
Serine-Driven Metabolic Plasticity Drives Adaptive Resilience in Pancreatic Cancer Cells

 ,
,  , , , ,
, , , , 
 ,
,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Proliferation Analysis
2.3. Metabolite Extraction from Cell Culture
2.4. Metabolites Quantification in the Media Samples
2.5. Metabolite Extraction from Media Samples
2.6. GC-MS Metabolic Profiling
2.7. LC-MS Metabolic Profiling
2.8. Metabolomics Statistical Data Analysis
2.9. Oxygen Consumption Rate Analysis
2.10. ROS Levels Measurement
2.11. Autophagy Assay
2.12. Lipid Peroxidation Measurement
2.13. Differential Correlation Analysis (DCA)
2.14. RNA Extraction and Real-Time PCR
2.15. cDNA Microarray Expression Analysis
2.16. Integration Analysis Between Transcriptomics and Metabolomics Data
3. Results
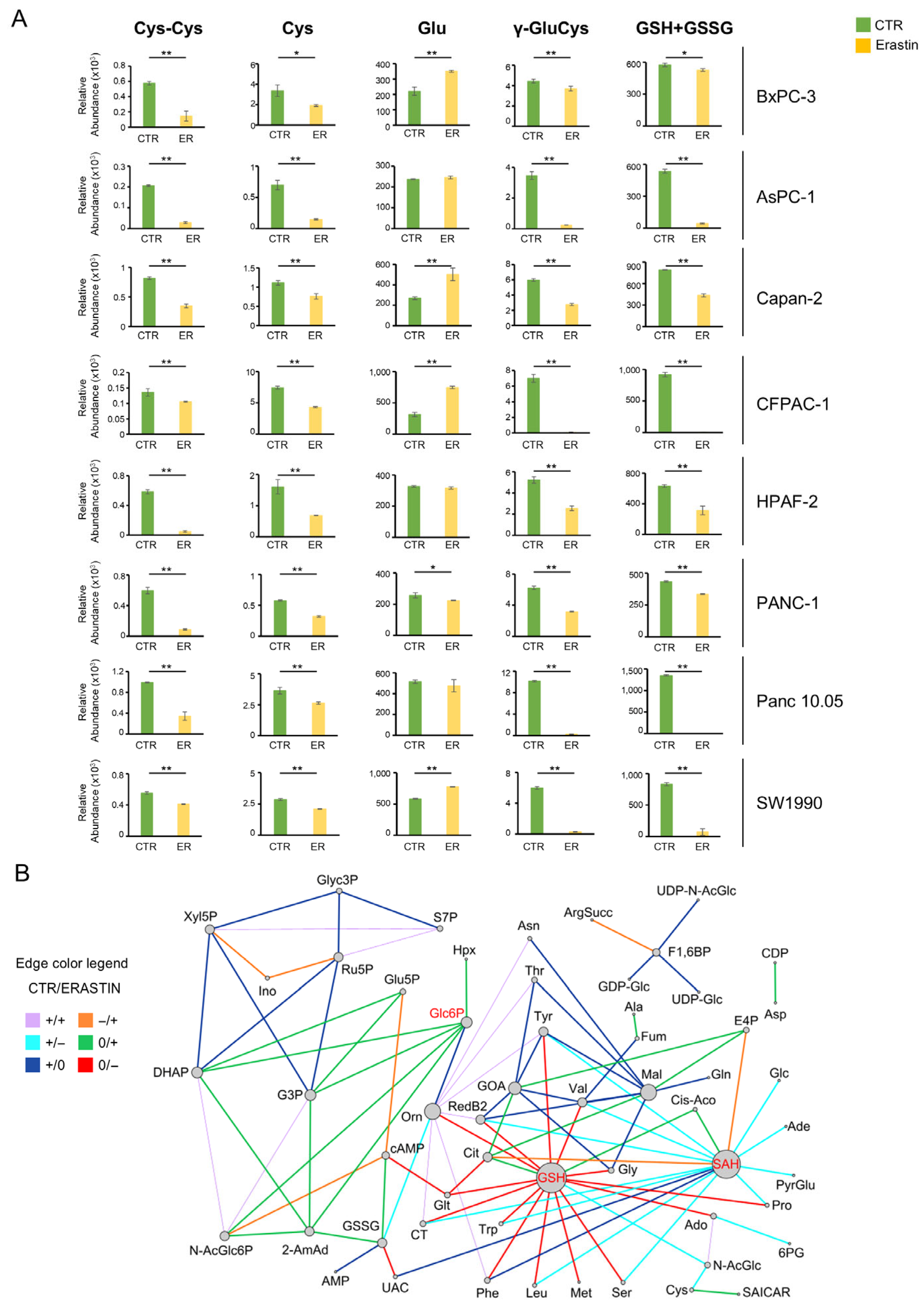
3.1. Pancreatic Cancer Cell Lines Exhibit Great Metabolic Heterogeneity
3.2. Pancreatic Cancer Metabolism Shows Sensitivity to Erastin, but It Is Not Enough
3.3. What Doesn’t Kill Makes Stronger: Metabolic Flexibility Biomarkers to Select Combinatorial Metabolic Drug-Targets
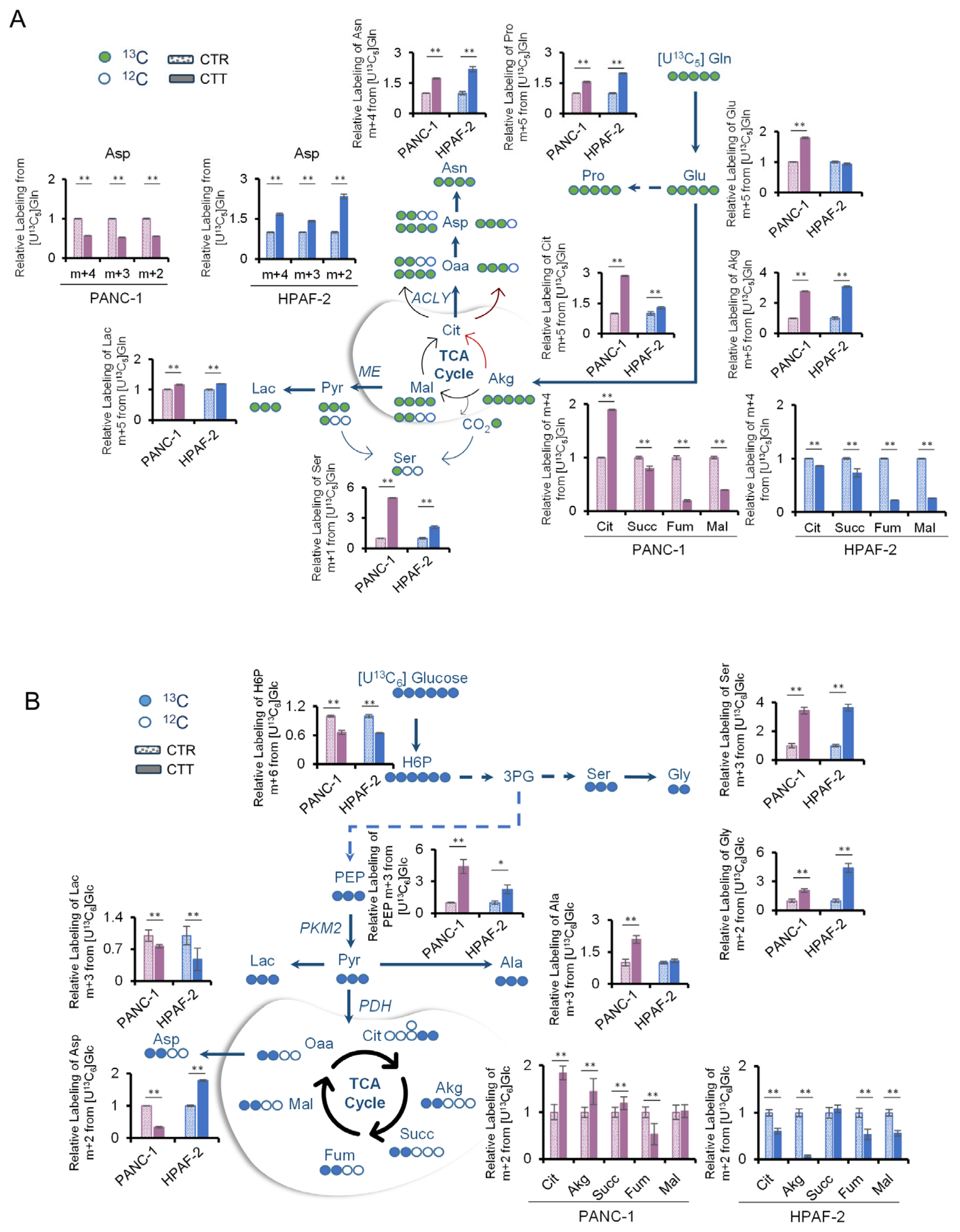
3.4. Combinatorial Drug Treatment Induces a New Metabolic Rewiring in Pancreatic Cancer Cells That Involves Serine Metabolism
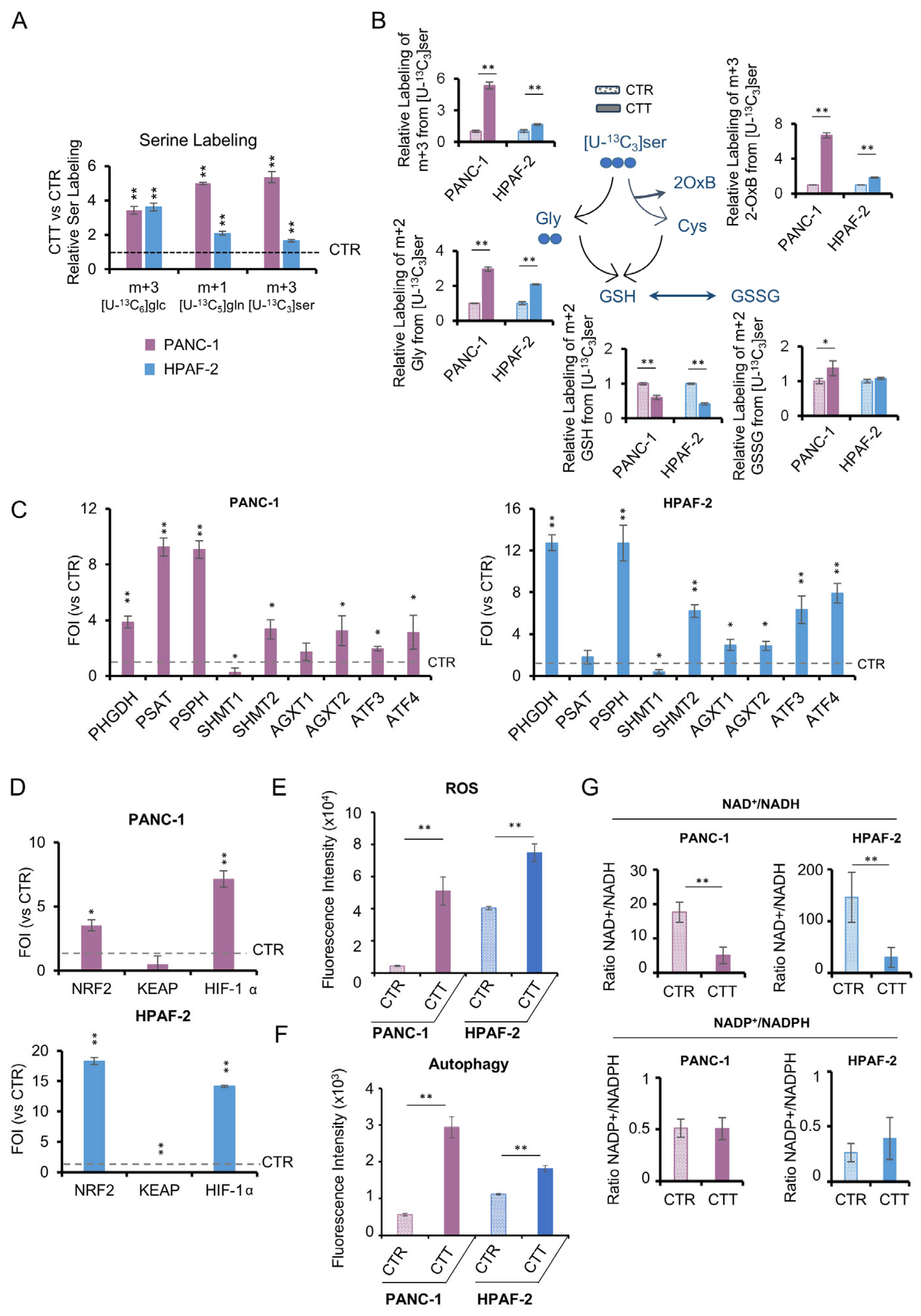
3.5. The Activation of De Novo Serine Synthesis Pathway (SSP) Reveals Pancreatic Cancer Resilience to Combinatorial Drug Treatments
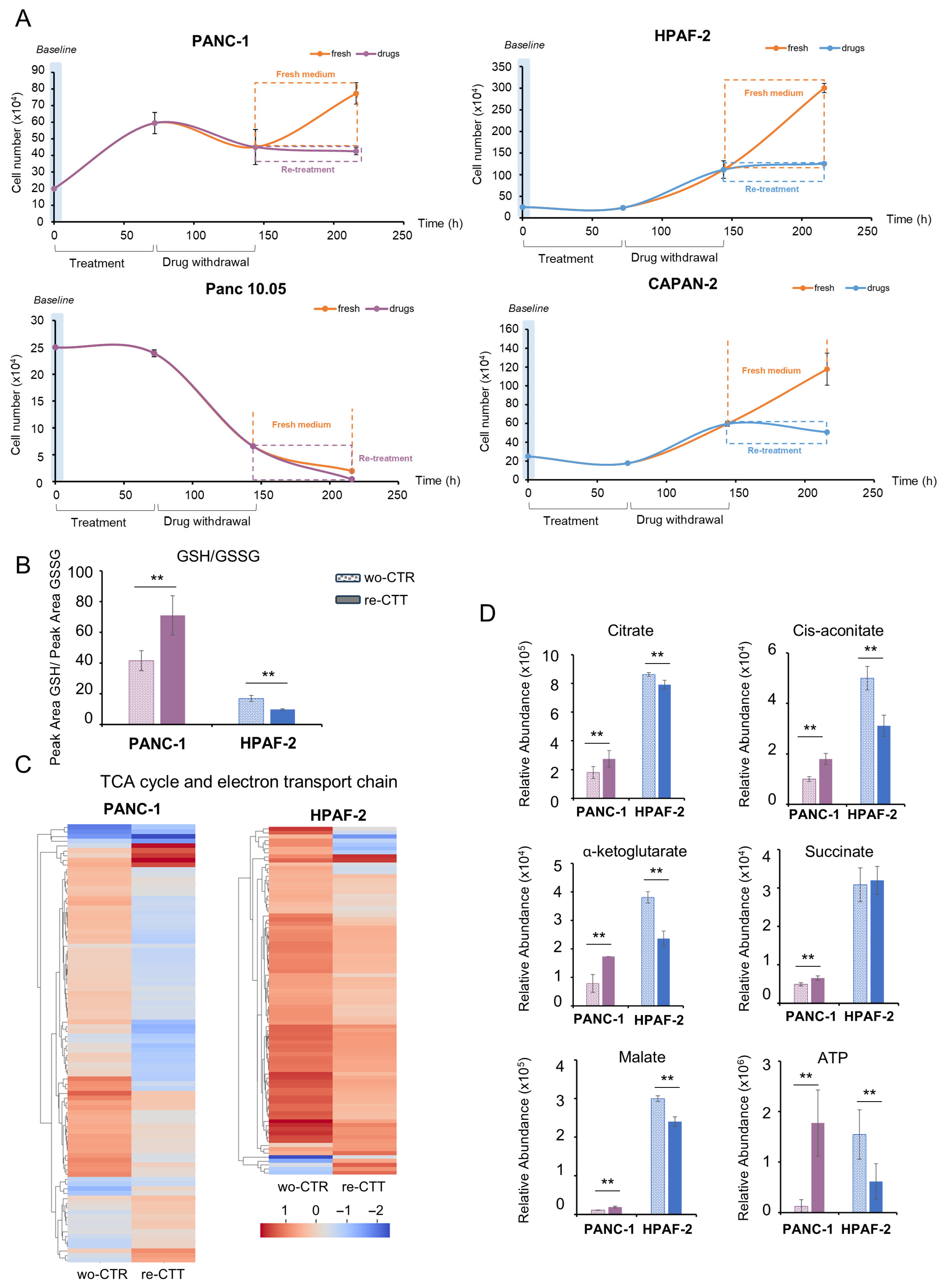
3.6. Metabolic and Transcriptional Rewiring Drives Adaptive Resilience to a Second Round of Treatment in PDAC Cells
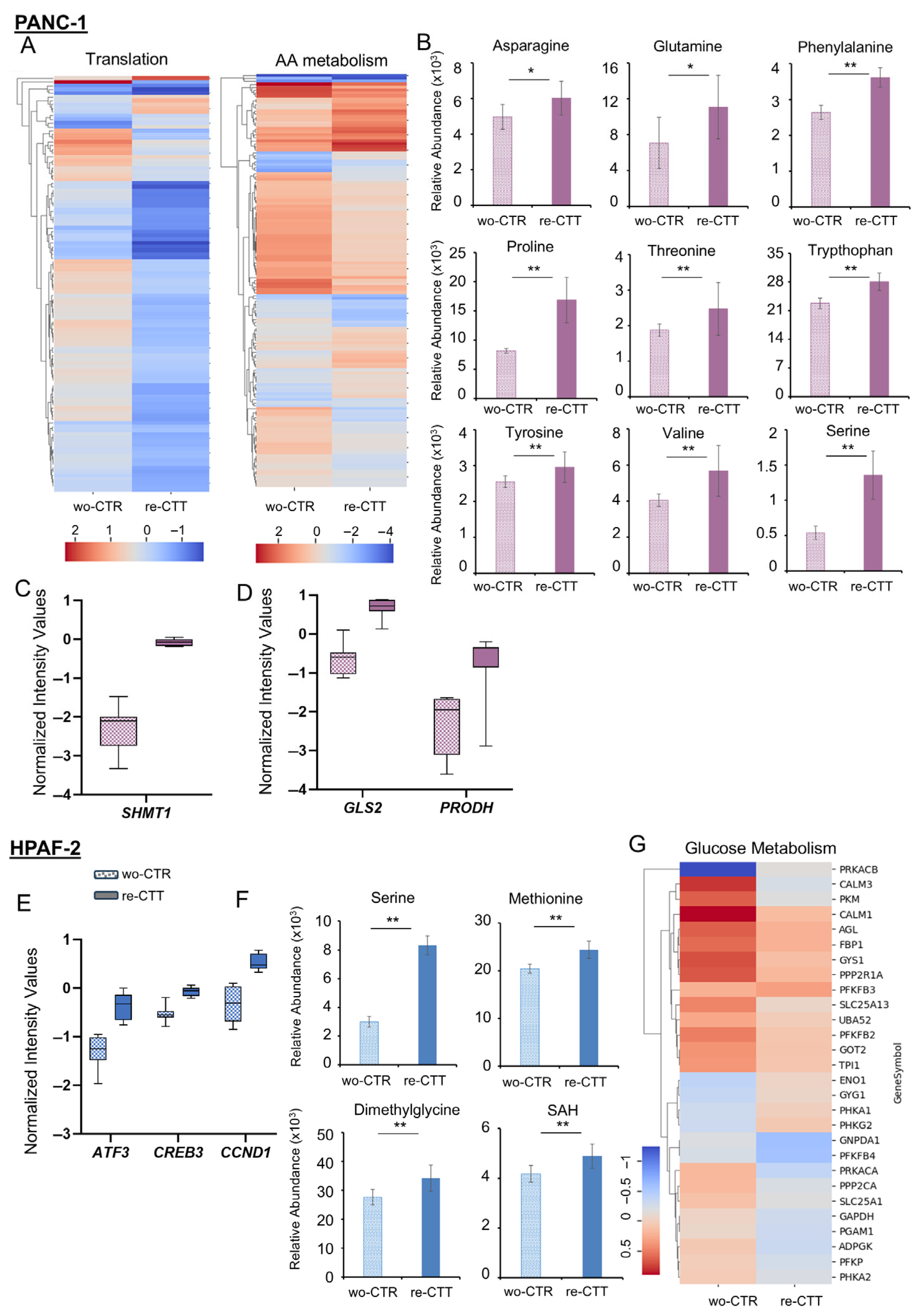
3.6.1. PANC-1: Metabolic Flexibility and Survival Signaling Under Drug Re-Treatment
3.6.2. HPAF-2 Cells: Metabolic Rewiring and Inflammatory Activation in Response to Drug Re-Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 2-AmAd | L-2-Aminoadipic acid |
| 2OxB | 2-oxobutanoate |
| 3PG | 3-Phosphoglyceric acid |
| 6PG | 6-phosphogluconic acid |
| ACLY | ATP citrate lyase |
| Ade | Adenine |
| Ado | Adenosine |
| ADP | Adenosine diphosphate |
| Akg | α-Ketoglutaric acid |
| Ala | L-Alanine |
| ALT | Aminotransferases |
| ANOVA | Analysis of variance |
| AMP | Adenosine monophosphate |
| ArgSucc | Arginosuccinic acid |
| Asn | L-Asparagine |
| Asp | L-aspartic acid |
| ATP | Adenosine triphosphate |
| BHT | Butylated hydroxytoluene |
| cAMP | Cyclic AMP |
| CCK-8 | Cell Counting Kit-8 |
| CDP | Cytidine diphosphate |
| Cis-Aco | Cis Aconitic acid |
| Cit | Citric acid |
| CO2 | Carbon dioxide |
| CT | Citrulline |
| CTR | Control |
| CTT | Combinatorial treatment |
| Cy3 | Cyanine3 |
| Cys | L-Cysteine |
| Cys-Cys | L-Cystine |
| DCA | Differential correlation analysis |
| DCFDA | Dichloro-dihydro-fluoresceine-diacetate |
| DHAP | Dihydroxyacetone phosphate |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| E4P | D-Erythrose-4-phosphate |
| ER | Erastin |
| F1,6BP | Fructose 1,6 bisphosphate |
| FBS | Fetal Bovine Serum |
| FCCP | Carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone |
| FDR | False discovery rate |
