
The Ferroptosis–Mitochondrial Axis in Depression: Unraveling the Feedforward Loop of Oxidative Stress, Metabolic Homeostasis Dysregulation, and Neuroinflammation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Relationship Between Ferroptosis and Depression
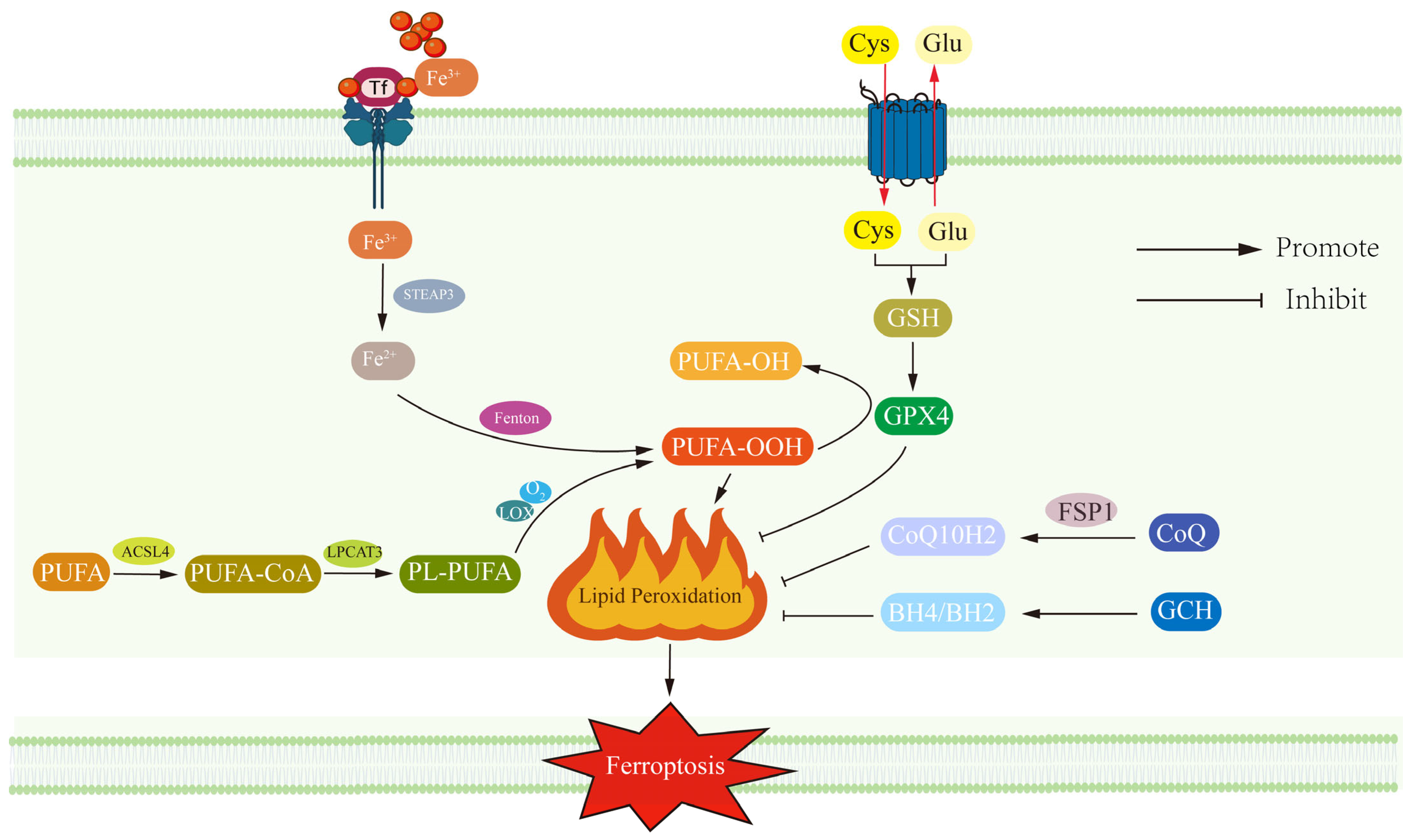
2.1. Ferroptosis Revealed: Lipid Peroxidation, Gpx4 Dysfunction and Neuronal Vulnerability in the Context of Neurodegeneration
2.2. Iron Overload in Depression: A Double-Edged Sword Linking Neurotransmitter Synthesis and Ferroptosis
3. Mitochondrial–Energy Metabolism Dysregulation in Depression
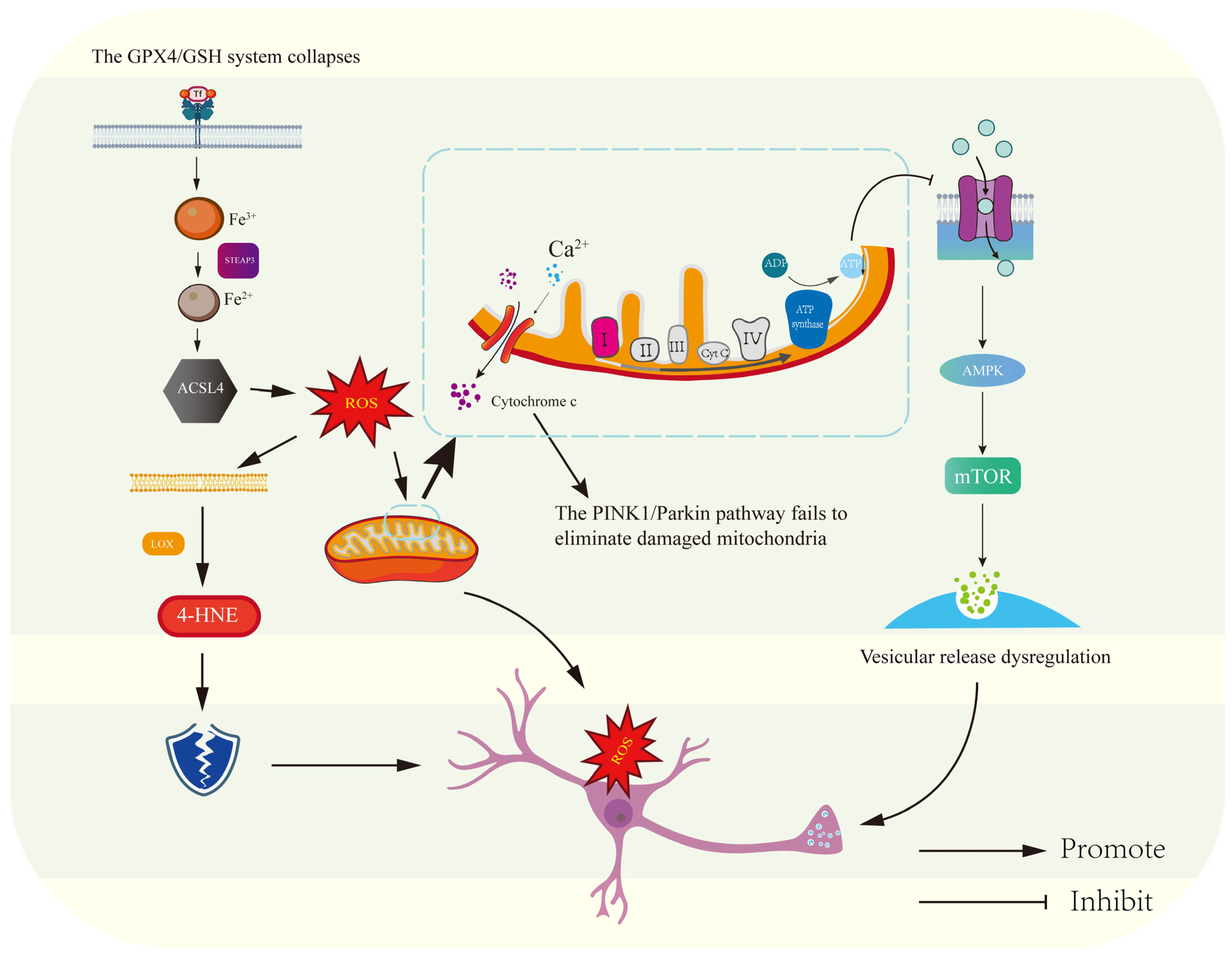
3.1. Mitochondria at the Central Hub: Bioenergetic Failure and ROS-Driven Synaptic Dysfunction in the Pathogenesis of Depression
3.2. Metabolic Crisis Meets Lipid Peroxidation: How Glucose and Fatty Acid Dysregulation Promotes Ferroptosis in Depression
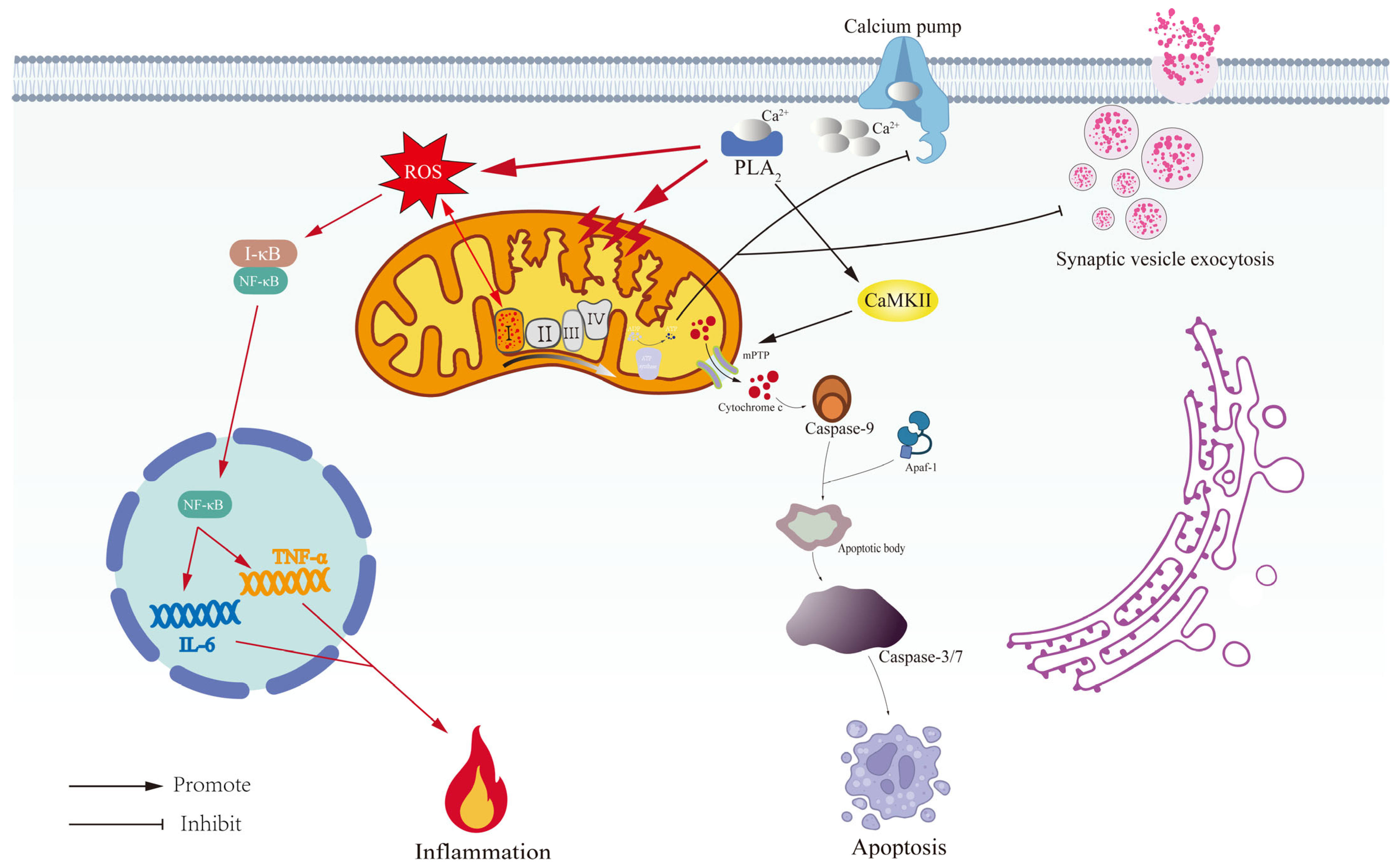
3.3. Mitochondrial Extinction in Depression: From Membrane Permeability Changes to Apoptosis
3.4. Broken Energy Reservoirs: Mitochondrial ROS, Dysregulated Calcium Homeostasis, and the Neuroinflammatory Cascade in Depression
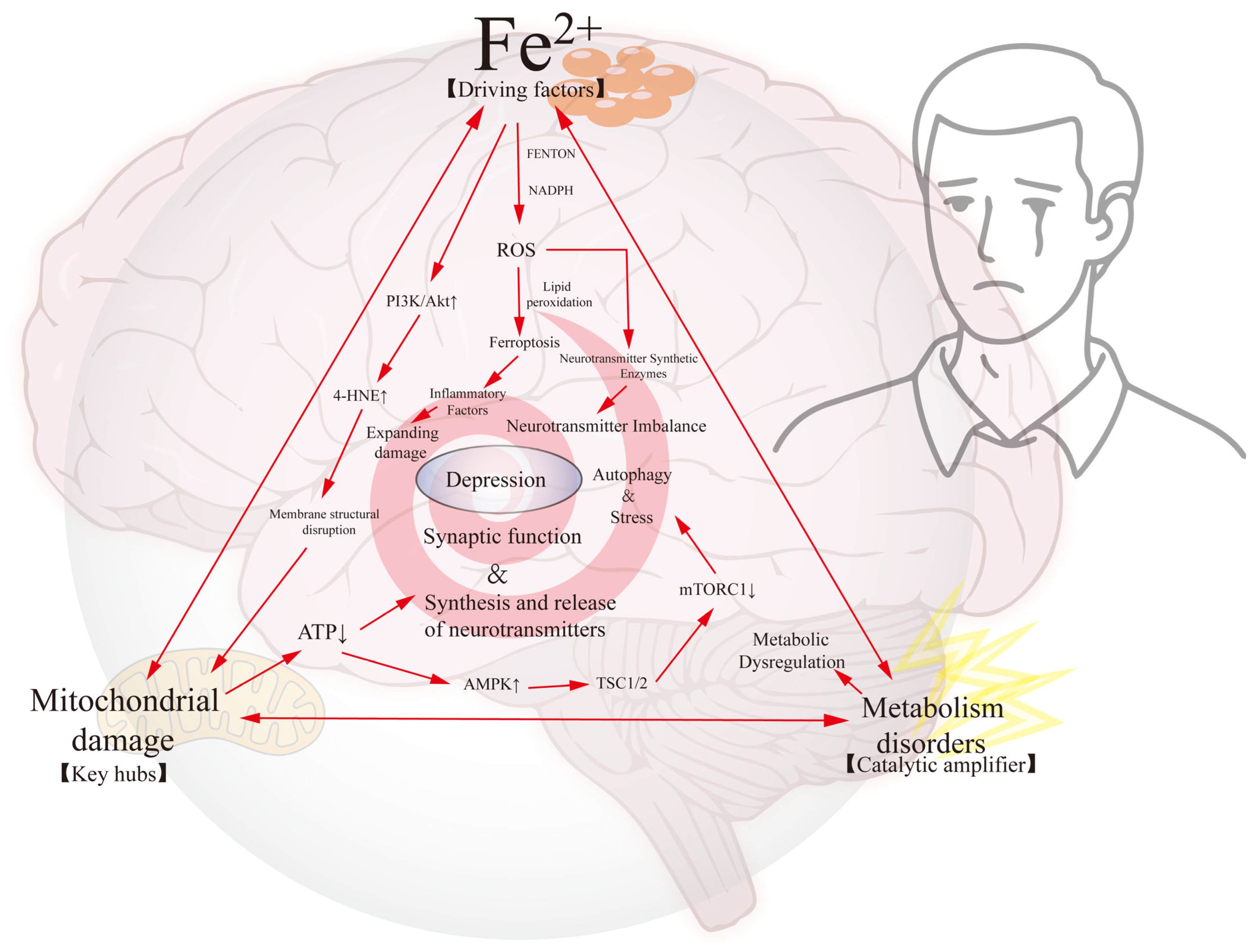
4. The Iron–Mitochondrial–Metabolic Triad: A Self-Enhancing Network Driving Neuronal Dysfunction in Depression
5. Outlook
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zuo, C.; Cao, H.; Song, Y.; Gu, Z.; Huang, Y.; Yang, Y.; Miao, J.; Zhu, L.; Chen, J.; Jiang, Y.; et al. Nrf2: An all-rounder in depression. Redox Biol. 2022, 58, 102522. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Yin, H.; Su, M.; Li, Q.; Zhao, Y.; Zhang, L.; Guo, J.; Lai, X.; Xue, X.; Tang, C. Inhibition of ferroptosis alleviates chronic unpredictable mild stress-induced depression in mice via tsRNA-3029b. Brain Res. Bull. 2023, 204, 110773. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Zuo, C.; Huang, Y.; Zhu, L.; Zhao, J.; Yang, Y.; Jiang, Y.; Wang, F. Hippocampal proteomic analysis reveals activation of necroptosis and ferroptosis in a mouse model of chronic unpredictable mild stress-induced depression. Behav. Brain Res. 2021, 407, 113261. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; You, J.; Xie, M.; Hu, Y.; Zhou, Q. Arginine Methylation of β-Catenin Induced by PRMT2 Aggravates LPS-Induced Cognitive Dysfunction and Depression-Like Behaviors by Promoting Ferroptosis. Mol. Neurobiol. 2024, 61, 7796–7813. [Google Scholar] [CrossRef]
- Shen, J.; Hao, C.; Yuan, S.; Chen, W.; Tong, T.; Chen, Y.; Shahzad Aslam, M.; Yan, S.; Li, J.; Zeng, J.; et al. Acupuncture alleviates CUMS-induced depression-like behaviors of rats by regulating oxidative stress, neuroinflammation and ferroptosis. Brain Res. 2024, 1826, 148715. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, F.; Zhai, M.; He, M.; Hu, Y.; Feng, L.; Li, Y.; Yang, J.; Wu, C. Hyperactive neuronal autophagy depletes BDNF and impairs adult hippocampal neurogenesis in a corticosterone-induced mouse model of depression. Theranostics 2023, 13, 1059–1075. [Google Scholar] [CrossRef]
- Drevets, W.C.; Wittenberg, G.M.; Bullmore, E.T.; Manji, H.K. Immune targets for therapeutic development in depression: Towards precision medicine. Nat. Rev. Drug Discov. 2022, 21, 224–244. [Google Scholar] [CrossRef]
- Wang, T.Z.; Wang, F.; Tian, Z.C.; Li, Z.Z.; Liu, W.N.; Ding, H.; Xie, T.T.; Cao, Z.X.; Li, H.T.; Sun, Z.C.; et al. Cingulate cGMP-dependent protein kinase I facilitates chronic pain and pain-related anxiety and depression. Pain 2023, 164, 2447–2462. [Google Scholar] [CrossRef]
- Lu, J.J.; Wu, P.F.; He, J.G.; Li, Y.K.; Long, L.H.; Yao, X.P.; Yang, J.H.; Chen, H.S.; Zhang, X.N.; Hu, Z.L.; et al. BNIP3L/NIX-mediated mitophagy alleviates passive stress-coping behaviors induced by tumor necrosis factor-α. Mol. Psychiatry 2023, 28, 5062–5076. [Google Scholar] [CrossRef]
- Wang, X.; Li, S.; Yu, J.; Wang, W.; Du, Z.; Gao, S.; Ma, Y.; Tang, R.; Liu, T.; Ma, S.; et al. Saikosaponin B2 ameliorates depression-induced microglia activation by inhibiting ferroptosis-mediated neuroinflammation and ER stress. J. Ethnopharmacol. 2023, 316, 116729. [Google Scholar] [CrossRef]
- Wang, D.; Wang, J.; Yu, Z.; Yao, R.; Zhang, J.; Zhao, X. Quercetin Alleviates Perimenopausal Depression Induced by Ovariectomy Combined with Chronic Unpredictable Mild Stress Through Regulating Serum Elements and Inhibiting Ferroptosis in Prefrontal Cortex of Rats. Biol. Trace Elem. Res. 2024, 202, 5596–5611. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xiao, A.; Yang, Y.; Zhao, Y.; Wang, C.C.; Wang, Y.; Han, J.; Wang, Z.; Wen, M. DHA and EPA Prevent Seizure and Depression-Like Behavior by Inhibiting Ferroptosis and Neuroinflammation via Different Mode-of-Actions in a Pentylenetetrazole-Induced Kindling Model in Mice. Mol. Nutr. Food Res. 2022, 66, e2200275. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Shimizu, J.; Murao, A.; Nofi, C.; Wang, P.; Aziz, M. Extracellular CIRP Promotes GPX4-Mediated Ferroptosis in Sepsis. Front. Immunol. 2022, 13, 903859. [Google Scholar] [CrossRef]
- Jiao, H.; Yang, H.; Yan, Z.; Chen, J.; Xu, M.; Jiang, Y.; Liu, Y.; Xue, Z.; Ma, Q.; Li, X.; et al. Traditional Chinese Formula Xiaoyaosan Alleviates Depressive-Like Behavior in CUMS Mice by Regulating PEBP1-GPX4-Mediated Ferroptosis in the Hippocampus. Neuropsychiatr. Dis. Treat. 2021, 17, 1001–1019. [Google Scholar] [CrossRef]
- Dang, R.; Wang, M.; Li, X.; Wang, H.; Liu, L.; Wu, Q.; Zhao, J.; Ji, P.; Zhong, L.; Licinio, J.; et al. Edaravone ameliorates depressive and anxiety-like behaviors via Sirt1/Nrf2/HO-1/Gpx4 pathway. J. Neuroinflamm. 2022, 19, 41. [Google Scholar] [CrossRef]
- Liu, Y.e.; Lu, S.; Wu, L.-l.; Yang, L.; Yang, L.; Wang, J. The diversified role of mitochondria in ferroptosis in cancer. Cell Death Dis. 2023, 14, 519. [Google Scholar] [CrossRef]
- Guo, T.; Yan, W.; Cui, X.; Liu, N.; Wei, X.; Sun, Y.; Fan, K.; Liu, J.; Zhu, Y.; Wang, Z.; et al. Liraglutide attenuates type 2 diabetes mellitus-associated non-alcoholic fatty liver disease by activating AMPK/ACC signaling and inhibiting ferroptosis. Mol. Med. 2023, 29, 132. [Google Scholar] [CrossRef]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol 2020, 22, 225–234. [Google Scholar] [CrossRef]
