
Hydrogen Sulfide (H2S) Metabolism, Iron Overload, and Apoptosis–Autophagy Equilibrium in Vascular Smooth Muscle Cells


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
2.1. Animals and Protocols
2.2. Aortic VSMC Culture
2.3. Iron Source
2.4. Cell Viability
2.5. Cell Proliferation
2.6. Ferrozine Assay
2.7. H2S Production Rate
2.8. Aconitase Activity Assay
2.9. ROS Production
2.10. Lipid Peroxidation
2.11. DNA Fragmentation (TUNEL) Assay
2.12. Caspase 3/7 Activity
2.13. Autophagy Assay
2.14. Nuclear and Cytoplasmic Extraction
2.15. Integrated Stress Response (ISR) Pathway Inhibition
2.16. Western Blot Analysis
2.17. Quantitative PCR
2.18. Statistical Analysis and Reproducibility
3. Results
3.1. Iron Loading Decreased Viability and Caused Iron Overload in CSE-KO Cells
3.2. H2S Promotes Ferritin Upregulation
3.3. H2S Inhibits Iron-Mediated Oxidative Stress
3.4. Iron-Treated CSE-KO Cells Undergo Apoptosis
3.5. Iron Upregulates CSE Expression via the ROS-eIF2α-ATF4 Pathway
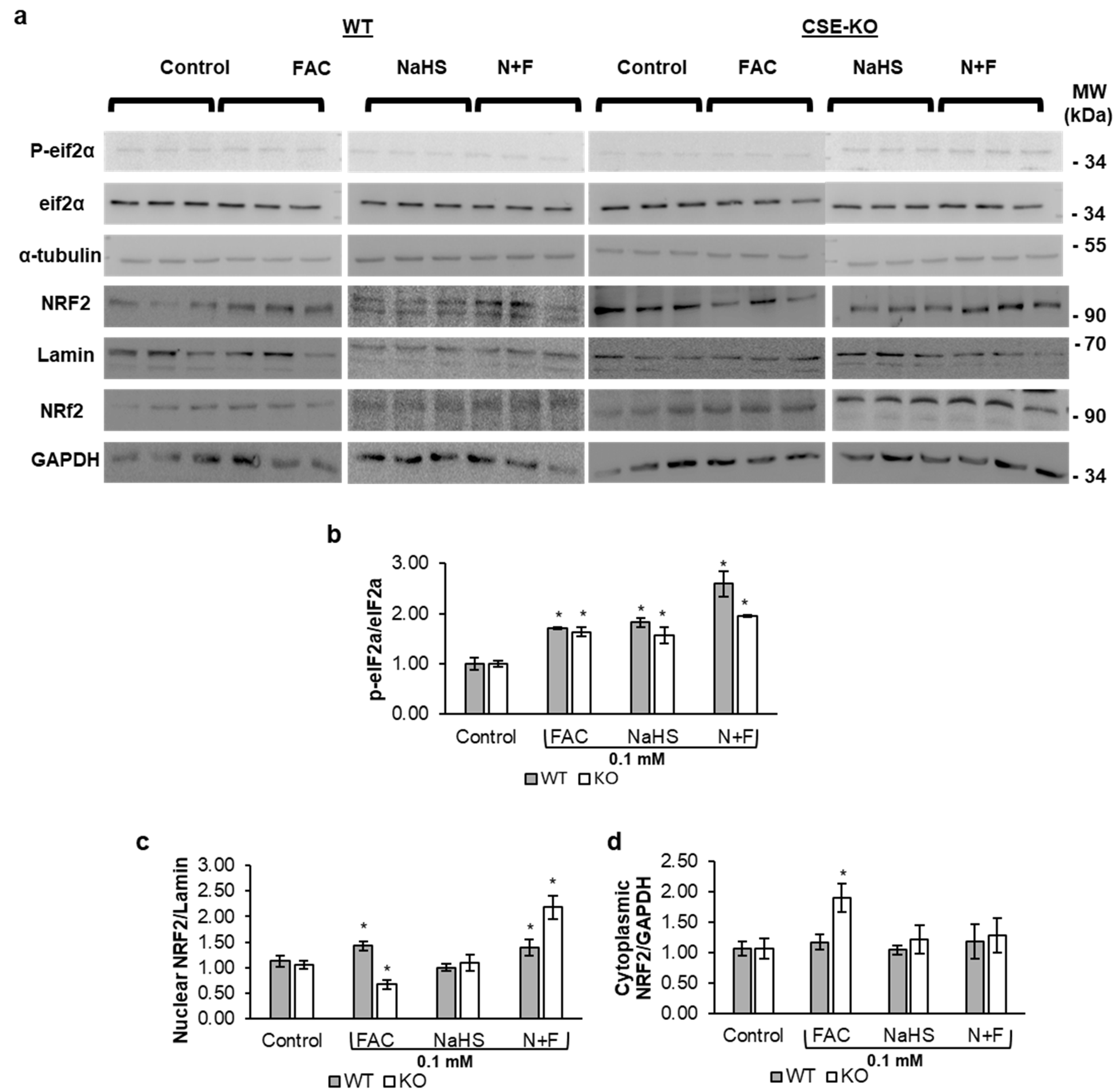
3.6. H2S Increases Nuclear Translocation of NRF2 in Response to ISR
3.7. H2S Protects Against Iron-Mediated Oxidative Stress via Upregulating AMPK-Mediated Autophagy
4. Discussion
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AMPK | Adenosine monophosphate-activated protein kinase |
| ANOVA | Analysis of variance |
| ATF4 | Activating transcription factor 4 |
| BrdU | Bromodeoxyuridine |
| CSE | Cystathionine gamma-lyase |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DMSO | Dimethyl sulfoxide |
| eIF2α | Eukaryotic translation initiation factor 2 |
| FAC | Ferric ammonium citrate |
| IRP1 | Iron regulatory proteins 1 |
| ISR | Integrated stress response |
| ISRIB | Integrated stress response inhibitor |
| KEAP1 | Kelch-like ECH-associated protein 1 |
| NCOA4 | Nuclear Receptor Coactivator 4 |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| PBS | Phosphate-buffered saline |
| ROS | Reactive oxygen species |
| TBS | Tris-buffered saline |
| VSMCs | Vascular smooth muscle cells |
References
- Lawen, A.; Lane, D.J.R. Mammalian Iron Homeostasis in Health and Disease: Uptake, Storage, Transport, and Molecular Mechanisms of Action. Antioxid. Redox Signal. 2013, 18, 2473–2507. [Google Scholar] [CrossRef] [PubMed]
- Arif, H.M.; Qian, Z.; Wang, R. Signaling Integration of Hydrogen Sulfide and Iron on Cellular Functions. Antioxid. Redox Signal. 2022, 36, 275–293. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.J.; Oudit, G.Y. Iron-Overload Cardiomyopathy: Pathophysiology, Diagnosis, and Treatment. J. Card. Fail. 2010, 16, 888–900. [Google Scholar] [CrossRef] [PubMed]
- Piperno, A.; Pelucchi, S.; Mariani, R. Inherited Iron Overload Disorders. Transl. Gastroenterol. Hepatol. 2020, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Vinchi, F.; Porto, G.; Simmelbauer, A.; Altamura, S.; Passos, S.T.; Garbowski, M.; Silva, A.M.N.; Spaich, S.; Seide, S.E.; Sparla, R. Atherosclerosis Is Aggravated by Iron Overload and Ameliorated by Dietary and Pharmacological Iron Restriction. Eur. Heart J. 2020, 41, 2681–2695. [Google Scholar] [CrossRef]
