Oral Sulforaphane Intervention Protects Against Diabetic Cardiomyopathy in db/db Mice: Focus on Cardiac Lipotoxicity and Substrate Metabolism


Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. SFN-I Improves Systemic Metabolism but Not the Three Common Diabetic Symptoms in db/db Mice
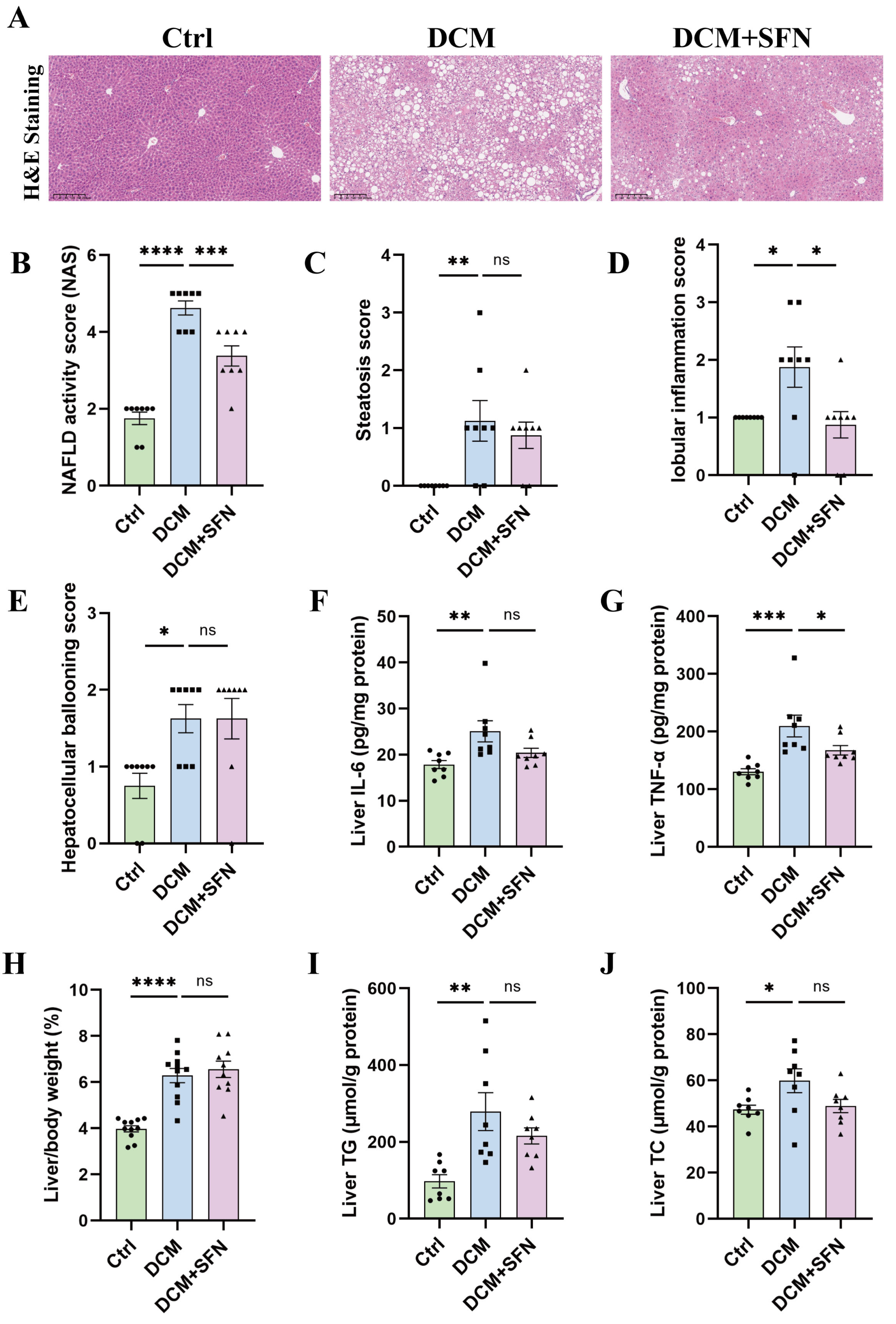
3.2. SFN-I Reduces Liver Inflammation Without Significantly Affecting Liver Lipid Deposition in db/db Mice
3.3. SFN-I Attenuates Diastolic Dysfunction in db/db Mice
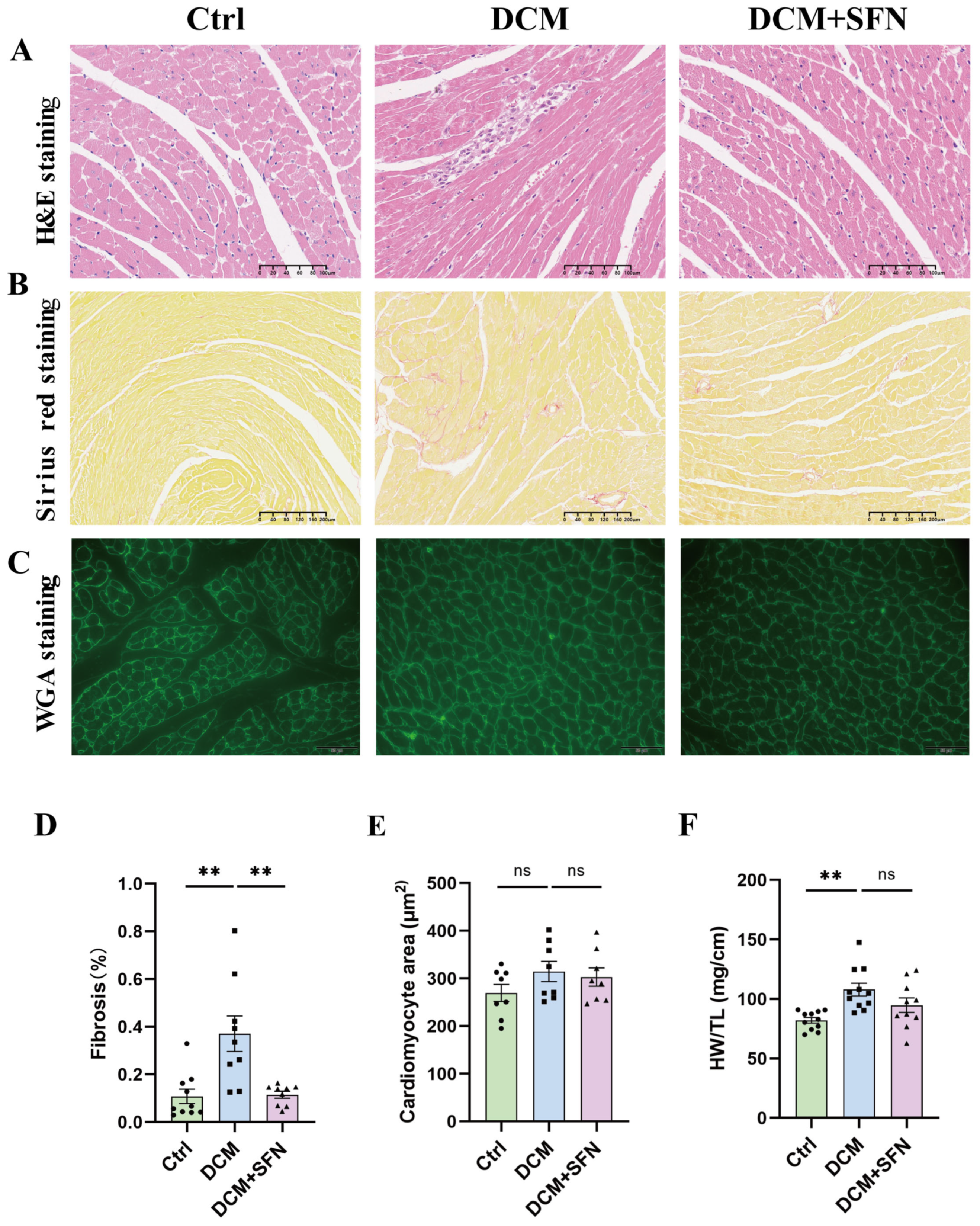
3.4. SFN-I Mitigates Cardiac Injury and Fibrosis in db/db Mice
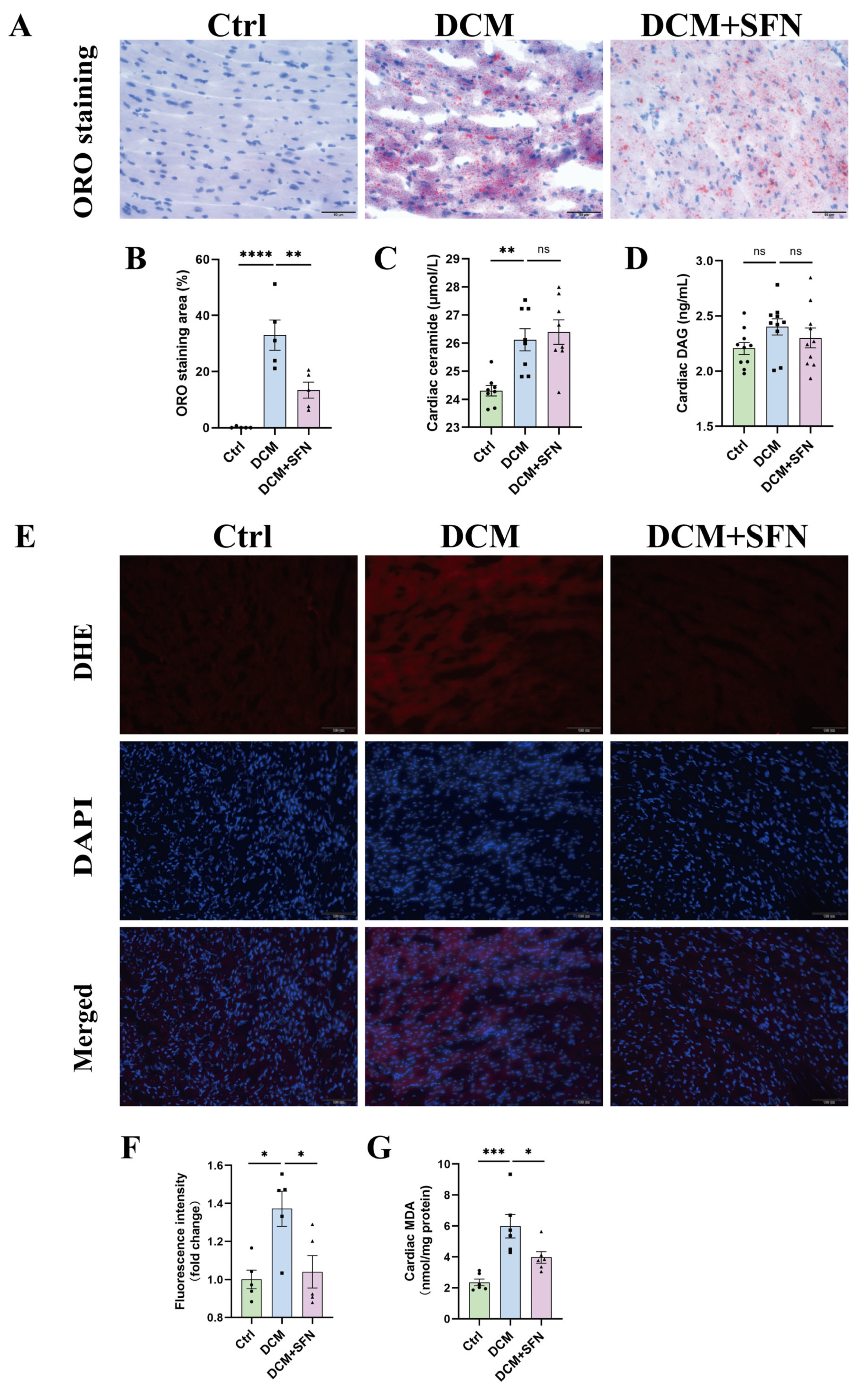
3.5. SFN-I Reduces Cardiac Lipid Accumulation and Oxidative Stress Without Significantly Altering Ceramide and DAG Levels in db/db Mice
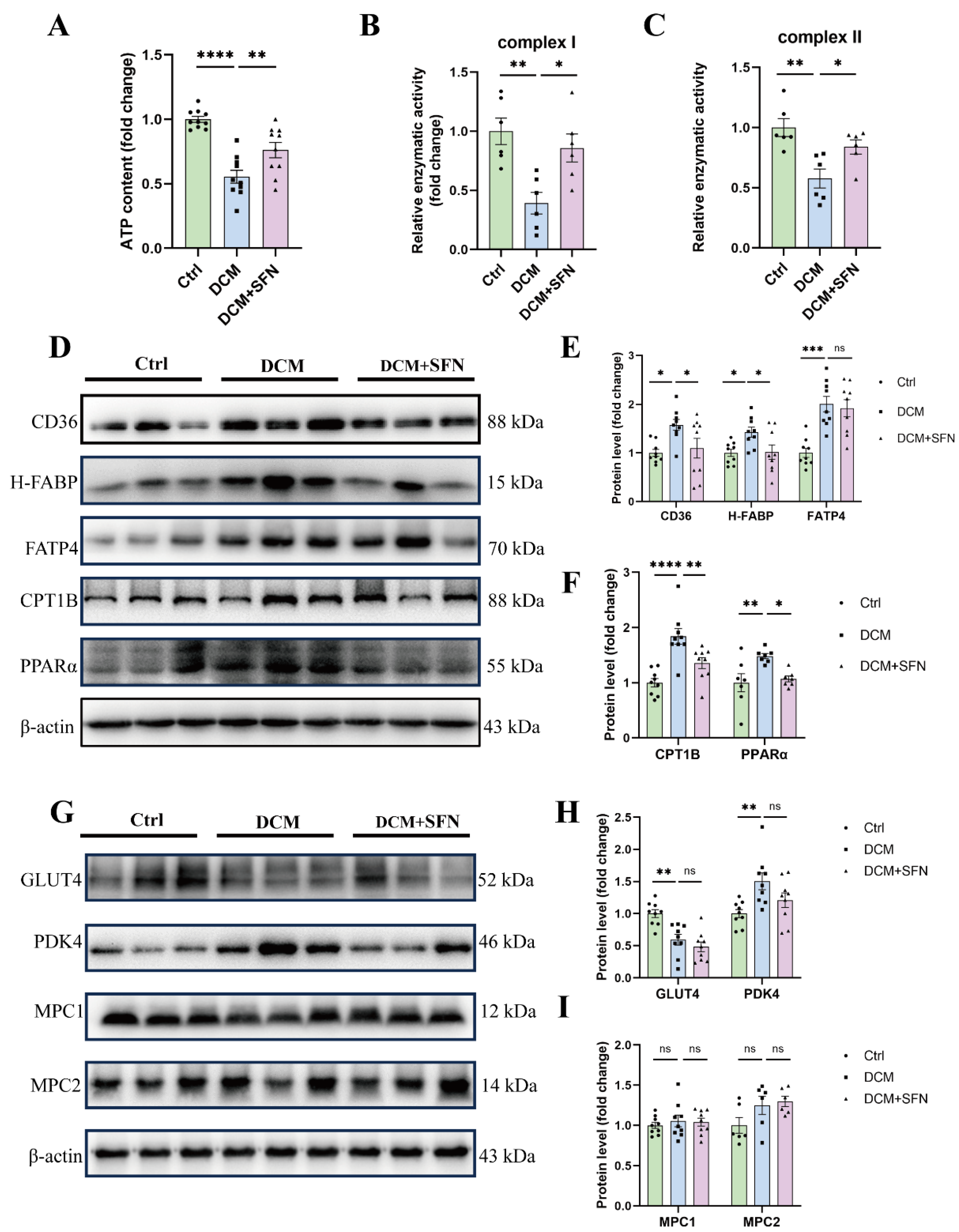
3.6. SFN-I Improves Cardiac Mitochondrial Function and Modulates Key Regulators of FA Metabolism, Rather than Glucose Metabolism, in db/db Mice
4. Discussion
Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SFN | Sulforaphane |
| SFN-I | Sulforaphane intervention |
| ITC | Isothiocyanate |
| DCM | Diabetic cardiomyopathy |
| T2DM | Type 2 diabetes mellitus |
| WT | Wild-type |
| MAFLD | Metabolic dysfunction-associated fatty liver disease |
| NAFLD | Nonalcoholic fatty liver disease |
| CMD | Cardiometabolic diseases |
| NAS | Nonalcoholic fatty liver disease activity score |
| DM | Diabetes mellitus |
| HF | Heart failure |
| IR | Insulin resistance |
| HOMA-IR | Homeostasis model assessment of insulin resistance |
| ER | Endoplasmic reticulum |
| MC | Metabolic cardiomyopathy |
| FA | Fatty acid |
| CD36 | Cluster of differentiation 36 |
| H-FABP | Heart-type fatty acid binding protein |
| FATP4 | Fatty acid transport protein 4 |
| CPT1B | Carnitine palmitoyltransferase 1B |
| PPARα | Peroxisome proliferator-activated receptor alpha |
| PDK4 | Pyruvate dehydrogenase kinase 4 |
| GLUT4 | Glucose transporter type 4 |
| MPC1 | Mitochondrial pyruvate carrier 1 |
| MPC2 | Mitochondrial pyruvate carrier 2 |
| PDH | Pyruvate dehydrogenase |
| FAO | Fatty acid beta oxidation |
| FGF21 | Fibroblast growth factor 21 |
| WAT | White adipose tissue |
| Nrf2 | Nuclear factor E2-related factor 2 |
| AMPK | AMP-activated protein kinase |
| BSE | Broccoli Sprout Extract |
| DAG | Diacylglycerol |
| ROS | Reactive oxygen species |
| ATP | Adenosine triphosphate |
| EC | Extracellular matrix |
| CVDs | Cardiovascular diseases |
| LV | Left ventricle |
| HFpEF | HF with preserved EF |
| HFrEF | HF with reduced EF |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator-1 alpha |
| ETC | Electron transport chain |
| HFD | High-fat diet |
| FFAs | Free fatty acids |
| TC | Total cholesterol |
| TG | Triglyceride |
| HDL-C | High-density lipoprotein-cholesterol |
| LDL-C | Low-density lipoprotein-cholesterol |
| IL-6 | Interleukin-6 |
| TNF-α | Tumor necrosis factor Alpha |
| MDA | Malondialdehyde |
| INS | Insulin |
| WGA | Wheat germ agglutinin |
| H&E | Hematoxylin and eosin |
| DHE | Dihydroethidium |
| HW/TL | Heart weight to tibia length |
| BW | Body weight |
| OCT | Optimum cutting temperature |
| ELISA | Enzyme-linked immunosorbent assay |
| HR | Heart rate |
| SBP | Systolic blood pressure |
| DBP | Diastolic blood pressure |
| MBP | Mean blood pressure |
| LVIDs/d | Left ventricular internal diameter at end-systole and end-diastole |
