1. Introduction
Coronary artery disease (CAD) remains a pressing global health issue that is responsible for a substantial number of worldwide fatalities [
1]. Myocardial infarction (MI), a primary complication of CAD, contributes significantly to morbidity and mortality rates, and there is a rising trend in related deaths. MI arises when the coronary artery, supplying blood to the heart muscle, becomes obstructed, resulting in cardiomyocyte death and impaired cardiac function. Timely treatment, ideally within the first hour of symptom onset, is critical for improving patient outcomes, while reperfusion therapy, aimed at restoring blood flow to the affected heart region, is a crucial emergency intervention that is achieved through thrombolysis or primary percutaneous coronary intervention (PCI) [
2]. However, reperfusion can paradoxically lead to myocardial ischemia/reperfusion injury (MIRI) and thus exacerbate tissue damage.
Various strategies, such as ischemic preconditioning, where the heart is exposed to short periods of ischemia before an MI occurs, have been extensively investigated for the prevention of MIRI [
3]. Although promising results were obtained from preclinical studies using animal models, these strategies have limited clinical value due to the unpredictable nature of ischemia and potential secondary damage from invasive procedures. Pharmacological interventions during reperfusion have also shown potential for cardioprotection, yet the clinical utility of these approaches is limited since the availability of drugs for co-administration in MI patients is restricted [
4]. Hence, exploring alternative therapeutic avenues is of great significance to enhancing outcomes in clinical patients with MIRI.
During reperfusion, the restoration of blood flow leads to mitochondrial damage and electrolyte imbalances, resulting in the rapid production of reactive oxygen species (ROS), such as hydrogen peroxide (H
2O
2) and hydroxyl radical (•OH), which contain unpaired electrons [
5]. The excessive accumulation of ROS can cause damage to cell and organelle membranes, which leads to programmed cell death through apoptosis or autophagy [
6]. ROS can also induce lipid peroxidation, which disrupts the membrane structure and releases toxic substances. Given the adverse role of ROS in MIRI, researchers have intensively explored the application of antioxidants as potential therapeutic agents. However, despite evidence that indicates cardioprotection from many natural and synthetic antioxidants, the clinical use of antioxidase-like drugs such as trimetazidine and superoxide dismutase (SOD) has not significantly improved heart function in patients [
7]. This underscores the importance of identifying potent antioxidants that are capable of efficiently eliminating ROS to provide timely cardioprotection during MIRI.
Recent studies have focused on phenolic compounds, as they are promising antioxidants due to their ability to chelate ions and scavenge free radicals [
8]. Extracts of leaves from
ginnala, a species of amur maple tree, in particular, have been reported to have exceptional antioxidative activities, offering biological benefits in attenuating inflammation and tissue damage caused by oxidative stress [
9,
10].
Ginnalin A, a major component of
ginnala leaf extracts, exhibited the rapid inhibition of lipid peroxidation, the scavenging of hydrogen peroxide, and the elimination of free radicals in a few organs but has never been studied in the cardiovascular system [
11].
Ginnalin A contains two galloyl derivatives, which allows it to efficiently and rapidly sense and diminish environmental ROS. These findings highlight the remarkable antioxidative properties of
ginnala extracts, particularly those that are due to the action of
Ginnalin A. The rapid and efficient elimination of ROS could offer promising prospects for readily available and valid therapeutics for combating MIRI and other conditions that involve excessive oxidative stress.
Thus, in the present study, we aimed to investigate the cardioprotective effects of ginnala leaf extracts (hereafter referred to as ginnala extracts) against MIRI by harnessing their remarkable antioxidative properties. Our results demonstrated that responsive components within ginnala extracts rapidly and effectively neutralize ROS surges within seconds in a sequential manner, safeguarding cardiomyocytes from MIRI. Through our investigation, which involved ROS measurement, we observed a significant and rapid decrease in intracellular, mitochondrial, and extracellular ROS levels upon concurrent treatment with ginnala extracts and various ROS inducers. Moreover, experiments conducted in a rat model of MIRI showcased notable enhancements in cardiac function, reduced infarct sizes, and the mitigation of fibrosis and cardiac remodeling following the administration of ginnala extracts. These results highlight the antioxidative and cardioprotective capabilities of ginnala extracts, particularly emphasizing Ginnalin A’s proficiency in countering ROS.
2. Materials and Methods
2.1. Leaf Materials
Acer tataricum L. subsp. ginnala (Maxim.) Wesm. were cultivated in the reservatory of forest biomaterials of the Korea Forest Research Institute, Jinju, Korea (TM coordinates; X: 305,331, Y: 191,057) and identified at Forest Biomaterials Research Center (Jinju, Korea). Voucher specimens were preserved by the Korea Forest Service (NF-A2-ATg-L). The leaves were collected and shade-dried for 10 days.