| FOI | Fold of induction |
| Fum | Fumaric acid |
| G3P | Glyceraldehyde 3-phosphate |
| G6P/Glc6P | Glucose 6-phosphate |
| GC-MS | Gas chromatography–mass spectrometry |
| GDH | Glutamate dehydrogenase |
| GDP-Glc | GDP-glucose |
| Glc | Glucose |
| Gln | L-Glutamine |
| GLS | Glutaminase |
| Glt | Glutaric acid |
| Glu | L-Glutamic acid |
| Glu5P | L-Glutamic acid 5-phosphate |
| Gly | Glycine |
| Glyc3P | Glycerol 3-phosphate |
| GMP | Guanosine monophosphate |
| GOA | Glyoxylic acid |
| GOT1/GOT2 | Glutamic oxaloacetic transaminases |
| GSH | Reduced glutathione |
| GSSG | Oxidized glutathione |
| H6P | Hexose 6-phosphate |
| HBP | Hexosamine biosynthesis pathway |
| HIF1α | Hypoxia Inducible Factor 1 Subunit Alpha |
| Hpx | Hypoxanthine |
| ICit | Isocitric acid |
| IMP | Inosine 5′-monophosphate |
| Ino | Inosine |
| Lac | Lactic acid |
| LC-MS | Liquid chromatography–mass spectrometry |
| LDH | Lactate dehydrogenase |
| Leu | L-Leucine |
| Mal | Malic acid |
| MDA | Malondialdehyde |
| MDH1 | Malate dehydrogenase |
| ME1 | Malic enzyme |
| Met | L-Methionine |
| MID | Mass Isotopologue Distribution |
| MTBSTFA | N-tert-Butyldimethylsilyl-N-methyltrifluoroacetamide |
| mTOR | Mammalian target of rapamycin |
| MTX | Methotrexate |
| N-AcGlc | N-Acetylglucosammine |
| N-AcGlc6P | N-Acetylglucosammine 6-phosphate |
| NAD | Oxidized nicotinammide adenine dinucleotide |
| NADH | Reduced nicotinammide adenine dinucleotide |
| NADP | Oxidized nicotinammide adenine dinucleotide phosphate |
| NADPH | Reduced nicotinammide adenine dinucleotide phosphate |
| NEAA | Non-essential amino acids |
| OAA | Oxalacetic acid |
| OCR | Oxygen consumption rate |
| Orn | Ornithine |
| PBS | Phosphate-buffered saline |
| PC | Pyruvate carboxylase |
| PDAC | Pancreatic ductal adenocarcinoma |
| PDH | Pyruvate dehydrogenase |
| PEP | Phosphoenolpyruvate |
| PFP | Pentafluorophenyl |
| Phe | L-Phenylalanine |
| PI3K | Phosphatidylinositol-3 kinase |
| PKM2 | Pyruvate kinase M2 subtype |
| PPP | Pentose phosphate pathway |
| Pro | L-Proline |
| Pyr | Pyruvic acid |
| PyrGlu | Pyroglutamic acid |
| QTOF | Quadrupole time-of-flight |
| RedB2 | Reduced riboflavin |
| RIN | RNA integrity number |
| ROS | Reactive oxygen species |
| RPMI | Roswell Park Memorial Institute |
| Ru5P | Ribulose 5-phosphate |
| S7P | Sedoheptulose 7-phosphate |
| SAH | S-Adenosylhomocysteine |
| SAICAR | 1-(phosphoribosyl)imidazolecarboxamide |
| sCoA | Succinyl-CoA |
| Ser | L-Serine |
| SS | Succinyl-CoA synthetase |
| SSP | Serine synthesis pathway |
| Succ | Succinic acid |
| TBDMCS | tert-Butyldimethylchlorosilane |
| TCA | Tricarboxylic acid |
| Thr | L-Threonine |
| Trp | L-Triptophan |
| Tyr | L-Tyrosine |
| UAC | Uric acid |
| UDP-Glc | UDP-Glucose |
| UDP-N-AcGlc | UDP-N-Acetylglucosammine |
| UHPLC | Ultra-High pressure liquid chromatography |
| UDP | Uridine diphosphate |
| UMP | Uridine monophosphate |
| UTP | Uridine triphosphate |
| Val | L-Valine |
| VDAC | Voltage-dependent anion channel |
| Xyl5P | Xylulose 5-phosphate |
| γ-Glu_Cys | γ-glutamylcysteine |
| ΔΨm | Mitochondrial membrane potential |