- Conrad, M.; Angeli, J.P.; Vandenabeele, P.; Stockwell, B.R. Regulated necrosis: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2016, 15, 348–366. [Google Scholar] [CrossRef]
- Chen, F.; Kang, R.; Tang, D.; Liu, J. Ferroptosis: Principles and significance in health and disease. J. Hematol. Oncol. 2024, 17, 41. [Google Scholar] [CrossRef] [PubMed]
- Onukwufor, J.O.; Dirksen, R.T.; Wojtovich, A.P. Iron Dysregulation in Mitochondrial Dysfunction and Alzheimer’s Disease. Antioxidants 2022, 11, 692. [Google Scholar] [CrossRef]
- Zhang, X.; Fang, Z.; Guo, X.; Li, Y.; Fan, Z.; Wu, Y.; Du, L. The Role of Ferroptosis in Nervous System Disorders. J. Integr. Neurosci. 2023, 22, 19. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, X.; Xiong, X.; Zhu, H.; Chen, R.; Zhang, S.; Chen, G.; Jian, Z. Nrf2 Regulates Oxidative Stress and Its Role in Cerebral Ischemic Stroke. Antioxidants 2022, 11, 2377. [Google Scholar] [CrossRef]
- Xu, Y.; Jia, B.; Li, J.; Li, Q.; Luo, C. The Interplay between Ferroptosis and Neuroinflammation in Central Neurological Disorders. Antioxidants 2024, 13, 395. [Google Scholar] [CrossRef]
- Win, S.; Than, T.A.; Kaplowitz, N. Mitochondrial P-JNK target, SAB (SH3BP5), in regulation of cell death. Front. Cell Dev. Biol. 2024, 12, 1359152. [Google Scholar] [CrossRef]
- Xue, Q.; Kang, R.; Klionsky, D.J.; Tang, D.; Liu, J.; Chen, X. Copper metabolism in cell death and autophagy. Autophagy 2023, 19, 2175–2195. [Google Scholar] [CrossRef]
- Singh, K.; Sethi, P.; Datta, S.; Chaudhary, J.S.; Kumar, S.; Jain, D.; Gupta, J.K.; Kumar, S.; Guru, A.; Panda, S.P. Advances in gene therapy approaches targeting neuro-inflammation in neurodegenerative diseases. Ageing Res. Rev. 2024, 98, 102321. [Google Scholar] [CrossRef]
- Xu, M.; Zhong, W.; Yang, C.; Liu, M.; Yuan, X.; Lu, T.; Li, D.; Zhang, G.; Liu, H.; Zeng, Y.; et al. Tiliroside disrupted iron homeostasis and induced ferroptosis via directly targeting calpain-2 in pancreatic cancer cells. Phytomedicine 2024, 127, 155392. [Google Scholar] [CrossRef]
- Qiao, L.; Yang, G.; Wang, P.; Xu, C. The potential role of mitochondria in the microbiota-gut-brain axis: Implications for brain health. Pharmacol. Res. 2024, 209, 107434. [Google Scholar] [CrossRef]
- Chen, B.; de Launoit, E.; Meseguer, D.; Garcia Caceres, C.; Eichmann, A.; Renier, N.; Schneeberger, M. The interactions between energy homeostasis and neurovascular plasticity. Nat. Rev. Endocrinol. 2024, 20, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, S.; Sun, Y.; Chen, C.; Hu, Z.; Li, Q.; Long, J.; Yan, Q.; Liang, J.; Lin, Y.; et al. Target modulation of glycolytic pathways as a new strategy for the treatment of neuroinflammatory diseases. Ageing Res. Rev. 2024, 101, 102472. [Google Scholar] [CrossRef]
- Ahola, S.; Langer, T. Ferroptosis in mitochondrial cardiomyopathy. Trends Cell Biol 2024, 34, 150–160. [Google Scholar] [CrossRef]
- Ma, H.; Dong, Y.; Chu, Y.; Guo, Y.; Li, L. The mechanisms of ferroptosis and its role in alzheimer’s disease. Front. Mol. Biosci. 2022, 9, 965064. [Google Scholar] [CrossRef]
- Tang, Z.; Zhao, P.; Wang, H.; Liu, Y.; Bu, W. Biomedicine Meets Fenton Chemistry. Chem. Rev. 2021, 121, 1981–2019. [Google Scholar] [CrossRef]
- Chen, Q.M. Nrf2 for cardiac protection: Pharmacological options against oxidative stress. Trends Pharmacol. Sci. 2021, 42, 729–744. [Google Scholar] [CrossRef]
- Yu, H.; Huang, X.; Zhu, H.H.; Wang, N.; Xie, C.; Zhou, Y.L.; Shi, H.L.; Chen, M.M.; Wu, Y.R.; Ruan, Z.H.; et al. Apigenin ameliorates non-eosinophilic inflammation, dysregulated immune homeostasis and mitochondria-mediated airway epithelial cell apoptosis in chronic obese asthma via the ROS-ASK1-MAPK pathway. Phytomedicine 2023, 111, 154646. [Google Scholar] [CrossRef]
- Chen, H.; Lu, M.; Lyu, Q.; Shi, L.; Zhou, C.; Li, M.; Feng, S.; Liang, X.; Zhou, X.; Ren, L. Mitochondrial dynamics dysfunction: Unraveling the hidden link to depression. Biomed. Pharmacother. 2024, 175, 116656. [Google Scholar] [CrossRef]
- Song, Y.; Cao, H.; Zuo, C.; Gu, Z.; Huang, Y.; Miao, J.; Fu, Y.; Guo, Y.; Jiang, Y.; Wang, F. Mitochondrial dysfunction: A fatal blow in depression. Biomed. Pharmacother. 2023, 167, 115652. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Jiang, X.; Gu, W. Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell Biol. 2020, 30, 478–490. [Google Scholar] [CrossRef]
- Uzungil, V.; Tran, H.; Aitken, C.; Wilson, C.; Opazo, C.M.; Li, S.; Payet, J.M.; Mawal, C.H.; Bush, A.I.; Hale, M.W.; et al. Novel Antidepressant-Like Properties of the Iron Chelator Deferiprone in a Mouse Model of Depression. Neurother. J. Am. Soc. Exp. Neurother. 2022, 19, 1662–1685. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Hwang, N.; Seok, B.G.; Lee, S.; Lee, S.J.; Chung, S.W. Autophagy mediates an amplification loop during ferroptosis. Cell Death Dis 2023, 14, 464. [Google Scholar] [CrossRef] [PubMed]
- Eagle, H. The specific amino acid requirements of a human carcinoma cell (Stain HeLa) in tissue culture. J. Exp. Med. 1955, 102, 37–48. [Google Scholar] [CrossRef]
- Eagle, H. Amino acid metabolism in mammalian cell cultures. Science 1959, 130, 432–437. [Google Scholar] [CrossRef]
- Schwarz, K. Production of dietary necrotic liver degeneration using American torula yeast. Proceedings of the Society for Experimental Biology and Medicine. Soc. Exp. Biol. Med. 1951, 77, 818–823. [Google Scholar] [CrossRef]
- Schwarz, K.; Foltz, C.M. Factor 3 activity of selenium compounds. J. Biol. Chem. 1958, 233, 245–251. [Google Scholar] [CrossRef]
- Golberg, L.; Smith, J.P. Changes associated with the accumulation of excessive amounts of iron in certain organs of the rat. Br. J. Exp. Pathol. 1958, 39, 59–73. [Google Scholar]
- Tai, P.; Chen, X.; Jia, G.; Chen, G.; Gong, L.; Cheng, Y.; Li, Z.; Wang, H.; Chen, A.; Zhang, G.; et al. WGX50 mitigates doxorubicin-induced cardiotoxicity through inhibition of mitochondrial ROS and ferroptosis. J. Transl. Med. 2023, 21, 823. [Google Scholar] [CrossRef]
