin, F.; Volpato, L.K.; Piovezan, A.P.; Graham, G.J.; et al. Placental Inflammation Leads to Abnormal Embryonic Heart Development. Circulation 2023, 147, 956–972. [Google Scholar] [CrossRef]
- Dai, J.; Wang, G.; Wu, C.; Pan, Z.; Li, H.; Shen, L.; Wu, Y. Exposure to Endocrine-Disrupting Chemicals and Congenital Heart Diseases: The Pooled Results Based on the Current Evidence. Pediatr. Cardiol. 2024, 46, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Escarda-Castro, E.; Herráez, M.P.; Lombó, M. Effects of Bisphenol A Exposure during Cardiac Cell Differentiation. Environ. Pollut. 2021, 286, 117567. [Google Scholar] [CrossRef] [PubMed]
- Khalili Sadrabad, E.; Hashemi, S.A.; Nadjarzadeh, A.; Askari, E.; Akrami Mohajeri, F.; Ramroudi, F. Bisphenol A Release from Food and Beverage Containers—A Review. Food Sci. Nutr. 2023, 11, 3718–3728. [Google Scholar] [CrossRef] [PubMed]
- Manzoor, M.F.; Tariq, T.; Fatima, B.; Sahar, A.; Tariq, F.; Munir, S.; Khan, S.; Nawaz Ranjha, M.M.A.; Sameen, A.; Zeng, X.-A.; et al. An Insight into Bisphenol A, Food Exposure and Its Adverse Effects on Health: A Review. Front. Nutr. 2022, 9, 1047827. [Google Scholar] [CrossRef]
- Vandenberg, L.N.; Hauser, R.; Marcus, M.; Olea, N.; Welshons, W.V. Human Exposure to Bisphenol A (BPA). Reprod. Toxicol. 2007, 24, 139–177. [Google Scholar] [CrossRef]
- Lee, J.; Choi, K.; Park, J.; Moon, H.-B.; Choi, G.; Lee, J.J.; Suh, E.; Kim, H.-J.; Eun, S.-H.; Kim, G.-H.; et al. Bisphenol A Distribution in Serum, Urine, Placenta, Breast Milk, and Umbilical Cord Serum in a Birth Panel of Mother-Neonate Pairs. Sci. Total Environ. 2018, 626, 1494–1501. [Google Scholar] [CrossRef]
- Nishikawa, M.; Iwano, H.; Yanagisawa, R.; Koike, N.; Inoue, H.; Yokota, H. Placental Transfer of Conjugated Bisphenol A and Subsequent Reactivation in the Rat Fetus. Environ. Health Perspect. 2010, 118, 1196–1203. [Google Scholar] [CrossRef]
- Nachman, R.M.; Hartle, J.C.; Lees, P.S.J.; Groopman, J.D. Early Life Metabolism of Bisphenol A: A Systematic Review of the Literature. Curr. Environ. Health Rep. 2014, 1, 90–100. [Google Scholar] [CrossRef]
- Hafezi, S.A.; Abdel-Rahman, W.M. The Endocrine Disruptor Bisphenol A (BPA) Exerts a Wide Range of Effects in Carcinogenesis and Response to Therapy. Curr. Mol. Pharmacol. 2019, 12, 230–238. [Google Scholar] [CrossRef]
- Fonseca, M.I.; Lorigo, M.; Cairrao, E. Endocrine-Disrupting Effects of Bisphenol A on the Cardiovascular System: A Review. J. Xenobiotics 2022, 12, 181–213. [Google Scholar] [CrossRef]
- Kang, J.-H.; Asai, D.; Toita, R. Bisphenol A (BPA) and Cardiovascular or Cardiometabolic Diseases. J. Xenobiotics 2023, 13, 775–810. [Google Scholar] [CrossRef] [PubMed]
- Matuszczak, E.; Komarowska, M.D.; Debek, W.; Hermanowicz, A. The Impact of Bisphenol A on Fertility, Reproductive System, and Development: A Review of the Literature. Int. J. Endocrinol. 2019, 2019, 4068717. [Google Scholar] [CrossRef] [PubMed]
- Colorado-Yohar, S.M.; Castillo-González, A.C.; Sánchez-Meca, J.; Rubio-Aparicio, M.; Sánchez-Rodríguez, D.; Salamanca-Fernández, E.; Ardanaz, E.; Amiano, P.; Fernández, M.F.; Mendiola, J.; et al. Concentrations of Bisphenol-A in Adults from the General Population: A Systematic Review and Meta-Analysis. Sci. Total Environ. 2021, 775, 145755. [Google Scholar] [CrossRef]