References
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for Functional Genomics Data Sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Mercanti, L.; Sindaco, M.; Mazzone, M.; Di Marcantonio, M.C.; Piscione, M.; Muraro, R.; Mincione, G. PDAC, the Influencer Cancer: Cross-Talk with Tumor Microenvironment and Connected Potential Therapy Strategies. Cancers 2023, 15, 2923. [Google Scholar] [CrossRef] [PubMed]
- De Santis, M.C.; Bockorny, B.; Hirsch, E.; Cappello, P.; Martini, M. Exploiting Pancreatic Cancer Metabolism: Challenges and Opportunities. Trends Mol. Med. 2024, 30, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Li, W.; Li, X.; Wang, Z.; Chen, Z.; Shi, H.; He, R.; Chen, C.; Zhou, W. Glucose Metabolism and Tumour Microenvironment in Pancreatic Cancer: A Key Link in Cancer Progression. Front. Immunol. 2022, 13, 1038650. [Google Scholar] [CrossRef]
- Jin, H.-R.; Wang, J.; Wang, Z.-J.; Xi, M.-J.; Xia, B.-H.; Deng, K.; Yang, J.-L. Lipid Metabolic Reprogramming in Tumor Microenvironment: From Mechanisms to Therapeutics. J. Hematol. Oncol. 2023, 16, 103. [Google Scholar] [CrossRef]
- Yousef, A.; Yousef, M.; Chowdhury, S.; Abdilleh, K.; Knafl, M.; Edelkamp, P.; Alfaro-Munoz, K.; Chacko, R.; Peterson, J.; Smaglo, B.G.; et al. Impact of KRAS Mutations and Co-Mutations on Clinical Outcomes in Pancreatic Ductal Adenocarcinoma. NPJ Precis. Oncol. 2024, 8, 27. [Google Scholar] [CrossRef]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS–RAF–MEK-Dependent Oxidative Cell Death Involving Voltage-Dependent Anion Channels. Nature 2007, 447, 865–869. [Google Scholar] [CrossRef]
- Parker, J.L.; Deme, J.C.; Kolokouris, D.; Kuteyi, G.; Biggin, P.C.; Lea, S.M.; Newstead, S. Molecular Basis for Redox Control by the Human Cystine/Glutamate Antiporter System Xc−. Nat. Commun. 2021, 12, 7147. [Google Scholar] [CrossRef]
- Sato, H.; Tamba, M.; Ishii, T.; Bannai, S. Cloning and Expression of a Plasma Membrane Cystine/Glutamate Exchange Transporter Composed of Two Distinct Proteins. J. Biol. Chem. 1999, 274, 11455–11458. [Google Scholar] [CrossRef]
- Ghasemitarei, M.; Yusupov, M.; Razzokov, J.; Shokri, B.; Bogaerts, A. Transport of Cystine across xC− Antiporter. Arch. Biochem. Biophys. 2019, 664, 117–126. [Google Scholar] [CrossRef]
- Kennedy, L.; Sandhu, J.K.; Harper, M.-E.; Cuperlovic-Culf, M. Role of Glutathione in Cancer: From Mechanisms to Therapies. Biomolecules 2020, 10, 1429. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, E.N.; Sheldon, K.L.; DeHart, D.N.; Patnaik, J.; Manevich, Y.; Townsend, D.M.; Bezrukov, S.M.; Rostovtseva, T.K.; Lemasters, J.J. Voltage-Dependent Anion Channels Modulate Mitochondrial Metabolism in Cancer Cells: Regulation by Free Tubulin and Erastin. J. Biol. Chem. 2013, 288, 11920–11929. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Bai, Y.; Jia, Y.; Zhao, Y.; Kang, R.; Tang, D.; Dai, E. Ferroptosis Is a Lysosomal Cell Death Process. Biochem. Biophys. Res. Commun. 2018, 503, 1550–1556. [Google Scholar] [CrossRef]