- Wang, Y.; Qi, H.; Liu, Y.; Duan, C.; Liu, X.; Xia, T.; Chen, D.; Piao, H.-l.; Liu, H.-X. The double-edged roles of ROS in cancer prevention and therapy. Theranostics 2021, 11, 4839–4857. [Google Scholar] [CrossRef]
- D’Alessandro, A.; Keele, G.R.; Hay, A.; Nemkov, T.; Earley, E.J.; Stephenson, D.; Vincent, M.; Deng, X.; Stone, M.; Dzieciatkowska, M.; et al. Ferroptosis regulates hemolysis in stored murine and human red blood cells. Blood 2025, 145, 765–783. [Google Scholar] [CrossRef]
- Zou, Y.; Zheng, S.; Xie, X.; Ye, F.; Hu, X.; Tian, Z.; Yan, S.M.; Yang, L.; Kong, Y.; Tang, Y.; et al. N6-methyladenosine regulated FGFR4 attenuates ferroptotic cell death in recalcitrant HER2-positive breast cancer. Nat. Commun. 2022, 13, 2672. [Google Scholar] [CrossRef] [PubMed]
- Qiu, B.; Zandkarimi, F.; Bezjian, C.T.; Reznik, E.; Soni, R.K.; Gu, W.; Jiang, X.; Stockwell, B.R. Phospholipids with two polyunsaturated fatty acyl tails promote ferroptosis. Cell 2024, 187, 1177–1190.e1118. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, W.K.; Bae, K.H.; Lee, S.C.; Lee, E.W. Lipid Metabolism and Ferroptosis. Biology 2021, 10, 184. [Google Scholar] [CrossRef]
- Broos, J.Y.; van der Burgt, R.T.M.; Konings, J.; Rijnsburger, M.; Werz, O.; de Vries, H.E.; Giera, M.; Kooij, G. Arachidonic acid-derived lipid mediators in multiple sclerosis pathogenesis: Fueling or dampening disease progression? J. Neuroinflamm. 2024, 21, 21. [Google Scholar] [CrossRef]
- Zheng, Q.; Xing, J.; Li, X.; Tang, X.; Zhang, D. PRDM16 suppresses ferroptosis to protect against sepsis-associated acute kidney injury by targeting the NRF2/GPX4 axis. Redox Biol. 2024, 78, 103417. [Google Scholar] [CrossRef]
- Lin, Z.; Liu, Z.; Yang, X.; Pan, Z.; Feng, Y.; Zhang, Y.; Chen, H.; Lao, L.; Chen, J.; Shi, F.; et al. Simeprevir induces ferroptosis through β-TrCP/Nrf2/GPX4 axis in triple-negative breast cancer cells. Biomed. Pharmacother. 2024, 180, 117558. [Google Scholar] [CrossRef]
- Li, S.; Liu, H.; Hu, H.; Ha, E.; Prasad, P.; Jenkins, B.C.; Das, U.S.; Mukherjee, S.; Shishikura, K.; Hu, R.; et al. Human genetics identify convergent signals in mitochondrial LACTB-mediated lipid metabolism in cardiovascular-kidney-metabolic syndrome. Cell Metab. 2025, 37, 154–168.e157. [Google Scholar] [CrossRef]
- Beharier, O.; Tyurin, V.A.; Goff, J.P.; Guerrero-Santoro, J.; Kajiwara, K.; Chu, T.; Tyurina, Y.Y.; St Croix, C.M.; Wallace, C.T.; Parry, S.; et al. PLA2G6 guards placental trophoblasts against ferroptotic injury. Proc. Natl. Acad. Sci. USA 2020, 117, 27319–27328. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Jacquemyn, J.; Ralhan, I.; Ioannou, M.S. Driving factors of neuronal ferroptosis. Trends Cell Biol. 2024, 34, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Jiao, Q.; Du, X.; Jia, F.; Chen, X.; Yan, C.; Jiang, H. Ferroptosis in Parkinson’s disease—The iron-related degenerative disease. Ageing Res. Rev. 2024, 101, 102477. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.K.; Ugalde, C.L.; Rolland, A.S.; Skidmore, J.; Devos, D.; Hammond, T.R. Therapeutic inhibition of ferroptosis in neurodegenerative disease. Trends Pharmacol. Sci. 2023, 44, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.S.; Gao, L.; Han, Z.; Eleuteri, S.; Shi, W.; Shen, Y.; Song, Z.Y.; Su, M.; Yang, Q.; Qu, Y.; et al. Ferroptosis in Parkinson’s disease: Molecular mechanisms and therapeutic potential. Ageing Res. Rev. 2023, 91, 102077. [Google Scholar] [CrossRef]
- Shah, H.E.; Bhawnani, N.; Ethirajulu, A.; Alkasabera, A.; Onyali, C.B.; Anim-Koranteng, C.; Mostafa, J.A. Iron Deficiency-Induced Changes in the Hippocampus, Corpus Striatum, and Monoamines Levels That Lead to Anxiety, Depression, Sleep Disorders, and Psychotic Disorders. Cureus 2021, 13, e18138. [Google Scholar] [CrossRef]
- Wang, L.; Xu, R.; Huang, C.; Yi, G.; Li, Z.; Zhang, H.; Ye, R.; Qi, S.; Huang, G.; Qu, S. Targeting the ferroptosis crosstalk: Novel alternative strategies for the treatment of major depressive disorder. Gen. Psychiatry 2023, 36, e101072. [Google Scholar] [CrossRef]
- Baj, J.; Bargieł, J.; Cabaj, J.; Skierkowski, B.; Hunek, G.; Portincasa, P.; Flieger, J.; Smoleń, A. Trace Elements Levels in Major Depressive Disorder-Evaluation of Potential Threats and Possible Therapeutic Approaches. Int. J. Mol. Sci. 2023, 24, 15071. [Google Scholar] [CrossRef]
- Kulaszyńska, M.; Kwiatkowski, S.; Skonieczna-Żydecka, K. The Iron Metabolism with a Specific Focus on the Functioning of the Nervous System. Biomedicines 2024, 12, 595. [Google Scholar] [CrossRef]
- Liu, D.; Liang, C.H.; Huang, B.; Zhuang, X.; Cui, W.; Yang, L.; Yang, Y.; Zhang, Y.; Fu, X.; Zhang, X.; et al. Tryptophan Metabolism Acts as a New Anti-Ferroptotic Pathway to Mediate Tumor Growth. Adv. Sci. 2023, 10, e2204006. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet. Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
- Ishii, T.; Warabi, E.; Mann, G.E. Circadian control of BDNF-mediated Nrf2 activation in astrocytes protects dopaminergic neurons from ferroptosis. Free Radic. Biol. Med. 2019, 133, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jia, B.; Cheng, Y.; Song, Y.; Li, Q.; Luo, C.; Birla, H. Targeting Molecular Mediators of Ferroptosis and Oxidative Stress for Neurological Disorders. Oxidative Med. Cell. Longev. 2022, 2022, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Guo, J.; Zhang, B.; Chen, J.; Ou, H.; He, R.-R.; So, K.-F.; Zhang, L. Lycium barbarum (Wolfberry) glycopeptide prevents stress-induced anxiety disorders by regulating oxidative stress and ferroptosis in the medial prefrontal cortex. Phytomedicine 2023, 116, 154864. [Google Scholar] [CrossRef]
- Sui, X.; Wang, J.; Zhao, Z.; Liu, B.; Liu, M.; Liu, M.; Shi, C.; Feng, X.; Fu, Y.; Shi, D.; et al. Phenolic compounds induce ferroptosis-like death by promoting hydroxyl radical generation in the Fenton reaction. Commun. Biol. 2024, 7, 199. [Google Scholar] [CrossRef]
- Ahmed, H.H.; Essam, R.M.; El-Yamany, M.F.; Ahmed, K.A.; El-Sahar, A.E. Unleashing lactoferrin’s antidepressant potential through the PI3K/Akt/mTOR pathway in chronic restraint stress rats. Food Funct. 2023, 14, 9265–9278. [Google Scholar] [CrossRef]
