
Scutellarin Alleviates Cuprizone-Induced Demyelination by Improving Mitochondrial Dysfunction, Reducing Lipid Oxidation and Inhibiting the p38 MAPK Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Experiment Groups and Drug Administration
2.3. Cell Culture and Treatment
2.4. Y-Maze
2.5. Open Field Test
2.6. Western Blot Analysis
2.7. Immunofluorescence Staining
2.8. Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
2.9. Detection of Intracellular Copper Content
2.10. Transmission Electron Microscopy
2.11. Mitochondria Isolation
2.12. ELISA
2.13. Mitochondrial Function Testing
2.14. Statistical Analyses
3. Result
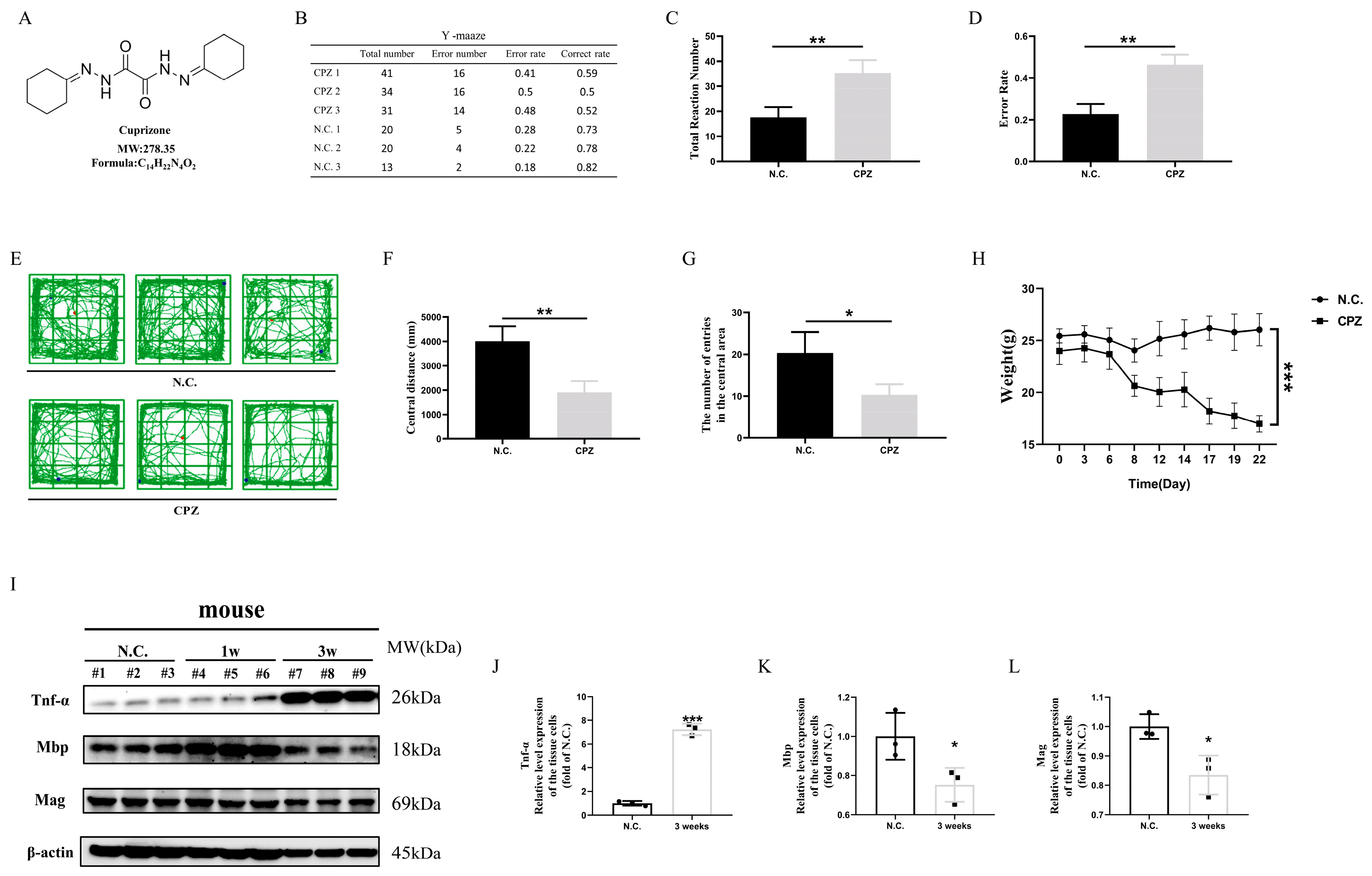
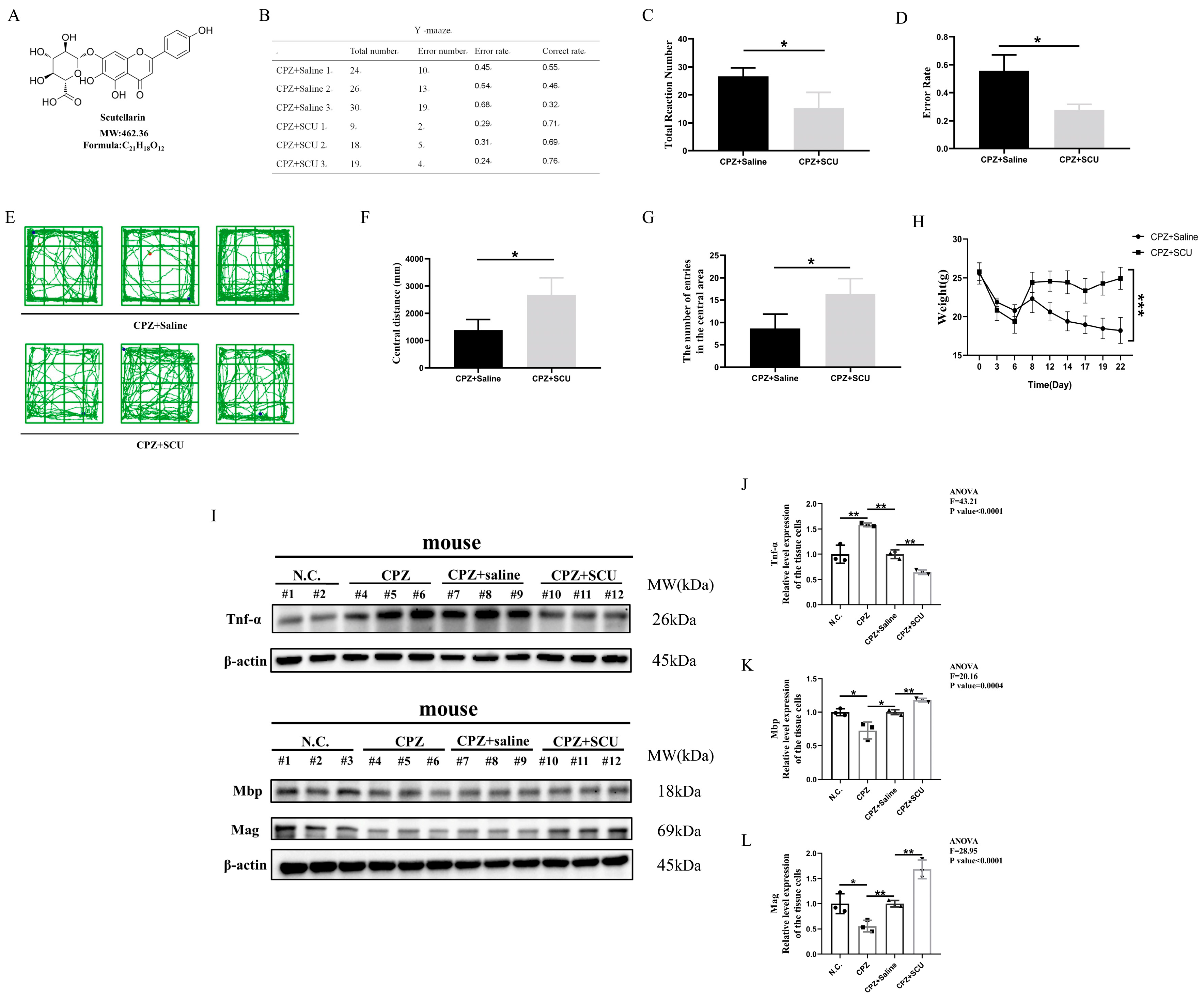
3.1. In Vivo Experiments Demonstrate That Scutellarin Alleviates Cuprizone-Induced Demyelination in Mice
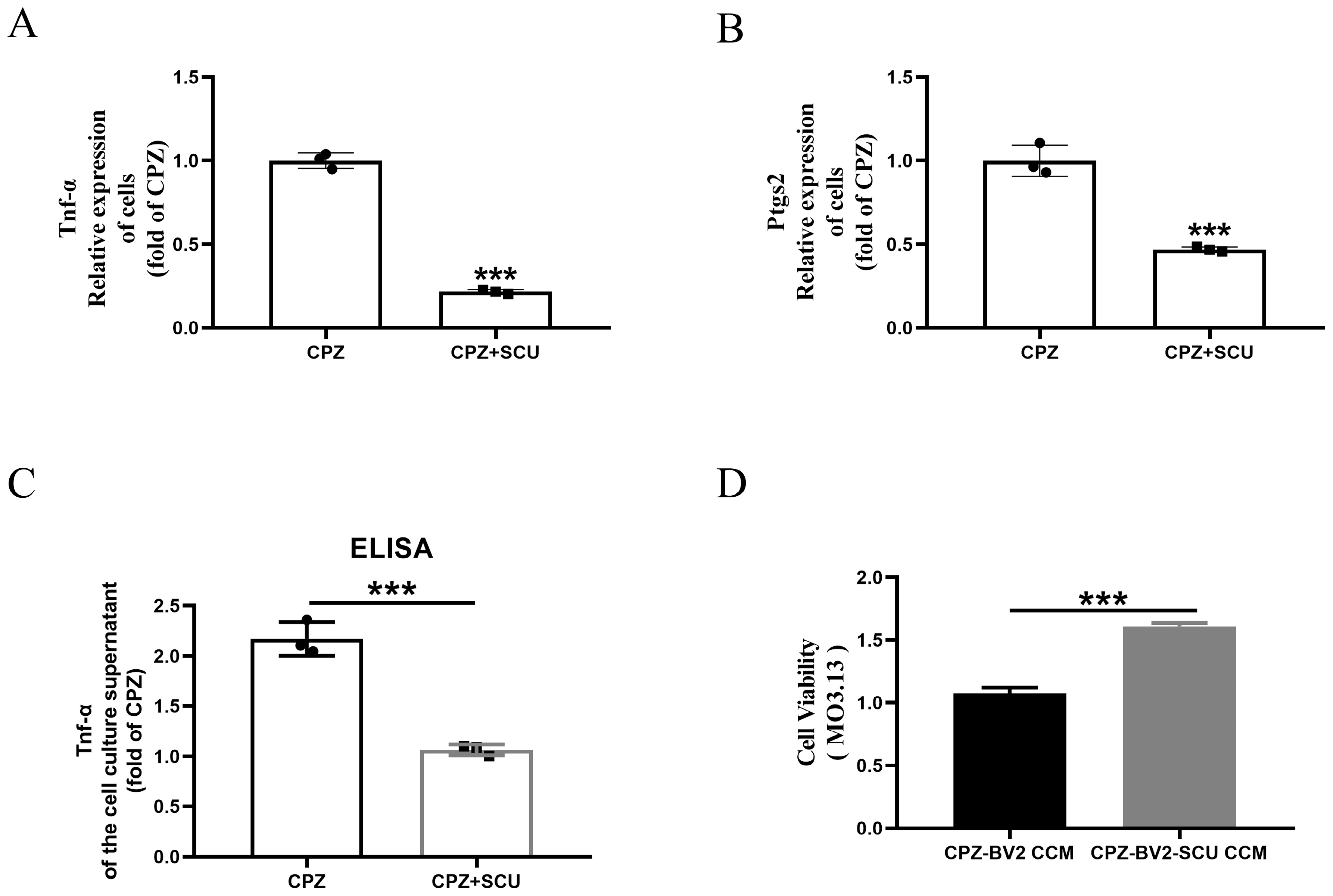
3.2. In Vitro Experiments Reveal That Scutellarin Alleviates Myelin Cell Damage by Inhibiting Cuprizone-Induced Pro-Inflammatory Microglial Activation
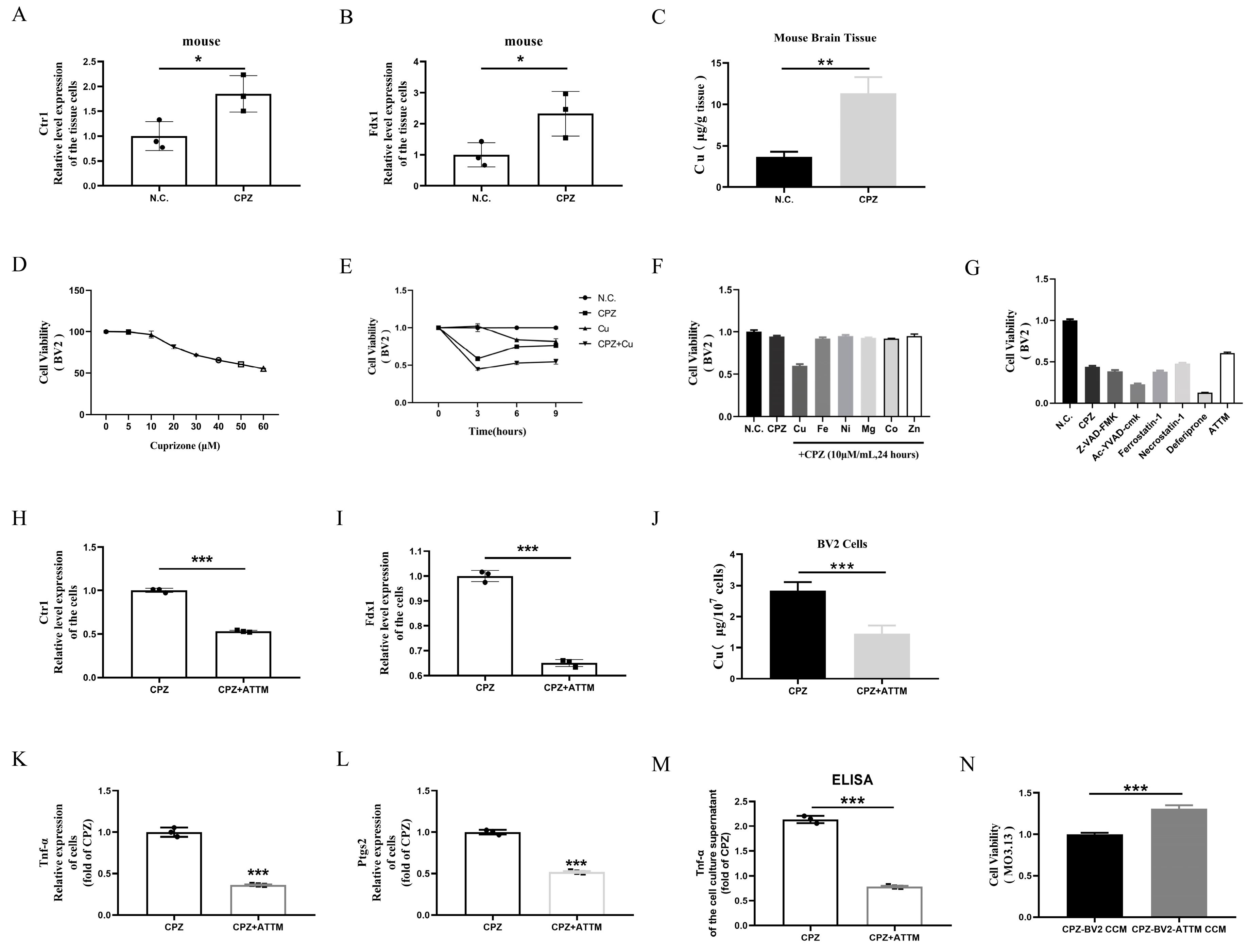
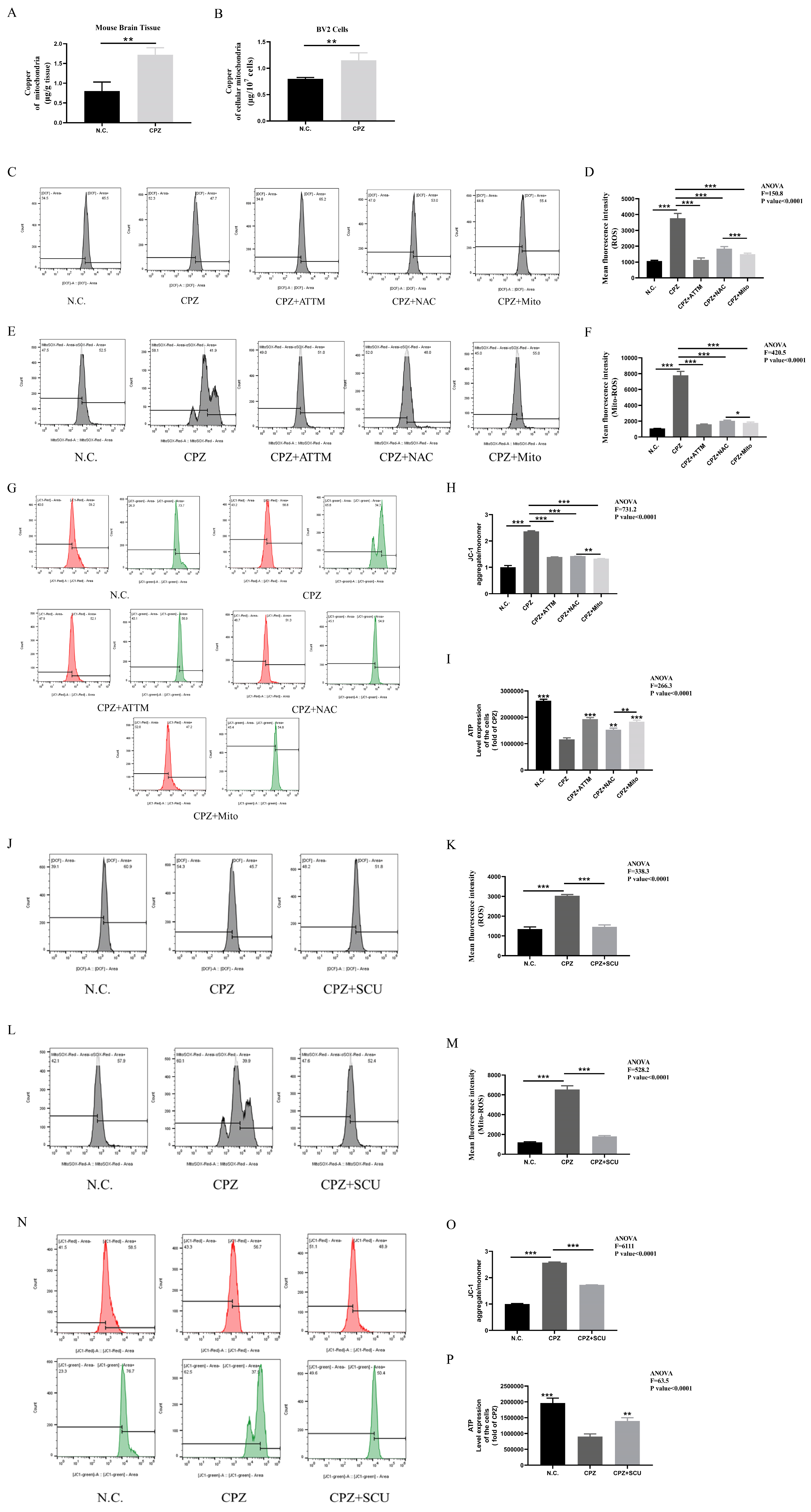
3.3. Cuprizone Forms Complexes with Copper, Inducing Cellular Copper Intoxication and Promoting Pro-Inflammatory Microglial Activation
3.4. Scutellarin Treatment Restores Mitochondrial Dysfunction in BV2 Cells Induced by Cuprizone–Copper(II) Complexes
3.5. Cuprizone–Copper(II) Complexes Promote Pro-Inflammatory Microglial Activation by Increasing Mitochondrial ROS
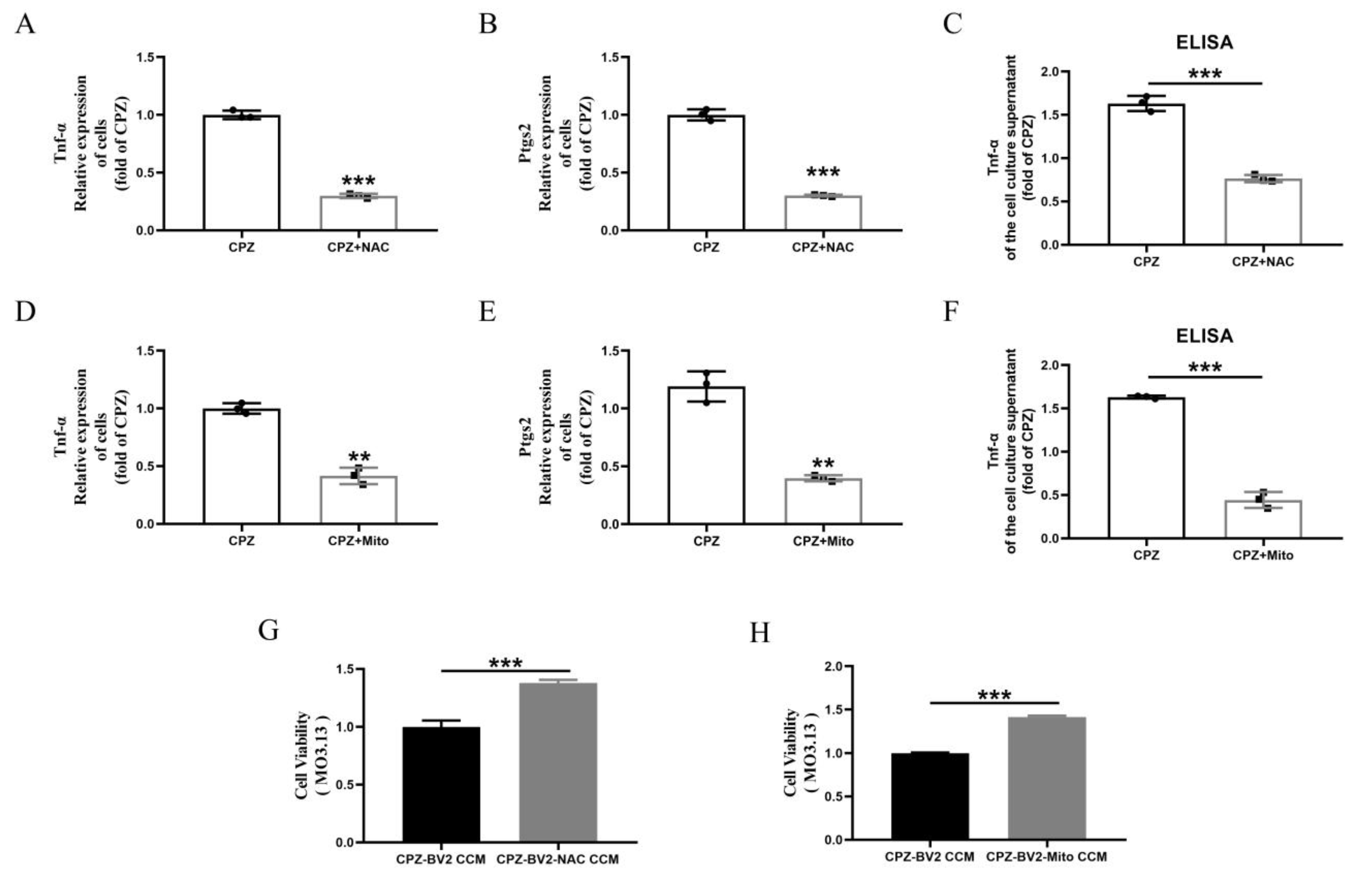
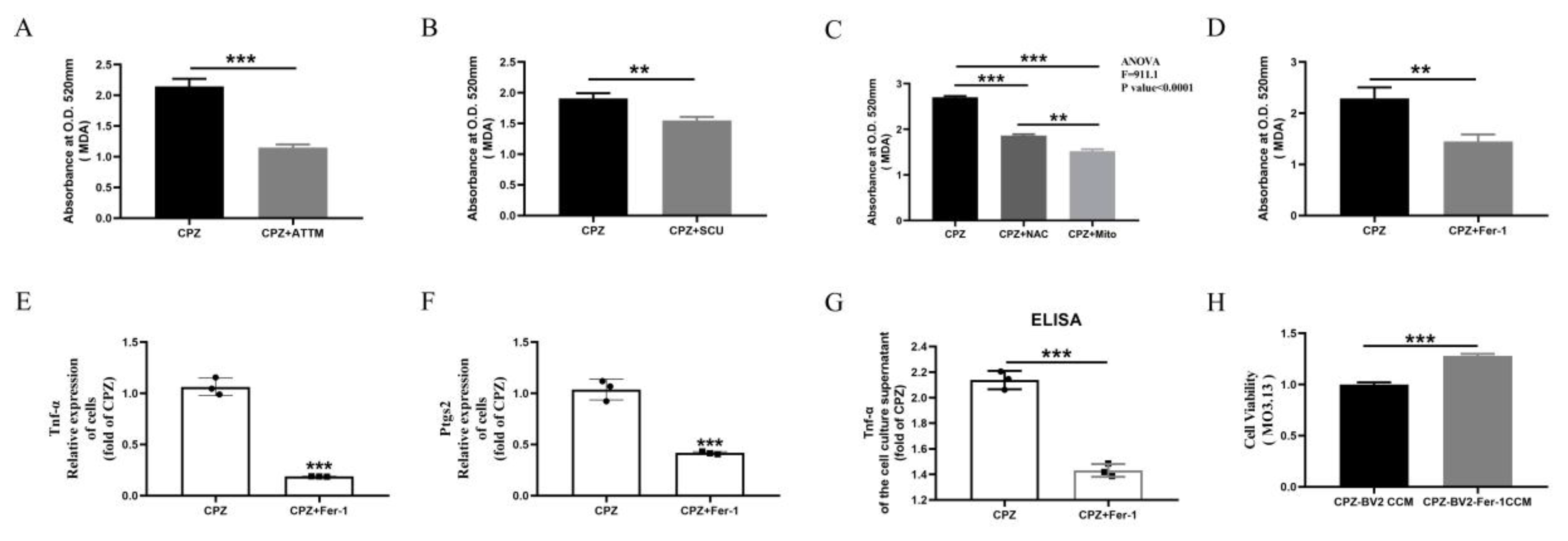
3.6. Scutellarin Inhibits Pro-Inflammatory Microglial Responses by Reducing Cuprizone–Copper Complex-Induced Mitochondrial Damage and Lipid Peroxidation
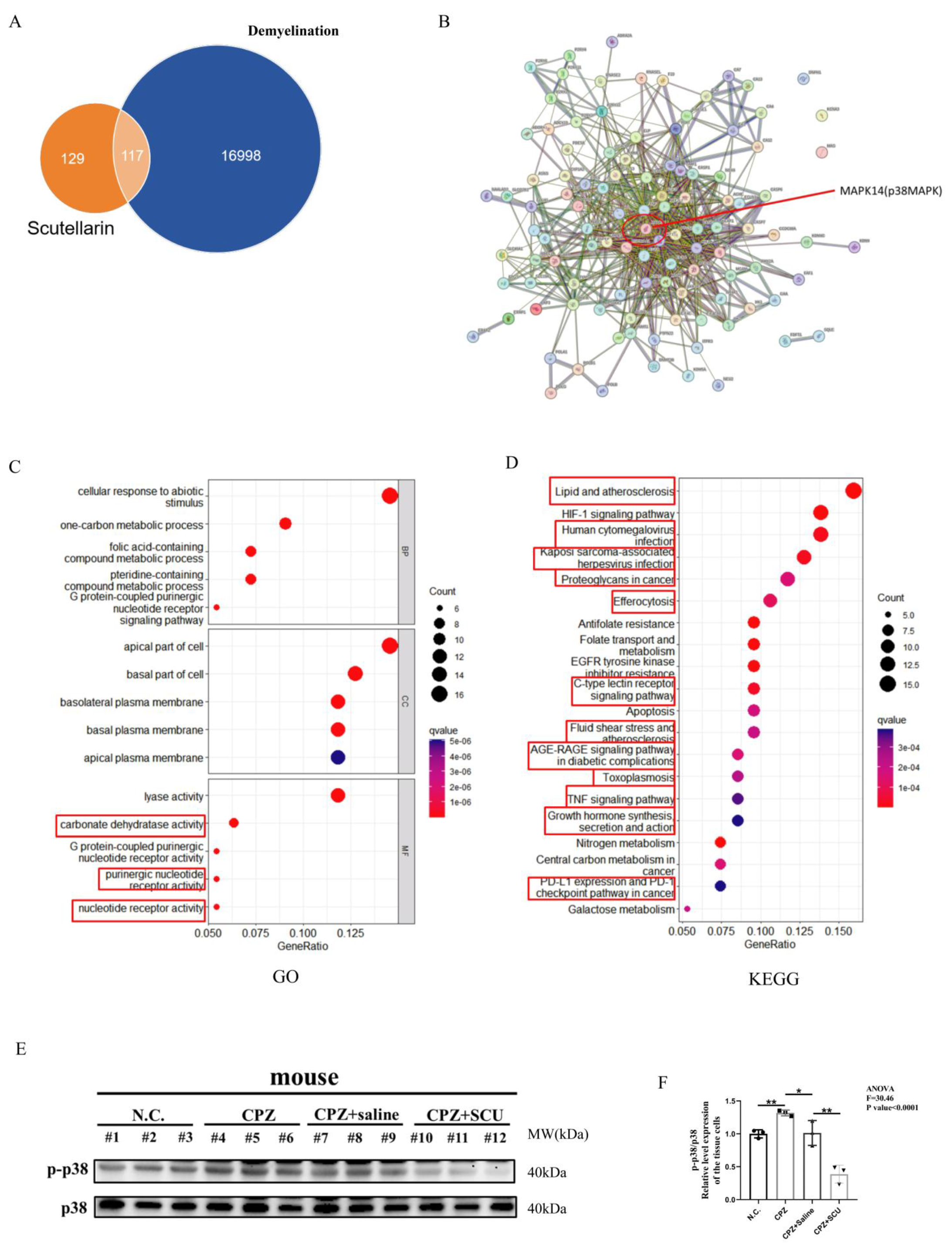
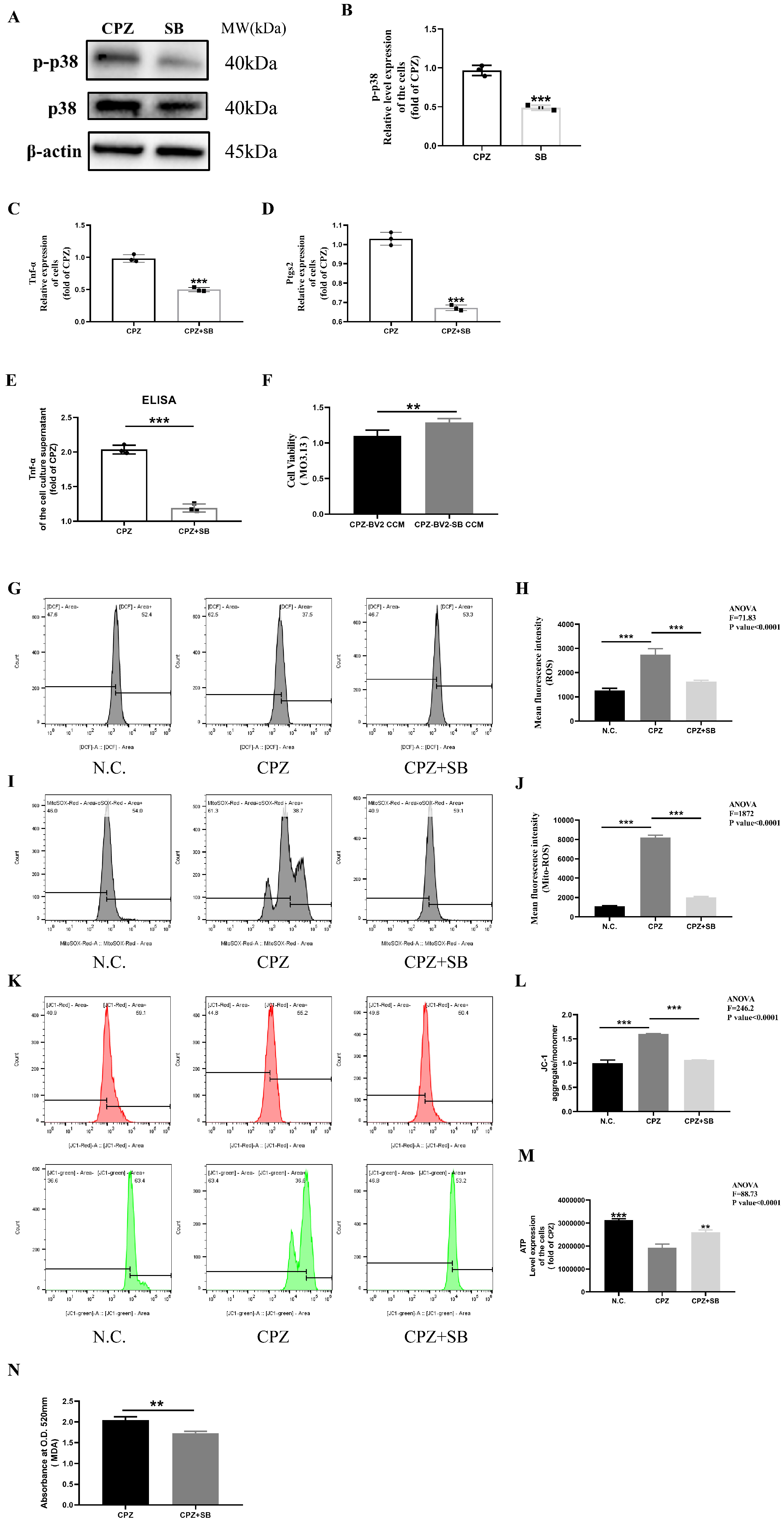
3.7. Scutellarin Inhibits Cuprizone–Copper Complex-Induced Pro-Inflammatory Microglial Activation via the p38MAPK/TNF-α Signaling Pathway
3.8. Reduced p38MAPK Phosphorylation Inhibits Pro-Inflammatory Cell Formation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- de Faria, O., Jr.; Pivonkova, H.; Varga, B.; Timmler, S.; Evans, K.A.; Káradóttir, R.T. Periods of synchronized myelin changes shape brain function and plasticity. Nat. Neurosci. 2021, 24, 1508–1521. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.N.; Appel, B. Oligodendrocytes express synaptic proteins that modulate myelin sheath formation. Nat. Commun. 2019, 10, 4125. [Google Scholar] [CrossRef] [PubMed]