- Rocca, C.; Boukhzar, L.; Granieri, M.C.; Alsharif, I.; Mazza, R.; Lefranc, B.; Tota, B.; Leprince, J.; Cerra, M.C.; Anouar, Y.; et al. A Selenoprotein T-derived Peptide Protects the Heart against Ischaemia/Reperfusion Injury through Inhibition of Apoptosis and Oxidative Stress. Acta Physiol. 2018, 223, e13067. [Google Scholar] [CrossRef] [PubMed]
- Rocca, C.; Scavello, F.; Colombo, B.; Gasparri, A.M.; Dallatomasina, A.; Granieri, M.C.; Amelio, D.; Pasqua, T.; Cerra, M.C.; Tota, B.; et al. Physiological Levels of Chromogranin A Prevent Doxorubicin-induced Cardiotoxicity Without Impairing Its Anticancer Activity. FASEB J. 2019, 33, 7734–7747. [Google Scholar] [CrossRef]
- De Bartolo, A.; Romeo, N.; Marrone, A.; Rago, V.; Granieri, M.C.; Vommaro, M.L.; Cupelli, A.; Cerra, M.C.; Indiveri, C.; Ronca, R.; et al. A Recombinant Fragment Antigen-Binding (Fab) of Trastuzumab Displays Low Cytotoxic Profile in Adult Human Cardiomyocytes: First Evidence and the Key Implication of FcγRIIA Receptor. Acta Pharmacol. Sin. 2024, 46, 618–631. [Google Scholar] [CrossRef]
- De Bartolo, A.; Pasqua, T.; Romeo, N.; Rago, V.; Perrotta, I.; Giordano, F.; Granieri, M.C.; Marrone, A.; Mazza, R.; Cerra, M.C.; et al. The Redox-Active Defensive Selenoprotein T as a Novel Stress Sensor Protein Playing a Key Role in the Pathophysiology of Heart Failure. J. Transl. Med. 2024, 22, 375. [Google Scholar] [CrossRef]
- Rocca, C.; De Bartolo, A.; Grande, F.; Rizzuti, B.; Pasqua, T.; Giordano, F.; Granieri, M.C.; Occhiuzzi, M.A.; Garofalo, A.; Amodio, N.; et al. Cateslytin Abrogates Lipopolysaccharide-Induced Cardiomyocyte Injury by Reducing Inflammation and Oxidative Stress through Toll like Receptor 4 Interaction. Int. Immunopharmacol. 2021, 94, 107487. [Google Scholar] [CrossRef]
- Li, T.; Danelisen, I.; Belló-Klein, A.; Singal, P.K. Effects of Probucol on Changes of Antioxidant Enzymes in Adriamycin-Induced Cardiomyopathy in Rats. Cardiovasc. Res. 2000, 46, 523–530. [Google Scholar] [CrossRef]
- Rocca, C.; De Bartolo, A.; Granieri, M.C.; Rago, V.; Amelio, D.; Falbo, F.; Malivindi, R.; Mazza, R.; Cerra, M.C.; Boukhzar, L.; et al. The Antioxidant Selenoprotein T Mimetic, PSELT, Induces Preconditioning-like Myocardial Protection by Relieving Endoplasmic-Reticulum Stress. Antioxidants 2022, 11, 571. [Google Scholar] [CrossRef] [PubMed]
- Reznick, A.Z.; Packer, L. Oxidative Damage to Proteins: Spectrophotometric Method for Carbonyl Assay. Methods Enzymol. 1994, 233, 357–363. [Google Scholar] [CrossRef]
- Weber, D.; Davies, M.J.; Grune, T. Determination of Protein Carbonyls in Plasma, Cell Extracts, Tissue Homogenates, Isolated Proteins: Focus on Sample Preparation and Derivatization Conditions. Redox Biol. 2015, 5, 367–380. [Google Scholar] [CrossRef]
- Alomari, E.; Bruno, S.; Ronda, L.; Paredi, G.; Bettati, S.; Mozzarelli, A. Protein Carbonylation Detection Methods: A Comparison. Data Br. 2018, 19, 2215–2220. [Google Scholar] [CrossRef]