| LVAWs/d | Left ventricular anterior wall thickness at end-systole and end-diastole |
| LVPWs/d | Left ventricular posterior wall thickness at end-systole and end-diastole |
| EF | Ejection fraction |
| FS | Fractional shortening |
| E/A | Early-to-atrial filling ratio |
| AET | Aortic ejection time |
| IVCT | Isovolumic contraction time |
| IVRT | Isovolumic relaxation time |
| MPI | Myocardial performance index |
References
- Ritchie, R.H.; Abel, E.D. Basic Mechanisms of Diabetic Heart Disease. Circ. Res. 2020, 126, 1501–1525. [Google Scholar] [CrossRef]
- Tan, Y.; Zhang, Z.; Zheng, C.; Wintergerst, K.A.; Keller, B.B.; Cai, L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: Preclinical and clinical evidence. Nat. Rev. Cardiol. 2020, 17, 585–607. [Google Scholar] [CrossRef]
- Seferović, P.M.; Paulus, W.J. Clinical diabetic cardiomyopathy: A two-faced disease with restrictive and dilated phenotypes. Eur. Heart J. 2015, 36, 1718–1727c. [Google Scholar] [CrossRef] [PubMed]
- van de Weijer, T.; Schrauwen-Hinderling, V.B.; Schrauwen, P. Lipotoxicity in type 2 diabetic cardiomyopathy. Cardiovasc. Res. 2011, 92, 10–18. [Google Scholar] [CrossRef]
- Shu, H.; Peng, Y.; Hang, W.; Nie, J.; Zhou, N.; Wang, D.W. The role of CD36 in cardiovascular disease. Cardiovasc. Res. 2022, 118, 115–129. [Google Scholar] [CrossRef]
- Schwenk, R.W.; Luiken, J.J.; Bonen, A.; Glatz, J.F. Regulation of sarcolemmal glucose and fatty acid transporters in cardiac disease. Cardiovasc. Res. 2008, 79, 249–258. [Google Scholar] [CrossRef]
- Ritterhoff, J.; Tian, R. Metabolism in cardiomyopathy: Every substrate matters. Cardiovasc. Res. 2017, 113, 411–421. [Google Scholar] [CrossRef]
- Schulze, P.C.; Drosatos, K.; Goldberg, I.J. Lipid Use and Misuse by the Heart. Circ. Res. 2016, 118, 1736–1751. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef]
- Gibb, A.A.; Hill, B.G. Metabolic Coordination of Physiological and Pathological Cardiac Remodeling. Circ. Res. 2018, 123, 107–128. [Google Scholar] [CrossRef]
- Shao, D.; Tian, R. Glucose Transporters in Cardiac Metabolism and Hypertrophy. Compr. Physiol. 2015, 6, 331–351. [Google Scholar] [CrossRef]
- Koliaki, C.; Roden, M. Alterations of Mitochondrial Function and Insulin Sensitivity in Human Obesity and Diabetes Mellitus. Annu. Rev. Nutr. 2016, 36, 337–367. [Google Scholar] [CrossRef]
- Bozi, L.H.M.; Campos, J.C.; Zambelli, V.O.; Ferreira, N.D.; Ferreira, J.C.B. Mitochondrially-targeted treatment strategies. Mol. Asp. Med. 2020, 71, 100836. [Google Scholar] [CrossRef]
- Gollmer, J.; Zirlik, A.; Bugger, H. Mitochondrial Mechanisms in Diabetic Cardiomyopathy. Diabetes Metab. J. 2020, 44, 33–53. [Google Scholar] [CrossRef]
- Taylor, E.B. Functional Properties of the Mitochondrial Carrier System. Trends Cell Biol. 2017, 27, 633–644. [Google Scholar] [CrossRef]
- Zangari, J.; Petrelli, F.; Maillot, B.; Martinou, J.C. The Multifaceted Pyruvate Metabolism: Role of the Mitochondrial Pyruvate Carrier. Biomolecules 2020, 10, 1068. [Google Scholar] [CrossRef]
- Judge, A.; Dodd, M.S. Metabolism. Essays Biochem. 2020, 64, 607–647. [Google Scholar] [CrossRef]
- Montaigne, D.; Butruille, L.; Staels, B. PPAR control of metabolism and cardiovascular functions. Nat. Rev. Cardiol. 2021, 18, 809–823. [Google Scholar] [CrossRef]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; De Bosscher, K. Molecular Actions of PPARα in Lipid Metabolism and Inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef]