- Bergles, D.E.; Richardson, W.D. Oligodendrocyte Development and Plasticity. Cold Spring Harb. Perspect. Biol. 2015, 8, a020453. [Google Scholar] [CrossRef]
- You, Y.; Joseph, C.; Wang, C.; Gupta, V.; Liu, S.; Yiannikas, C.; Chua, B.E.; Chitranshi, N.; Shen, T.; Dheer, Y.; et al. Demyelination precedes axonal loss in the transneuronal spread of human neurodegenerative disease. Brain 2019, 142, 426–442. [Google Scholar] [CrossRef]
- Granberg, T.; Fan, Q.; Treaba, C.A.; Ouellette, R.; Herranz, E.; Mangeat, G.; Louapre, C.; Cohen-Adad, J.; Klawiter, E.C.; Sloane, J.A.; et al. In vivo characterization of cortical and white matter neuroaxonal pathology in early multiple sclerosis. Brain 2017, 140, 2912–2926. [Google Scholar] [CrossRef]
- Saporta, M.A.; Katona, I.; Zhang, X.; Roper, H.P.; McClelland, L.; Macdonald, F.; Brueton, L.; Blake, J.; Suter, U.; Reilly, M.M.; et al. Neuropathy in a human without the PMP22 gene. Arch. Neurol. 2011, 68, 814–821. [Google Scholar] [CrossRef]
- Croxford, J.L.; Anger, H.A.; Miller, S.D. Viral delivery of an epitope from Haemophilus influenzae induces central nervous system autoimmune disease by molecular mimicry. J. Immunol. 2005, 174, 907–917. [Google Scholar] [CrossRef]
- Karsai, G.; Kraft, F.; Haag, N.; Korenke, G.C.; Hänisch, B.; Othman, A.; Suriyanarayanan, S.; Steiner, R.; Knopp, C.; Mull, M.; et al. DEGS1-associated aberrant sphingolipid metabolism impairs nervous system function in humans. J. Clin. Investig. 2019, 129, 1229–1239. [Google Scholar] [CrossRef]
- McLaughlin, K.A.; Wucherpfennig, K.W. B cells and autoantibodies in the pathogenesis of multiple sclerosis and related inflammatory demyelinating diseases. Adv. Immunol. 2008, 98, 121–149. [Google Scholar] [CrossRef]
- Höftberger, R.; Lassmann, H. Inflammatory demyelinating diseases of the central nervous system. Handb. Clin. Neurol. 2017, 45, 263–283. [Google Scholar]
- Lloyd, A.F.; Miron, V.E. The pro-remyelination properties of microglia in the central nervous system. Nat. Rev. Neurol. 2019, 15, 447–458. [Google Scholar] [CrossRef]
- Okano, H.; Sawamoto, K. Neural stem cells: Involvement in adult neurogenesis and CNS repair. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 2111–2122. [Google Scholar] [CrossRef]
- Blakemore, W.F. Demyelination of the superior cerebellar peduncle in the mouse induced by Cuprizone. J. Neurol. Sci. 1973, 20, 63–72. [Google Scholar] [CrossRef]
