
Oxidative Stress Defense Module in Lung Cancers: Molecular Pathways and Therapeutic Approaches



Abstract
1. Introduction
2. Methodology
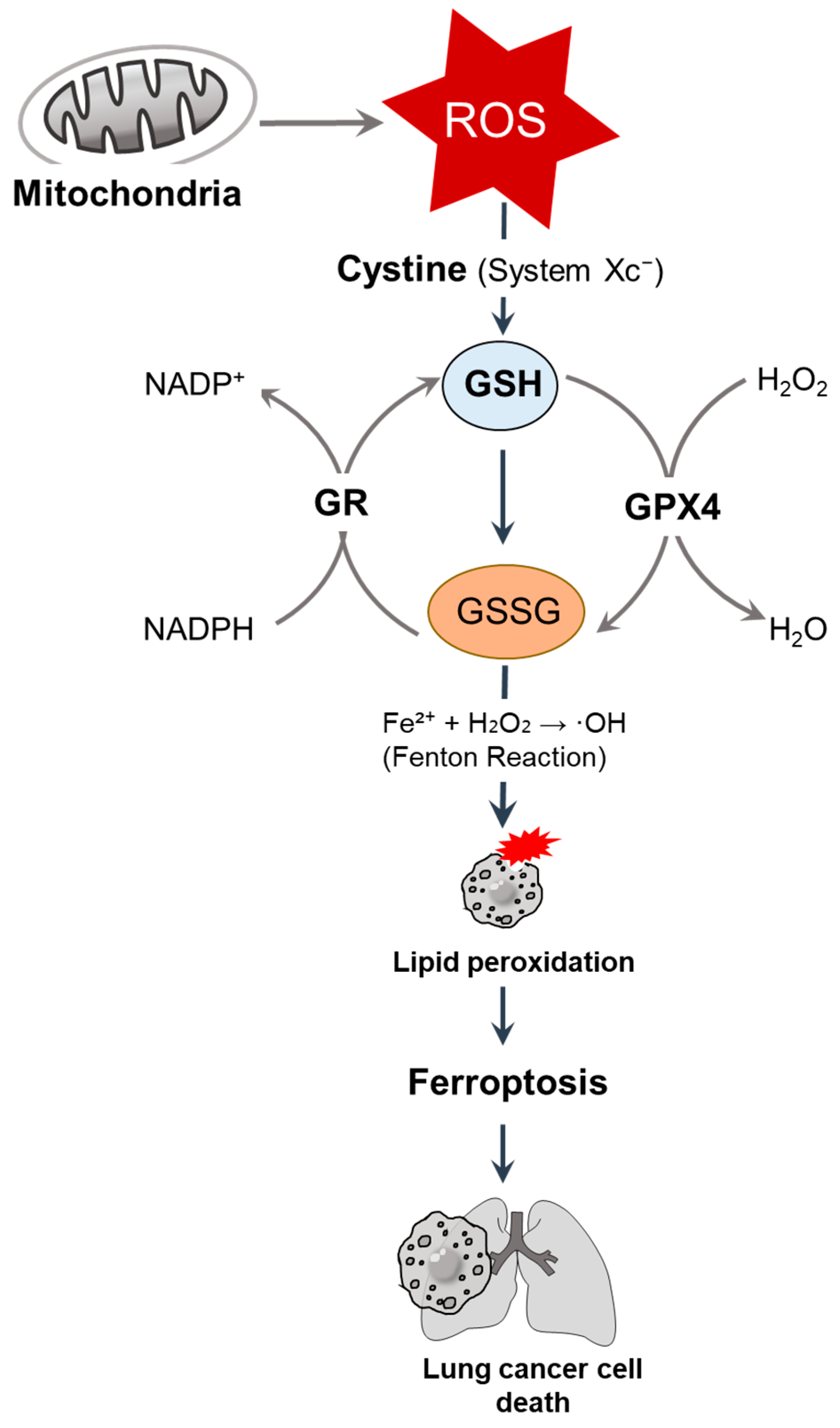
3. GSH
3.1. GSH-Driven Redox Regulation
3.2. Nrf2–GSH Axis
3.3. GSH Depletion Strategy in the Lung Cancer System
3.4. GSH Rebound Mediates Potential Awareness
4. Nrf2
4.1. Endogenous Anti-Oxidative Role of Nrf2
4.2. Nrf2/Keap1-Associated Lung Cancer Therapies
5. SOD
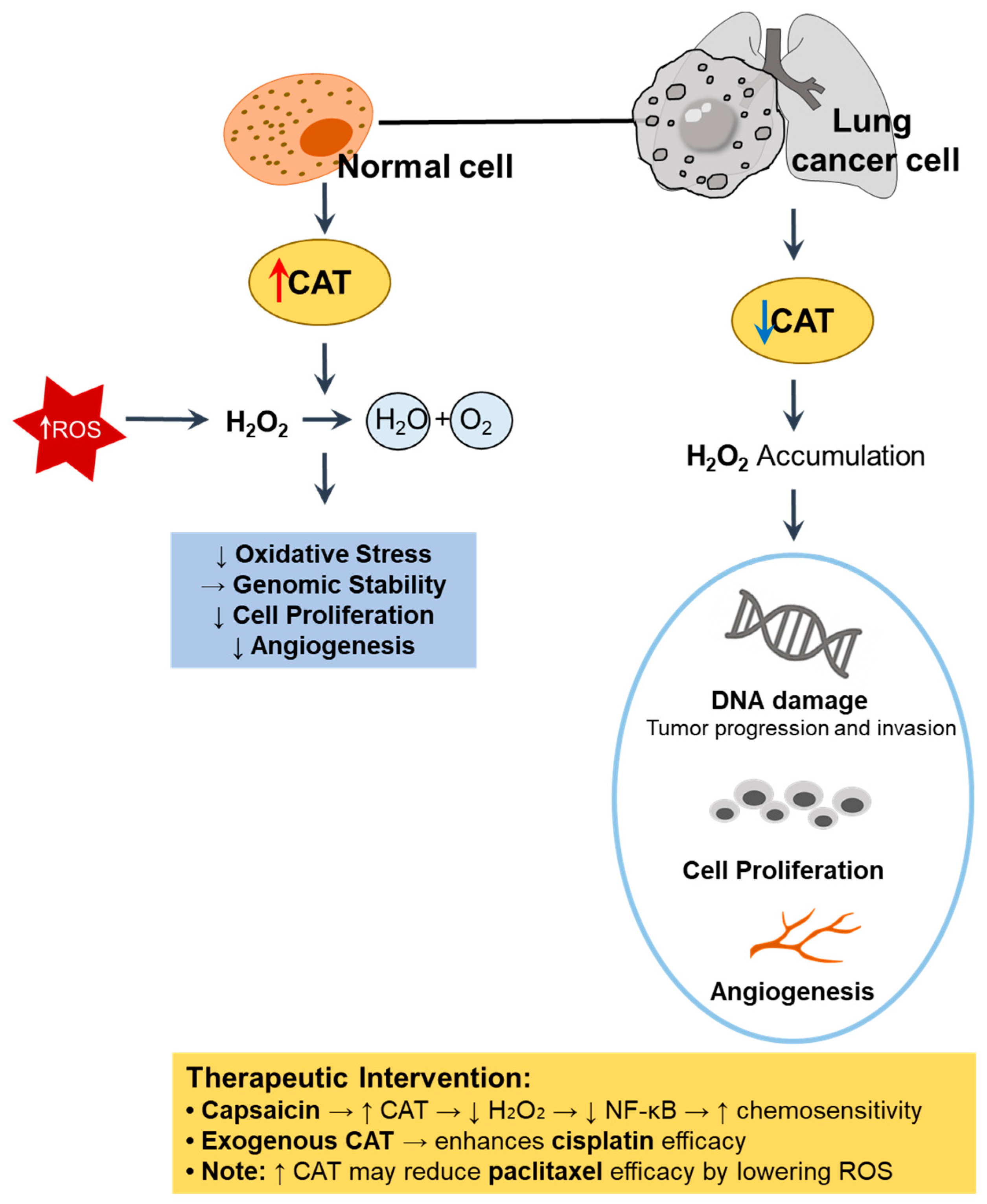
6. CAT
7. HO-1
8. PRXs
9. GRXs
10. Thioredoxin and Thioredoxin Reductases
11. 8-oxodG
12. Mitochondrial Citrate Carrier (SLC25A1)
13. Summary and Perspectives
13.1. Combination Therapies
13.2. Biomarkers and Patient Stratification
13.3. Drug Delivery Innovations
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jomova, K.; Raptova, R.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Reactive oxygen species, toxicity, oxidative stress, and antioxidants: Chronic diseases and aging. Arch. Toxicol. 2023, 97, 2499–2574. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Several lines of antioxidant defense against oxidative stress: Antioxidant enzymes, nanomaterials with multiple enzyme-mimicking activities, and low-molecular-weight antioxidants. Arch. Toxicol. 2024, 98, 1323–1367. [Google Scholar] [CrossRef] [PubMed]
- Raval, C.M.; Lee, P.J. Heme oxygenase-1 in lung disease. Curr. Drug Targets 2010, 11, 1532–1540. [Google Scholar] [CrossRef] [PubMed]
- Demirci-Cekic, S.; Ozkan, G.; Avan, A.N.; Uzunboy, S.; Capanoglu, E.; Apak, R. Biomarkers of Oxidative Stress and Antioxidant Defense. J. Pharm. Biomed. Anal. 2022, 209, 114477. [Google Scholar] [CrossRef]
- Singh, A.; Bodas, M.; Wakabayashi, N.; Bunz, F.; Biswal, S. Gain of Nrf2 function in non-small-cell lung cancer cells confers radioresistance. Antioxid. Redox Signal. 2010, 13, 1627–1637. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Pompella, A.; Visvikis, A.; Paolicchi, A.; De Tata, V.; Casini, A.F. The changing faces of glutathione, a cellular protagonist. Biochem. Pharmacol. 2003, 66, 1499–1503. [Google Scholar] [CrossRef]
- Townsend, D.M.; Tew, K.D.; Tapiero, H. The importance of glutathione in human disease. Biomed. Pharmacother. 2003, 57, 145–155. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Kachadourian, R.; Leitner, H.M.; Day, B.J. Selected flavonoids potentiate the toxicity of cisplatin in human lung adenocarcinoma cells: A role for glutathione depletion. Int. J. Oncol. 2007, 31, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shi, J.; Wang, X.; Zhou, L.; Wang, Q.; Xie, Y.; Peng, C.; Kuang, L.; Yang, D.; Yang, J.; et al. An antioxidant feedforward cycle coordinated by linker histone variant H1.2 and NRF2 that drives nonsmall cell lung cancer progression. Proc. Natl. Acad. Sci. USA 2023, 120, e2306288120. [Google Scholar] [CrossRef] [PubMed]
- Hori, R.; Yamaguchi, K.; Sato, H.; Watanabe, M.; Tsutsumi, K.; Iwamoto, S.; Abe, M.; Onodera, H.; Nakamura, S.; Nakai, R. The discovery and characterization of K-563, a novel inhibitor of the Keap1/Nrf2 pathway produced by Streptomyces sp. Cancer Med. 2019, 8, 1157–1168. [Google Scholar] [CrossRef] [PubMed]
- Gai, C.; Liu, C.; Wu, X.; Yu, M.; Zheng, J.; Zhang, W.; Lv, S.; Li, W. MT1DP loaded by folate-modified liposomes sensitizes erastin-induced ferroptosis via regulating miR-365a-3p/NRF2 axis in non-small cell lung cancer cells. Cell Death Dis. 2020, 11, 751. [Google Scholar] [CrossRef]
- Silva, M.M.; Rocha, C.R.R.; Kinker, G.S.; Pelegrini, A.L.; Menck, C.F.M. The balance between NRF2/GSH antioxidant mediated pathway and DNA repair modulates cisplatin resistance in lung cancer cells. Sci. Rep. 2019, 9, 17639. [Google Scholar] [CrossRef]
- Niu, B.; Liao, K.; Zhou, Y.; Wen, T.; Quan, G.; Wu, C.; Pan, X. Cellular defense system-destroying nanoparticles as a platform for enhanced chemotherapy against drug-resistant cancer. Mater. Sci. Eng. C Mater. Biol. Appl. 2021, 131, 112494. [Google Scholar] [CrossRef]
- Zhou, C.; Yu, T.; Zhu, R.; Lu, J.; Ouyang, X.; Zhang, Z.; Chen, Q.; Li, J.; Cui, J.; Jiang, F.; et al. Timosaponin AIII promotes non-small-cell lung cancer ferroptosis through targeting and facilitating HSP90 mediated GPX4 ubiquitination and degradation. Int. J. Biol. Sci. 2023, 19, 1471–1489. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Kanaan, M.N.; Pileggi, C.A.; Karam, C.Y.; Kennedy, L.S.; Fong-McMaster, C.; Cuperlovic-Culf, M.; Harper, M.E. Cystine/glutamate antiporter xCT controls skeletal muscle glutathione redox, bioenergetics and differentiation. Redox Biol. 2024, 73, 103213. [Google Scholar] [CrossRef]