- Long, Q.; Li, T.; Zhu, Q.; He, L.; Zhao, B. SuanZaoRen decoction alleviates neuronal loss, synaptic damage and ferroptosis of AD via activating DJ-1/Nrf2 signaling pathway. J. Ethnopharmacol. 2024, 323, 117679. [Google Scholar] [CrossRef]
- Long, H.; Zhu, W.; Wei, L.; Zhao, J. Iron homeostasis imbalance and ferroptosis in brain diseases. MedComm 2023, 4, e298. [Google Scholar] [CrossRef]
- Chen, H.; Wu, J.; Zhu, X.; Ma, Y.; Li, Z.; Lu, L.; Aschner, M.; Su, P.; Luo, W. Manganese-induced miR-125b-2-3p promotes anxiety-like behavior via TFR1-mediated ferroptosis. Environ. Pollut. 2024, 344, 123255. [Google Scholar] [CrossRef]
- Chen, L.; Yang, Y.; Zhang, N.; Che, H.; Wang, Z.; Han, J.; Wen, M. DHA and EPA alleviate depressive-like behaviors in chronic sleep-deprived mice: Involvement of iron metabolism, oligodendrocyte-lipids peroxidation and the LCN2-NLRP3 signaling axis. Free Radic. Biol. Med. 2024, 225, 654–664. [Google Scholar] [CrossRef]
- Liu, P.; Chen, W.; Kang, Y.; Wang, C.; Wang, X.; Liu, W.; Hayashi, T.; Qiu, Z.; Mizuno, K.; Hattori, S.; et al. Silibinin ameliorates STING-mediated neuroinflammation via downregulation of ferroptotic damage in a sporadic Alzheimer’s disease model. Arch. Biochem. Biophys. 2023, 744, 109691. [Google Scholar] [CrossRef]
- Tarnacka, B.; Jopowicz, A.; Maślińska, M. Copper, Iron, and Manganese Toxicity in Neuropsychiatric Conditions. Int. J. Mol. Sci. 2021, 22, 7820. [Google Scholar] [CrossRef] [PubMed]
- Bellavite, P. Neuroprotective Potentials of Flavonoids: Experimental Studies and Mechanisms of Action. Antioxidants 2023, 12, 280. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, P.; Xu, Y.; Feng, L.; Fang, Y.; Song, G.; Xu, L.; Zhu, Z.; Wang, W.; Mei, Q.; et al. Lactate metabolism and histone lactylation in the central nervous system disorders: Impacts and molecular mechanisms. J. Neuroinflamm. 2024, 21, 308. [Google Scholar] [CrossRef]
- Bonvento, G.; Bolaños, J.P. Astrocyte-neuron metabolic cooperation shapes brain activity. Cell Metab. 2021, 33, 1546–1564. [Google Scholar] [CrossRef]
- Feng, Y.; Chen, J.; Wang, X.; Long, C.; Wang, W.; Lin, J.; He, Y.; Wang, Y.; Luo, F.; Li, Z.; et al. Reprogramming metabolic microenvironment for nerve regeneration via waterborne polylactic acid-polyurethane copolymer scaffolds. Biomaterials 2024, 315, 122942. [Google Scholar] [CrossRef]
- Tiwari, A.; Myeong, J.; Hashemiaghdam, A.; Stunault, M.I.; Zhang, H.; Niu, X.; Laramie, M.A.; Sponagel, J.; Shriver, L.P.; Patti, G.J.; et al. Mitochondrial pyruvate transport regulates presynaptic metabolism and neurotransmission. Sci. Adv. 2024, 10, eadp7423. [Google Scholar] [CrossRef]
- Díaz-Castro, F.; Morselli, E.; Claret, M. Interplay between the brain and adipose tissue: A metabolic conversation. EMBO Rep. 2024, 25, 5277–5293. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Li, Y.; Pang, J.; Höhn, A.; Dong, W.; Gao, R.; Liu, Y.; Wang, D.; She, Y.; et al. Methionine restriction alleviates diabetes-associated cognitive impairment via activation of FGF21. Redox Biol. 2024, 77, 103390. [Google Scholar] [CrossRef]
- Heffernan, Á.B.; Steinruecke, M.; Dempsey, G.; Chandran, S.; Selvaraj, B.T.; Jiwaji, Z.; Stavrou, M. Role of glia in delirium: Proposed mechanisms and translational implications. Mol. Psychiatry 2024, 30, 1138–1147. [Google Scholar] [CrossRef]
- Murali Mahadevan, H.; Hashemiaghdam, A.; Ashrafi, G.; Harbauer, A.B. Mitochondria in Neuronal Health: From Energy Metabolism to Parkinson’s Disease. Adv. Biol. 2021, 5, e2100663. [Google Scholar] [CrossRef]
- Ryu, K.W.; Fung, T.S.; Baker, D.C.; Saoi, M.; Park, J.; Febres-Aldana, C.A.; Aly, R.G.; Cui, R.; Sharma, A.; Fu, Y.; et al. Cellular ATP demand creates metabolically distinct subpopulations of mitochondria. Nature 2024, 635, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Fiorenza, M.; Onslev, J.; Henríquez-Olguín, C.; Persson, K.W.; Hesselager, S.A.; Jensen, T.E.; Wojtaszewski, J.F.P.; Hostrup, M.; Bangsbo, J. Reducing the mitochondrial oxidative burden alleviates lipid-induced muscle insulin resistance in humans. Sci. Adv. 2024, 10, eadq4461. [Google Scholar] [CrossRef] [PubMed]
- Glover, H.L.; Schreiner, A.; Dewson, G.; Tait, S.W.G. Mitochondria and cell death. Nat. Cell Biol. 2024, 26, 1434–1446. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Cao, N.; Zhang, D.; Wang, M. The effect of ferroptosis-related mitochondrial dysfunction in the development of temporal lobe epilepsy. Ageing Res. Rev. 2024, 96, 102248. [Google Scholar] [CrossRef]
- Talukdar, P.D.; Pramanik, K.; Gatti, P.; Mukherjee, P.; Ghosh, D.; Roy, H.; Germain, M.; Chatterji, U. Precise targeting of transcriptional co-activators YAP/TAZ annihilates chemoresistant brCSCs by alteration of their mitochondrial homeostasis. Signal Transduct. Target. Ther. 2025, 10, 61. [Google Scholar] [CrossRef]
- Court, A.C.; Vega-Letter, A.M.; Parra-Crisóstomo, E.; Velarde, F.; García, C.; Ortloff, A.; Vernal, R.; Pradenas, C.; Luz-Crawford, P.; Khoury, M.; et al. Mitochondrial transfer balances cell redox, energy and metabolic homeostasis in the osteoarthritic chondrocyte preserving cartilage integrity. Theranostics 2024, 14, 6471–6486. [Google Scholar] [CrossRef]
- Gao, Q.; Tian, R.; Han, H.; Slone, J.; Wang, C.; Ke, X.; Zhang, T.; Li, X.; He, Y.; Liao, P.; et al. PINK1-mediated Drp1(S616) phosphorylation modulates synaptic development and plasticity via promoting mitochondrial fission. Signal Transduct. Target. Ther. 2022, 7, 103. [Google Scholar] [CrossRef]
- Yao, S.; Xu, M.D.; Wang, Y.; Zhao, S.T.; Wang, J.; Chen, G.F.; Chen, W.B.; Liu, J.; Huang, G.B.; Sun, W.J.; et al. Astrocytic lactate dehydrogenase A regulates neuronal excitability and depressive-like behaviors through lactate homeostasis in mice. Nat. Commun. 2023, 14, 729. [Google Scholar] [CrossRef]
- Chen, B.; Jin, K.; Dong, J.; Cheng, S.; Kong, L.; Hu, S.; Chen, Z.; Lu, J. Hypocretin-1/Hypocretin Receptor 1 Regulates Neuroplasticity and Cognitive Function through Hippocampal Lactate Homeostasis in Depressed Model. Adv. Sci. 2024, 11, e2405354. [Google Scholar] [CrossRef]
- Fu, C.; Cao, N.; Zeng, S.; Zhu, W.; Fu, X.; Liu, W.; Fan, S. Role of mitochondria in the regulation of ferroptosis and disease. Front. Med. 2023, 10, 1301822. [Google Scholar] [CrossRef]