- Akaike, T.; Ida, T.; Wei, F.-Y.; Nishida, M.; Kumagai, Y.; Alam, M.M.; Ihara, H.; Sawa, T.; Matsunaga, T.; Kasamatsu, S. Cysteinyl-TRNA Synthetase Governs Cysteine Polysulfidation and Mitochondrial Bioenergetics. Nat. Commun. 2017, 8, 1177. [Google Scholar] [CrossRef]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a Physiologic Vasorelaxant: Hypertension in Mice with Deletion of Cystathionine γ-Lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, R. H2S-Induced Vasorelaxation and Underlying Cellular and Molecular Mechanisms. Am. J. Physiol. Circ. Physiol. 2002, 283, H474–H480. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, R.; Wu, L.; Yang, G. Hydrogen Sulfide Guards Myoblasts from Ferroptosis by Inhibiting ALOX12 Acetylation. Cell. Signal. 2021, 78, 109870. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, Y.; Rivett, A.; Yang, G. H2S Regulation of Iron Homeostasis by IRP1 Improves Vascular Smooth Muscle Cell Functions. Cell. Signal. 2023, 110, 110826. [Google Scholar] [CrossRef]
- Linjacki, S.; Wang, Y.; Baath, N.; Mantle, D.; Yang, G. H2S Protects from Rotenone-Induced Ferroptosis by Stabilizing Fe-S Clusters in Rat Cardiac Cells. Cells 2024, 13, 371. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Bithi, N.; Link, C.; Saunthararajah, Y.; Hine, C. Iron Catalyzed Hydrogen Sulfide and Its Prevalence during Oxidative Events in Sickle Cell Disease. Blood 2019, 134, 3534. [Google Scholar] [CrossRef]
- Sung, H.K.; Murugathasan, M.; Abdul-Sater, A.A.; Sweeney, G. Autophagy Deficiency Exacerbates Iron Overload Induced Reactive Oxygen Species Production and Apoptotic Cell Death in Skeletal Muscle Cells. Cell Death Dis. 2023, 14, 252. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wu, L.; Wang, R. Pro-apoptotic Effect of Endogenous H2S on Human Aorta Smooth Muscle Cells. FASEB J. 2006, 20, 553–555. [Google Scholar] [CrossRef]
- Yang, G.; Wu, L.; Bryan, S.; Khaper, N.; Mani, S.; Wang, R. Cystathionine Gamma-Lyase Deficiency and Overproliferation of Smooth Muscle Cells. Cardiovasc. Res. 2010, 86, 487–495. [Google Scholar] [CrossRef]
- Liu, J.; Wu, J.; Sun, A.; Sun, Y.; Yu, X.; Liu, N.; Dong, S.; Yang, F.; Zhang, L.; Zhong, X. Hydrogen Sulfide Decreases High Glucose/Palmitate-Induced Autophagy in Endothelial Cells by the Nrf2-ROS-AMPK Signaling Pathway. Cell Biosci. 2016, 6, 33. [Google Scholar] [CrossRef]
- Wu, D.; Wang, H.; Teng, T.; Duan, S.; Ji, A.; Li, Y. Hydrogen Sulfide and Autophagy: A Double Edged Sword. Pharmacol. Res. 2018, 131, 120–127. [Google Scholar] [CrossRef]