- Deng, J.; Lin, X.; Qin, J.; Li, Q.; Zhang, Y.; Zhang, Q.; Ji, C.; Shen, S.; Li, Y.; Zhang, B.; et al. SPTBN2 suppresses ferroptosis in NSCLC cells by facilitating SLC7A11 membrane trafficking and localization. Redox Biol. 2024, 70, 103039. [Google Scholar] [CrossRef]
- Kogami, M.; Abe, S.; Nakamura, H.; Aoshiba, K. Fenofibrate attenuates the cytotoxic effect of cisplatin on lung cancer cells by enhancing the antioxidant defense system in vitro. Oncol. Lett. 2023, 26, 313. [Google Scholar] [CrossRef] [PubMed]
- Cloer, E.W.; Goldfarb, D.; Schrank, T.P.; Weissman, B.E.; Major, M.B. NRF2 Activation in Cancer: From DNA to Protein. Cancer Res. 2019, 79, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Zimta, A.A.; Cenariu, D.; Irimie, A.; Magdo, L.; Nabavi, S.M.; Atanasov, A.G.; Berindan-Neagoe, I. The Role of Nrf2 Activity in Cancer Development and Progression. Cancers 2019, 11, 1755. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Wang, Z.Y.; Li, Y.K.; Ye, D.M.; Zeng, J.; Hu, J.L.; Chen, P.F.; Xiao, J.; Zou, J.; Li, Z.H. Nuclear factor erythroid 2 (NF-E2)-related factor 2 (Nrf2) in non-small cell lung cancer. Life Sci. 2020, 254, 117325. [Google Scholar] [CrossRef]
- Li, B.; Ming, H.; Qin, S.; Nice, E.C.; Dong, J.; Du, Z.; Huang, C. Redox regulation: Mechanisms, biology and therapeutic targets in diseases. Signal Transduct. Target. Ther. 2025, 10, 72. [Google Scholar] [CrossRef]
- Homma, S.; Ishii, Y.; Morishima, Y.; Yamadori, T.; Matsuno, Y.; Haraguchi, N.; Kikuchi, N.; Satoh, H.; Sakamoto, T.; Hizawa, N.; et al. Nrf2 enhances cell proliferation and resistance to anticancer drugs in human lung cancer. Clin. Cancer Res. 2009, 15, 3423–3432. [Google Scholar] [CrossRef]
- Li, Q.K.; Singh, A.; Biswal, S.; Askin, F.; Gabrielson, E. KEAP1 gene mutations and NRF2 activation are common in pulmonary papillary adenocarcinoma. J. Hum. Genet. 2011, 56, 230–234. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Spentzos, D.; Fountzilas, E.; Francoeur, N.; Sanisetty, S.; Grammatikos, A.P.; Hecht, J.L.; Cannistra, S.A. Keap1 mutations and Nrf2 pathway activation in epithelial ovarian cancer. Cancer Res. 2011, 71, 5081–5089. [Google Scholar] [CrossRef]
- Zhan, L.; Zhang, H.; Zhang, Q.; Woods, C.G.; Chen, Y.; Xue, P.; Dong, J.; Tokar, E.J.; Xu, Y.; Hou, Y.; et al. Regulatory role of KEAP1 and NRF2 in PPARgamma expression and chemoresistance in human non-small-cell lung carcinoma cells. Free Radic. Biol. Med. 2012, 53, 758–768. [Google Scholar] [CrossRef]
- Satoh, H.; Moriguchi, T.; Saigusa, D.; Baird, L.; Yu, L.; Rokutan, H.; Igarashi, K.; Ebina, M.; Shibata, T.; Yamamoto, M. NRF2 Intensifies Host Defense Systems to Prevent Lung Carcinogenesis, but After Tumor Initiation Accelerates Malignant Cell Growth. Cancer Res. 2016, 76, 3088–3096. [Google Scholar] [CrossRef]
- Ji, L.; Li, H.; Gao, P.; Shang, G.; Zhang, D.D.; Zhang, N.; Jiang, T. Nrf2 pathway regulates multidrug-resistance-associated protein 1 in small cell lung cancer. PLoS ONE 2013, 8, e63404. [Google Scholar] [CrossRef] [PubMed]
- Gegotek, A.; Niklinski, J.; Zarkovic, N.; Zarkovic, K.; Waeg, G.; Luczaj, W.; Charkiewicz, R.; Skrzydlewska, E. Lipid mediators involved in the oxidative stress and antioxidant defence of human lung cancer cells. Redox Biol. 2016, 9, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.C.; Lin, R.J.; Cheng, J.Y.; Wang, S.H.; Yu, J.C.; Wu, J.C.; Liang, Y.J.; Hsu, H.M.; Yu, J.; Yu, A.L. FAM129B, an antioxidative protein, reduces chemosensitivity by competing with Nrf2 for Keap1 binding. EBioMedicine 2019, 45, 25–38. [Google Scholar] [CrossRef]
- de Miranda Ramos, V.; Gasparotto, J.; Figueiro, F.; de Fraga Dias, A.; Rostirolla, D.C.; Somensi, N.; da Rosa, H.T.; Grun, L.K.; Barbe-Tuana, F.M.; Gelain, D.P.; et al. Retinoic acid downregulates thiol antioxidant defences and homologous recombination while promotes A549 cells sensitization to cisplatin. Cell Signal. 2019, 62, 109356. [Google Scholar] [CrossRef]
- Yang, H.; Xiang, S.; Kazi, A.; Sebti, S.M. The GTPase KRAS suppresses the p53 tumor suppressor by activating the NRF2-regulated antioxidant defense system in cancer cells. J. Biol. Chem. 2020, 295, 3055–3063. [Google Scholar] [CrossRef]