- Pasqua, T.; Rocca, C.; Lupi, F.R.; Baldino, N.; Amelio, D.; Parisi, O.I.; Granieri, M.C.; De Bartolo, A.; Lauria, A.; Dattilo, M.; et al. Cardiac and Metabolic Impact of Functional Foods with Antioxidant Properties Based on Whey Derived Proteins Enriched with Hemp Seed Oil. Antioxidants 2020, 9, 1066. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-ΚB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Dhalla, N.S. The Role of Pro-Inflammatory Cytokines in the Pathogenesis of Cardiovascular Disease. Int. J. Mol. Sci. 2024, 25, 1082. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, H.M.; Wanderer, A.A. Inflammasome and IL-1β-Mediated Disorders. Curr. Allergy Asthma Rep. 2010, 10, 229–235. [Google Scholar] [CrossRef]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in Ferroptosis and Its Pharmacological Implication. Free Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 Dictates Ferroptosis Sensitivity by Shaping Cellular Lipid Composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Martins, D.; Garcia, L.R.; Queiroz, D.A.R.; Lazzarin, T.; Tonon, C.R.; da Balin, P.S.; Polegato, B.F.; de Paiva, S.A.R.; Azevedo, P.S.; Minicucci, M.F.; et al. Oxidative Stress as a Therapeutic Target of Cardiac Remodeling. Antioxidants 2022, 11, 2371. [Google Scholar] [CrossRef] [PubMed]
- Kologrivova, I.; Shtatolkina, M.; Suslova, T.; Ryabov, V. Cells of the Immune System in Cardiac Remodeling: Main Players in Resolution of Inflammation and Repair After Myocardial Infarction. Front. Immunol. 2021, 12, 664457. [Google Scholar] [CrossRef] [PubMed]
- Felkin, L.E.; Lara-Pezzi, E.; George, R.; Yacoub, M.H.; Birks, E.J.; Barton, P.J.R. Expression of Extracellular Matrix Genes During Myocardial Recovery from Heart Failure After Left Ventricular Assist Device Support. J. Hear. Lung Transplant. 2009, 28, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Accornero, F.; van Berlo, J.H.; Correll, R.N.; Elrod, J.W.; Sargent, M.A.; York, A.; Rabinowitz, J.E.; Leask, A.; Molkentin, J.D. Genetic Analysis of Connective Tissue Growth Factor as an Effector of Transforming Growth Factor β Signaling and Cardiac Remodeling. Mol. Cell. Biol. 2015, 35, 2154–2164. [Google Scholar] [CrossRef]
- McLendon, P.M.; Robbins, J. Desmin-Related Cardiomyopathy: An Unfolding Story. Am. J. Physiol. Circ. Physiol. 2011, 301, H1220–H1228. [Google Scholar] [CrossRef]
- Sidney, L.E.; Branch, M.J.; Dunphy, S.E.; Dua, H.S.; Hopkinson, A. Concise Review: Evidence for CD34 as a Common Marker for Diverse Progenitors. Stem Cells 2014, 32, 1380–1389. [Google Scholar] [CrossRef]
- Uxa, S.; Castillo-Binder, P.; Kohler, R.; Stangner, K.; Müller, G.A.; Engeland, K. Ki-67 Gene Expression. Cell Death Differ. 2021, 28, 3357–3370. [Google Scholar] [CrossRef]
- Rocca, C.; Angelone, T. Emerging Molecular Determinants and Protective Strategies in Heart Disease: What’s New in the Journal of Clinical Medicine? Outlook to the Future. J. Clin. Med. 2023, 12, 4564. [Google Scholar] [CrossRef]
- Ventura-Clapier, R.; Piquereau, J.; Veksler, V.; Garnier, A. Estrogens, Estrogen Receptors Effects on Cardiac and Skeletal Muscle Mitochondria. Front. Endocrinol. 2019, 10, 557. [Google Scholar] [CrossRef]
- Iorga, A.; Cunningham, C.M.; Moazeni, S.; Ruffenach, G.; Umar, S.; Eghbali, M. The Protective Role of Estrogen and Estrogen Receptors in Cardiovascular Disease and the Controversial Use of Estrogen Therapy. Biol. Sex Differ. 2017, 8, 33. [Google Scholar] [CrossRef]