- Yarmohammadi, F.; Hayes, A.W.; Karimi, G. Natural and chemical compounds as protective agents against cardiac lipotoxicity. Biomed. Pharmacother. 2022, 145, 112413. [Google Scholar] [CrossRef]
- Axelsson, A.S.; Tubbs, E.; Mecham, B.; Chacko, S.; Nenonen, H.A.; Tang, Y.; Fahey, J.W.; Derry, J.M.J.; Wollheim, C.B.; Wierup, N.; et al. Sulforaphane reduces hepatic glucose production and improves glucose control in patients with type 2 diabetes. Sci. Transl. Med. 2017, 9, eaah4477. [Google Scholar] [CrossRef] [PubMed]
- Bahadoran, Z.; Tohidi, M.; Nazeri, P.; Mehran, M.; Azizi, F.; Mirmiran, P. Effect of broccoli sprouts on insulin resistance in type 2 diabetic patients: A randomized double-blind clinical trial. Int. J. Food Sci. Nutr. 2012, 63, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Bahadoran, Z.; Mirmiran, P.; Hosseinpanah, F.; Rajab, A.; Asghari, G.; Azizi, F. Broccoli sprouts powder could improve serum triglyceride and oxidized LDL/LDL-cholesterol ratio in type 2 diabetic patients: A randomized double-blind placebo-controlled clinical trial. Diabetes Res. Clin. Pract. 2012, 96, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Lei, Y.; Zhao, F.; Che, J.; Wu, Y.; Lei, P.; Kang, Y.E.; Shan, Y. Improving insulin resistance by sulforaphane via activating the Bacteroides and Lactobacillus SCFAs-GPR-GLP1 signal axis. Food Funct. 2024, 15, 8644–8660. [Google Scholar] [CrossRef]
- López-Chillón, M.T.; Carazo-Díaz, C.; Prieto-Merino, D.; Zafrilla, P.; Moreno, D.A.; Villaño, D. Effects of long-term consumption of broccoli sprouts on inflammatory markers in overweight subjects. Clin. Nutr. 2019, 38, 745–752. [Google Scholar] [CrossRef]
- Kikuchi, M.; Ushida, Y.; Shiozawa, H.; Umeda, R.; Tsuruya, K.; Aoki, Y.; Suganuma, H.; Nishizaki, Y. Sulforaphane-rich broccoli sprout extract improves hepatic abnormalities in male subjects. World J. Gastroenterol. 2015, 21, 12457–12467. [Google Scholar] [CrossRef]
- Armah, C.N.; Derdemezis, C.; Traka, M.H.; Dainty, J.R.; Doleman, J.F.; Saha, S.; Leung, W.; Potter, J.F.; Lovegrove, J.A.; Mithen, R.F. Diet rich in high glucoraphanin broccoli reduces plasma LDL cholesterol: Evidence from randomised controlled trials. Mol. Nutr. Food Res. 2015, 59, 918–926. [Google Scholar] [CrossRef]
- Baralić, K.; Živanović, J.; Marić, Đ.; Bozic, D.; Grahovac, L.; Antonijević Miljaković, E.; Ćurčić, M.; Buha Djordjevic, A.; Bulat, Z.; Antonijević, B.; et al. Sulforaphane-A Compound with Potential Health Benefits for Disease Prevention and Treatment: Insights from Pharmacological and Toxicological Experimental Studies. Antioxidants 2024, 13, 147. [Google Scholar] [CrossRef]
- Wang, Y.; He, X.; Cheng, N.; Huang, K. Unveiling the Nutritional Veil of Sulforaphane: With a Major Focus on Glucose Homeostasis Modulation. Nutrients 2024, 16, 1877. [Google Scholar] [CrossRef]
- Martins, T.; Colaço, B.; Venâncio, C.; Pires, M.J.; Oliveira, P.A.; Rosa, E.; Antunes, L.M. Potential effects of sulforaphane to fight obesity. J. Sci. Food Agric. 2018, 98, 2837–2844. [Google Scholar] [CrossRef]
- Mthembu, S.X.H.; Mazibuko-Mbeje, S.E.; Moetlediwa, M.T.; Muvhulawa, N.; Silvestri, S.; Orlando, P.; Nkambule, B.B.; Muller, C.J.F.; Ndwandwe, D.; Basson, A.K.; et al. Sulforaphane: A nutraceutical against diabetes-related complications. Pharmacol. Res. 2023, 196, 106918. [Google Scholar] [CrossRef] [PubMed]