- Rai, A.; Patwardhan, R.S.; Jayakumar, S.; Pachpatil, P.; Das, D.; Panigrahi, G.C.; Gota, V.; Patwardhan, S.; Sandur, S.K. Clobetasol propionate, a Nrf-2 inhibitor, sensitizes human lung cancer cells to radiation-induced killing via mitochondrial ROS-dependent ferroptosis. Acta Pharmacol. Sin. 2024, 45, 1506–1519. [Google Scholar] [CrossRef]
- Huang, S.; He, T.; Yang, S.; Sheng, H.; Tang, X.; Bao, F.; Wang, Y.; Lin, X.; Yu, W.; Cheng, F.; et al. Metformin reverses chemoresistance in non-small cell lung cancer via accelerating ubiquitination-mediated degradation of Nrf2. Transl. Lung Cancer Res. 2020, 9, 2337–2355. [Google Scholar] [CrossRef]
- Ahmadian, S.; Sabzichi, M.; Rashidi, M.; Mohammadian, J.; Mahmoudi, S.; Maroufi, N.F.; Ramezani, F.; Ghorbani, M.; Mohammadi, M.; Pirouzpanah, M.; et al. Sensitization of A-549 lung cancer cells to Cisplatin by Quinacrine-loaded lipidic nanoparticles via suppressing Nrf2 mediated defense mechanism. Naunyn Schmiedebergs Arch. Pharmacol. 2021, 394, 1521–1528. [Google Scholar] [CrossRef]
- Kim, H.K.; Jeong, H.; Jeong, M.G.; Won, H.Y.; Lee, G.; Bae, S.H.; Nam, M.; Lee, S.H.; Hwang, G.S.; Hwang, E.S. TAZ deficiency impairs the autophagy-lysosomal pathway through NRF2 dysregulation and lysosomal dysfunction. Int. J. Biol. Sci. 2024, 20, 2592–2606. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noe, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Martin-Mateo, M.C.; Molpeceres, L.M.; Ramos, G. Assay for erythrocyte superoxide dismutase activity in patients with lung cancer and effects on pollution and smoke trace elements. Biol. Trace Elem. Res. 1997, 60, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Svensk, A.M.; Soini, Y.; Paakko, P.; Hiravikoski, P.; Kinnula, V.L. Differential expression of superoxide dismutases in lung cancer. Am. J. Clin. Pathol. 2004, 122, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Chung-man Ho, J.; Zheng, S.; Comhair, S.A.; Farver, C.; Erzurum, S.C. Differential expression of manganese superoxide dismutase and catalase in lung cancer. Cancer Res. 2001, 61, 8578–8585. [Google Scholar] [PubMed]
- Jarvinen, K.; Pietarinen-Runtti, P.; Linnainmaa, K.; Raivio, K.O.; Krejsa, C.M.; Kavanagh, T.; Kinnula, V.L. Antioxidant defense mechanisms of human mesothelioma and lung adenocarcinoma cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 278, L696–L702. [Google Scholar] [CrossRef]
- Zheng, Y.; Yang, S.; Si, J.; Zhao, Y.; Zhao, M.; Ji, E. Shashen-Maidong Decoction inhibited cancer growth under intermittent hypoxia conditions by suppressing oxidative stress and inflammation. J. Ethnopharmacol. 2022, 299, 115654. [Google Scholar] [CrossRef]
- Concato-Lopes, V.M.; Goncalves-Lens, M.D.; Tomiotto-Pellissier, F.; Detoni, M.B.; Cruz, E.M.S.; Bortoleti, B.; Carloto, A.C.M.; Rodrigues, A.C.J.; Silva, T.F.; Siqueira, E.D.S.; et al. Trilobolide-6-O-isobutyrate from Sphagneticola trilobata acts by inducing oxidative stress, metabolic changes and apoptosis-like processes by caspase 3/7 activation of human lung cancer cell lines. Phytomedicine 2024, 128, 155536. [Google Scholar] [CrossRef]
- Pulliam, C.F.; Fath, M.A.; Sho, S.; Johnson, S.T.; Wagner, B.A.; Singhania, M.; Kalen, A.L.; Bayanbold, K.; Solst, S.R.; Allen, B.G.; et al. Pharmacological ascorbate combined with rucosopasem selectively radio-chemo-sensitizes NSCLC via generation of H(2)O(2). Redox Biol. 2025, 80, 103505. [Google Scholar] [CrossRef]
- Zhuang, Y.; Wu, H.; Wang, X.; He, J.; He, S.; Yin, Y. Resveratrol Attenuates Oxidative Stress-Induced Intestinal Barrier Injury through PI3K/Akt-Mediated Nrf2 Signaling Pathway. Oxid. Med. Cell Longev. 2019, 2019, 7591840. [Google Scholar] [CrossRef]
- Glorieux, C.; Buc Calderon, P. Targeting catalase in cancer. Redox Biol. 2024, 77, 103404. [Google Scholar] [CrossRef]
- Anandakumar, P.; Kamaraj, S.; Jagan, S.; Ramakrishnan, G.; Vinodhkumar, R.; Devaki, T. Capsaicin modulates pulmonary antioxidant defense system during benzo(a)pyrene-induced lung cancer in Swiss albino mice. Phytother. Res. 2008, 22, 529–533. [Google Scholar] [CrossRef]