- Arnett, H.A.; Mason, J.; Marino, M.; Suzuki, K.; Matsushima, G.K.; Ting, J.P. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat. Neurosci. 2001, 4, 1116–1122. [Google Scholar] [CrossRef]
- Blakemore, W.F. Observations on oligodendrocyte degeneration, the resolution of status spongiosus and remyelination in cuprizone intoxication in mice. J. Neurocytol. 1972, 1, 413–426. [Google Scholar] [CrossRef]
- Hiremath, M.M.; Saito, Y.; Knapp, G.W.; Ting, J.P.; Suzuki, K.; Matsushima, G.K. Microglial/macrophage accumulation during cuprizone-induced demyelination in C57BL/6 mice. J. Neuroimmunol. 1998, 92, 38–49. [Google Scholar] [CrossRef]
- Mason, J.L.; Suzuki, K.; Chaplin, D.D.; Matsushima, G.K. Interleukin-1beta promotes repair of the CNS. J. Neurosci. 2001, 21, 7046–7052. [Google Scholar] [CrossRef]
- Jurevics, H.; Hostettler, J.; Muse, E.D.; Sammond, D.W.; Matsushima, G.K.; Toews, A.D.; Morell, P. Cerebroside synthesis as a measure of the rate of remyelination following cuprizone-induced demyelination in brain. J. Neurochem. 2001, 77, 1067–1076. [Google Scholar] [CrossRef]
- Norkute, A.; Hieble, A.; Braun, A.; Johann, S.; Clarner, T.; Baumgartner, W.; Beyer, C.; Kipp, M. Cuprizone treatment induces demyelination and astrocytosis in the mouse hippocampus. J. Neurosci. Res. 2009, 87, 1343–1355. [Google Scholar] [CrossRef]
- Suzuki, K.; Kikkawa, Y. Status spongiosus of CNS and hepatic changes induced by cuprizone (biscyclohexanone oxalyldihydrazone). Am. J. Pathol. 1969, 54, 307–325. [Google Scholar]
- Zirngibl, M.; Assinck, P.; Sizov, A.; Caprariello, A.V.; Plemel, J.R. Oligodendrocyte death and myelin loss in the cuprizone model: An updated overview of the intrinsic and extrinsic causes of cuprizone demyelination. Mol. Neurodegener 2022, 17, 34. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.L.; Teo, W.; Hernandez, Y.; Brideau, C.; Cummins, K.; Kuipers, H.F.; Stys, P.K. Cuprizone-induced Demyelination in Mouse Brain is not due to Depletion of Copper. ASN Neuro 2022, 14, 17590914221126367. [Google Scholar] [CrossRef]
- Layton, W.; Sutherland, J.M. Geochemistry and multiple sclerosis: A hypothesis. Med. J. Aust. 1975, 1, 73–77. [Google Scholar] [CrossRef]
- Åström, M.E.; Roos, P.M. Geochemistry of multiple sclerosis in Finland. Sci. Total Environ. 2022, 841, 156672. [Google Scholar] [CrossRef]