- Paul, B.T.; Tesfay, L.; Winkler, C.R.; Torti, F.M.; Torti, S.V. Sideroflexin 4 affects Fe-S cluster biogenesis, iron metabolism, mitochondrial respiration and heme biosynthetic enzymes. Sci. Rep. 2019, 9, 19634. [Google Scholar] [CrossRef] [PubMed]
- Terada, T.; Obi, T.; Bunai, T.; Matsudaira, T.; Yoshikawa, E.; Ando, I.; Futatsubashi, M.; Tsukada, H.; Ouchi, Y. In vivo mitochondrial and glycolytic impairments in patients with Alzheimer disease. Neurology 2020, 94, e1592–e1604. [Google Scholar] [CrossRef]
- Abeysekera, M.V.; Ni, D.; Gilbert, L.; Hibbert, E.; Nanan, R. Linking the reversal of gestational insulin resistance to postpartum depression. BMC Med. 2024, 22, 433. [Google Scholar] [CrossRef]
- McNay, E.C.; Pearson-Leary, J. GluT4: A central player in hippocampal memory and brain insulin resistance. Exp. Neurol. 2020, 323, 113076. [Google Scholar] [CrossRef]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflug. Arch. Eur. J. Physiol. 2020, 472, 1299–1343. [Google Scholar] [CrossRef]
- Liu, J.; Quan, L.; Wang, J.; Zhang, G.; Cai, L.; Pan, Z.; Liu, S.; Zhu, C.; Wu, R.; Wang, L.; et al. Knockdown of VEGF-B improves HFD-induced insulin resistance by enhancing glucose uptake in vascular endothelial cells via the PI3K/Akt pathway. Int. J. Biol. Macromol. 2024, 285, 138279. [Google Scholar] [CrossRef]
- Khawagi, W.Y.; Al-Kuraishy, H.M.; Hussein, N.R.; Al-Gareeb, A.I.; Atef, E.; Elhussieny, O.; Alexiou, A.; Papadakis, M.; Jabir, M.S.; Alshehri, A.A.; et al. Depression and type 2 diabetes: A causal relationship and mechanistic pathway. Diabetes Obes. Metab. 2024, 26, 3031–3044. [Google Scholar] [CrossRef]
- Li, S.; Yang, D.; Zhou, X.; Chen, L.; Liu, L.; Lin, R.; Li, X.; Liu, Y.; Qiu, H.; Cao, H.; et al. Neurological and metabolic related pathophysiologies and treatment of comorbid diabetes with depression. CNS Neurosci. Ther. 2024, 30, e14497. [Google Scholar] [CrossRef]
- Xu, W.; Tian, S.; Mao, G.; Li, Y.; Qian, H.; Tao, W. Sini San ameliorates lipid metabolism in hyperprolactinemia rat with liver-depression. Curr. Res. Food Sci. 2024, 9, 100853. [Google Scholar] [CrossRef]
- Song, Y.; Lai, M.; Liao, Z.; Zhang, Z.; Zhu, G.; Yang, M.; Ai, Z.; Zheng, Q.; Su, D. Saikosaponin antidepressant mechanism: Improving the sphingolipid metabolism in the cortex via Apolipoprotein E and triggering neurovascular coupling. Phytomedicine 2024, 132, 155829. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, W.; Wang, L.; Zhu, C.; Cui, S.; Wang, T.; Gu, X.; Liu, Y.; Qiu, P. Unraveling the role and mechanism of mitochondria in postoperative cognitive dysfunction: A narrative review. J. Neuroinflamm. 2024, 21, 293. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Yang, C.; Jia, X.; Yu, Z.; Wang, C.; Zhao, J.; Chen, Y.; Xie, B.; Zhuang, H.; Sun, C.; et al. High-fat diet consumption promotes adolescent neurobehavioral abnormalities and hippocampal structural alterations via microglial overactivation accompanied by an elevated serum free fatty acid concentration. Brain Behav. Immun. 2024, 119, 236–250. [Google Scholar] [CrossRef]
- Pinna, G. Role of PPAR-Allopregnanolone Signaling in Behavioral and Inflammatory Gut-Brain Axis Communications. Biol. Psychiatry 2023, 94, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Matrisciano, F.; Pinna, G. PPAR-α Hypermethylation in the Hippocampus of Mice Exposed to Social Isolation Stress Is Associated with Enhanced Neuroinflammation and Aggressive Behavior. Int. J. Mol. Sci. 2021, 22, 10678. [Google Scholar] [CrossRef]
- Flannery, L.E.; Kerr, D.M.; Hughes, E.M.; Kelly, C.; Costello, J.; Thornton, A.M.; Humphrey, R.M.; Finn, D.P.; Roche, M. N-acylethanolamine regulation of TLR3-induced hyperthermia and neuroinflammatory gene expression: A role for PPARα. J. Neuroimmunol. 2021, 358, 577654. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, M.; Cao, J.; Wang, F.; Han, J.R.; Wu, T.W.; Li, L.; Yu, J.; Fan, Y.; Xie, G.; et al. ACSL4 and polyunsaturated lipids support metastatic extravasation and colonization. Cell 2025, 188, 412–429.e427. [Google Scholar] [CrossRef]
- Chen, G.H.; Song, C.C.; Pantopoulos, K.; Wei, X.L.; Zheng, H.; Luo, Z. Mitochondrial oxidative stress mediated Fe-induced ferroptosis via the NRF2-ARE pathway. Free Radic. Biol. Med. 2022, 180, 95–107. [Google Scholar] [CrossRef]
- Esteves, A.R.; Munoz-Pinto, M.F.; Nunes-Costa, D.; Candeias, E.; Silva, D.F.; Magalhães, J.D.; Pereira-Santos, A.R.; Ferreira, I.L.; Alarico, S.; Tiago, I.; et al. Footprints of a microbial toxin from the gut microbiome to mesencephalic mitochondria. Gut 2023, 72, 73–89. [Google Scholar] [CrossRef]
- Sun, M.; Liu, M.; Li, Q.; Liu, S.; Yang, H.; Song, Y.; Qu, M.; Zhang, X.; Ma, Y.; Mi, W. Insulin attenuates LPS-induced cognitive impairment and ferroptosis through regulation of glucose metabolism in hippocampus. CNS Neurosci. Ther. 2024, 30, e14887. [Google Scholar] [CrossRef]
- Qin, X.; Tan, Z.; Li, Q.; Zhang, S.; Hu, D.; Wang, D.; Wang, L.; Zhou, B.; Liao, R.; Wu, Z.; et al. Rosiglitazone attenuates Acute Kidney Injury from hepatic ischemia-reperfusion in mice by inhibiting arachidonic acid metabolism through the PPAR-γ/NF-κB pathway. Inflamm. Res. 2024, 73, 1765–1780. [Google Scholar] [CrossRef]
- Sałaciak, K.; Koszałka, A.; Żmudzka, E.; Pytka, K. The Calcium/Calmodulin-Dependent Kinases II and IV as Therapeutic Targets in Neurodegenerative and Neuropsychiatric Disorders. Int. J. Mol. Sci. 2021, 22, 4307. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Wu, Y.; Dabas, H.; Hammarlund, M. Activation of the CaMKII-Sarm1-ASK1-p38 MAP kinase pathway protects against axon degeneration caused by loss of mitochondria. eLife 2022, 11, e73557. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Li, S.; Wang, S.; Wu, X.; Liu, Y.; Yu, W.; Wang, Y.; Tang, Y.; Xia, M.; Li, B. Major depressive disorder: Hypothesis, mechanism, prevention and treatment. Signal Transduct. Target. Ther. 2024, 9, 30. [Google Scholar] [CrossRef]
- Panda, S.K.; Peng, V.; Sudan, R.; Ulezko Antonova, A.; Di Luccia, B.; Ohara, T.E.; Fachi, J.L.; Grajales-Reyes, G.E.; Jaeger, N.; Trsan, T.; et al. Repression of the aryl-hydrocarbon receptor prevents oxidative stress and ferroptosis of intestinal intraepithelial lymphocytes. Immunity 2023, 56, 797–812.e794. [Google Scholar] [CrossRef]