- de Oliveira, V.A.; da Motta, L.L.; De Bastiani, M.A.; Lopes, F.M.; Muller, C.B.; Gabiatti, B.P.; Franca, F.S.; Castro, M.A.; Klamt, F. In vitro evaluation of antitumoral efficacy of catalase in combination with traditional chemotherapeutic drugs against human lung adenocarcinoma cells. Tumor Biol. 2016, 37, 10775–10784. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Kim, I.K.; Lee, H.I.; Lee, H.Y.; Kang, H.S.; Yeo, C.D.; Kang, H.H.; Moon, H.S.; Lee, S.H. Combination of carboplatin and intermittent normobaric hyperoxia synergistically suppresses benzo[a]pyrene-induced lung cancer. Korean J. Intern. Med. 2018, 33, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.L.; Dong, X.; Yang, S.C.; Lai, X.; Liu, H.J.; Gao, Y.; Feng, H.Y.; Zhu, M.H.; Yuan, Y.; Lu, Q.; et al. Biomimetic Liposomal Nanoplatinum for Targeted Cancer Chemophototherapy. Adv. Sci. 2021, 8, 2003679. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.Y.; Liou, H.C.; Fu, W.M. The mechanism of heme oxygenase-1 action involved in the enhancement of neurotrophic factor expression. Neuropharmacology 2010, 58, 321–329. [Google Scholar] [CrossRef]
- Boschetto, P.; Zeni, E.; Mazzetti, L.; Miotto, D.; Lo Cascio, N.; Maestrelli, P.; Marian, E.; Querzoli, P.; Pedriali, M.; Murer, B.; et al. Decreased heme-oxygenase (HO)-1 in the macrophages of non-small cell lung cancer. Lung Cancer 2008, 59, 192–197. [Google Scholar] [CrossRef]
- Degese, M.S.; Mendizabal, J.E.; Gandini, N.A.; Gutkind, J.S.; Molinolo, A.; Hewitt, S.M.; Curino, A.C.; Coso, O.A.; Facchinetti, M.M. Expression of heme oxygenase-1 in non-small cell lung cancer (NSCLC) and its correlation with clinical data. Lung Cancer 2012, 77, 168–175. [Google Scholar] [CrossRef]
- Tertil, M.; Skrzypek, K.; Florczyk, U.; Weglarczyk, K.; Was, H.; Collet, G.; Guichard, A.; Gil, T.; Kuzdzal, J.; Jozkowicz, A.; et al. Regulation and novel action of thymidine phosphorylase in non-small cell lung cancer: Crosstalk with Nrf2 and HO-1. PLoS ONE 2014, 9, e97070. [Google Scholar] [CrossRef]
- Tsai, J.R.; Wang, H.M.; Liu, P.L.; Chen, Y.H.; Yang, M.C.; Chou, S.H.; Cheng, Y.J.; Yin, W.H.; Hwang, J.J.; Chong, I.W. High expression of heme oxygenase-1 is associated with tumor invasiveness and poor clinical outcome in non-small cell lung cancer patients. Cell. Oncol. 2012, 35, 461–471. [Google Scholar] [CrossRef]
- Jo, E.J.; Park, S.J.; Kim, B.C. Propyl gallate sensitizes human lung cancer cells to cisplatin-induced apoptosis by targeting heme oxygenase-1 for TRC8-mediated degradation. Eur. J. Pharmacol. 2016, 788, 321–327. [Google Scholar] [CrossRef]
- Do, M.T.; Kim, H.G.; Khanal, T.; Choi, J.H.; Kim, D.H.; Jeong, T.C.; Jeong, H.G. Metformin inhibits heme oxygenase-1 expression in cancer cells through inactivation of Raf-ERK-Nrf2 signaling and AMPK-independent pathways. Toxicol. Appl. Pharmacol. 2013, 271, 229–238. [Google Scholar] [CrossRef]
- Yu, C.; Jiao, Y.; Xue, J.; Zhang, Q.; Yang, H.; Xing, L.; Chen, G.; Wu, J.; Zhang, S.; Zhu, W.; et al. Metformin Sensitizes Non-small Cell Lung Cancer Cells to an Epigallocatechin-3-Gallate (EGCG) Treatment by Suppressing the Nrf2/HO-1 Signaling Pathway. Int. J. Biol. Sci. 2017, 13, 1560–1569. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Qiao, T.; Zha, L. Inhibition of heme oxygenase-1 enhances the radiosensitivity in human nonsmall cell lung cancer a549 cells. Cancer Biother. Radiopharm. 2011, 26, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Spampinato, M.; Sferrazzo, G.; Pittala, V.; Di Rosa, M.; Vanella, L.; Salerno, L.; Sorrenti, V.; Carota, G.; Parrinello, N.; Raffaele, M.; et al. Non-competitive heme oxygenase-1 activity inhibitor reduces non-small cell lung cancer glutathione content and regulates cell proliferation. Mol. Biol. Rep. 2020, 47, 1949–1964. [Google Scholar] [CrossRef]
- Li, C.G.; Pu, M.F.; Li, C.Z.; Gao, M.; Liu, M.X.; Yu, C.Z.; Yan, H.; Peng, C.; Zhao, Y.; Li, Y.; et al. MicroRNA-1304 suppresses human non-small cell lung cancer cell growth in vitro by targeting heme oxygenase-1. Acta Pharmacol. Sin. 2017, 38, 110–119. [Google Scholar] [CrossRef]
- Jeon, W.K.; Hong, H.Y.; Seo, W.C.; Lim, K.H.; Lee, H.Y.; Kim, W.J.; Song, S.Y.; Kim, B.C. Smad7 sensitizes A549 lung cancer cells to cisplatin-induced apoptosis through heme oxygenase-1 inhibition. Biochem. Biophys. Res. Commun. 2012, 420, 288–292. [Google Scholar] [CrossRef]
- Zhang, L.; Qu, Z.; Song, A.; Yang, J.; Yu, J.; Zhang, W.; Zhuang, C. Garlic oil blocks tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)-induced lung tumorigenesis by inducing phase II drug-metabolizing enzymes. Food Chem. Toxicol. 2021, 157, 112581. [Google Scholar] [CrossRef]