2.2. Extraction, Isolation, and of Acertannin
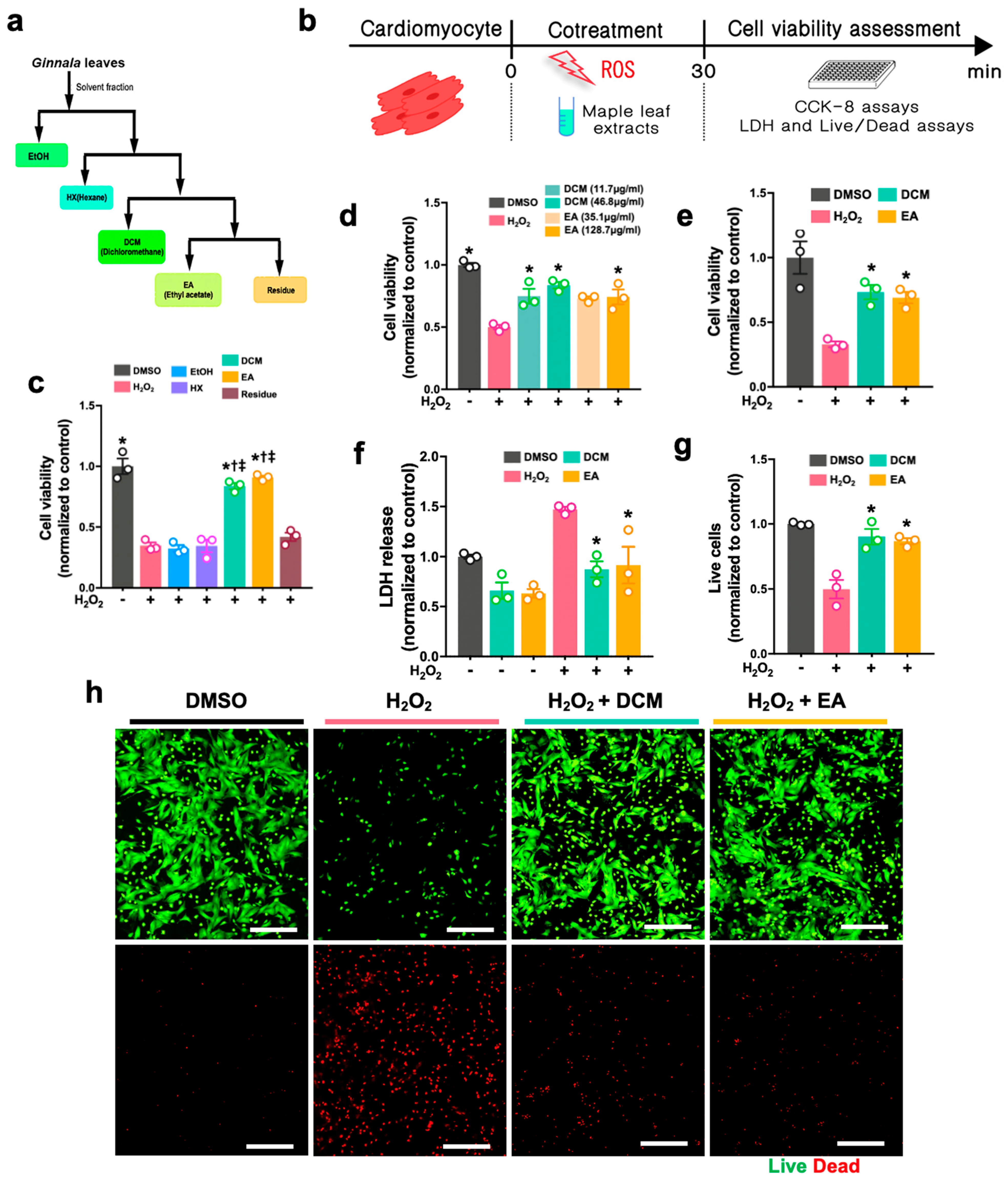
Extraction of the dried leaves (1 kg) was conducted three times, for two days each time, using EtOH (10 L) at room temperature (RT), followed by evaporation under reduced pressure to yield approximately 400 g extracts. A part of the extract (~167.0 g) was then processed on Sephadex LH-20 (10–25 μm, 10 cm × 80 cm, GE Healthcare Bio-Science AB, Uppsala, Sweden) and eluted successively with equal volume of chloroform and methanol to produce 9 fractions (1~9,
Figure S1), where fraction 2 (2.6 g, 40% eluate) was put through ODS-A (30–50% MeOH) and ODS-A (25–35% ACN). Fraction 2-6-11 (2.4 g, 60% eluate) was determined to be acertannin (740 mg). The solvents, such as DCM and EA, were fully evaporated, and DMSO was used to dissolve the dried extracts prior to cell treatment and in vivo administration to rat hearts.
2.3. Neonatal Rat Cardiomyocytes (NRCMs) Isolation
NRCMs were isolated from neonatal Sprague-Dawley (SD) rat (2–4 days) hearts in accordance with institutional guidelines. The procedure was approved by the Institutional Animal Care and Use Committee of The Catholic University of Korea (approval no. CUMC-2024-0279-03; approval date: 1 December 2024). Generally, rat heart ventricles were collected, chopped, and digested using 0.1% trypsin solution (Gibco, 25200056, Grand Island, CA, USA) with gentle agitation at 4 °C in a fridge for 16 h. The tissues were further incubated with 1 mg/mL rat type 2 collagenase (Worthington, LS004174, Lakewood, NJ, USA) at 37 °C for 20 min, and this step was repeated 3 times in total. After digestion, the supernatant was collected and transferred to cold DPBS (Gibco, 14190144, USA). A Percoll (Cytiva, Marlborough, MA, USA) density-gradient centrifugation was performed for 30 min at 1800× g at 4 °C to remove non-cardiomyocytes, and obtained NRCMs were seeded in gelatin (Sigma, G1393, Darmstadt, Germany)-coated cell culture dishes with a seeding density of 1 × 105 per cm2.
2.4. Cell Culture
Rat cardiac myoblast H9c2 cell line (ATCC, Manassas, VA, CRL-1446, USA) and NRCMs were used in all experiments. H9c2 cells were cultured with high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco, 11965092, USA) supplemented with 10% fetal bovine serum (FBS, ExCell, FSP500, Shanghai, China) (Gibco, 26140079, USA), 1% non-essential amino acid (Gibco, 11140050, USA), 1% antibiotics (Gibco, 15240062, USA), and 1% sodium pyruvate (Gibco, 11360070, USA). Prior to ginnala leaf extract treatment, H9c2 cells were plated on 96-well plates with a seeding density of 1 × 105 per cm2 and cultured overnight to allow cell attachment. NRCMs were maintained with low-glucose DMEM (Gibco, 10567014, USA), together with 5% horse serum (HS, Gibco, 16050122, USA), 1% non-essential amino acid, and 1% antibiotics. Both H9c2 cells and NRCMs were cultured 37 °C under 5% CO2.