- Wang, H.; Liu, C.; Zhao, Y.; Zhang, W.; Xu, K.; Li, D.; Zhou, Y.; Li, H.; Xiao, G.; Lu, B.; et al. Inhibition of LONP1 Protects Against Erastin-Induced Ferroptosis in Pancreatic Ductal Adenocarcinoma PANC1 Cells. Biochem. Biophys. Res. Commun. 2020, 522, 1063–1068. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, P.; Li, Z.; Xu, C.; Wang, H.; Jia, B.; Gong, A.; Xu, M. Vitamin C Sensitizes Pancreatic Cancer Cells to Erastin-Induced Ferroptosis by Activating the AMPK/Nrf2/HMOX1 Pathway. Oxid. Med. Cell. Longev. 2022, 2022, 5361241. [Google Scholar] [CrossRef]
- Biancur, D.E.; Paulo, J.A.; Małachowska, B.; Quiles Del Rey, M.; Sousa, C.M.; Wang, X.; Sohn, A.S.W.; Chu, G.C.; Gygi, S.P.; Harper, J.W.; et al. Compensatory Metabolic Networks in Pancreatic Cancers upon Perturbation of Glutamine Metabolism. Nat. Commun. 2017, 8, 15965. [Google Scholar] [CrossRef]
- Gaglio, D.; Valtorta, S.; Ripamonti, M.; Bonanomi, M.; Damiani, C.; Todde, S.; Negri, A.S.; Sanvito, F.; Mastroianni, F.; Campli, A.D.; et al. Divergent in Vitro/in Vivo Responses to Drug Treatments of Highly Aggressive NIH-Ras Cancer Cells: A PET Imaging and Metabolomics-Mass-Spectrometry Study. Oncotarget 2016, 7, 52017–52031. [Google Scholar] [CrossRef]
- Bonanomi, M.; Salmistraro, N.; Porro, D.; Pinsino, A.; Colangelo, A.M.; Gaglio, D. Polystyrene Micro and Nano-Particles Induce Metabolic Rewiring in Normal Human Colon Cells: A Risk Factor for Human Health. Chemosphere 2022, 303, 134947. [Google Scholar] [CrossRef]
- Antoniewicz, M.R. A Guide to 13C Metabolic Flux Analysis for the Cancer Biologist. Exp. Mol. Med. 2018, 50, 1–13. [Google Scholar] [CrossRef]
- Bonanomi, M.; Salmistraro, N.; Fiscon, G.; Conte, F.; Paci, P.; Bravatà, V.; Forte, G.I.; Volpari, T.; Scorza, M.; Mastroianni, F.; et al. Transcriptomics and Metabolomics Integration Reveals Redox-Dependent Metabolic Rewiring in Breast Cancer Cells. Cancers 2021, 13, 5058. [Google Scholar] [CrossRef] [PubMed]
- McDermaid, A.; Monier, B.; Zhao, J.; Liu, B.; Ma, Q. Interpretation of Differential Gene Expression Results of RNA-Seq Data: Review and Integration. Brief. Bioinform. 2019, 20, 2044–2054. [Google Scholar] [CrossRef]
- McKenzie, A.T.; Katsyv, I.; Song, W.-M.; Wang, M.; Zhang, B. DGCA: A Comprehensive R Package for Differential Gene Correlation Analysis. BMC Syst. Biol. 2016, 10, 106. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Garapati, P.; Gillespie, M.; Hausmann, K.; Haw, R.; Jassal, B.; Jupe, S.; Korninger, F.; McKay, S.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2016, 44, D481–D487. [Google Scholar] [CrossRef]
- Waskom, M.L. Seaborn: Statistical Data Visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- Elizarraras, J.M.; Liao, Y.; Shi, Z.; Zhu, Q.; Pico, A.R.; Zhang, B. WebGestalt 2024: Faster Gene Set Analysis and New Support for Metabolomics and Multi-Omics. Nucleic Acids Res. 2024, 52, W415–W421. [Google Scholar] [CrossRef]
- Mehla, K.; Singh, P.K. Metabolic Subtyping for Novel Personalized Therapies Against Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 6–8. [Google Scholar] [CrossRef]