- Tertil, M.; Golda, S.; Skrzypek, K.; Florczyk, U.; Weglarczyk, K.; Kotlinowski, J.; Maleszewska, M.; Czauderna, S.; Pichon, C.; Kieda, C.; et al. Nrf2-heme oxygenase-1 axis in mucoepidermoid carcinoma of the lung: Antitumoral effects associated with down-regulation of matrix metalloproteinases. Free Radic. Biol. Med. 2015, 89, 147–157. [Google Scholar] [CrossRef]
- Zhou, Y.; Du, Q.; Zhao, Q.; Zhang, M.; Qin, X.; Jiang, Y.; Luan, Y. A heme-regulatable chemodynamic nanodrug harnessing transcription factor Bach1 against lung cancer metastasis. J. Colloid Interface Sci. 2022, 610, 698–708. [Google Scholar] [CrossRef]
- Ma, J.; Yu, K.N.; Cheng, C.; Ni, G.; Shen, J.; Han, W. Targeting Nrf2-mediated heme oxygenase-1 enhances non-thermal plasma-induced cell death in non-small-cell lung cancer A549 cells. Arch. Biochem. Biophys. 2018, 658, 54–65. [Google Scholar] [CrossRef]
- Kuo, K.T.; Lin, C.H.; Wang, C.H.; Pikatan, N.W.; Yadav, V.K.; Fong, I.H.; Yeh, C.T.; Lee, W.H.; Huang, W.C. HNMT Upregulation Induces Cancer Stem Cell Formation and Confers Protection against Oxidative Stress through Interaction with HER2 in Non-Small-Cell Lung Cancer. Int. J. Mol. Sci. 2022, 23, 1663. [Google Scholar] [CrossRef]
- Man, S.; Bi, J.; Liu, F.; Xie, W.; Ma, L. Vitamin C Inhibited Pulmonary Metastasis through Activating Nrf2/HO-1 Pathway. Mol. Nutr. Food Res. 2024, 68, e2300706. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Feng, L.; Jin, W.; Chang, J.; Li, J.; Li, H. Identification of the Key Active Pharmaceutical Ingredients of Yishen Qutong Granule, A Chinese Medicine Formula, In The Treatment of Primary Lung Cancer. Comb. Chem. High Throughput Screen. 2023, 26, 1594–1608. [Google Scholar] [CrossRef] [PubMed]
- Lehtonen, S.T.; Svensk, A.M.; Soini, Y.; Paakko, P.; Hirvikoski, P.; Kang, S.W.; Saily, M.; Kinnula, V.L. Peroxiredoxins, a novel protein family in lung cancer. Int. J. Cancer 2004, 111, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Guo, X.; Nakamura, Y.; Zhou, X.; Yamaguchi, R.; Zhang, J.; Ishigaki, Y.; Uramoto, H.; Yamada, S. Overexpression of PRDX4 Modulates Tumor Microenvironment and Promotes Urethane-Induced Lung Tumorigenesis. Oxid. Med. Cell. Longev. 2020, 2020, 8262730. [Google Scholar] [CrossRef]
- Fernandes, A.P.; Holmgren, A. Glutaredoxins: Glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system. Antioxid. Redox Signal. 2004, 6, 63–74. [Google Scholar] [CrossRef]
- Chai, Y.C.; Mieyal, J.J. Glutathione and Glutaredoxin-Key Players in Cellular Redox Homeostasis and Signaling. Antioxidants 2023, 12, 1553. [Google Scholar] [CrossRef]
- Cha, M.K.; Kim, I.H. Preferential overexpression of glutaredoxin3 in human colon and lung carcinoma. Cancer Epidemiol. 2009, 33, 281–287. [Google Scholar] [CrossRef]
- Lonn, M.E.; Hudemann, C.; Berndt, C.; Cherkasov, V.; Capani, F.; Holmgren, A.; Lillig, C.H. Expression pattern of human glutaredoxin 2 isoforms: Identification and characterization of two testis/cancer cell-specific isoforms. Antioxid. Redox Signal. 2008, 10, 547–557. [Google Scholar] [CrossRef]
- Hudemann, C.; Lonn, M.E.; Godoy, J.R.; Zahedi Avval, F.; Capani, F.; Holmgren, A.; Lillig, C.H. Identification, expression pattern, and characterization of mouse glutaredoxin 2 isoforms. Antioxid. Redox Signal. 2009, 11, 1–14. [Google Scholar] [CrossRef]
- Fernandes, A.P.; Capitanio, A.; Selenius, M.; Brodin, O.; Rundlof, A.K.; Bjornstedt, M. Expression profiles of thioredoxin family proteins in human lung cancer tissue: Correlation with proliferation and differentiation. Histopathology 2009, 55, 313–320. [Google Scholar] [CrossRef]
- Wang, L.; Liu, J.; Liu, J.; Chen, X.; Chang, M.; Li, J.; Zhou, J.; Bai, C.; Song, Y. GLRX inhibition enhances the effects of geftinib in EGFR-TKI-resistant NSCLC cells through FoxM1 signaling pathway. J. Cancer Res. Clin. Oncol. 2019, 145, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Pandya, P.; Jethva, M.; Rubin, E.; Birnbaum, R.Y.; Braiman, A.; Isakov, N. PICOT binding to chromatin-associated EED negatively regulates cyclin D2 expression by increasing H3K27me3 at the CCND2 gene promoter. Cell Death Dis. 2019, 10, 685. [Google Scholar] [CrossRef]