2.5. CCK-8 Assay
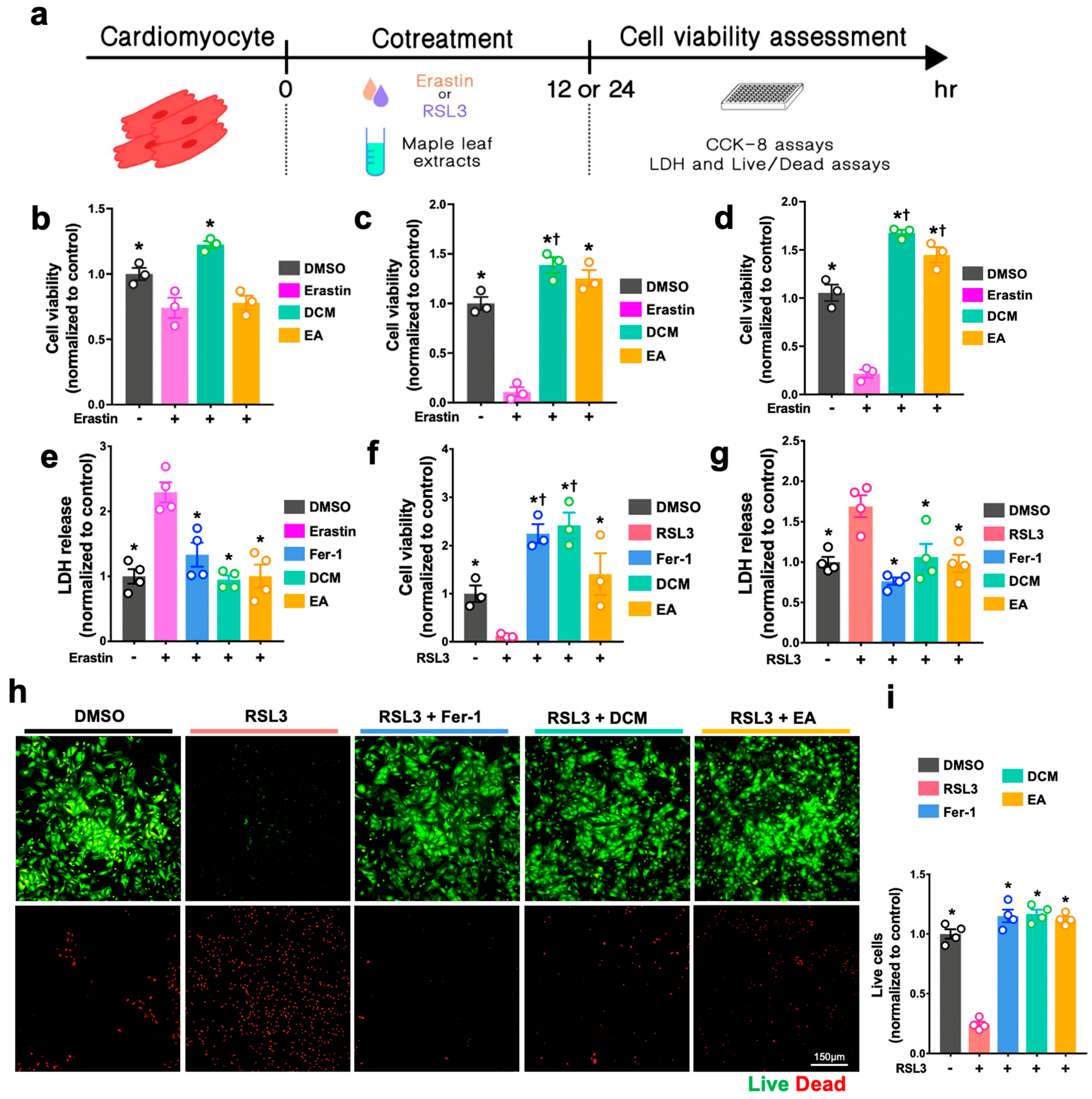
For the H2O2-induced cell injury, H9c2 cells and NRCMs were either co-treated with H2O2 (Sigma-Aldrich, H1009, Darmstadt, Germany) and ginnala leaf extract for 30 min or pre-treated with ginnala leaf extract candidates for 1 h prior to 30 min of exposure to H2O2. To induce ferroptosis, ferroptosis inducers, either Erastin (S7242, Selleck, Boston, MA, USA) or RSL3 (Sigma-Aldrich, SML2234, Germany), were co-administrated with ginnala leaf extract on the cells for 12 or 24 h. After administration, CCK-8 assay (Dojindo, CK04, Tokyo, Japan) was performed to evaluate the cell survival. In brief, the CCK-8 reagent was mixed with FBS-free culture medium and incubated with the cells at 37 °C in an incubator for 2 h. Subsequently, the absorbance was measured at 450 nm.
2.6. Lactate Dehydrogenase (LDH) Assay
Lactase dehydrogenase (LDH) assay (Cell Biolabs, CAB241, San Diego, CA, USA) was used to detect the cellular dagame by using culture medium. Briefly, after treatment, as mentioned previously, 90 µL of supernatant from various groups was mixed with 10 µL of LDH reagent. After 1 h incubation at 37 °C, the absorbance was measured at 450 nm. To obtain accurate measurements, the absorbance of the media-only controls was subtracted from the results of each group.
2.7. Live and Dead Assay
The live and dead assay (live/dead assay) was conducted according to the manufacturer’s instructions (Invitrogen, L3224, Waltham, MA, USA). Briefly, after treatment, calcein AM and EthD-1 were mixed with phenol-free DMEM (Sigma-Aldrich, D5921, Burlington, MA, USA) to final concentrations of 2 μM and 4 μM, respectively. Thirty minutes after incubation at RT, the stained cells were imaged and analyzed using a fluorescence microscope (Nikon, Tokyo, Japan). The number of live and dead cells from each group were quantified and normalized using the values of controls by ImageJ (NIH, Bethesda, MD, USA).