- Stanciu, S.; Ionita-Radu, F.; Stefani, C.; Miricescu, D.; Stanescu-Spinu, I.-I.; Greabu, M.; Ripszky Totan, A.; Jinga, M. Targeting PI3K/AKT/mTOR Signaling Pathway in Pancreatic Cancer: From Molecular to Clinical Aspects. Int. J. Mol. Sci. 2022, 23, 10132. [Google Scholar] [CrossRef]
- Cui, J.; Guo, Y.; Wu, H.; Xiong, J.; Peng, T. Everolimus Regulates the Activity of Gemcitabine-Resistant Pancreatic Cancer Cells by Targeting the Warburg Effect via PI3K/AKT/mTOR Signaling. Mol. Med. 2021, 27, 38. [Google Scholar] [CrossRef]
- Saunier, E.; Antonio, S.; Regazzetti, A.; Auzeil, N.; Laprévote, O.; Shay, J.W.; Coumoul, X.; Barouki, R.; Benelli, C.; Huc, L.; et al. Resveratrol Reverses the Warburg Effect by Targeting the Pyruvate Dehydrogenase Complex in Colon Cancer Cells. Sci. Rep. 2017, 7, 6945. [Google Scholar] [CrossRef]
- Filippone, M.G.; Gaglio, D.; Bonfanti, R.; Tucci, F.A.; Ceccacci, E.; Pennisi, R.; Bonanomi, M.; Jodice, G.; Tillhon, M.; Montani, F.; et al. CDK12 Promotes Tumorigenesis but Induces Vulnerability to Therapies Inhibiting Folate One-Carbon Metabolism in Breast Cancer. Nat. Commun. 2022, 13, 2642. [Google Scholar] [CrossRef] [PubMed]
- Scalise, M.; Pochini, L.; Galluccio, M.; Indiveri, C. Glutamine Transport. From Energy Supply to Sensing and Beyond. Biochim. Biophys. Acta (BBA)-Bioenerg. 2016, 1857, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Cooper, D.E.; Scheidemantle, G.; Locasale, J.W.; Kirsch, D.G.; Liu, X. 13C Tracer Analysis Suggests Extensive Recycling of Endogenous CO2 in Vivo. Cancer Metab. 2022, 10, 11. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gracilla, D.; Cai, L.; Zhang, M.; Yu, X.; Chen, X.; Zhang, J.; Long, X.; Ding, H.-F.; Yan, C. ATF3 Promotes the Serine Synthesis Pathway and Tumor Growth under Dietary Serine Restriction. Cell Rep. 2021, 36, 109706. [Google Scholar] [CrossRef]
- Gaglio, D.; Bonanomi, M.; Valtorta, S.; Bharat, R.; Ripamonti, M.; Conte, F.; Fiscon, G.; Righi, N.; Napodano, E.; Papa, F.; et al. Disruption of Redox Homeostasis for Combinatorial Drug Efficacy in K-Ras Tumors as Revealed by Metabolic Connectivity Profiling. Cancer Metab. 2020, 8, 22. [Google Scholar] [CrossRef]
- Zhang, G.; Yang, Y.; Hu, H.; Liu, K.; Li, B.; Zhu, Y.; Wang, Z.; Wu, Q.; Mei, Y. Energy Stress-Induced Linc01564 Activates the Serine Synthesis Pathway and Facilitates Hepatocellular Carcinogenesis. Oncogene 2021, 40, 2936–2951. [Google Scholar] [CrossRef]
- Sun, H.; Ma, H.; Zhang, H.; Ji, M. Up-Regulation of MELK by E2F1 Promotes the Proliferation in Cervical Cancer Cells. Int. J. Biol. Sci. 2021, 17, 3875–3888. [Google Scholar] [CrossRef]
- Li, X.; Wang, F.; Xu, X.; Zhang, J.; Xu, G. The Dual Role of STAT1 in Ovarian Cancer: Insight Into Molecular Mechanisms and Application Potentials. Front. Cell Dev. Biol. 2021, 9, 636595. [Google Scholar] [CrossRef]
- Pérez-Benavente, B.; Fathinajafabadi, A.; de la Fuente, L.; Gandía, C.; Martínez-Férriz, A.; Pardo-Sánchez, J.M.; Milián, L.; Conesa, A.; Romero, O.A.; Carretero, J.; et al. New Roles for AP-1/JUNB in Cell Cycle Control and Tumorigenic Cell Invasion via Regulation of Cyclin E1 and TGF-Β2. Genome Biol. 2022, 23, 252. [Google Scholar] [CrossRef]