- Hao, C.C.; Luo, J.N.; Xu, C.Y.; Zhao, X.Y.; Zhong, Z.B.; Hu, X.N.; Jin, X.M.; Ge, X. TRIAP1 knockdown sensitizes non-small cell lung cancer to ionizing radiation by disrupting redox homeostasis. Thorac. Cancer 2020, 11, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- You, B.R.; Shin, H.R.; Park, W.H. PX-12 inhibits the growth of A549 lung cancer cells via G2/M phase arrest and ROS-dependent apoptosis. Int. J. Oncol. 2014, 44, 301–308. [Google Scholar] [CrossRef]
- You, B.R.; Shin, H.R.; Han, B.R.; Park, W.H. PX-12 induces apoptosis in Calu-6 cells in an oxidative stress-dependent manner. Tumor Biol. 2015, 36, 2087–2095. [Google Scholar] [CrossRef]
- Onodera, T.; Momose, I.; Kawada, M. Potential Anticancer Activity of Auranofin. Chem. Pharm. Bull. 2019, 67, 186–191. [Google Scholar] [CrossRef]
- Chupakhin, E.; Krasavin, M. Thioredoxin reductase inhibitors: Updated patent review (2017-present). Expert Opin. Ther. Pat. 2021, 31, 745–758. [Google Scholar] [CrossRef]
- Jayakumar, S.; Patwardhan, R.S.; Pal, D.; Sharma, D.; Sandur, S.K. Dimethoxycurcumin, a metabolically stable analogue of curcumin enhances the radiosensitivity of cancer cells: Possible involvement of ROS and thioredoxin reductase. Biochem. Biophys. Res. Commun. 2016, 478, 446–454. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, S.; Xu, W.; Yang, R.; Yang, Y.; Guo, J.; Ma, K.; Xu, J. Thioredoxin reductase 1 inhibitor shikonin promotes cell necroptosis via SecTRAPs generation and oxygen-coupled redox cycling. Free Radic. Biol. Med. 2022, 180, 52–62. [Google Scholar] [CrossRef]
- Cui, X.Y.; Park, S.H.; Park, W.H. Anti-Cancer Effects of Auranofin in Human Lung Cancer Cells by Increasing Intracellular ROS Levels and Depleting GSH Levels. Molecules 2022, 27, 5207. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Xia, Y.; Shen, X.; Lu, H.; Chen, Y.; Xu, C.; Qiu, C.; Zhang, Y.; Zou, P.; Cui, R.; et al. Combination of TrxR1 inhibitor and lenvatinib triggers ROS-dependent cell death in human lung cancer cells. Int. J. Biol. Sci. 2024, 20, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.B.; Xu, C.; Wang, W.; Zhang, Y.Z.; Huang, J.M.; Xie, Y.J.; Wang, Q.Q.; Fan, X.X.; Yao, X.J.; Xie, C.; et al. Plumbagin suppresses non-small cell lung cancer progression through downregulating ARF1 and by elevating CD8(+) T cells. Pharmacol. Res. 2021, 169, 105656. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhang, Y.; Xu, W.; Yang, R.; Yang, Y.; Guo, J.; Ma, Q.; Ma, K.; Zhang, J.; Xu, J. Plumbagin reduction by thioredoxin reductase 1 possesses synergy effects with GLUT1 inhibitor on KEAP1-mutant NSCLC cells. Biomed. Pharmacother. 2022, 146, 112546. [Google Scholar] [CrossRef]
- Ni, Y.; Luo, Z.; Lv, Y.; Ma, S.; Luo, C.; Du, D. Thimerosal, a competitive thioredoxin reductase 1 (TrxR1) inhibitor discovered via high-throughput screening. Biochem. Biophys. Res. Commun. 2023, 650, 117–122. [Google Scholar] [CrossRef]
- Wang, R.; Zhong, L.; Wang, T.; Sun, T.; Yang, J.; Liu, X.; Wu, Y.; Guo, Q.; Gao, Y.; Zhao, K. Inducing ubiquitination and degradation of TrxR1 protein by LW-216 promotes apoptosis in non-small cell lung cancer via triggering ROS production. Neoplasia 2024, 53, 101004. [Google Scholar] [CrossRef]
- Ren, H.; Wang, Y.J.; Wang, X.Y.; Li, X.; Han, Z.; Zhang, G.; Gu, L.; Bai, M.; Yao, G.D.; Liu, Q.; et al. Design of ROS-Triggered Sesquiterpene Lactone SC Prodrugs as TrxR1 Covalent Inhibitors for the Treatment of Non-Small Cell Lung Cancer. J. Med. Chem. 2025, 68, 3088–3122. [Google Scholar] [CrossRef]
- Wang, J.; Wang, P.; Dong, C.; Zhao, Y.; Zhou, J.; Yuan, C.; Zou, L. Mechanisms of ebselen as a therapeutic and its pharmacology applications. Future Med. Chem. 2020, 12, 2141–2160. [Google Scholar] [CrossRef]
- Park, W.H. Ebselen Inhibits the Growth of Lung Cancer Cells via Cell Cycle Arrest and Cell Death Accompanied by Glutathione Depletion. Molecules 2023, 28, 6472. [Google Scholar] [CrossRef]
- Ames, B.N. Endogenous oxidative DNA damage, aging, and cancer. Free Radic. Res. Commun. 1989, 7, 121–128. [Google Scholar] [CrossRef]