- Zhu, L.; Duan, W.; Wu, G.; Zhang, D.; Wang, L.; Chen, D.; Chen, Z.; Yang, B. Protective Effect of Hydrogen Sulfide on Endothelial Cells through Sirt1-FoxO1-Mediated Autophagy. Ann. Transl. Med. 2020, 8, 1586. [Google Scholar] [CrossRef]
- Shuang, T.; Fu, M.; Yang, G.; Huang, Y.; Qian, Z.; Wu, L.; Wang, R. Interaction among Estrogen, IGF-1, and H2S on Smooth Muscle Cell Proliferation. J. Endocrinol. 2021, 248, 17–30. [Google Scholar] [CrossRef]
- Li, Z.; Erion, D.M.; Maurer, T.S. Model-based Assessment of Plasma Citrate Flux into the Liver: Implications for NaCT as a Therapeutic Target. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 132–139. [Google Scholar] [CrossRef]
- Sweeney, G.; Sung, H.K.; Song, E.; Pantopoulos, K.; Jahng, J.W.S. Iron Induces Insulin Resistance in Cardiomyocytes via Regulation of Oxidative Stress. Sci. Rep. 2019, 9, 4668. [Google Scholar]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Zhang, X.; Li, W.; Wang, T.; Cui, S.; Li, T.; Wang, Y.; Xu, W.; Ma, Y.; Yang, B. EIF2α-Mediated Integrated Stress Response Links Multiple Intracellular Signaling Pathways to Reprogram Vascular Smooth Muscle Cell Fate in Carotid Artery Plaque. Heliyon 2024, 10, e26904. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Mani, S.; Cao, W.; Yang, G.; Lai, C.; Wu, L.; Wang, R. Interaction of Hydrogen Sulfide and Estrogen on the Proliferation of Vascular Smooth Muscle Cells. PLoS ONE 2012, 7, e41614. [Google Scholar] [CrossRef]
- Balla, G.; Jacob, H.S.; Balla, J.; Rosenberg, M.; Nath, K.; Apple, F.; Eaton, J.W.; Vercellotti, G.M. Ferritin: A Cytoprotective Antioxidant Strategem of Endothelium. J. Biol. Chem. 1992, 267, 18148–18153. [Google Scholar] [CrossRef]
- Rouault, T.A. The Role of Iron Regulatory Proteins in Mammalian Iron Homeostasis and Disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Mancias, J.D. The Role of NCOA4-Mediated ferritinophagy in Health and Disease. Pharmaceuticals 2018, 11, 114. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Gikandi, A.; Mancias, J.D. The Role of NCOA4-Mediated ferritinophagy in Ferroptosis. In Ferroptosis: Mechanism and Diseases; Springer: Cham, Switzerland, 2021; pp. 41–57. [Google Scholar]
- Yang, J.; Minkler, P.; Grove, D.; Wang, R.; Willard, B.; Dweik, R.; Hine, C. Non-Enzymatic Hydrogen Sulfide Production from Cysteine in Blood Is Catalyzed by Iron and Vitamin B 6. Commun. Biol. 2019, 2, 194. [Google Scholar] [CrossRef]
- Jahng, J.W.S.; Alsaadi, R.M.; Palanivel, R.; Song, E.; Hipolito, V.E.B.; Sung, H.K.; Botelho, R.J.; Russell, R.C.; Sweeney, G. Iron Overload Inhibits Late Stage Autophagic Flux Leading to Insulin Resistance. EMBO Rep. 2019, 20, e47911. [Google Scholar] [CrossRef]
- Nguyen, K.; Tang, J.; Cho, S.; Ying, F.; Sung, H.K.; Jahng, J.W.; Pantopoulos, K.; Sweeney, G. Salubrinal Promotes Phospho-EIF2α-Dependent Activation of UPR Leading to Autophagy-Mediated Attenuation of Iron-Induced Insulin Resistance. Mol. Metab. 2024, 83, 101921. [Google Scholar] [CrossRef]