- Li, R.; Ji, Q.; Fu, S.; Gu, J.; Liu, D.; Wang, L.; Yuan, X.; Wen, Y.; Dai, C.; Li, H. ITGA3 Promotes Pancreatic Cancer Progression Through HIF1α- and c-Myc-Driven Glycolysis in a Collagen I-Dependent Autocrine Manner. Cancer Gene Ther. 2025, 32, 240–253. [Google Scholar] [CrossRef]
- Sun, Q.; Ye, Z.; Qin, Y.; Fan, G.; Ji, S.; Zhuo, Q.; Xu, W.; Liu, W.; Hu, Q.; Liu, M.; et al. Oncogenic Function of TRIM2 in Pancreatic Cancer by Activating ROS-Related NRF2/ITGB7/FAK Axis. Oncogene 2020, 39, 6572–6588. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.U.; Cho, S.Y.; Jeong, H.; Han, J.; Chae, H.Y.; Yang, H.; Sung, C.O.; Choi, Y.-L.; Shin, Y.K.; Kwon, M.J. Matrix Metalloproteinase 11 (MMP11) in Macrophages Promotes the Migration of HER2-Positive Breast Cancer Cells and Monocyte Recruitment Through CCL2–CCR2 Signaling. Lab. Investig. 2022, 102, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.; Schäfer, J.; Rothe, M.; Bange, J.; Knyazev, P.; Ullrich, A. Identification of MMP-15 as an Anti-Apoptotic Factor in Cancer Cells. J. Biol. Chem. 2005, 280, 34123–34132. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Zhang, Z.; Cheng, Y.; Yang, J.; Fan, C.; Zhang, X.; Yang, J.; Wang, L.; Guo, D.; Yan, D. Hypoxia-Induced NFATc3 deSUMOylation Enhances Pancreatic Carcinoma Progression. Cell Death Dis. 2022, 13, 413. [Google Scholar] [CrossRef]
- Colloca, A.; Balestrieri, A.; Anastasio, C.; Balestrieri, M.L.; D’Onofrio, N. Mitochondrial Sirtuins in Chronic Degenerative Diseases: New Metabolic Targets in Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 3212. [Google Scholar] [CrossRef]
- Ianni, A.; Kumari, P.; Tarighi, S.; Braun, T.; Vaquero, A. SIRT7: A Novel Molecular Target for Personalized Cancer Treatment? Oncogene 2024, 43, 993–1006. [Google Scholar] [CrossRef]
- Gu, Y.; Ding, C.; Yu, T.; Liu, B.; Tang, W.; Wang, Z.; Tang, X.; Liang, G.; Peng, J.; Zhang, X.; et al. SIRT7 Promotes Hippo/YAP Activation and Cancer Cell Proliferation in Hepatocellular Carcinoma via Suppressing MST1. Cancer Sci. 2024, 115, 1209–1223. [Google Scholar] [CrossRef]
- Ferreira, J.M.; Gonçalves, C.S.; Costa, B.M. Emerging Roles and Biomarker Potential of WNT6 in Human Cancers. Cell Commun. Signal. 2024, 22, 538. [Google Scholar] [CrossRef]
- Wu, Y.; Ginther, C.; Kim, J.; Mosher, N.; Chung, S.; Slamon, D.; Vadgama, J.V. Expression of Wnt3 Activates Wnt/β-Catenin Pathway and Promotes EMT-like Phenotype in Trastuzumab Resistant HER2-Overexpressing Breast Cancer Cells. Mol. Cancer Res. 2012, 10, 1597–1606. [Google Scholar] [CrossRef]
- Shi, Q.; Xue, C.; Zeng, Y.; Yuan, X.; Chu, Q.; Jiang, S.; Wang, J.; Zhang, Y.; Zhu, D.; Li, L. Notch Signaling Pathway in Cancer: From Mechanistic Insights to Targeted Therapies. Signal Transduct. Target. Ther. 2024, 9, 128. [Google Scholar] [CrossRef]
- Landor, S.K.-J.; Mutvei, A.P.; Mamaeva, V.; Jin, S.; Busk, M.; Borra, R.; Grönroos, T.J.; Kronqvist, P.; Lendahl, U.; Sahlgren, C.M. Hypo- and Hyperactivated Notch Signaling Induce a Glycolytic Switch Through Distinct Mechanisms. Proc. Natl. Acad. Sci. USA 2011, 108, 18814–18819. [Google Scholar] [CrossRef]