- Kim, D.H.; Cho, I.H.; Kim, H.S.; Jung, J.E.; Kim, J.E.; Lee, K.H.; Park, T.; Yang, Y.M.; Seong, S.Y.; Ye, S.K.; et al. Anti-inflammatory effects of 8-hydroxydeoxyguanosine in LPS-induced microglia activation: Suppression of STAT3-mediated intercellular adhesion molecule-1 expression. Exp. Mol. Med. 2006, 38, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Choi, H.H.; Lee, S.H.; Ko, S.H.; You, H.J.; Ye, S.K.; Chung, M.H. Anti-inflammatory effects of 8-hydroxy-2′-deoxyguanosine on lipopolysaccharide-induced inflammation via Rac suppression in Balb/c mice. Free Radic. Biol. Med. 2007, 43, 1594–1603. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Li, Z.; Wang, S.; Wang, Z. Inhibition of (-)epigallocatechin gallate on dimethylarsinic acid promoting lung tumorigenesis through the induction of oxidative stress in mice. Wei Sheng Yan Jiu 2008, 37, 748–750. [Google Scholar]
- Naylor, S.L.; Johnson, B.E.; Minna, J.D.; Sakaguchi, A.Y. Loss of heterozygosity of chromosome 3p markers in small-cell lung cancer. Nature 1987, 329, 451–454. [Google Scholar] [CrossRef]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef]
- Nishimura, S. 8-Hydroxyguanine: A base for discovery. DNA Repair 2011, 10, 1078–1083. [Google Scholar] [CrossRef]
- Risom, L.; Dybdahl, M.; Bornholdt, J.; Vogel, U.; Wallin, H.; Moller, P.; Loft, S. Oxidative DNA damage and defence gene expression in the mouse lung after short-term exposure to diesel exhaust particles by inhalation. Carcinogenesis 2003, 24, 1847–1852. [Google Scholar] [CrossRef]
- Park, J.M.; Han, Y.M.; Jeong, M.; Chung, M.H.; Kwon, C.I.; Ko, K.H.; Hahm, K.B. Synthetic 8-hydroxydeoxyguanosine inhibited metastasis of pancreatic cancer through concerted inhibitions of ERM and Rho-GTPase. Free Radic. Biol. Med. 2017, 110, 151–161. [Google Scholar] [CrossRef]
- Ji, M.J.; Son, K.H.; Hong, J.H. Addition of oh8dG to Cardioplegia Attenuated Myocardial Oxidative Injury through the Inhibition of Sodium Bicarbonate Cotransporter Activity. Antioxidants 2022, 11, 1641. [Google Scholar] [CrossRef]
- Hlouschek, J.; Hansel, C.; Jendrossek, V.; Matschke, J. The Mitochondrial Citrate Carrier (SLC25A1) Sustains Redox Homeostasis and Mitochondrial Metabolism Supporting Radioresistance of Cancer Cells With Tolerance to Cycling Severe Hypoxia. Front. Oncol. 2018, 8, 170. [Google Scholar] [CrossRef]
- Fernandez, H.R.; Gadre, S.M.; Tan, M.; Graham, G.T.; Mosaoa, R.; Ongkeko, M.S.; Kim, K.A.; Riggins, R.B.; Parasido, E.; Petrini, I.; et al. The mitochondrial citrate carrier, SLC25A1, drives stemness and therapy resistance in non-small cell lung cancer. Cell Death Differ. 2018, 25, 1239–1258. [Google Scholar] [CrossRef] [PubMed]
- Xiang, K.; Kalthoff, C.; Munch, C.; Jendrossek, V.; Matschke, J. Accumulation of oncometabolite D-2-Hydroxyglutarate by SLC25A1 inhibition: A metabolic strategy for induction of HR-ness and radiosensitivity. Cell Death Dis. 2022, 13, 641. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Woo, H.A.; Kil, I.S.; Bae, S.H. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. 2012, 287, 4403–4410. [Google Scholar] [CrossRef]
- Brigelius-Flohe, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Arner, E.S.J. Targeting the Selenoprotein Thioredoxin Reductase 1 for Anticancer Therapy. Adv. Cancer Res. 2017, 136, 139–151. [Google Scholar] [CrossRef]
- Lee, J.M.; Calkins, M.J.; Chan, K.; Kan, Y.W.; Johnson, J.A. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J. Biol. Chem. 2003, 278, 12029–12038. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef]
- Liang, X.; Wang, Z.; Dai, Z.; Liu, J.; Zhang, H.; Wen, J.; Zhang, N.; Zhang, J.; Luo, P.; Liu, Z.; et al. Oxidative stress is involved in immunosuppression and macrophage regulation in glioblastoma. Clin. Immunol. 2024, 258, 109802. [Google Scholar] [CrossRef]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Li, D.; Wang, J.; Yang, X. Reactive oxygen species-sensitive polymeric nanocarriers for synergistic cancer therapy. Acta Biomater. 2021, 130, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Peng, R.; Jin, L.; Ma, J.; Yang, Q.; Sun, D.; Wu, W. Recent Advances in ROS-Sensitive Nano-Formulations for Atherosclerosis Applications. Pharmaceutics 2021, 13, 1452. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhou, Z.; Wang, X.; Deng, H.; Sun, L.; Lin, H.; Kang, F.; Zhang, Y.; Wang, Z.; Yang, W.; et al. Yolk-shell nanovesicles endow glutathione-responsive concurrent drug release and T(1) MRI activation for cancer theranostics. Biomaterials 2020, 244, 119979. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Strategies | Mechanisms | Outcomes | Experimental Models | Ref. |
|---|---|---|---|---|
| Disulfide-bridged organosilica NPs | ↓ GSH, ↑ Cisplatin release | ↑ DNA damage, ↑ apoptosis | A549/DDP, xenograft | [16] |