- Mayneris-Perxachs, J.; Castells-Nobau, A.; Arnoriaga-Rodríguez, M.; Martin, M.; de la Vega-Correa, L.; Zapata, C.; Burokas, A.; Blasco, G.; Coll, C.; Escrichs, A.; et al. Microbiota alterations in proline metabolism impact depression. Cell Metab. 2022, 34, 681–701.e610. [Google Scholar] [CrossRef]
- Newman-Tancredi, A.; Depoortère, R.Y.; Kleven, M.S.; Kołaczkowski, M.; Zimmer, L. Translating biased agonists from molecules to medications: Serotonin 5-HT(1A) receptor functional selectivity for CNS disorders. Pharmacol. Ther. 2022, 229, 107937. [Google Scholar] [CrossRef]
- Pearce, W.J. Mitochondrial influences on smooth muscle phenotype. Am. J. Physiol. Cell Physiol. 2024, 326, C442–C448. [Google Scholar] [CrossRef]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Mo, J.W.; Kong, P.L.; Ding, L.; Fan, J.; Ren, J.; Lu, C.L.; Guo, F.; Chen, L.Y.; Mo, R.; Zhong, Q.L.; et al. Lysosomal TFEB-TRPML1 Axis in Astrocytes Modulates Depressive-like Behaviors. Adv. Sci. 2024, 11, e2403389. [Google Scholar] [CrossRef]
- Gowda, P.; Reddy, P.H.; Kumar, S. Deregulated mitochondrial microRNAs in Alzheimer’s disease: Focus on synapse and mitochondria. Ageing Res. Rev. 2022, 73, 101529. [Google Scholar] [CrossRef]
- Pilotto, F.; Smeele, P.H.; Scheidegger, O.; Diab, R.; Schobesberger, M.; Sierra-Delgado, J.A.; Saxena, S. Kaempferol enhances ER-mitochondria coupling and protects motor neurons from mitochondrial dysfunction and ER stress in C9ORF72-ALS. Acta Neuropathol. Commun. 2025, 13, 21. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Xu, J.; Williams, K.; Easley, M.; Elder, J.B.; Lonser, R.; Lang, F.F.; Lapalombella, R.; Sampath, D.; Puduvalli, V.K. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme of the nicotinamide adenine dinucleotide (NAD) salvage pathway, to target glioma heterogeneity through mitochondrial oxidative stress. Neuro-Oncology 2022, 24, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Flores-Romero, H.; Dadsena, S.; García-Sáez, A.J. Mitochondrial pores at the crossroad between cell death and inflammatory signaling. Mol. Cell 2023, 83, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Vringer, E.; Tait, S.W.G. Mitochondria and cell death-associated inflammation. Cell Death Differ. 2023, 30, 304–312. [Google Scholar] [CrossRef]
- Katayama, H.; Hama, H.; Nagasawa, K.; Kurokawa, H.; Sugiyama, M.; Ando, R.; Funata, M.; Yoshida, N.; Homma, M.; Nishimura, T.; et al. Visualizing and Modulating Mitophagy for Therapeutic Studies of Neurodegeneration. Cell 2020, 181, 1176–1187.e1116. [Google Scholar] [CrossRef]
- Mary, A.; Eysert, F.; Checler, F.; Chami, M. Mitophagy in Alzheimer’s disease: Molecular defects and therapeutic approaches. Mol. Psychiatry 2023, 28, 202–216. [Google Scholar] [CrossRef]
- Akwa, Y.; Di Malta, C.; Zallo, F.; Gondard, E.; Lunati, A.; Diaz-de-Grenu, L.Z.; Zampelli, A.; Boiret, A.; Santamaria, S.; Martinez-Preciado, M.; et al. Stimulation of synaptic activity promotes TFEB-mediated clearance of pathological MAPT/Tau in cellular and mouse models of tauopathies. Autophagy 2023, 19, 660–677. [Google Scholar] [CrossRef]
- Ni, X.C.; Wang, H.F.; Cai, Y.Y.; Yang, D.; Alolga, R.N.; Liu, B.; Li, J.; Huang, F.Q. Ginsenoside Rb1 inhibits astrocyte activation and promotes transfer of astrocytic mitochondria to neurons against ischemic stroke. Redox Biol. 2022, 54, 102363. [Google Scholar] [CrossRef]
- Zhang, Q.; Song, Q.; Yu, R.; Wang, A.; Jiang, G.; Huang, Y.; Chen, J.; Xu, J.; Wang, D.; Chen, H.; et al. Nano-Brake Halts Mitochondrial Dysfunction Cascade to Alleviate Neuropathology and Rescue Alzheimer’s Cognitive Deficits. Adv. Sci. 2023, 10, e2204596. [Google Scholar] [CrossRef]
- Deng, D.; Cui, Y.; Gan, S.; Xie, Z.; Cui, S.; Cao, K.; Wang, S.; Shi, G.; Yang, L.; Bai, S.; et al. Sinisan alleviates depression-like behaviors by regulating mitochondrial function and synaptic plasticity in maternal separation rats. Phytomedicine 2022, 106, 154395. [Google Scholar] [CrossRef]
- Yang, Y.; Yu, L.; Zhu, T.; Xu, S.; He, J.; Mao, N.; Liu, Z.; Wang, D. Neuroprotective effects of Lycium barbarum polysaccharide on light-induced oxidative stress and mitochondrial damage via the Nrf2/HO-1 pathway in mouse hippocampal neurons. Int. J. Biol. Macromol. 2023, 251, 126315. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Arroum, T.; Luo, X.; Kang, R.; Lee, Y.J.; Tang, D.; Hüttemann, M.; Song, X. Diverse functions of cytochrome c in cell death and disease. Cell Death Differ. 2024, 31, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; Wang, S.; Yan, D.; Wu, J.; Zhang, Y.; Li, W.; Hu, J.; Liu, Z. The cGAS-STING pathway-dependent sensing of mitochondrial DNA mediates ocular surface inflammation. Signal Transduct. Target. Ther. 2023, 8, 371. [Google Scholar] [CrossRef]
- Petersen, O.H.; Gerasimenko, J.V.; Gerasimenko, O.V.; Gryshchenko, O.; Peng, S. The roles of calcium and ATP in the physiology and pathology of the exocrine pancreas. Physiol. Rev. 2021, 101, 1691–1744. [Google Scholar] [CrossRef]
- Lezmy, J.; Arancibia-Cárcamo, I.L.; Quintela-López, T.; Sherman, D.L.; Brophy, P.J.; Attwell, D. Astrocyte Ca(2+)-evoked ATP release regulates myelinated axon excitability and conduction speed. Science 2021, 374, eabh2858. [Google Scholar] [CrossRef]
- Huang, J.; He, J.; Wang, J.; Li, Y.; Xu, Z.; Zhang, L.; Kang, Y.; Xue, P. Calcium carbonate-actuated ion homeostasis perturbator for oxidative damage-augmented Ca(2+)/Mg(2+) interference therapy. Biomaterials 2023, 302, 122340. [Google Scholar] [CrossRef]
- Zhang, X.; Wei, M.; Fan, J.; Yan, W.; Zha, X.; Song, H.; Wan, R.; Yin, Y.; Wang, W. Ischemia-induced upregulation of autophagy preludes dysfunctional lysosomal storage and associated synaptic impairments in neurons. Autophagy 2021, 17, 1519–1542. [Google Scholar] [CrossRef]
- Jeong, S.J.; Stitham, J.; Evans, T.D.; Zhang, X.; Rodriguez-Velez, A.; Yeh, Y.S.; Tao, J.; Takabatake, K.; Epelman, S.; Lodhi, I.J.; et al. Trehalose causes low-grade lysosomal stress to activate TFEB and the autophagy-lysosome biogenesis response. Autophagy 2021, 17, 3740–3752. [Google Scholar] [CrossRef]