- Monti, M.C.; Guido, D.; Montomoli, C.; Sardu, C.; Sanna, A.; Pretti, S.; Lorefice, L.; Marrosu, M.G.; Valera, P.; Cocco, E.; et al. Is Geo-Environmental Exposure a Risk Factor for MultipleSclerosis? A Population-Based Cross-Sectional Study in South-Western Sardinia. PLoS ONE 2016, 11, e0163313. [Google Scholar] [CrossRef]
- Etemadifar, M.; Mehrabi, B.; Kiani-Peykani, R.; Abtahi, S.-H.; Nekouie-Isfahani, K.; Ramagopalan, S.V.; Fereidan-Esfahani, M. Soil heavy metals are associated with the distribution of multiple sclerosis in Isfahan, Iran. Acta Neurol. Scand. 2016, 134, 292–299. [Google Scholar] [CrossRef]
- Johnson, S. The possible role of gradual accumulation of copper, cadmium, lead and iron and gradual depletion of zinc, magnesium, selenium, vitamins B2, B6, D, and E and essential fatty acids in multiple sclerosis. Med. Hypotheses 2000, 55, 239–241. [Google Scholar] [CrossRef]
- Viquez, O.M.; Valentine, H.L.; Amarnath, K.; Milatovic, D.; Valentine, W.M. Copper accumulation and lipid oxidation precede inflammation and myelin lesions in N,N-diethyldithiocarbamate peripheral myelinopathy. Toxicol. Appl. Pharmacol. 2008, 229, 77–85. [Google Scholar] [CrossRef]
- Valentine, H.L.; Amarnath, K.; Amarnath, V.; Valentine, W.M. Dietary copper enhances the peripheral myelinopathy produced by oral pyrrolidine dithiocarbamate. Toxicol. Sci. Off. J. Soc. Toxicol. 2006, 89, 485–494. [Google Scholar] [CrossRef]
- Valentine, H.L.; Does, M.D.; Marshall, V.; Tonkin, E.G.; Valentine, W.M. Multicomponent T2 analysis of dithiocarbamate-mediated peripheral nerve demyelination. Neurotoxicology 2007, 28, 645–654. [Google Scholar] [CrossRef]
- Tonkin, E.G.; Valentine, H.L.; Milatovic, D.M.; Valentine, W.M. N,Ndiethyldithiocarbamate produces copper accumulation, lipid peroxidation, and myelin injury in rat peripheral nerve. Toxicol. Sci. Off. J. Soc. Toxicol. 2004, 81, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Viquez, O.M.; Lai, B.; Ahn, J.H.; Does, M.D.; Valentine, H.L.; Valentine, W.M. N,N-diethyldithiocarbamate promotes oxidative stress prior to myelin structural changes and increases myelin copper content. Toxicol. Appl. Pharmacol. 2009, 239, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Valentine, H.L.; Viquez, O.M.; Amarnath, K.; Amarnath, V.; Zyskowski, J.; Kassa, E.N.; Valentine, W.M. Nitrogen substituent polarity influences dithiocarbamate-mediated lipid oxidation, nerve copper accumulation, and myelin injury. Chem. Res. Toxicol. 2009, 22, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Valentine, H.L.; Viquez, O.M.; Valentine, W.M. Peripheral nerve and brain differ in their capacity to resolve N,Ndiethyldithiocarbamate-mediated elevations in copper and oxidative injury. Toxicology 2010, 274, 10–17. [Google Scholar] [CrossRef]