- Abbasi, U.; Abbina, S.; Gill, A.; Kizhakkedathu, J.N. Development of an Iron Overload HepG2 Cell Model Using Ferrous Ammonium Citrate. Sci. Rep. 2023, 13, 21915. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Z.; Niu, J.; Xu, Y.; Ma, L.; Lu, A.; Wang, X.; Qian, Z.; Huang, Z.; Jin, X. Antiviral Effects of Ferric Ammonium Citrate. Cell Discov. 2018, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Grootveld, M.; Bell, J.D.; Halliwell, B.; Aruoma, O.I.; Bomford, A.; Sadler, P.J. Non-Transferrin-Bound Iron in Plasma or Serum from Patients with Idiopathic Hemochromatosis: Characterization by High Performance Liquid Chromatography and Nuclear Magnetic Resonance Spectroscopy. J. Biol. Chem. 1989, 264, 4417–4422. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Ropert, M.; Le Lan, C.; Loréal, O. Non-Transferrin Bound Iron: A Key Role in Iron Overload and Iron Toxicity. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2012, 1820, 403–410. [Google Scholar] [CrossRef]
- Zhao, K.; Li, H.; Li, S.; Yang, G. Regulation of Cystathionine Gamma-Lyase/H2S System and Its Pathological Implication. Front. Biosci. (Landmark Ed.) 2014, 19, 1355–1369. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, X.; Wang, C.; Zhou, P.; Xiao, J.; Zheng, H.; Wang, L.; Yan, S.; Zhang, Y.; Ji, X. Deacetylated Sp1 Improves Β-glycerophosphate-induced Calcification in Vascular Smooth Muscle Cells. Exp. Ther. Med. 2021, 22, 1152. [Google Scholar] [CrossRef]
- Bao, W.-D.; Fan, Y.; Deng, Y.-Z.; Long, L.-Y.; Wang, J.; Guan, D.-X.; Qian, Z.-Y.; An, P.; Feng, Y.-Y.; He, Z.-Y. Iron Overload in Hereditary Tyrosinemia Type 1 Induces Liver Injury through the Sp1/Tfr2/Hepcidin Axis. J. Hepatol. 2016, 65, 137–145. [Google Scholar] [CrossRef]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen Sulfide Protects against Cellular Senescence via S-Sulfhydration of Keap1 and Activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar] [CrossRef]
- Khan, N.M.; Ahmad, I.; Haqqi, T.M. Nrf2/ARE Pathway Attenuates Oxidative and Apoptotic Response in Human Osteoarthritis Chondrocytes by Activating ERK1/2/ELK1-P70S6K-P90RSK Signaling Axis. Free Radic. Biol. Med. 2018, 116, 159–171. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Read, E.; Fu, M.; Pei, Y.; Wu, L.; Wang, R.; Yang, G. Golgi Stress Response, Hydrogen Sulfide Metabolism, and Intracellular Calcium Homeostasis. Antioxid. Redox Signal. 2020, 32, 583–601. [Google Scholar] [CrossRef]
- Rzymski, T.; Milani, M.; Singleton, D.C.; Harris, A.L. Role of ATF4 in Regulation of Autophagy and Resistance to Drugs and Hypoxia. Cell Cycle 2009, 8, 3838–3847. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, A. ROS-mediated PERK-eIF2α-ATF4 Pathway Plays an Important Role in Arsenite-induced L-02 Cells Apoptosis via Regulating CHOP-DR5 Signaling. Environ. Toxicol. 