- Li, S.; Sheng, Z.H. Energy matters: Presynaptic metabolism and the maintenance of synaptic transmission. Nat. Rev. Neurosci. 2022, 23, 4–22. [Google Scholar] [CrossRef]
- Gebara, E.; Zanoletti, O.; Ghosal, S.; Grosse, J.; Schneider, B.L.; Knott, G.; Astori, S.; Sandi, C. Mitofusin-2 in the Nucleus Accumbens Regulates Anxiety and Depression-like Behaviors Through Mitochondrial and Neuronal Actions. Biol. Psychiatry 2021, 89, 1033–1044. [Google Scholar] [CrossRef]
- Ye, J.; Duan, C.; Han, J.; Chen, J.; Sun, N.; Li, Y.; Yuan, T.; Peng, D. Peripheral mitochondrial DNA as a neuroinflammatory biomarker for major depressive disorder. Neural Regen. Res. 2025, 20, 1541–1554. [Google Scholar] [CrossRef] [PubMed]
- D’Acunzo, P.; Argyrousi, E.K.; Ungania, J.M.; Kim, Y.; DeRosa, S.; Pawlik, M.; Goulbourne, C.N.; Arancio, O.; Levy, E. Mitovesicles secreted into the extracellular space of brains with mitochondrial dysfunction impair synaptic plasticity. Mol. Neurodegener. 2024, 19, 34. [Google Scholar] [CrossRef]
- Bai, R.; Guo, J.; Ye, X.Y.; Xie, Y.; Xie, T. Oxidative stress: The core pathogenesis and mechanism of Alzheimer’s disease. Ageing Res. Rev. 2022, 77, 101619. [Google Scholar] [CrossRef]
- Amadio, P.; Sandrini, L.; Zarà, M.; Barbieri, S.S.; Ieraci, A. NADPH-oxidases as potential pharmacological targets for thrombosis and depression comorbidity. Redox Biol. 2024, 70, 103060. [Google Scholar] [CrossRef]
- Chang, L.; Wei, Y.; Qu, Y.; Zhao, M.; Zhou, X.; Long, Y.; Hashimoto, K. Role of oxidative phosphorylation in the antidepressant effects of arketamine via the vagus nerve-dependent spleen-brain axis. Neurobiol. Dis. 2024, 199, 106573. [Google Scholar] [CrossRef]
- Al-Shami, A.S.; Haroun, M.; Essawy, A.E.; Moussa, N.; Abd Elkader, H.A.E. Early-life bisphenol A exposure causes detrimental age-related changes in anxiety, depression, learning, and memory in juvenile and adult male rats: Involvement of NMDAR/PSD-95-PTEN/AKT signaling pathway. Neurotoxicology 2025, 106, 17–36. [Google Scholar] [CrossRef]
- Hopfner, K.P.; Hornung, V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef]
- Di Bona, M.; Chen, Y.; Agustinus, A.S.; Mazzagatti, A.; Duran, M.A.; Deyell, M.; Bronder, D.; Hickling, J.; Hong, C.; Scipioni, L.; et al. Micronuclear collapse from oxidative damage. Science 2024, 385, eadj8691. [Google Scholar] [CrossRef]
- Gedam, M.; Comerota, M.M.; Propson, N.E.; Chen, T.; Jin, F.; Wang, M.C.; Zheng, H. Complement C3aR depletion reverses HIF-1α-induced metabolic impairment and enhances microglial response to Aβ pathology. J. Clin. Investig. 2023, 133, e167501. [Google Scholar] [CrossRef]
- Ward, D.M.; Cloonan, S.M. Mitochondrial Iron in Human Health and Disease. Annu. Rev. Physiol. 2019, 81, 453–482. [Google Scholar] [CrossRef]
- Zhang, S.; Xin, W.; Anderson, G.J.; Li, R.; Gao, L.; Chen, S.; Zhao, J.; Liu, S. Double-edge sword roles of iron in driving energy production versus instigating ferroptosis. Cell Death Dis. 2022, 13, 40. [Google Scholar] [CrossRef]
- Cheng, G.; Li, Z.; Liu, Y.; Ma, R.; Chen, X.; Liu, W.; Song, Y.; Zhang, Y.; Yu, G.; Wu, Z.; et al. “Swiss Army Knife” black phosphorus-based nanodelivery platform for synergistic antiparkinsonian therapy via remodeling the brain microenvironment. J. Control. Release 2023, 353, 752–766. [Google Scholar] [CrossRef]
- Liu, H.; Wang, S.; Wang, J.; Guo, X.; Song, Y.; Fu, K.; Gao, Z.; Liu, D.; He, W.; Yang, L.L. Energy metabolism in health and diseases. Signal Transduct. Target. Ther. 2025, 10, 69. [Google Scholar] [CrossRef]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef]
- Hao, J.; Chen, Q.; Feng, Y.; Jiang, Q.; Sun, H.; Deng, B.; Huang, X.; Guan, J.; Chen, Q.; Liu, X.; et al. Combination treatment with FAAH inhibitors/URB597 and ferroptosis inducers significantly decreases the growth and metastasis of renal cell carcinoma cells via the PI3K-AKT signaling pathway. Cell Death Dis. 2023, 14, 247. [Google Scholar] [CrossRef]
- Wei, F.L.; Wang, T.F.; Wang, C.L.; Zhang, Z.P.; Zhao, J.W.; Heng, W.; Tang, Z.; Du, M.R.; Yan, X.D.; Li, X.X.; et al. Cytoplasmic Escape of Mitochondrial DNA Mediated by Mfn2 Downregulation Promotes Microglial Activation via cGas-Sting Axis in Spinal Cord Injury. Adv. Sci. 2024, 11, e2305442. [Google Scholar] [CrossRef]
- Ko, M.S.; Yun, J.Y.; Baek, I.J.; Jang, J.E.; Hwang, J.J.; Lee, S.E.; Heo, S.H.; Bader, D.A.; Lee, C.H.; Han, J.; et al. Mitophagy deficiency increases NLRP3 to induce brown fat dysfunction in mice. Autophagy 2021, 17, 1205–1221. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, Y.; Guo, C.; Li, P.; Cheng, Z.; Zheng, L.; Sha, B.; Xu, H.; Su, X.; Wang, Y. Chronic stress dysregulates the Hippo/YAP/14-3-3η pathway and induces mitochondrial damage in basolateral amygdala in a mouse model of depression. Theranostics 2024, 14, 3653–3673. [Google Scholar] [CrossRef]
- Luo, M.; Ma, X.; Ye, J. Reductive stress-a common metabolic feature of obesity and cancer. Acta Pharm. Sin. B 2024, 14, 5181–5185. [Google Scholar] [CrossRef]
- Medeiros, H.C.D.; Lunt, S.Y. The Warburg effect: Saturation of mitochondrial NADH shuttles triggers aerobic lactate fermentation. Mol. Cell 2022, 82, 3119–3121. [Google Scholar] [CrossRef]
- Carrard, A.; Cassé, F.; Carron, C.; Burlet-Godinot, S.; Toni, N.; Magistretti, P.J.; Martin, J.L. Role of adult hippocampal neurogenesis in the antidepressant actions of lactate. Mol. Psychiatry 2021, 26, 6723–6735. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xiong, G.J.; Huang, N.; Sheng, Z.H. The cross-talk of energy sensing and mitochondrial anchoring sustains synaptic efficacy by maintaining presynaptic metabolism. Nat. Metab. 2020, 2, 1077–1095. [Google Scholar] [CrossRef] [PubMed]
- Smiles, W.J.; Ovens, A.J.; Kemp, B.E.; Galic, S.; Petersen, J.; Oakhill, J.S. New developments in AMPK and mTORC1 cross-talk. Essays Biochem. 2024, 68, 321–336. [Google Scholar] [CrossRef]
- Dong, W.T.; Long, L.H.; Deng, Q.; Liu, D.; Wang, J.L.; Wang, F.; Chen, J.G. Mitochondrial fission drives neuronal metabolic burden to promote stress susceptibility in male mice. Nat. Metab. 2023, 5, 2220–2236. [Google Scholar] [CrossRef]