- Smith, D.K.; Feldman, E.B.; Feldman, D.S. Trace element status in multiple sclerosis. Am. J. Clin. Nutr. 1989, 50, 136–140. [Google Scholar] [CrossRef]
- Pomary, P.K.; Eichau, S.; Amigó, N.; Barrios, L.; Matesanz, F.; García-Valdecasas, M.; Hrom, I.; Sánchez, M.I.G.; Garcia-Martin, M.L. Multifaceted Analysis of Cerebrospinal Fluid and Serum from Progressive Multiple Sclerosis Patients: Potential Role of Vitamin C and Metal Ion Imbalance in the Divergence of Primary Progressive Multiple Sclerosis and Secondary Progressive Multiple Sclerosis. J. Proteome Res. 2023, 22, 743–757. [Google Scholar]
- Melo, T.M.; Larsen, C.; White, L.R.; Aasly, J.; Sjobakk, T.E.; Flaten, T.P.; Sonnewald, U.; Syversen, T. Manganese, copper, and zinc in cerebrospinal fluid from patients with multiple sclerosis. Biol. Trace Elem. Res. 2003, 93, 1–8. [Google Scholar] [CrossRef]
- Sarmadi, M.; Bidel, Z.; Najafi, F.; Ramakrishnan, R.; Teymoori, F.; Zarmehri, H.A.; Nazarzadeh, M. Copper concentration in multiple sclerosis: A systematic review and meta-analysis. Mult. Scler. Relat. Disord. 2020, 45, 102426. [Google Scholar] [CrossRef]
- De Riccardis, L.; Buccolieri, A.; Muci, M.; Pitotti, E.; De Robertis, F.; Trianni, G.; Manno, D.; Maffia, M. Copper and ceruloplasmin dyshomeostasis in serum and cerebrospinal fluid of multiple sclerosis subjects. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1828–1838. [Google Scholar] [CrossRef]
- Aspli, K.T.; Flaten, T.P.; Roos, P.M.; Holmøy, T.; Skogholt, J.H.; Aaseth, J. Iron and copper in progressive demyelination–New lessons from Skogholt’s disease. J. Trace Elem. Med. Biol. Organ Soc. Miner. Trace Elem. (GMS) 2015, 31, 183–187. [Google Scholar] [CrossRef]
- Aspli, K.T.; Holmøy, T.; Flaten, T.P.; Whist, J.E.; Aaseth, J.O. Skogholt’s disease-A tauopathy precipitated by iron and copper? J. Trace Elem. Med. Biol. Organ Soc. Miner. Trace Elem. (GMS) 2022, 70, 126915. [Google Scholar] [CrossRef] [PubMed]
- Miyakawa, T.; Murayama, E. An autopsy case of the “demyelinating type” of Wilson’s disease. Acta Neuropathol. 1976, 35, 235–241. [Google Scholar] [PubMed]