2.8. Measurement of Cellular and Mitochondrial ROS Level
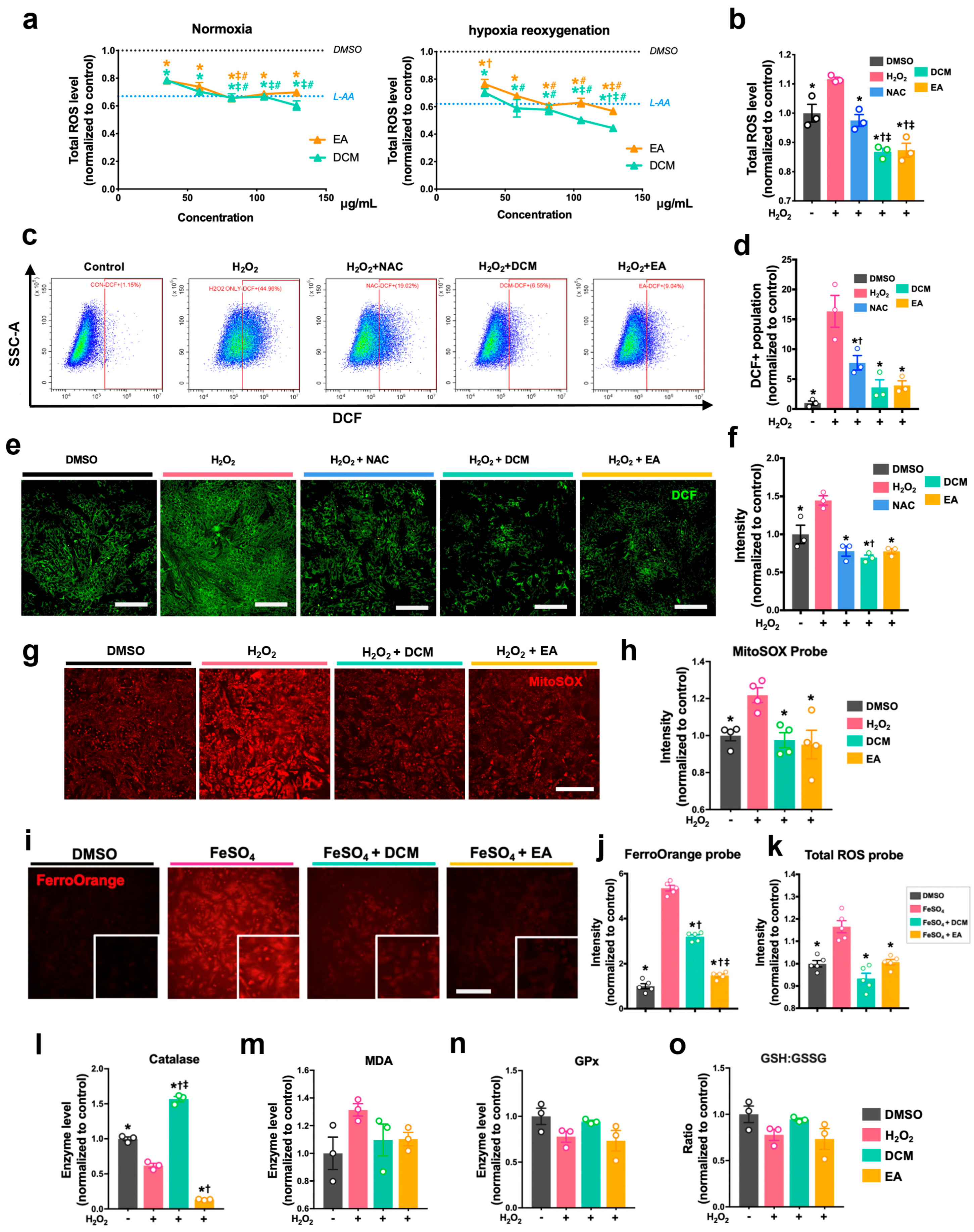
Prior to treatment, 5 µM DCFDA (Sigma-Aldrich, D6883, USA) solution was added to stain the H9c2 cells and NRCMs. After 45 min incubation at 37 °C in the dark, the solution was then removed and replaced with PBS. After treatment with H2O2 and ginnala leaf extracts, the cellular DCF fluorescence intensity of cells was measured immediately at 488/530 nm. The cellular ROS level was also examined using flow cytometry, for which treated cells were collected using 0.1% trypsin to conduct flow cytometer analysis. Mitochondrial ROS was measured and analyzed by a kit that indicated mitochondrial superoxide (the MitoSOXTM Red, Invitrogen, M36008, USA). In brief, cells were stained with 5 μM MitoSOXTM reagent for 10 min at 37 °C followed by rinsing with PBS twice. Subsequently, cells were treated with H2O2 and ginnala leaf extracts for 30 min before the fluorescent intensity was determined at 529/599 nm.
2.9. Enzymatic Assays
After treatment, NRCMs were harvested by cell scalper and lysed using lysis buffer (Beyotime, P0013, Nantong, China) with 1x Protease Inhibitor Cocktail (MCE, HY-K0010, New York, NY, USA). The protein concentration of cell lysates from each treatment group was determined by PierceTM BCA Protein Assay Kit (Thermo Scientific, 23227, Logan, UT, USA). The malondialdehyde (MDA, Beyotime, S0131, China) contents, enzymatic activities of glutathione peroxidase (GPx, Beyotime, S0056, China), superoxide dismutase contents (SOD, Beyotime, S0101, China), catalase (CAT, Beyotime, S0051, China), and total, reduced, and oxidized glutathione (GSH/GSSG, Beyotime, S0053, China) were detected following instructions from the reagents manufacturers.