- Waterman, C.; Rojas-Silva, P.; Tumer, T.B.; Kuhn, P.; Richard, A.J.; Wicks, S.; Stephens, J.M.; Wang, Z.; Mynatt, R.; Cefalu, W.; et al. Isothiocyanate-rich Moringa oleifera extract reduces weight gain, insulin resistance, and hepatic gluconeogenesis in mice. Mol. Nutr. Food Res. 2015, 59, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Lee, H.; Im, S.W.; Jung, C.H.; Ha, T.Y. Allyl isothiocyanate ameliorates insulin resistance through the regulation of mitochondrial function. J. Nutr. Biochem. 2014, 25, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Nagata, N.; Xu, L.; Kohno, S.; Ushida, Y.; Aoki, Y.; Umeda, R.; Fuke, N.; Zhuge, F.; Ni, Y.; Nagashimada, M.; et al. Glucoraphanin Ameliorates Obesity and Insulin Resistance Through Adipose Tissue Browning and Reduction of Metabolic Endotoxemia in Mice. Diabetes 2017, 66, 1222–1236. [Google Scholar] [CrossRef]
- Tian, S.; Lei, P.; Teng, C.; Sun, Y.; Song, X.; Li, B.; Shan, Y. Targeting PLIN2/PLIN5-PPARγ: Sulforaphane Disturbs the Maturation of Lipid Droplets. Mol. Nutr. Food Res. 2019, 63, e1900183. [Google Scholar] [CrossRef]
- Wu, Y.K.; Ren, Z.N.; Zhu, S.L.; Wu, Y.Z.; Wang, G.; Zhang, H.; Chen, W.; He, Z.; Ye, X.L.; Zhai, Q.X. Sulforaphane ameliorates non-alcoholic fatty liver disease in mice by promoting FGF21/FGFR1 signaling pathway. Acta Pharmacol. Sin. 2022, 43, 1473–1483. [Google Scholar] [CrossRef]
- Lei, P.; Hu, Y.; Gao, P.; Ding, Q.; Yan, J.; Zhao, J.; Li, B.; Shan, Y. Sulforaphane Ameliorates Hepatic Lipid Metabolism via Modulating Lipophagy In Vivo and In Vitro. J. Agric. Food Chem. 2022, 70, 15126–15133. [Google Scholar] [CrossRef]
- Savic, N.; Markelic, M.; Stancic, A.; Velickovic, K.; Grigorov, I.; Vucetic, M.; Martinovic, V.; Gudelj, A.; Otasevic, V. Sulforaphane prevents diabetes-induced hepatic ferroptosis by activating Nrf2 signaling axis. Biofactors. 2024, 50, 810–827. [Google Scholar] [CrossRef]
- Ma, S.; Pang, X.; Tian, S.; Sun, J.; Hu, Q.; Li, X.; Lu, Y. The protective effects of sulforaphane on high-fat diet-induced metabolic associated fatty liver disease in mice via mediating the FXR/LXRα pathway. Food Funct. 2022, 13, 12966–12982. [Google Scholar] [CrossRef]
- Tian, S.; Wang, Y.; Li, X.; Liu, J.; Wang, J.; Lu, Y. Sulforaphane Regulates Glucose and Lipid Metabolisms in Obese Mice by Restraining JNK and Activating Insulin and FGF21 Signal Pathways. J. Agric. Food Chem. 2021, 69, 13066–13079. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, Q.; Liu, J.; Zhang, Z.; Ma, X.; Zhang, Y.; Zhu, J.; Thring, R.W.; Wu, M.; Gao, Y.; et al. Sulforaphane alleviates high fat diet-induced insulin resistance via AMPK/Nrf2/GPx4 axis. Biomed. Pharmacother. 2022, 152, 113273. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Sun, S.; Liang, L.; Lou, C.; He, Q.; Ran, M.; Zhang, L.; Zhang, J.; Yan, C.; Yuan, H.; et al. Role of the Aryl Hydrocarbon Receptor and Gut Microbiota-Derived Metabolites Indole-3-Acetic Acid in Sulforaphane Alleviates Hepatic Steatosis in Mice. Front. Nutr. 2021, 8, 756565. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Q.; Chen, S.Y.; Wang, A.S.; Yao, A.J.; Fu, J.F.; Zhao, J.S.; Chen, F.; Zou, Z.Q.; Zhang, X.H.; Shan, Y.J.; et al. Sulforaphane induces adipocyte browning and promotes glucose and lipid utilization. Mol. Nutr. Food Res. 2016, 60, 2185–2197. [Google Scholar] [CrossRef]