- Dezortova, M.; Lescinskij, A.; Dusek, P.; Herynek, V.; Acosta-Cabronero, J.; Bruha, R.; Jiru, F.; Robinson, S.D.; Hajek, M. Multiparametric Quantitative Brain MRI in Neurological and Hepatic Forms of Wilson’s Disease. J. Magn. Reson. Imaging JMRI 2020, 51, 1829–1835. [Google Scholar] [CrossRef] [PubMed]
- Dzieżyc, K.; Litwin, T.; Członkowska, A. Multiple sclerosis in two patients with coexisting Wilson’s disease. Mult. Scler. Relat. Disord. 2014, 3, 387–390. [Google Scholar] [CrossRef]
- Dusek, P.; Litwin, T.; Członkowska, A. Neurologic impairment in Wilson disease. Ann. Transl. Med. 2019, 7, S64. [Google Scholar] [CrossRef]
- Despotov, K.; Klivényi, P.; Nagy, I.; Pálvölgyi, A.; Vécsei, L.; Rajda, C. Rare co-occurrence of multiple sclerosis and Wilson’s disease—Case report. BMC Neurol. 2022, 22, 178. [Google Scholar] [CrossRef]
- Meenakshi-Sundaram, S.; Mahadevan, A.; Taly, A.B.; Arunodaya, G.R.; Swamy, H.S.; Shankar, S.K. Wilson’s disease: A cliniconeuropathological autopsy study. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2008, 15, 409–417. [Google Scholar]
- Chen, J.; Jiang, Y.; Shi, H.; Peng, Y.; Fan, X.; Li, C. The molecular mechanisms of copper metabolism and its roles in human diseases, Pflueg. Arch. Eur. J. Physiol. 2020, 472, 1415–1429. [Google Scholar] [CrossRef]
- Cobine, P.A.; Moore, S.A.; Leary, S.C. Getting out what you put in: Copper in mitochondria and its impacts on human disease, Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118867. [Google Scholar] [CrossRef]
- Bogie, J.F.J.; Stinissen, P.; Hendriks, J.J.A. Macrophage subsets and microglia in multiple sclerosis. Acta Neuropathol. 2014, 128, 191–213. [Google Scholar] [CrossRef]
- Prineas, J.W.; Parratt, J.D.E. Multiple Sclerosis: Microglia, Monocytes, and Macrophage-Mediated Demyelination. J. Neuropathol. Exp. Neurol. 2021, 80, 975–996. [Google Scholar] [CrossRef] [PubMed]
- Corrales, A.V.G.; Verberk, S.G.S.; Haidar, M.; Grajchen, E.; Dehairs, J.; Vanherle, S.; Loix, M.; Weytjens, T.; Gervois, P.; Matsuzaka, T.; et al. Fatty acid elongation by ELOVL6 hampers remyelination by promoting inflammatory foam cell formation during demyelination. Proc. Natl. Acad. Sci. USA 2023, 120, e2301030120. [Google Scholar] [CrossRef] [PubMed]