- Chapalamadugu, K.C.; VandeVoort, C.A.; Settles, M.L.; Robison, B.D.; Murdoch, G.K. Maternal Bisphenol A Exposure Impacts the Fetal Heart Transcriptome. PLoS ONE 2014, 9, e89096. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Li, B.; Yan, Y.; Zhang, N.; Shao, S.; Yang, L.; Ouyang, L.; Wu, P.; Duan, H.; Zhou, K.; et al. Maternal Exposure to Bisphenol A Induces Congenital Heart Disease through Mitochondrial Dysfunction. FASEB J. 2025, 39, e70351. [Google Scholar] [CrossRef]
- vom Saal, F.S.; Hughes, C. An Extensive New Literature Concerning Low-Dose Effects of Bisphenol A Shows the Need for a New Risk Assessment. Environ. Health Perspect. 2005, 113, 926–933. [Google Scholar] [CrossRef]
- Vandenberg, L.N. Non-Monotonic Dose Responses in Studies of Endocrine Disrupting Chemicals: Bisphenol a as a Case Study. Dose Response 2014, 12, 259–276. [Google Scholar] [CrossRef]
- Lamberto, F.; Shashikadze, B.; Elkhateib, R.; Lombardo, S.D.; Horánszky, A.; Balogh, A.; Kistamás, K.; Zana, M.; Menche, J.; Fröhlich, T.; et al. Low-Dose Bisphenol A Exposure Alters the Functionality and Cellular Environment in a Human Cardiomyocyte Model. Environ. Pollut. 2023, 335, 122359. [Google Scholar] [CrossRef] [PubMed]
- Lombó, M.; González-Rojo, S.; Fernández-Díez, C.; Herráez, M.P. Cardiogenesis Impairment Promoted by Bisphenol A Exposure Is Successfully Counteracted by Epigallocatechin Gallate. Environ. Pollut. 2019, 246, 1008–1019. [Google Scholar] [CrossRef]
- Chen, L.; Wang, Z.; Gu, W.; Zhang, X.; Ren, H.; Wu, B. Single-Cell Sequencing Reveals Heterogeneity Effects of Bisphenol A on Zebrafish Embryonic Development. Environ. Sci. Technol. 2020, 54, 9537–9546. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Adachi, Y.; Liu, P.; Fukuma, N.; Takimoto, E. Regulatory Actions of Estrogen Receptor Signaling in the Cardiovascular System. Front. Endocrinol. 2020, 10, 909. [Google Scholar] [CrossRef]
- Meyer, M.R.; Prossnitz, E.R.; Barton, M. The G Protein-Coupled Estrogen Receptor GPER/GPR30 as a Regulator of Cardiovascular Function. Vascul. Pharmacol. 2011, 55, 17–25. [Google Scholar] [CrossRef]
- Aryan, L.; Younessi, D.; Zargari, M.; Banerjee, S.; Agopian, J.; Rahman, S.; Borna, R.; Ruffenach, G.; Umar, S.; Eghbali, M. The Role of Estrogen Receptors in Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 4314. [Google Scholar] [CrossRef]
- Sakamoto, T.; Matsuura, T.R.; Wan, S.; Ryba, D.M.; Kim, J.U.; Won, K.J.; Lai, L.; Petucci, C.; Petrenko, N.; Musunuru, K.; et al. A Critical Role for Estrogen-Related Receptor Signaling in Cardiac Maturation. Circ. Res. 2020, 126, 1685–1702. [Google Scholar] [CrossRef]
- Mahmoodzadeh, S.; Eder, S.; Nordmeyer, J.; Ehler, E.; Huber, O.; Martus, P.; Weiske, J.; Pregla, R.; Hetzer, R.; Regitz-Zagrosek, V. Estrogen Receptor Alpha Up-regulation and Redistribution in Human Heart Failure. FASEB J. 2006, 20, 926–934. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Rocca, C.; Scavello, F.; Amelio, D.; Pasqua, T.; Rigiracciolo, D.C.; Scarpelli, A.; Avino, S.; Cirillo, F.; Amodio, N.; et al. Protective Role of GPER Agonist G-1 on Cardiotoxicity Induced by Doxorubicin. J. Cell. Physiol. 2017, 232, 1640–1649. [Google Scholar] [CrossRef]
- Kalaitzidis, D.; Gilmore, T.D. Transcription Factor Cross-Talk: The Estrogen Receptor and NF-KappaB. Trends Endocrinol. Metab. 2005, 16, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Goswami, S.K.; Ranjan, P.; Dutta, R.K.; Verma, S.K. Management of Inflammation in Cardiovascular Diseases. Pharmacol. Res. 2021, 173, 105912. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Liu, K.; Zhu, L. Emerging Roles of Inflammasomes in Cardiovascular Diseases. Front. Immunol. 2022, 13, 834289. [Google Scholar] [CrossRef]