| Tim-AIII | ↓ GPX4, ↓GSH, ↑ lipid peroxidation | ↑ Ferroptosis, ↓ cancer growth | A549, H1299, xenograft | [17] |
| SPTBN2 loss/Abrine | ↑ SLC7A11 mislocalization, ↓ GSH | ↑ Drug sensitivity, ↑ ferroptosis | A549, H358, H1299 | [20] |
| Strategies and Targets | Nrf2-Related Pathway | Redox Regulation | Effects | Ref. |
|---|---|---|---|---|
| FAM129B | ↓ Ubiquitination (via Keap1) | – | ↑ Redox balance | [33] |
| K563 | ↓ Nrf2 gene expression | ↑ ROS, ↓ GSH | ↑ Oxidative stress sensitivity | [13] |
| Retinoic acid + cisplatin | ↓ DNA repair support (via Nrf2) | ↑ ROS | ↑ Cisplatin sensitivity | [34] |
| KRas depletion | ↓ Nrf2, ↓ NQO1 | ↓ GSH | ↓ Tumor survival | [35] |
| MT1DP/miR-365a-3p | ↓ Nrf2 mRNA | ↑ ROS | ↑ Ferroptosis in NSCLC | [14] |
| Clobetasol + Radiation | ↓ Nrf2 | ↑ ROS, ↑ Fe2+ | ↑ Ferroptosis, ↑ cytotoxicity | [36] |
| Metformin + Cisplatin | ↓ Nrf2 (via ERK1/2 degradation) | ↑ ROS | ↓ Antioxidant defense | [37] |
| Quinacrine (via LNPs) | ↓ Nrf2 signaling | ↑ ROS | ↑ Cisplatin efficacy | [38] |
| TAZ (loss of function) | ↑ Nrf2 dysregulation | ↑ ROS, ↑ Autophagy | ↑ Cell damage and death | [39] |
| Strategy | Mechanism | Outcome | Experimental Models | Ref. |
|---|---|---|---|---|
| Propyl gallate | ↓ HO-1 | ↑ Cisplatin sensitivity, ↑ Apoptvvosis | A549 | [59] |
| Metformin + EGCG | ↓ HO-1, ↓ SIRT1 | ↑ ROS, ↑ apoptosis | A549 | [60,61] |
| ZnPPIX + irradiation | ↓ HO-1 | ↑ Radiosensitivity, ↑ apoptosis | A549 | [62] |
| VP13/47 | ↓ HO-1 activity | ↑ Apoptosis, ↑ mitochondrial dysfunction | A549 | [63] |
| miR-1304 overexpression | ↓ HO-1 | ↓ Viability, ↑ cell cycle arrest | A549, H1975 | [64] |
| Smad7 activation | ↓ HO-1/Akt | ↑ Cisplatin sensitivity | A549 | [65] |
| Garlic oil | ↑ HO-1, ↑ GSTA1, ↑ NQO1 | ↓ Tumor formation | NNK-induced A/J mice | [66] |
| HO-1 overexpression | ↑ HO-1 | ↓ MMPs, ↓ inflammation, ↓ tumor growth | NCI-H292 xenograft | [67] |
| TinPPIX/Fc nanodrug | ↓ HO-1, ↑ Heme | ↓ Metastasis via Bach1 degradation | A549, SCID mouse xenograft | [68] |
| NTP + ZnPPIX | ↓ HO-1 via Nrf2 | ↑ ROS, ↑ apoptosis | A549, H322, H1299 | [69] |
| HNMT inhibition | ↓ HO-1/HER2 axis | ↑ Cisplatin sensitivity, ↓ CSC properties | H441 | [70] |
| Vitamin C | ↑ HO-1 | ↓ Metastasis (p53-related ROS) | H22, BEAS-2B, H1299, xenograft | [71] |
| Yishen Qutong Granules | ↓ HO-1 | ↓ Tumor burden | NSCLC xenograft-bearing mice | [72] |
| Strategies | Mechanisms | Outcomes | Experimental Models | Ref. |
|---|---|---|---|---|
| Dimethoxycurcumin + radiation | TrxR1 inhibition, ↑ ROS, ↓ GSH/GSSG | ↑ Apoptosis, ↑ radiosensitivity | A549 | [89] |
| Shikonin + BAY876/6-AN | TrxR1 (SeC498) inhibition, ↓ NADPH | ↑ Necroptosis, ↓ drug resistance | Keap1-mutant NSCLC | [90] |
| Auranofin | TrxR1 inhibition, ↑ ROS, ↓ GSH | ↑ Cell death, ↓ MMP | Calu-6, A549, H1299 | [91] |
| Auranofin + Lenvatinib | TrxR1 inhibition, ↑ ER stress, ↑ JNK | ↑ Synergistic effect | H1299, H520, A549 | [92] |
| Plumbagin | TrxR1 modification, ↑ ROS | ↑ Apoptosis, redox imbalance | NSCLC cells, xenograft | [93,94] |
| Plumbagin + BAY876 /6-AN | TrxR1 modification, ↑ ROS | ↑ Apoptosis, ↓ resistance | Keap1-mutant NSCLC | [94] |
| Thimerosal | TrxR1 inhibition, ↑ ROS | ↑ Apoptosis | A549 | [95] |
| LW-216 | TrxR1 binding (R371, G442) | ↑ TrxR1, ↑ DNA damage | NSCLC mouse model | [96] |
| 5u (Covalent prodrug) | Covalent binding (C475, SeC498) | ↑ Redox imbalance, apoptosis, ferroptosis | NSCLC cells, xenograft | [97] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, E.; Hong, J.H. Oxidative Stress Defense Module in Lung Cancers: Molecular Pathways and Therapeutic Approaches. Antioxidants 2025, 14, 857. https://doi.org/10.3390/antiox14070857
Lee E, Hong JH. Oxidative Stress Defense Module in Lung Cancers: Molecular Pathways and Therapeutic Approaches. Antioxidants. 2025; 14(7):857. https://doi.org/10.3390/antiox14070857
Chicago/Turabian StyleLee, Eunsun, and Jeong Hee Hong. 2025. "Oxidative Stress Defense Module in Lung Cancers: Molecular Pathways and Therapeutic Approaches" Antioxidants 14, no. 7: 857. https://doi.org/10.3390/antiox14070857
APA StyleLee, E., & Hong, J. H. (2025). Oxidative Stress Defense Module in Lung Cancers: Molecular Pathways and Therapeutic Approaches. Antioxidants, 14(7), 857. https://doi.org/10.3390/antiox14070857