- Bogie, J.F.J.; Grajchen, E.; Wouters, E.; Corrales, A.G.; Dierckx, T.; Vanherle, S. Stearoyl-CoA desaturase-1 impairs the reparative properties of macrophages and microglia in the brain. J. Exp. Med. 2020, 217, e20191660. [Google Scholar] [CrossRef]
- Inojosa, H.; Schriefer, D.; Ziemssen, T. Clinical outcome measures in multiple sclerosis: A review. Autoimmun. Rev. 2020, 19, 102512. [Google Scholar] [CrossRef]
- Liu, M.; Li, H.; Luo, G.; Liu, Q.; Wang, Y. Pharmacokinetics and biodistribution of surface modification polymeric nanoparticles. Arch. Pharmacal Res. 2008, 31, 547–554. [Google Scholar] [CrossRef]
- Yuan, Y.; Zha, H.; Rangarajan, P.; Ling, E.A.; Wu, C. Anti-inflammatory effects of Edaravone and Scutellarin in activated microglia in experimentally induced ischemia injury in rats and in BV-2 microglia. BMC Neurosci. 2014, 15, 125. [Google Scholar] [CrossRef]
- Hu, X.M.; Zhou, M.M.; Hu, X.M.; Zeng, F.D. Neuroprotective effects of scutellarin on rat neuronal damage induced by cerebral ischemia/reperfusion. Acta Pharmacol. Sin. 2005, 26, 1454–1459. [Google Scholar] [CrossRef]
- Liu, H.; Yang, X.; Tang, R.; Liu, J.; Xu, H. Effect of scutellarin on nitric oxide production in early stages of neuron damage induced by hydrogen peroxide. Pharmacol. Res. 2005, 51, 205–210. [Google Scholar] [CrossRef]
- Yang, J.; Wu, X.; Yu, H.; Liao, X.; Teng, L. NMDA receptor-mediated neuroprotective effect of the Scutellaria baicalensis Georgi extract on the excitotoxic neuronal cell death in primary rat cortical cell cultures. Sci. World J. 2014, 2014, 459549. [Google Scholar]
- Guo, L.L.; Guan, Z.Z.; Huang, Y.; Wang, Y.L.; Shi, J.S. The neurotoxicity of beta-amyloid peptide toward rat brain is associated with enhanced oxidative stress, inflammation and apoptosis, all of which can be attenuated by scutellarin. Exp. Toxicol. Pathol. 2013, 65, 579–584. [Google Scholar] [CrossRef]
- Wang, S.; Wang, H.; Guo, H.; Kang, L.; Gao, X.; Hu, L. Neuroprotection of Scutellarin is mediated by inhibition of microglial inflammatory activation. Neuroscience 2011, 185, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-W.; Lu, L.; Bao, T.-H.; Zhang, H.-M.; Yuan, J.; Miao, W.; Wang, S.-F.; Xiao, Z.-C. Scutellarin Alleviates Behavioral Deficits in a Mouse Model of Multiple Sclerosis, Possibly Through Protecting Neural Stem Cells. J. Mol. Neurosci. 2016, 58, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Yang, H.; Gu, Q.; Wu, Z.; Zou, N.; Wang, C.-Z.; Wan, C.; Yuan, C.-S. The Small-Molecule compound baicalein alleviates experimental autoimmune encephalomyelitis by suppressing pathogenetic CXCR6+ CD4 cells. Int. Immunopharmacol. 2023, 114, 109562. [Google Scholar] [CrossRef]