- Liu, Y.; Fu, X.; Chen, Z.; Luo, T.; Zhu, C.; Ji, Y.; Bian, Z. The Protective Effects of Sulforaphane on High-Fat Diet-Induced Obesity in Mice Through Browning of White Fat. Front. Pharmacol. 2021, 12, 665894. [Google Scholar] [CrossRef]
- Men, X.; Han, X.; Lee, S.J.; Oh, G.; Park, K.T.; Han, J.K.; Choi, S.I.; Lee, O.H. Anti-Obesogenic Effects of Sulforaphane-Rich Broccoli (Brassica oleracea var. italica) Sprouts and Myrosinase-Rich Mustard (Sinapis alba L.) Seeds In Vitro and In Vivo. Nutrients 2022, 14, 3814. [Google Scholar] [CrossRef]
- Choi, K.M.; Lee, Y.S.; Kim, W.; Kim, S.J.; Shin, K.O.; Yu, J.Y.; Lee, M.K.; Lee, Y.M.; Hong, J.T.; Yun, Y.P.; et al. Sulforaphane attenuates obesity by inhibiting adipogenesis and activating the AMPK pathway in obese mice. J. Nutr. Biochem. 2014, 25, 201–207. [Google Scholar] [CrossRef]
- Piragine, E.; Flori, L.; Di Cesare Mannelli, L.; Ghelardini, C.; Pagnotta, E.; Matteo, R.; Lazzeri, L.; Martelli, A.; Miragliotta, V.; Pirone, A.; et al. Eruca sativa Mill. seed extract promotes anti-obesity and hypoglycemic effects in mice fed with a high-fat diet. Phytother. Res. 2021, 35, 1983–1990. [Google Scholar] [CrossRef]
- Bai, Y.; Cui, W.; Xin, Y.; Miao, X.; Barati, M.T.; Zhang, C.; Chen, Q.; Tan, Y.; Cui, T.; Zheng, Y.; et al. Prevention by sulforaphane of diabetic cardiomyopathy is associated with up-regulation of Nrf2 expression and transcription activation. J. Mol. Cell Cardiol. 2013, 57, 82–95. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, S.; Zhou, S.; Yan, X.; Wang, Y.; Chen, J.; Mellen, N.; Kong, M.; Gu, J.; Tan, Y.; et al. Sulforaphane prevents the development of cardiomyopathy in type 2 diabetic mice probably by reversing oxidative stress-induced inhibition of LKB1/AMPK pathway. J. Mol. Cell Cardiol. 2014, 77, 42–52. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, S.; Ji, H.; Zhang, Z.; Chen, J.; Tan, Y.; Wintergerst, K.; Zheng, Y.; Sun, J.; Cai, L. Broccoli sprout extract prevents diabetic cardiomyopathy via Nrf2 activation in db/db T2DM mice. Sci. Rep. 2016, 6, 30252. [Google Scholar] [CrossRef]
- Gu, J.; Cheng, Y.; Wu, H.; Kong, L.; Wang, S.; Xu, Z.; Zhang, Z.; Tan, Y.; Keller, B.B.; Zhou, H.; et al. Metallothionein Is Downstream of Nrf2 and Partially Mediates Sulforaphane Prevention of Diabetic Cardiomyopathy. Diabetes 2017, 66, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, S.; Wang, W.; Chen, J.; Zhang, Z.; Zheng, Q.; Liu, Q.; Cai, L. Protection against diabetic cardiomyopathy is achieved using a combination of sulforaphane and zinc in type 1 diabetic OVE26 mice. J. Cell Mol. Med. 2019, 23, 6319–6330. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhou, S.; Guo, H.; Zhang, J.; Ma, T.; Zheng, Y.; Zhang, Z.; Cai, L. Protective effects of sulforaphane on type 2 diabetes-induced cardiomyopathy via AMPK-mediated activation of lipid metabolic pathways and NRF2 function. Metabolism 2020, 102, 154002. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, X.; Zhou, W.; Men, H.; Bao, T.; Sun, Y.; Wang, Q.; Tan, Y.; Keller, B.B.; Tong, Q.; et al. Ferroptosis is essential for diabetic cardiomyopathy and is prevented by sulforaphane via AMPK/NRF2 pathways. Acta Pharm. Sin. B. 2022, 12, 708–722. [Google Scholar] [CrossRef]