2020, 35, 1100–1113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Ling, X.; Liu, K.; Wang, Z.; Zou, P.; Gao, J.; Cao, J.; Ao, L. The P-EIF2α/ATF4 Pathway Links Endoplasmic Reticulum Stress to Autophagy Following the Production of Reactive Oxygen Species in Mouse Spermatocyte-Derived Cells Exposed to Dibutyl Phthalate. Free Radic. Res. 2016, 50, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Hartwick Bjorkman, S.; Oliveira Pereira, R. The Interplay between Mitochondrial Reactive Oxygen Species, Endoplasmic Reticulum Stress, and Nrf2 Signaling in Cardiometabolic Health. Antioxid. Redox Signal. 2021, 35, 252–269. [Google Scholar] [CrossRef]
- Sarcinelli, C.; Dragic, H.; Piecyk, M.; Barbet, V.; Duret, C.; Barthelaix, A.; Ferraro-Peyret, C.; Fauvre, J.; Renno, T.; Chaveroux, C. ATF4-Dependent NRF2 Transcriptional Regulation Promotes Antioxidant Protection during Endoplasmic Reticulum Stress. Cancers 2020, 12, 569. [Google Scholar] [CrossRef]
- Liu, Z.-W.; Zhu, H.-T.; Chen, K.-L.; Dong, X.; Wei, J.; Qiu, C.; Xue, J.-H. Protein Kinase RNA-like Endoplasmic Reticulum Kinase (PERK) Signaling Pathway Plays a Major Role in Reactive Oxygen Species (ROS)-Mediated Endoplasmic Reticulum Stress-Induced Apoptosis in Diabetic Cardiomyopathy. Cardiovasc. Diabetol. 2013, 12, 158. [Google Scholar] [CrossRef]
- Kotla, N.K.; Dutta, P.; Parimi, S.; Das, N.K. The Role of Ferritin in Health and Disease: Recent Advances and Understandings. Metabolites 2022, 12, 609. [Google Scholar] [CrossRef]
- Zhao, H.; Lu, Y.; Zhang, J.; Sun, Z.; Cheng, C.; Liu, Y.; Wu, L.; Zhang, M.; He, W.; Hao, S. NCOA4 Requires a [3Fe-4S] to Sense and Maintain the Iron Homeostasis. J. Biol. Chem. 2024, 300, 105612. [Google Scholar] [CrossRef]
- Vásquez-Vivar, J.; Kalyanaraman, B.; Kennedy, M.C. Mitochondrial Aconitase Is a Source of Hydroxyl Radical: An Electron Spin Resonance Investigation. J. Biol. Chem. 2000, 275, 14064–14069. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell Death: A Review of the Major Forms of Apoptosis, Necrosis and Autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a New Form of Cell Death: Opportunities and Challenges in Cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Ding, X.; Zheng, J.; Wang, B.; Li, Y.; Xiang, H.; Dou, M.; Qiao, Y.; Tian, P.; Xue, W. MiR-182-5p and MiR-378a-3p Regulate Ferroptosis in I/R-Induced Renal Injury. Cell Death Dis. 2020, 11, 929. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kang, S.-W.; Joo, J.; Han, S.H.; Shin, H.; Nam, B.Y.; Park, J.; Yoo, T.-H.; Kim, G.; Lee, P. Characterization of Ferroptosis in Kidney Tubular Cell Death under Diabetic Conditions. Cell Death Dis. 2021, 12, 160. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, D.G.; Kim, B.; Park, S.-J.; Kim, J.-H.; Lee, S.-R.; Chang, K.-T.; Lee, H.-S.; Lee, D.-S. Iron Overload Triggers Mitochondrial Fragmentation via Calcineurin-Sensitive Signals in HT-22 Hippocampal Neuron Cells. Toxicology 2015, 337, 39–46. [Google Scholar] [CrossRef]