2.10. DPPH Assay
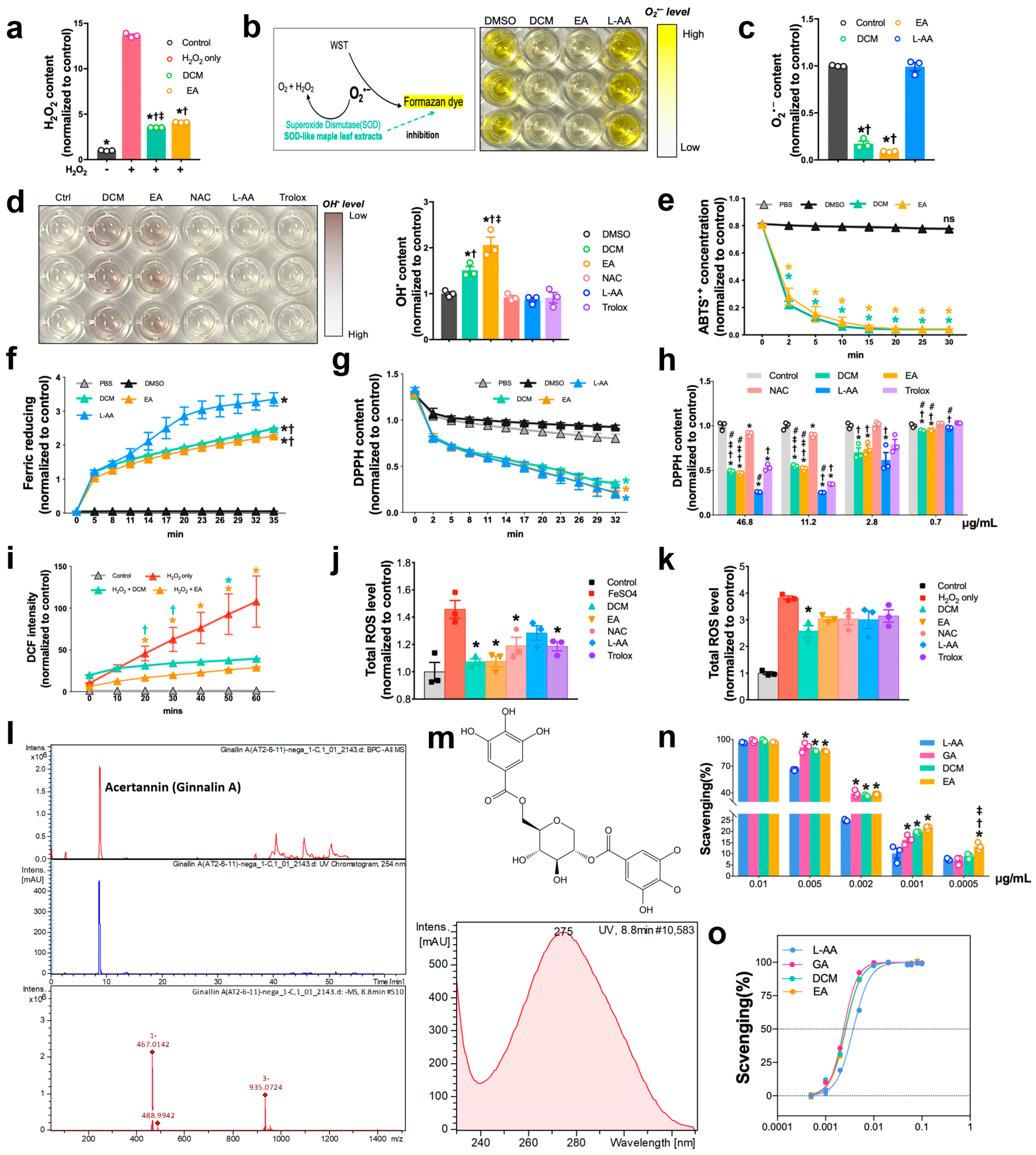
The protocol for determining the scavenging activities of extracts by using 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay was modified as previously reported. Briefly, DPPH (Sigma-Aldrich, D9132, USA) working compound was freshly prepared after 2 mg DPPH was dissolved in 54 mL methanol. DCM, EA extracts, L-ascorbic acid (Sigma-Aldrich, A4544, Germany), and NAC (Sigma-Aldrich, A7250, Germany) were mixed with DPPH solution. Beginning immediately after the reaction, the absorbance was measured at 515 nm every 3 min until 30 min.
2.11. Hydrogen Peroxide (H2O2) Assay
Hydrogen peroxide was quantified using hydrogen peroxide assay (Elabscience, E-BC-F001, Wuhan, China). In brief, a standard curve was produced using H2O2 solution in quantities in the range of 0.5–10 µM. DCM and EA at selected concentrations were mixed with 700 µM H2O2 solution, followed by 30 min reaction, after which the fluorescence intensity of each group was determined at 535/587 nm.
2.12. Total Antioxidant Capacity Assay
Total antioxidative activity was measured with two total antioxidant capacity assay kits and using the ferric reducing ability of plasma (FRAP) (Beyotime, S0116, China) and [2,2′-azinobis (3-ethyl-benzothiazoline-6-sulphonate)] (ABTS) (Beyotime, S0119, China) methods, following the manufacturer’s instructions. For FRAP assay, quantities of FeSO4 solution ranging from 0.15 to 1.5 mM were included to yield the standard curve. DCM and EA at selected concentrations were mixed with 180 μL of FRAP working solution. Immediately after reaction, the absorbance was measured at 593 nm every 3 min for 35 min. FRAP activities of each sample were determined according to the equivalent value of FeSO4 concentration calculated and obtained based on the standard curve. For ABTS assay, quantities of Trolox ranging from 0.15 to 1.5 mM were employed to generate the standard curve. DCM and EA at selected concentrations were mixed with 200 μL of ABTS working solution. Immediately after reaction, the absorbance of each group was acquired at 734 nm every 3 min for 30 min. ABTS activities were assessed based on the equivalent value of Trolox concentrations.