- de Souza, C.G.; da Motta, L.L.; de Assis, A.M.; Rech, A.; Bruch, R.; Klamt, F.; Souza, D.O. Sulforaphane ameliorates the insulin responsiveness and the lipid profile but does not alter the antioxidant response in diabetic rats. Food Funct. 2016, 7, 2060–2065. [Google Scholar] [CrossRef]
- Waterman, C.; Graham, J.L.; Arnold, C.D.; Stanhope, K.L.; Tong, J.H.; Jaja-Chimedza, A.; Havel, P.J. Moringa Isothiocyanate-rich Seed Extract Delays the Onset of Diabetes in UC Davis Type-2 Diabetes Mellitus Rats. Sci. Rep. 2020, 10, 8861. [Google Scholar] [CrossRef]
- Souza, C.G.; Riboldi, B.P.; Hansen, F.; Moreira, J.D.; Souza, D.G.; de Assis, A.M.; Brum, L.M.; Perry, M.L.; Souza, D.O. Chronic sulforaphane oral treatment accentuates blood glucose impairment and may affect GLUT3 expression in the cerebral cortex and hypothalamus of rats fed with a highly palatable diet. Food Funct. 2013, 4, 1271–1276. [Google Scholar] [CrossRef]
- Wang, M.; Pu, D.; Zhao, Y.; Chen, J.; Zhu, S.; Lu, A.; Liao, Z.; Sun, Y.; Xiao, Q. Sulforaphane protects against skeletal muscle dysfunction in spontaneous type 2 diabetic db/db mice. Life Sci. 2020, 255, 117823. [Google Scholar] [CrossRef]
- Çakır, I.; Lining Pan, P.; Hadley, C.K.; El-Gamal, A.; Fadel, A.; Elsayegh, D.; Mohamed, O.; Rizk, N.M.; Ghamari-Langroudi, M. Sulforaphane reduces obesity by reversing leptin resistance. eLife 2022, 11, e67368. [Google Scholar] [CrossRef]
- Tian, S.; Li, X.; Wang, Y.; Lu, Y. The protective effect of sulforaphane on type II diabetes induced by high-fat diet and low-dosage streptozotocin. Food Sci. Nutr. 2020, 9, 747–756. [Google Scholar] [CrossRef]
- Teng, W.; Li, Y.; Du, M.; Lei, X.; Xie, S.; Ren, F. Sulforaphane Prevents Hepatic Insulin Resistance by Blocking Serine Palmitoyltransferase 3-Mediated Ceramide Biosynthesis. Nutrients 2019, 11, 1185. [Google Scholar] [CrossRef]
- Li, J.; Xie, S.; Teng, W. Sulforaphane Attenuates Nonalcoholic Fatty Liver Disease by Inhibiting Hepatic Steatosis and Apoptosis. Nutrients 2021, 14, 76. [Google Scholar] [CrossRef]
- Wang, G.; Nie, J.H.; Bao, Y.; Yang, X. Sulforaphane Rescues Ethanol-Suppressed Angiogenesis through Oxidative and Endoplasmic Reticulum Stress in Chick Embryos. J. Agric. Food Chem. 2018, 66, 9522–9533, Erratum in J. Agric. Food Chem. 2018, 66, 10662. [Google Scholar] [CrossRef]
- Yan, X.; Shen, Z.; Yu, D.; Zhao, C.; Zou, H.; Ma, B.; Dong, W.; Chen, W.; Huang, D.; Yu, Z. Nrf2 contributes to the benefits of exercise interventions on age-related skeletal muscle disorder via regulating Drp1 stability and mitochondrial fission. Free Radic. Biol. Med. 2022, 178, 59–75. [Google Scholar] [CrossRef]
- Singh, P.; Sharma, R.; McElhanon, K.; Allen, C.D.; Megyesi, J.K.; Beneš, H.; Singh, S.P. Sulforaphane protects the heart from doxorubicin-induced toxicity. Free Radic. Biol. Med. 2015, 86, 90–101. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88 Pt B, 179–188. [Google Scholar] [CrossRef]
- Lei, P.; Tian, S.; Teng, C.; Huang, L.; Liu, X.; Wang, J.; Zhang, Y.; Li, B.; Shan, Y. Sulforaphane Improves Lipid Metabolism by Enhancing Mitochondrial Function and Biogenesis In Vivo and In Vitro. Mol. Nutr. Food Res. 2021, 65, e2170023. [Google Scholar] [CrossRef]