- Totaro, A.; Castellan, M.; Di Biagio, D.; Piccolo, S. Crosstalk between YAP/TAZ and Notch Signaling. Trends Cell Biol. 2018, 28, 560–573. [Google Scholar] [CrossRef]
- Singhi, A.D.; Koay, E.J.; Chari, S.T.; Maitra, A. Early Detection of Pancreatic Cancer: Opportunities and Challenges. Gastroenterology 2019, 156, 2024–2040. [Google Scholar] [CrossRef]
- McDonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic Reprogramming During Pancreatic Cancer Progression Links Anabolic Glucose Metabolism to Distant Metastasis. Nat. Genet. 2017, 49, 367–376. [Google Scholar] [CrossRef]
- Sullivan, M.R.; Danai, L.V.; Lewis, C.A.; Chan, S.H.; Gui, D.Y.; Kunchok, T.; Dennstedt, E.A.; Vander Heiden, M.G.; Muir, A. Quantification of Microenvironmental Metabolites in Murine Cancers Reveals Determinants of Tumor Nutrient Availability. eLife 2019, 8, e44235. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, X.; Wang, W.; Li, X.; Sun, X.; Zhao, Y.; Wang, Q.; Li, Y.; Hu, F.; Ren, H. Metabolic Reprogramming and Therapeutic Resistance in Primary and Metastatic Breast Cancer. Mol. Cancer 2024, 23, 261. [Google Scholar] [CrossRef]
- Yamamoto, K.; Iwadate, D.; Kato, H.; Nakai, Y.; Tateishi, K.; Fujishiro, M. Targeting the Metabolic Rewiring in Pancreatic Cancer and Its Tumor Microenvironment. Cancers 2022, 14, 4351. [Google Scholar] [CrossRef]
- Serrano, J.J.; Medina, M.Á. Metabolic Reprogramming at the Edge of Redox: Connections Between Metabolic Reprogramming and Cancer Redox State. Int. J. Mol. Sci. 2025, 26, 498. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine Supports Pancreatic Cancer Growth Through a KRAS-Regulated Metabolic Pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Islam, M.M.; Goertzen, A.; Singh, P.K.; Saha, R. Exploring the Metabolic Landscape of Pancreatic Ductal Adenocarcinoma Cells Using Genome-Scale Metabolic Modeling. iScience 2022, 25, 104483. [Google Scholar] [CrossRef]
- Chen, H.; Wang, C.; Liu, Z.; He, X.; Tang, W.; He, L.; Feng, Y.; Liu, D.; Yin, Y.; Li, T. Ferroptosis and Its Multifaceted Role in Cancer: Mechanisms and Therapeutic Approach. Antioxidants 2022, 11, 1504. [Google Scholar] [CrossRef] [PubMed]
- Conger, K.O.; Chidley, C.; Ozgurses, M.E.; Zhao, H.; Kim, Y.; Semina, S.E.; Burns, P.; Rawat, V.; Lietuvninkas, L.; Sheldon, R.; et al. ASCT2 Is a Major Contributor to Serine Uptake in Cancer Cells. Cell Rep. 2024, 43, 114552. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W. Serine, Glycine and One-Carbon Units: Cancer Metabolism in Full Circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef]
- Maddocks, O.D.K.; Berkers, C.R.; Mason, S.M.; Zheng, L.; Blyth, K.; Gottlieb, E.; Vousden, K.H. Serine Starvation Induces Stress and P53-Dependent Metabolic Remodelling in Cancer Cells. Nature 2013, 493, 542–546. [Google Scholar] [CrossRef]
- Vettore, L.; Westbrook, R.L.; Tennant, D.A. New Aspects of Amino Acid Metabolism in Cancer. Br. J. Cancer 2020, 122, 150–156. [Google Scholar] [CrossRef]
- Mattaini, K.R.; Sullivan, M.R.; Vander Heiden, M.G. The Importance of Serine Metabolism in Cancer. J. Cell Biol. 2016, 214, 249–257. [Google Scholar] [CrossRef]