- Shen, Z.; Zhao, L.; Yoo, S.; Lin, Z.; Zhang, Y.; Yang, W.; Piao, J. Emodin Induces Ferroptosis in Colorectal Cancer through NCOA4-Mediated ferritinophagy and NF-Κb Pathway Inactivation. Apoptosis 2024, 29, 1810–1823. [Google Scholar] [CrossRef]
- Mani, S.; Li, H.; Untereiner, A.; Wu, L.; Yang, G.; Austin, R.C.; Dickhout, J.G.; Lhoták, Š.; Meng, Q.H.; Wang, R. Decreased Endogenous Production of Hydrogen Sulfide Accelerates Atherosclerosis. Circulation 2013, 127, 2523–2534. [Google Scholar] [CrossRef]
- Greenough, M.A.; Camakaris, J.; Bush, A.I. Metal Dyshomeostasis and Oxidative Stress in Alzheimer’s Disease. Neurochem. Int. 2013, 62, 540–555. [Google Scholar] [CrossRef]
- Calvert, J.W.; Jha, S.; Gundewar, S.; Elrod, J.W.; Ramachandran, A.; Pattillo, C.B.; Kevil, C.G.; Lefer, D.J. Hydrogen Sulfide Mediates Cardioprotection through NRF2 Signaling. Circ. Res. 2009, 105, 365–374. [Google Scholar] [CrossRef]
- Kasai, S.; Yamazaki, H.; Tanji, K.; Engler, M.J.; Matsumiya, T.; Itoh, K. Role of the ISR-ATF4 Pathway and Its Cross Talk with Nrf2 in Mitochondrial Quality Control. J. Clin. Biochem. Nutr. 2019, 64, 1–12. [Google Scholar] [CrossRef]
- Hou, X.-O.; Tu, H.-Y.; Qian, H.-C.; Li, Q.; Yang, Y.-P.; Xu, G.-Q.; Wang, F.; Liu, C.-F.; Wang, Y.-L.; Hu, L.-F. AMPK S-Sulfuration Contributes to H2S Donors-Induced AMPK Phosphorylation and Autophagy Activation in Dopaminergic Cells. Neurochem. Int. 2021, 150, 105187. [Google Scholar] [CrossRef]
- Wang, H.; Zhong, P.; Sun, L. Exogenous Hydrogen Sulfide Mitigates NLRP3 Inflammasome-Mediated Inflammation through Promoting Autophagy via the AMPK-MTOR Pathway. Biol. Open 2019, 8, bio043653. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, Y. AMPK and Autophagy. In Autophagy: Biology and Diseases; Springer: Singapore, 2019; pp. 85–108. [Google Scholar]
- Yang, F.; Zhang, L.; Gao, Z.; Sun, X.; Yu, M.; Dong, S.; Wu, J.; Zhao, Y.; Xu, C.; Zhang, W. Exogenous H2S Protects against Diabetic Cardiomyopathy by Activating Autophagy via the AMPK/MTOR Pathway. Cell. Physiol. Biochem. 2017, 43, 1168–1187. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.R.; Yang, J.Q.; Liu, F.; Shen, X.Q.; Zhou, Y.J. Melatonin Attenuates Vascular Calcification by Activating Autophagy via an AMPK/MTOR/ULK1 Signaling Pathway. Exp. Cell Res. 2020, 389, 111883. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Qiu, Y.; Wu, N.; Chen, A.-D.; Zhou, H.; Chen, Q.; Kang, Y.-M.; Li, Y.-H.; Zhu, G.-Q. FNDC5 Attenuates Oxidative Stress and NLRP3 Inflammasome Activation in Vascular Smooth Muscle Cells via Activating the AMPK-SIRT1 Signal Pathway. Oxid. Med. Cell. Longev. 2020, 2020, 6384803. [Google Scholar] [CrossRef]
- Duan, X.-H.; Chang, J.-R.; Zhang, J.; Zhang, B.-H.; Li, Y.-L.; Teng, X.; Zhu, Y.; Du, J.; Tang, C.-S.; Qi, Y.-F. Activating Transcription Factor 4 Is Involved in Endoplasmic Reticulum Stress-Mediated Apoptosis Contributing to Vascular Calcification. Apoptosis 2013, 18, 1132–1144. [Google Scholar] [CrossRef]