- Tian, S.; Lei, P.; Zhang, J.; Sun, Y.; Li, B.; Shan, Y. Sulforaphane Balances Ca2+ Homeostasis Injured by Excessive Fat via Mitochondria-Associated Membrane (MAM). Mol. Nutr. Food Res. 2021, 65, e2001076. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, F.; Zhang, Y.; Zheng, X.; Liu, S.; Tang, M.; Wang, Z.; Wang, P.; Bao, Y.; Li, D. Biphasic effect of sulforaphane on angiogenesis in hypoxia via modulation of both Nrf2 and mitochondrial dynamics. Food Funct. 2022, 13, 2884–2898. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, Y.; Zheng, X.; Wang, Z.; Wang, P.; Zhang, M.; Shen, M.; Bao, Y.; Li, D. Sulforaphane Inhibits Foam Cell Formation and Atherosclerosis via Mechanisms Involving the Modulation of Macrophage Cholesterol Transport and the Related Phenotype. Nutrients 2023, 15, 2117. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Tapia, E.; Sánchez-Lozada, L.G.; García-Arroyo, F.E.; Amador-Martínez, I.; Orozco-Ibarra, M.; Fernández-Valverde, F.; Pedraza-Chaverri, J. Sulforaphane Protects against Unilateral Ureteral Obstruction-Induced Renal Damage in Rats by Alleviating Mitochondrial and Lipid Metabolism Impairment. Antioxid 2022, 11, 1854. [Google Scholar] [CrossRef]
- Xu, J.; Kulkarni, S.R.; Donepudi, A.C.; More, V.R.; Slitt, A.L. Enhanced Nrf2 activity worsens insulin resistance, impairs lipid accumulation in adipose tissue, and increases hepatic steatosis in leptin-deficient mice. Diabetes 2012, 61, 3208–3218. [Google Scholar] [CrossRef]
- Li, X.; Wu, Y.; Zhao, J.; Wang, H.; Tan, J.; Yang, M.; Li, Y.; Deng, S.; Gao, S.; Li, H.; et al. Distinct cardiac energy metabolism and oxidative stress adaptations between obese and non-obese type 2 diabetes mellitus. Theranostics 2020, 10, 2675–2695. [Google Scholar] [CrossRef]
- Chuang, W.T.; Yen, C.C.; Huang, C.S.; Chen, H.W.; Lii, C.K. Benzyl Isothiocyanate Ameliorates High-Fat Diet-Induced Hyperglycemia by Enhancing Nrf2-Dependent Antioxidant Defense-Mediated IRS-1/AKT/TBC1D1 Signaling and GLUT4 Expression in Skeletal Muscle. J. Agric. Food Chem. 2020, 68, 15228–15238. [Google Scholar] [CrossRef]
- Chiba, M.; Ito, Y.; Nagasawa, T. Phenethyl isothiocyanate stimulates glucose uptake through the Akt pathway in C2C12 myotubes. Biosci. Biotechnol. Biochem. 2019, 83, 1319–1328. [Google Scholar] [CrossRef]
- Xu, Y.; Fu, J.F.; Chen, J.H.; Zhang, Z.W.; Zou, Z.Q.; Han, L.Y.; Hua, Q.H.; Zhao, J.S.; Zhang, X.H.; Shan, Y.J. Sulforaphane ameliorates glucose intolerance in obese mice via the upregulation of the insulin signaling pathway. Food Funct. 2018, 9, 4695–4701. [Google Scholar] [CrossRef]
- Luo, J.; Alkhalidy, H.; Jia, Z.; Liu, D. Sulforaphane Ameliorates High-Fat-Diet-Induced Metabolic Abnormalities in Young and Middle-Aged Obese Male Mice. Foods 2024, 13, 1055. [Google Scholar] [CrossRef]
- Nagami, M.; Ito, Y.; Nagasawa, T. Phenethyl isothiocyanate protects against H2O2-induced insulin resistance in 3T3-L1 adipocytes. Biosci. Biotechnol. Biochem. 2017, 81, 2195–2203. [Google Scholar] [CrossRef]
- Liang, Y.; Sasaki, I.; Takeda, Y.; Zhu, B.; Munemasa, S.; Nakamura, T.; Murata, Y.; Nakamura, Y. Benzyl isothiocyanate ameliorates lipid accumulation in 3T3-L1 preadipocytes during adipocyte differentiation. Biosci. Biotechnol. Biochem. 2018, 82, 2130–2139. [Google Scholar] [CrossRef]
- Heiss, E.H.; Schachner, D.; Zimmermann, K.; Dirsch, V.M. Glucose availability is a decisive factor for Nrf2-mediated gene expression. Redox Biol. 2013, 1, 359–365. [Google Scholar] [CrossRef]