- Zhu, J.; Thompson, C.B. Metabolic Regulation of Cell Growth and Proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef]
- Tufail, M.; Jiang, C.-H.; Li, N. Altered Metabolism in Cancer: Insights into Energy Pathways and Therapeutic Targets. Mol. Cancer 2024, 23, 203. [Google Scholar] [CrossRef]
- Ni, C.; Li, J. Take Metabolic Heterogeneity into Consideration When Applying Dietary Interventions to Cancer Therapy: A Review. Heliyon 2023, 9, e22814. [Google Scholar] [CrossRef]
- Colombo, M.; Mirandola, L.; Chiriva-Internati, M.; Basile, A.; Locati, M.; Lesma, E.; Chiaramonte, R.; Platonova, N. Cancer Cells Exploit Notch Signaling to Redefine a Supportive Cytokine Milieu. Front. Immunol. 2018, 9, 1823. [Google Scholar] [CrossRef] [PubMed]
- Pikman, Y.; Ocasio-Martinez, N.; Alexe, G.; Dimitrov, B.; Kitara, S.; Diehl, F.F.; Robichaud, A.L.; Conway, A.S.; Ross, L.; Su, A.; et al. Targeting Serine Hydroxymethyltransferases 1 and 2 for T-Cell Acute Lymphoblastic Leukemia Therapy. Leukemia 2022, 36, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Chen, Y.; Wang, Y.; Zhao, X.; Zi, R.; Hao, J.; Ding, Q.; Jiang, H.; Wang, X.; Lu, F.; et al. Cancer-Associated Fibroblasts Derived Fibronectin Extra Domain A Promotes Sorafenib Resistance in Hepatocellular Carcinoma Cells by Activating SHMT1. Genes Dis. 2024, 11, 101330. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhou, H.; Tan, L.; Siu, K.T.H.; Guan, X.-Y. Exploring Treatment Options in Cancer: Tumor Treatment Strategies. Signal Transduct. Target. Ther. 2024, 9, 175. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Vander Heiden, M.G.; McCormick, F. The Metabolic Landscape of RAS-Driven Cancers from Biology to Therapy. Nat. Cancer 2021, 2, 271–283. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonanomi, M.; Mallia, S.; Scalise, M.; Aramini, T.; Baldassari, F.; Brivio, E.; Conte, F.; Lo Dico, A.; Bonas, M.; Porro, D.; et al. Serine-Driven Metabolic Plasticity Drives Adaptive Resilience in Pancreatic Cancer Cells. Antioxidants 2025, 14, 833. https://doi.org/10.3390/antiox14070833
Bonanomi M, Mallia S, Scalise M, Aramini T, Baldassari F, Brivio E, Conte F, Lo Dico A, Bonas M, Porro D, et al. Serine-Driven Metabolic Plasticity Drives Adaptive Resilience in Pancreatic Cancer Cells. Antioxidants. 2025; 14(7):833. https://doi.org/10.3390/antiox14070833
Chicago/Turabian StyleBonanomi, Marcella, Sara Mallia, Mariafrancesca Scalise, Tecla Aramini, Federica Baldassari, Elisa Brivio, Federica Conte, Alessia Lo Dico, Matteo Bonas, Danilo Porro, and et al. 2025. "Serine-Driven Metabolic Plasticity Drives Adaptive Resilience in Pancreatic Cancer Cells" Antioxidants 14, no. 7: 833. https://doi.org/10.3390/antiox14070833
APA StyleBonanomi, M., Mallia, S., Scalise, M., Aramini, T., Baldassari, F., Brivio, E., Conte, F., Lo Dico, A., Bonas, M., Porro, D., Indiveri, C., Metallo, C. M., & Gaglio, D. (2025). Serine-Driven Metabolic Plasticity Drives Adaptive Resilience in Pancreatic Cancer Cells. Antioxidants, 14(7), 833. https://doi.org/10.3390/antiox14070833