- Jiang, M.; Wang, L.; Sheng, H. Mitochondria in depression: The dysfunction of mitochondrial energy metabolism and quality control systems. CNS Neurosci. Ther. 2024, 30, e14576. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, R.; Fan, J.; Chen, Y.; Wang, H.; Ge, Y.; Liang, H.; Li, W.; Liu, H.; Lv, Z.; et al. The role of ROS/p38 MAPK/NLRP3 inflammasome cascade in arsenic-induced depression-/anxiety-like behaviors of mice. Ecotoxicol. Environ. Saf. 2023, 261, 115111. [Google Scholar] [CrossRef]
- Ugbode, C.; Garnham, N.; Fort-Aznar, L.; Evans, G.J.O.; Chawla, S.; Sweeney, S.T. JNK signalling regulates antioxidant responses in neurons. Redox Biol. 2020, 37, 101712. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, X.; Wu, T.; Li, B.; Liu, T.; Wang, R.; Liu, Q.; Liu, Z.; Gong, Y.; Shao, C. Isoliensinine induces apoptosis in triple-negative human breast cancer cells through ROS generation and p38 MAPK/JNK activation. Sci. Rep. 2015, 5, 12579. [Google Scholar] [CrossRef]
- Chen, J.; Ren, Y.; Gui, C.; Zhao, M.; Wu, X.; Mao, K.; Li, W.; Zou, F. Phosphorylation of Parkin at serine 131 by p38 MAPK promotes mitochondrial dysfunction and neuronal death in mutant A53T α-synuclein model of Parkinson’s disease. Cell Death Dis. 2018, 9, 700. [Google Scholar] [CrossRef]
- Caruso, G.; Benatti, C.; Blom, J.M.C.; Caraci, F.; Tascedda, F. The Many Faces of Mitochondrial Dysfunction in Depression: From Pathology to Treatment. Front. Pharmacol. 2019, 10, 995. [Google Scholar] [CrossRef]
- Ding, Y.; Chen, X.; Liu, C.; Ge, W.; Wang, Q.; Hao, X.; Wang, M.; Chen, Y.; Zhang, Q. Identification of a small molecule as inducer of ferroptosis and apoptosis through ubiquitination of GPX4 in triple negative breast cancer cells. J. Hematol. Oncol. 2021, 14, 19. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liang, L.; Liu, S.; Yi, H.; Zhou, Y. FSP1: A key regulator of ferroptosis. Trends Mol. Med. 2023, 29, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Huynh, C.; Ryu, J.; Lee, J.; Inoki, A.; Inoki, K. Nutrient-sensing mTORC1 and AMPK pathways in chronic kidney diseases. Nat. Rev. Nephrol. 2023, 19, 102–122. [Google Scholar] [CrossRef]
- Zhang, J.; Hughes, R.N.; Kim, N.; Fallon, I.P.; Bakhurin, K.; Kim, J.; Severino, F.P.U.; Yin, H.H. A one-photon endoscope for simultaneous patterned optogenetic stimulation and calcium imaging in freely behaving mice. Nat. Biomed. Eng. 2023, 7, 499–510. [Google Scholar] [CrossRef]
- Simon, G.E. Adding mirtazapine to ongoing SNRIs or SSRIs did not improve symptoms of treatment-resistant depression. Ann. Intern. Med. 2019, 170, Jc20. [Google Scholar] [CrossRef]
- Rosa, A.C.; Bruni, N.; Meineri, G.; Corsi, D.; Cavi, N.; Gastaldi, D.; Dosio, F. Strategies to expand the therapeutic potential of superoxide dismutase by exploiting delivery approaches. Int. J. Biol. Macromol. 2021, 168, 846–865. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, W.; Li, C.; Li, L.; Yang, M.; Jiang, N.; Luo, S.; Xi, Y.; Liu, C.; Han, Y.; et al. DsbA-L interacting with catalase in peroxisome improves tubular oxidative damage in diabetic nephropathy. Redox Biol. 2023, 66, 102855. [Google Scholar] [CrossRef]
- Dikalova, A.E.; Pandey, A.; Xiao, L.; Arslanbaeva, L.; Sidorova, T.; Lopez, M.G.; Billings, F.T.t.; Verdin, E.; Auwerx, J.; Harrison, D.G.; et al. Mitochondrial Deacetylase Sirt3 Reduces Vascular Dysfunction and Hypertension While Sirt3 Depletion in Essential Hypertension Is Linked to Vascular Inflammation and Oxidative Stress. Circ. Res. 2020, 126, 439–452. [Google Scholar] [CrossRef]
- Zhang, J.; Xiang, H.; Liu, J.; Chen, Y.; He, R.R.; Liu, B. Mitochondrial Sirtuin 3: New emerging biological function and therapeutic target. Theranostics 2020, 10, 8315–8342. [Google Scholar] [CrossRef]
- Zhang, D.; Liu, X.; Xiao, Q.; Han, L.; Yang, J.; Li, X.; Xu, J.; Zheng, Q.; Ma, J.; Chen, J.; et al. Co-Exposure to Bisphenols, Parabens, and Antimicrobials and Association with Coronary Heart Disease: Oxidative Stress as a Potential Mediating Factor? Environ. Sci. Technol. 2023, 57, 531–538. [Google Scholar] [CrossRef]
- Zhou, Q.; Rizzo, S.; Oetjen, J.; Fülöp, A.; Rittner, M.; Gillandt, H.; Hopf, C. A Caged In-Source Laser-Cleavable MALDI Matrix with High Vacuum Stability for Extended MALDI-MS Imaging. Angew. Chem. 2023, 62, e202217047. [Google Scholar] [CrossRef] [PubMed]
- Qiu, P.; Jiang, J.; Liu, Z.; Cai, Y.; Huang, T.; Wang, Y.; Liu, Q.; Nie, Y.; Liu, F.; Cheng, J.; et al. BMAL1 knockout macaque monkeys display reduced sleep and psychiatric disorders. Natl. Sci. Rev. 2019, 6, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Vermilyea, S.C.; Zammit, M.; Lu, J.; Olsen, M.; Metzger, J.M.; Yao, L.; Chen, Y.; Phillips, S.; Holden, J.E.; et al. Autologous transplant therapy alleviates motor and depressive behaviors in parkinsonian monkeys. Nat. Med. 2021, 27, 632–639. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Luo, Q.; Zhao, Y.; Ren, P.; Jin, Y.; Zhou, J. The Ferroptosis–Mitochondrial Axis in Depression: Unraveling the Feedforward Loop of Oxidative Stress, Metabolic Homeostasis Dysregulation, and Neuroinflammation. Antioxidants 2025, 14, 613. https://doi.org/10.3390/antiox14050613
Liu X, Luo Q, Zhao Y, Ren P, Jin Y, Zhou J. The Ferroptosis–Mitochondrial Axis in Depression: Unraveling the Feedforward Loop of Oxidative Stress, Metabolic Homeostasis Dysregulation, and Neuroinflammation. Antioxidants. 2025; 14(5):613. https://doi.org/10.3390/antiox14050613
Chicago/Turabian StyleLiu, Xu, Qiang Luo, Yulong Zhao, Peng Ren, Yu Jin, and Junjie Zhou. 2025. "The Ferroptosis–Mitochondrial Axis in Depression: Unraveling the Feedforward Loop of Oxidative Stress, Metabolic Homeostasis Dysregulation, and Neuroinflammation" Antioxidants 14, no. 5: 613. https://doi.org/10.3390/antiox14050613
APA StyleLiu, X., Luo, Q., Zhao, Y., Ren, P., Jin, Y., & Zhou, J. (2025). The Ferroptosis–Mitochondrial Axis in Depression: Unraveling the Feedforward Loop of Oxidative Stress, Metabolic Homeostasis Dysregulation, and Neuroinflammation. Antioxidants, 14(5), 613. https://doi.org/10.3390/antiox14050613