- Zhao, Q.; Chen, L.; Ma, Y.; Wang, S. Scutellarin Attenuates Pro-Inflammatory Foam Cell Formation and Facilitates M2 Polarization in Microglia during Copper Homeostasis Imbalance via the MAPK Signaling Pathway. Front. Biosci. (Landmark Ed.) 2025, 30, 36255. [Google Scholar] [CrossRef]
- Hou, G.; Abrams, G.D.; Dick, R.; Brewer, G.J. Efficacy of tetrathiomolybdate in a mouse model of multiple sclerosis. Transl. Res. 2008, 152, 239–244. [Google Scholar] [CrossRef]
- Fan, J.; Han, Y.; Sun, H.; Sun, S.; Wang, Y.; Guo, R.; Guo, J.; Tian, X.; Wang, J.; Wang, J. Mesenchymal stem cell-derived exosomal microRNA-367-3p alleviates experimental autoimmune encephalomyelitis via inhibition of microglial ferroptosis by targeting EZH2. Biomed. Pharmacother. 2023, 162, 114593. [Google Scholar] [CrossRef]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef]
- Guo, H.; Hu, L.M.; Wang, S.X.; Wang, Y.L.; Shi, F.; Li, H.; Liu, Y.; Kang, L.Y.; Gao, X.M. Neuroprotective effects of scutellarin against hypoxic-ischemicinduced cerebral injury via augmentation of antioxidant defense capacity. Chin. J. Physiol. 2011, 54, 399–405. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Q.; Ma, Y.; Wang, S. Scutellarin Alleviates Cuprizone-Induced Demyelination by Improving Mitochondrial Dysfunction, Reducing Lipid Oxidation and Inhibiting the p38 MAPK Pathway. Antioxidants 2025, 14, 723. https://doi.org/10.3390/antiox14060723
Zhao Q, Ma Y, Wang S. Scutellarin Alleviates Cuprizone-Induced Demyelination by Improving Mitochondrial Dysfunction, Reducing Lipid Oxidation and Inhibiting the p38 MAPK Pathway. Antioxidants. 2025; 14(6):723. https://doi.org/10.3390/antiox14060723
Chicago/Turabian StyleZhao, Qiting, Yantuanjin Ma, and Shufen Wang. 2025. "Scutellarin Alleviates Cuprizone-Induced Demyelination by Improving Mitochondrial Dysfunction, Reducing Lipid Oxidation and Inhibiting the p38 MAPK Pathway" Antioxidants 14, no. 6: 723. https://doi.org/10.3390/antiox14060723
APA StyleZhao, Q., Ma, Y., & Wang, S. (2025). Scutellarin Alleviates Cuprizone-Induced Demyelination by Improving Mitochondrial Dysfunction, Reducing Lipid Oxidation and Inhibiting the p38 MAPK Pathway. Antioxidants, 14(6), 723. https://doi.org/10.3390/antiox14060723