- Zhang, R.; Jiang, M.; Zhang, J.; Qiu, Y.; Li, D.; Li, S.; Liu, J.; Liu, C.; Fang, Z.; Cao, F. Regulation of the Cerebrovascular Smooth Muscle Cell Phenotype by Mitochondrial Oxidative Injury and Endoplasmic Reticulum Stress in Simulated Microgravity Rats via the PERK-EIF2α-ATF4-CHOP Pathway. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165799. [Google Scholar] [CrossRef]
- Jain, S.K.; Bull, R.; Rains, J.L.; Bass, P.F.; Levine, S.N.; Reddy, S.; McVie, R.; Bocchini, J.A., Jr. Low Levels of Hydrogen Sulfide in the Blood of Diabetes Patients and Streptozotocin-Treated Rats Causes Vascular Inflammation? Antioxid. Redox Signal. 2010, 12, 1333–1337. [Google Scholar] [CrossRef]
- Liu, J.; Li, Q.; Yang, Y.; Ma, L. Iron Metabolism and Type 2 Diabetes Mellitus: A Meta-analysis and Systematic Review. J. Diabetes Investig. 2020, 11, 946–955. [Google Scholar] [CrossRef]
- Hine, C.; Harputlugil, E.; Zhang, Y.; Ruckenstuhl, C.; Lee, B.C.; Brace, L.; Longchamp, A.; Treviño-Villarreal, J.H.; Mejia, P.; Ozaki, C.K. Endogenous Hydrogen Sulfide Production Is Essential for Dietary Restriction Benefits. Cell 2015, 160, 132–144. [Google Scholar] [CrossRef]
- Wiliński, B.; Wiliński, J.; Somogyi, E.; Piotrowska, J.; Opoka, W. Metformin Raises Hydrogen Sulfide Tissue Concentrations in Various Mouse Organs. Pharmacol. Rep. 2013, 65, 737–742. [Google Scholar] [CrossRef]
- Yue, F.; Shi, Y.; Wu, S.; Xing, L.; He, D.; Wei, L.; Qiu, A.; Russell, R.; Zhang, D. Metformin Alleviates Hepatic Iron Overload and Ferroptosis through AMPK-Ferroportin Pathway in HFD-Induced NAFLD. Iscience 2023, 26, 108560. [Google Scholar] [CrossRef]
- Liu, J.-L.; Fan, Y.-G.; Yang, Z.-S.; Wang, Z.-Y.; Guo, C. Iron and Alzheimer’s Disease: From Pathogenesis to Therapeutic Implications. Front. Neurosci. 2018, 12, 632. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arif, H.M.; Fu, M.; Wang, R. Hydrogen Sulfide (H2S) Metabolism, Iron Overload, and Apoptosis–Autophagy Equilibrium in Vascular Smooth Muscle Cells. Antioxidants 2025, 14, 560. https://doi.org/10.3390/antiox14050560
Arif HM, Fu M, Wang R. Hydrogen Sulfide (H2S) Metabolism, Iron Overload, and Apoptosis–Autophagy Equilibrium in Vascular Smooth Muscle Cells. Antioxidants. 2025; 14(5):560. https://doi.org/10.3390/antiox14050560
Chicago/Turabian StyleArif, Hassan Mustafa, Ming Fu, and Rui Wang. 2025. "Hydrogen Sulfide (H2S) Metabolism, Iron Overload, and Apoptosis–Autophagy Equilibrium in Vascular Smooth Muscle Cells" Antioxidants 14, no. 5: 560. https://doi.org/10.3390/antiox14050560
APA StyleArif, H. M., Fu, M., & Wang, R. (2025). Hydrogen Sulfide (H2S) Metabolism, Iron Overload, and Apoptosis–Autophagy Equilibrium in Vascular Smooth Muscle Cells. Antioxidants, 14(5), 560. https://doi.org/10.3390/antiox14050560