- Angelone, T.; Rocca, C.; Lionetti, V.; Penna, C.; Pagliaro, P. Expanding the Frontiers of Guardian Antioxidant Selenoproteins in Cardiovascular Pathophysiology. Antioxid. Redox Signal. 2024, 40, 369–432. [Google Scholar] [CrossRef]
- Rocca, C.; Pasqua, T.; Boukhzar, L.; Anouar, Y.; Angelone, T. Progress in the Emerging Role of Selenoproteins in Cardiovascular Disease: Focus on Endoplasmic Reticulum-Resident Selenoproteins. Cell. Mol. Life Sci. 2019, 76, 3969–3985. [Google Scholar] [CrossRef]
- Boukhzar, L.; Hamieh, A.; Cartier, D.; Tanguy, Y.; Alsharif, I.; Castex, M.; Arabo, A.; El Hajji, S.; Bonnet, J.-J.; Errami, M.; et al. Selenoprotein T Exerts an Essential Oxidoreductase Activity That Protects Dopaminergic Neurons in Mouse Models of Parkinson’s Disease. Antioxid. Redox Signal. 2016, 24, 557–574. [Google Scholar] [CrossRef]
- Meagher, E.A.; FitzGerald, G.A. Indices of Lipid Peroxidation in Vivo: Strengths and Limitations. Free Radic. Biol. Med. 2000, 28, 1745–1750. [Google Scholar] [CrossRef]
- Chen, Y.; Meng, Z.; Li, S.; Wang, W.; Wang, Y.; Yin, C. Exposure to Bisphenol A Induces Abnormal Fetal Heart Development by Promoting Ferroptosis. Ecotoxicol. Environ. Saf. 2023, 255, 114753. [Google Scholar] [CrossRef]
- Sergeeva, I.A.; Christoffels, V.M. Regulation of Expression of Atrial and Brain Natriuretic Peptide, Biomarkers for Heart Development and Disease. Biochim. Biophys. Acta -Mol. Basis Dis. 2013, 1832, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Kerkelä, R.; Ulvila, J.; Magga, J. Natriuretic Peptides in the Regulation of Cardiovascular Physiology and Metabolic Events. J. Am. Heart Assoc. 2015, 4, e002423. [Google Scholar] [CrossRef]
- Bergman, M.R.; Teerlink, J.R.; Mahimkar, R.; Li, L.; Zhu, B.-Q.; Nguyen, A.; Dahi, S.; Karliner, J.S.; Lovett, D.H. Cardiac Matrix Metalloproteinase-2 Expression Independently Induces Marked Ventricular Remodeling and Systolic Dysfunction. Am. J. Physiol. Circ. Physiol. 2007, 292, H1847–H1860. [Google Scholar] [CrossRef] [PubMed]
- Buckley, L.F.; Agha, A.M.; Dorbala, P.; Claggett, B.L.; Yu, B.; Hussain, A.; Nambi, V.; Chen, L.Y.; Matsushita, K.; Hoogeveen, R.C.; et al. MMP-2 Associates with Incident Heart Failure and Atrial Fibrillation: The ARIC Study. Circ. Hear. Fail. 2023, 16, e010849. [Google Scholar] [CrossRef]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β Signaling in Fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Rasdi, Z.; Kamaludin, R.; Rahim, A.S.; Syed Ahmad Fuad, S.B.; Othman, M.H.D.; Siran, R.; Mohd Nor, N.S.; Abdul Hamid Hasani, N.; Sheikh Abdul Kadir, S.H. The Impacts of Intrauterine Bisphenol A Exposure on Pregnancy and Expression of MiRNAs Related to Heart Development and Diseases in Animal Model. Sci. Rep. 2020, 10, 5882. [Google Scholar] [CrossRef]
- Ghazal, R.; Wang, M.; Liu, D.; Tschumperlin, D.J.; Pereira, N.L. Cardiac Fibrosis in the Multi-Omics Era: Implications for Heart Failure. Circ. Res. 2025, 136, 773–802. [Google Scholar] [CrossRef]