2.13. Seahorse Metabolic Analysis
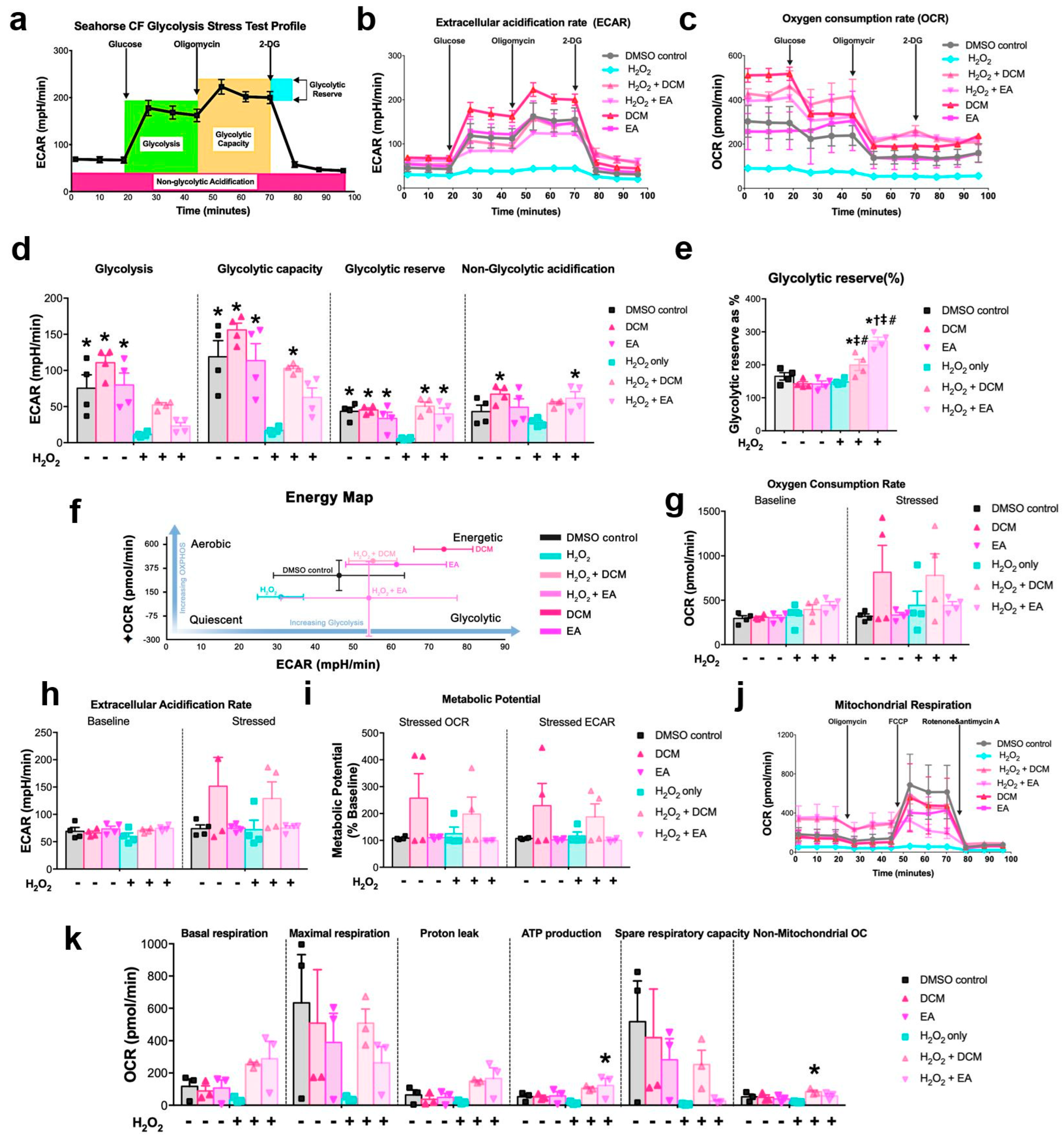
XF glycolysis stress, cell energy phenotype, and cell mitochondrial stress tests were determined to demonstrate the metabolic profile of ginnala extract-treated NRCMs. It should be noted that, unlike the 24 h incubation time used for drugs, to closely mimic the cell state from previous cell viability assays, the 30 min co-treatment of ginnala extracts with or without H2O2 solution was used as the protocol for cell treatment. One day prior to treatment, Agilent Seahorse XFe Analyzer (Agilent Technologies, Santa Clara, CA, USA) was turned on to warm it up, the cartridge was hydrated with XF Calibrant (1 mL/well), and placed in a 37 °C incubator without supply of CO2. Wave program was used to design the experimental flow. Different assay media were formulated for different tests. For glycolysis stress, assay medium was prepared using Base Medium supplemented with 1 mM glutamine. The medium containing 1 mM pyruvate, 2 mM glutamine, and 10 mM glucose was used for profiling cell energy phenotype and cell mitochondrial stress tests. A total of 0.1 N NaOH was required for pH adjustment of assay media to 7.4, after which assay media would be incubated in a 37 °C water bath prior to use. After treatment and before the assays were run, NRCMs from XF 24-well cell culture microplate were refreshed with warmed assay medium and placed into a 37 °C incubator without supply of CO2 for 1 h. Specific compounds were prepared and loaded into the appropriate ports of the hydrated sensor cartridges as follows: for glycolysis stress test: 56 µL glucose (10 mM), 62 µL oligomycin (1 µM), and 69 µL 2-DG were injected into ports A, B, and C, respectively; for cell energy test: 20 µL stressor mix containing 50 µM oligomycin and FCCP were injected into port A; for mitochondrial stress test: 56 µL oligomycin (15 µM), 62 µL FCCP (5 µM), and 69 µL Rot/AA (5 µM) were injected into ports A, B, and C, respectively. After loading compounds, sensor cartridges were be plated on the instrument for 15 min calibration, after which the designated program for each assay would start.