- Lee, I.K. The role of pyruvate dehydrogenase kinase in diabetes and obesity. Diabetes Metab. J. 2014, 38, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Jeoung, N.H. Pyruvate Dehydrogenase Kinases: Therapeutic Targets for Diabetes and Cancers. Diabetes Metab. J. 2015, 39, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Caggiano, M.; Eaton, P. Heart failure-emerging roles for the mitochondrial pyruvate carrier. Cell Death Differ. 2021, 28, 1149–1158. [Google Scholar] [CrossRef]
- Fernandez-Caggiano, M.; Kamynina, A.; Francois, A.A.; Prysyazhna, O.; Eykyn, T.R.; Krasemann, S.; Crespo-Leiro, M.G.; Vieites, M.G.; Bianchi, K.; Morales, V.; et al. Mitochondrial pyruvate carrier abundance mediates pathological cardiac hypertrophy. Nat. Metab. 2020, 2, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Taufalele, P.V.; Cochran, J.D.; Robillard-Frayne, I.; Marx, J.M.; Soto, J.; Rauckhorst, A.J.; Tayyari, F.; Pewa, A.D.; Gray, L.R.; et al. Mitochondrial pyruvate carriers are required for myocardial stress adaptation. Nat. Metab. 2020, 2, 1248–1264, Erratum in Nat. Metab. 2020, 2, 1498. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Ctrl | DCM | DCM + SFN |
|---|---|---|---|
| FFAs (mmol/L) | 0.44 ± 0.03 | 1.11 ± 0.08 **** | 0.80 ± 0.08 ** |
| TC (mmol/L) | 2.25 ± 0.21 | 4.85 ± 0.63 ** | 2.97 ± 0.47 * |
| TG (mmol/L) | 0.52 ± 0.04 | 1.30 ± 0.10 **** | 1.10 ± 0.09 |
| LDL-C (mmol/L) | 0.49 ± 0.05 | 1.45 ± 0.25 ** | 1.07 ± 0.26 |
| HDL-C (mmol/L) | 1.06 ± 0.26 | 0.96 ± 0.60 | 1.66 ± 0.54 * |
| MDA (μM) | 6.42 ± 0.37 | 13.31 ± 0.62 **** | 9.46 ± 0.64 *** |
| IL-6 (pg/mL) | 112.6 ± 6.28 | 109.0 ± 5.64 | 112.7 ± 4.23 |
| TNF-α (pg/mL) | 743.9 ±25.40 | 748.5 ± 40.90 | 793.6 ± 9.71 |
| Groups | Ctrl | DCM | DCM + SFN |
|---|---|---|---|
| LVIDs (mm) | 2.23 ± 0.10 | 1.91 ± 0.14 | 2.04 ± 0.11 |
| LVIDd (mm) | 3.67 ± 0.09 | 3.53 ± 0.11 | 3.80 ± 0.12 |
| LVAWs (mm) | 1.61 ± 0.08 | 1.75 ± 0.10 | 1.71 ± 0.06 |
| LVAWd (mm) | 1.08 ± 0.04 | 1.10 ± 0.05 | 1.02 ± 0.05 |
| LVPWs (mm) | 1.54 ± 0.07 | 1.66 ± 0.12 | 1.55 ± 0.07 |
| LVPWd (mm) | 1.00 ± 0.06 | 1.11 ± 0.09 | 1.01 ± 0.06 |
| Groups | Ctrl | DCM | DCM + SFN |
|---|---|---|---|
| HR (bpm) | 670 ± 29 | 562 ± 43 | 606 ± 15 |
| SBP (mmHg) | 125 ± 4 | 122 ± 8 | 124 ± 4 |
| DBP (mmHg) | 79 ± 3 | 72 ± 9 | 85 ± 7 |
| MBP (mmHg) | 95 ± 3 | 89 ± 9 | 98 ± 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, P.; Wang, Z.; Jin, X.; Zhang, M.; Shen, M.; Li, D. Oral Sulforaphane Intervention Protects Against Diabetic Cardiomyopathy in db/db Mice: Focus on Cardiac Lipotoxicity and Substrate Metabolism. Antioxidants 2025, 14, 603. https://doi.org/10.3390/antiox14050603
Wang P, Wang Z, Jin X, Zhang M, Shen M, Li D. Oral Sulforaphane Intervention Protects Against Diabetic Cardiomyopathy in db/db Mice: Focus on Cardiac Lipotoxicity and Substrate Metabolism. Antioxidants. 2025; 14(5):603. https://doi.org/10.3390/antiox14050603
Chicago/Turabian StyleWang, Pan, Ziling Wang, Xinyuan Jin, Mengdi Zhang, Mengfan Shen, and Dan Li. 2025. "Oral Sulforaphane Intervention Protects Against Diabetic Cardiomyopathy in db/db Mice: Focus on Cardiac Lipotoxicity and Substrate Metabolism" Antioxidants 14, no. 5: 603. https://doi.org/10.3390/antiox14050603
APA StyleWang, P., Wang, Z., Jin, X., Zhang, M., Shen, M., & Li, D. (2025). Oral Sulforaphane Intervention Protects Against Diabetic Cardiomyopathy in db/db Mice: Focus on Cardiac Lipotoxicity and Substrate Metabolism. Antioxidants, 14(5), 603. https://doi.org/10.3390/antiox14050603