- Pasqua, T.; Rocca, C.; Giglio, A.; Angelone, T. Cardiometabolism as an Interlocking Puzzle between the Healthy and Diseased Heart: New Frontiers in Therapeutic Applications. J. Clin. Med. 2021, 10, 721. [Google Scholar] [CrossRef]
- Ermini, L.; Mandalà, M.; Cresti, L.; Passaponti, S.; Patrussi, L.; Paulesu, L.; Thornburg, K.; Ietta, F. Fetal Myocardial Expression of GLUT1: Roles of BPA Exposure and Cord Blood Exosomes in a Rat Model. Cells 2022, 11, 3195. [Google Scholar] [CrossRef]
- ul Haq, M.E.; Akash, M.S.H.; Rehman, K.; Mahmood, M.H. Chronic Exposure of Bisphenol A Impairs Carbohydrate and Lipid Metabolism by Altering Corresponding Enzymatic and Metabolic Pathways. Environ. Toxicol. Pharmacol. 2020, 78, 103387. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Abe, S.; Rodríguez-Vázquez, J.F.; Fujimiya, M.; Murakami, G.; Ide, Y. Immunohistochemical Distribution of Desmin in the Human Fetal Heart. J. Anat. 2011, 219, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Connors, L.T.; Zhu, H.-L.; Gill, M.; Walsh, E.; Singh, R.D.; Easson, S.; Ahmed, S.B.; Habibi, H.R.; Cole, W.C.; Thompson, J.A. Prenatal Exposure to a Low Dose of BPS Causes Sex-Dependent Alterations to Vascular Endothelial Function in Adult Offspring. Front. Toxicol. 2022, 4, 933572. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Accession Number | Forward Primer 5′-3′ | Reverse Primer 5′-3′ |
|---|---|---|
| NM_031512 (Il-1β) | -CCCAGGACATGCTAGGGAGCC- | -AGGCAGGGAGGGAAACACACG- |
| NM_012675.3 (Tnf-α) | -CACCACGCTCTTCTGTCTACTG- | -GCTACGGGCTTGTCACTCG- |
| NM_012612.2 (NppA) | -GGAAGTCAACCCGTCTCAGA- | -TGGGCTCCAATCCTGTCAAT- |
| NM_031545.1 (NppB) | -CCAGAACAATCCACGATGCA- | -GCAGCTTGAACTATGTGCCA- |
| NM_053304.1 (Col1A1) | -GACATGTTCAGCTTTGTGGACCT- | -AGGGACCCTTAGGCCATTGTGTA- |
| NM_032085.1 (Col3A1) | -TTTGGCACAGCAGTCCAATGTA- | -GACAGATCCCGAGTCGCAGA- |
| NM_021578.2 (Tgf-β) | -AACCGACCCTTCCTGCTCCT- | -TCCACTTCCAACCCAGGTCCT- |
| NR_046237.2 (18s rRNA) | -CATTCGAACGTCTGCCCTAT- | -GTTTCTCAGGCTCCCTCTCC- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marrone, A.; De Bartolo, A.; Rago, V.; Conforti, F.; Urlandini, L.; Angelone, T.; Mazza, R.; Mandalà, M.; Rocca, C. Prenatal Bisphenol A Exposure Impairs Fetal Heart Development: Molecular and Structural Alterations with Sex-Specific Differences. Antioxidants 2025, 14, 863. https://doi.org/10.3390/antiox14070863
Marrone A, De Bartolo A, Rago V, Conforti F, Urlandini L, Angelone T, Mazza R, Mandalà M, Rocca C. Prenatal Bisphenol A Exposure Impairs Fetal Heart Development: Molecular and Structural Alterations with Sex-Specific Differences. Antioxidants. 2025; 14(7):863. https://doi.org/10.3390/antiox14070863
Chicago/Turabian StyleMarrone, Alessandro, Anna De Bartolo, Vittoria Rago, Francesco Conforti, Lidia Urlandini, Tommaso Angelone, Rosa Mazza, Maurizio Mandalà, and Carmine Rocca. 2025. "Prenatal Bisphenol A Exposure Impairs Fetal Heart Development: Molecular and Structural Alterations with Sex-Specific Differences" Antioxidants 14, no. 7: 863. https://doi.org/10.3390/antiox14070863
APA StyleMarrone, A., De Bartolo, A., Rago, V., Conforti, F., Urlandini, L., Angelone, T., Mazza, R., Mandalà, M., & Rocca, C. (2025). Prenatal Bisphenol A Exposure Impairs Fetal Heart Development: Molecular and Structural Alterations with Sex-Specific Differences. Antioxidants, 14(7), 863. https://doi.org/10.3390/antiox14070863