2.14. Structural Identification of Acertannin
Purity of acertannin was determined by a LC-q-ToF mass spectroscopy (Impact II, Bruker, Billerica, MA, USA) and ultra-performance liquid chromatography (Waters H-class system, Milford, MA, USA), which determined it to have approximately 98% purity. Spectral analyses were conducted to identify the structure of acertannin. Avance DRX500 (Bruker, USA) was employed to acquire the 1-D nuclear magnetic resonance (NMR) findings, such as 1H (500 MHz) and 13C (125 MHz) NMR, in Center for Research Facilities of Gyeongsang National University, Jinju, Korea. A Waters H-class UHPLC system interfaced with a Bruker Impact II hybrid quadrupole time of flight mass spectrometer (Bruker, USA) was used to analyze the samples. A Waters Acquity UPLC® BEH C18 (150 × 2.1 mm 1.7 µm, Waters Corporation, Milford, MA, USA) was included to perform the chromatographic separations. This step was carried out at 35 °C with gradient elutions containing 0.1% formic acid in water (A) and 0.1% formic acid in methanol (B) as mobile phase with a flow rate of 0.2 mL/min. The elutions used included 20%, 0–3 min; 20–100%, 3–50 min; 100%, 50–535 min. The volume for injection of each sample was 5 μL. Mass spectrometry was conducted in negative electrospray ionization mode and the spectra were obtained via scanning the mass range from m/z 50 to 1500 using MS and MS/MS modes. Nitrogen was applied as a gas for drying, nebulizing, and collision and the drying gas flow rate was set as 8.0 L/min. Heated capillary temperature was at 200 °C while 0.8 bar was set as the nebulizer pressure. The source parameters, the capillary voltage (VCap) and source plate offset, were set at 3000 V and 500 V, respectively.
2.15. Experimental Animals
All procedures involving animals adhered to institutional and international ethical standards, including NIH regulations and the European Parliament’s 2010/63/EU directive. Prior approval for the study was granted by the IACUC of The Catholic University of Korea (Approval No. CUMC-2023-0234-01; Approval date: 1 October 2023).
2.16. Ischemic/Reperfusion Injury Model and In Vivo Drug Administration
To establish ischemia/reperfusion (I/R) injury, male Fischer 344 rats (age: 8 weeks; weight: 160–180 g) sourced from Central Lab. Animal Inc. were anesthetized with 2% isoflurane and ventilated following intubation via an 18-gauge catheter. The procedure was carried out on a preheated platform (37 °C). After thoracotomy and LAD exposure, the artery was ligated with a 7-0 suture for one hour. Five minutes prior to reperfusion, extracts (EA: 168 µg/kg; DCM: 84 µg/kg in 50 µL volume) were injected at two peri-infarct zones. Reperfusion was achieved by releasing the ligature, and the chest was closed aseptically.
2.17. Measurement of Myocardial Infarct Size
Twenty-four hours post-reperfusion, myocardial damage was evaluated using Evans blue and TTC dyes. Following LAD re-occlusion and intravenous administration of 8% Evans blue, the heart was excised and sliced into transverse sections. The tissue was stained with 2% TTC at 37 °C for 10 min, then fixed with 4% paraformaldehyde. Infarct demarcation was visualized as blue (non-perfused), red (viable), and white (necrotic). ImageJ software (version 1.54g) was utilized to quantify infarct extent and the area at risk.
2.18. Evaluation of Heart Function by Echocardiography
The rats were lightly anesthetized with 2% isoflurane (Hana Pharm, Seongnam, Republic of Korea). Echocardiography was performed using a 15 MHz probe (Philips Affiniti 50G, Eindhoven, Dutch) under mild anesthesia. Recordings were obtained at 4 h, 1 week, and 2 weeks after surgery. Investigators were blinded to group allocation. Ejection fraction and fractional shortening were computed using the following equation:
2.19. Hemodynamic Measurements
At 2 weeks post-injury, invasive cardiac function analysis was conducted under anesthesia. After thoracotomy, a conductance catheter (2F, Millar SPR-838, Pearland, TX, USA) was inserted into the left ventricle. Pressure-volume loops were monitored using a system from Emka Technologies and converted via a PowerLab interface (ADInstruments, Colorado Springs, CO, USA). Load-independent indices such as ESPVR and EDPVR were derived by transient IVC occlusion. Parallel conductance calibration involved injection of 20% NaCl (60 µL) through the jugular vein, and LV blood samples were collected for volume determination.
2.20. Measurement of Capillary Density by Immunohistochemical Staining
Following overnight fixation in 4% paraformaldehyde, hearts were embedded in paraffin and sectioned at 5 µm thicknesses (Leica RM2255, Wetzlar, Germany). After dewaxing and hydration, heat-induced epitope retrieval was performed. Slides were incubated overnight at 4 °C with CHP (20 μM), anti-cTnT (1:200, Abcam, Cambridge, UK), and anti-CD31 (1:200, R&D). Secondary antibodies included Alexa Fluor 488 donkey anti-mouse IgG and Alexa Fluor 594 rabbit anti-goat IgG (1:400, Invitrogen, Waltham, MA, USA). DAPI-containing mounting medium was applied for nuclear staining. Cardiomyocyte and capillary counts, as well as collagen denaturation areas, were measured in five randomly selected regions per sample.
2.21. Measurement of Myocardial Infarct Size by Masson’s Trichrome Staining
Masson’s trichrome stain (Sigma HT15) was employed to assess fibrotic remodeling. Sections were fixed in Bouin’s solution at 56 °C, then sequentially stained using Weigert’s iron hematoxylin for 10 min, Biebrich scarlet acid fuchsin for 20 min, and aniline blue for 15 min. Blue and red staining indicated fibrotic and viable myocardium, respectively. Scanned images (Pannoramic MIDI) were analyzed using ImageJ to quantify fibrotic area.
2.22. Data Analysis
Statistical analyses were performed using GraphPad Prism 9 (GraphPad Software, La Jolla, CA, USA). Quantitative data are presented as mean ± standard error of the mean (SEM), unless otherwise specified. Depending on the data distribution and comparison groups, statistical significance was assessed using two-tailed Student’s t-tests or one-way ANOVA followed by Tukey’s post hoc test. Detailed information regarding the statistical methods used is provided in the respective figure legends.
4. Discussion
Although several antioxidants, including recombinant human SOD, have undergone preliminary clinical trials for their potential therapeutic roles in eliminating ROS and managing MIRI, these treatments have demonstrated limited efficacy in improving heart function recovery in patients undergoing coronary angioplasty for myocardial ischemia. This limited effectiveness stems from the sudden surge in oxygen levels and the restricted capacity of these drugs to regulate oxidative stress. ROS, triggered by the surge in oxygen upon blood flow restoration, is the primary culprit behind MIRI. Hence, it is imperative to identify potent ROS scavengers that are capable of effectively mitigating MIRI.
In the present study, we aimed to pinpoint a natural antioxidase known as Ginnalin A, which is sourced from the leaves of Acer tataricum L. subsp. ginnala (Maxim.) Wesm, a variety of amur maple trees. By subjecting various fractions with different concentrations to cardiomyocytes under oxidative stress, we showcased the swift and comprehensive ROS scavenging attributes of Ginnalin A. It efficiently neutralizes diverse ROS and free radicals, including O2−, H2O2, OH•, and Fe2+, and thus delivers prompt and robust cardioprotection when introduced into both in vitro and in vivo models.
Contrary to the usual protocol of administering drugs before percutaneous coronary intervention (PCI), our study primarily employed a co-administration method, where ginnala extracts were concomitantly administered with ROS inducers. This approach aimed to closely replicate the clinical urgency during heart reperfusion. Within a concise 30 min window, the ginnala extracts exhibited ultrafast suppression of ROS production and safeguarded cardiomyocyte viability. Through a series of assays assessing antioxidative prowess, we observed that the ginnala extracts demonstrated rapid and sequential ROS scavenging characteristics. These extracts showcased behaviors akin to SOD and CAT within minutes. Notably, they effectively targeted a broad spectrum of ROS species, including O2•− and OH•, outperforming common antioxidants like ascorbic acid, NAC, and Trolox, even at equivalent concentrations. Furthermore, the ginnala extracts significantly shielded cardiomyocytes against oxidative harm induced by H2O2 and ferroptosis, which underscores their potential to salvage impaired myocardium, fortify vasculature, and enhance cardiac functions. To delve deeper into the antioxidative attributes of these extracts, we conducted a comparative analysis with three conventional antioxidants—ascorbic acid, NAC, and Trolox. Various ROS species were employed to evaluate their scavenging efficacy. While ascorbic acid exhibited superior effects at higher concentrations, when tested at identical doses to the extracts, only the effects of the ginnala extracts persisted rapidly. Leveraging the ABTS assay, which reflects of anti-radical activity by gauging the remaining ABTS•+ radicals, we determined that the ginnala extracts exhibited superior scavenging effects with a lower IC50 in comparison to ascorbic acid. Taken together, these results suggest that ginnala extracts confer ultrafast and cascade-like biocatalytic activities through chemically derived functional groups that effectively mimic intracellular ROS defense mechanisms, such as those of SOD and CAT. Unlike regulatory effects that involve modulating molecular pathways within cells, these catalytic actions enable ginnala extracts to serve as a timely and effective approach for sensing and neutralizing bursts of ROS, and their quick action suits the clinical urgency encountered during myocardial IR injury.
The antioxidative effects of ginnala extracts transcend their capacity to scavenge extracellular ROS from the environment. The direct application of the extracts on NRCMs under normoxic conditions resulted in a reduction in cellular ROS levels, indicating a proactive effect upon cellular entry. Rather than eliciting cellular antioxidative enzymes such as SOD, CAT, and GPx, the extracts embedded themselves within the cells, acting as potent antioxidases that efficiently regulated cellular oxidative stress. Consequently, when NRCMs were exposed to the ferroptosis inducers RSL3 and Erastin, which typically necessitate prolonged incubation times for their effects to manifest, for 12 and 24 h, the protection conferred by the ginnala extracts remained significant. This underscores the comprehensive and sustained rapid ROS-eliminating effects of the studied extracts.
Mass spectrometry analysis unveiled Ginnalin A (GA) as a pivotal polyphenolic component that was present in the ginnala extracts, which aligns with prior reports. GA, characterized by two gallic acid groups situated at each end of the molecule, is recognized for its robust antioxidant activity compared to pyrogallol. The chemical structure of GA, a galloyl ester derivative of 3,4,5-trihydroxybenzoic acid, has exhibited the highest efficacy in inhibiting lipid peroxidation, scavenging hydrogen peroxide, and neutralizing DPPH radicals, surpassing other phenolic acids. One standout advantage of natural antioxidants like phenolic acids is their therapeutic efficacy coupled with their favorable safety profile. In assessing the toxicity of the ginnala extracts, we extended the treatment duration to three days and observed that the release of lactate dehydrogenase (LDH) from cells treated with the extracts was comparable to or even lower than that of the control group, highlighting the excellent biosafety of these natural extracts. Furthermore, by assessing the size of cardiomyocytes, we demonstrated the anti-hypertrophy potential of the extracts, which hints at long-term beneficial effects in heart repair in addition to their immediate protective attributes. Interestingly, 30 min treatment with the extracts induced an energetic state in NRCMs, with the extracts stimulating increased glycolysis and mitochondrial respiration. Furthermore, hemodynamic pressure and volume (PV) measurements showcased a substantial enhancement in cardiac contractility post the administration of the ginnala extracts, signifying their capability to sustain myocardial energy levels during MIRI.
Despite these promising results, there are some limitations that require further efforts to achieve the widespread therapeutic application of ginnala extracts. Given that myocardial IR injury impairs different kinds of cells in the heart, the selection of cardiomyocytes as the major target during heart therapy could disregard the indispensable contributions from other cell types, including endothelial cells, immune cells, and cardiac fibroblasts. For instance, although the in vivo results demonstrated higher density of the capillary, it could be more significant to investigate and confirm the direct effects of ginnala extracts on the cellular behavior of endothelial cells, as they could potentially assist in angiogenesis. In addition, intramyocardial injection, while offering precise delivery, could induce unneglectable damage to the myocardium due to mechanical invasiveness. Thereby, identifying other approaches such as intrapericardial administration and epicardial patch implantation could greatly prevent undesired risk and allow the sustained release of ginnala extracts to achieve more effective and safe treatment. Thirdly, regarding the long-term benefits of ginnala extracts in limiting cardiac hypertrophy, a comprehensive transcriptomic analysis of extracts treated with cardiomyocytes and myocardia is essential to further unravel the underlying mechanism that confers clinical relevance to the generation ginnala extract-based heart care products as precautions against hypertrophic cardiomyopathy (HCM).


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}