Abstract
Background: Oxidative stress, defined as an imbalance between reactive oxygen species and antioxidant defenses, plays a pivotal role in the pathogenesis of sex chromosome aneuploidies (SCAs), such as Turner syndrome (TS) and Klinefelter syndrome (KS). Pediatric patients with SCAs are particularly susceptible due to hormonal deficiencies, metabolic disturbances, and systemic complications. Methods: A comprehensive literature search was conducted in November 2024 using PubMed, Scopus, and Web of Science. Keywords included “antioxidants”, “oxidative stress”, “pediatrics”, “Turner syndrome”, “Klinefelter syndrome”, and “sex chromosome aneuploidies”. English-language articles were included without publication year restrictions. Relevant data on oxidative stress mechanisms and antioxidant interventions were systematically extracted. Results: The relationship between oxidative stress and SCAs can be described as bidirectional, where oxidative stress both contributes to and is exacerbated by aneuploidies. TS is marked by estrogen deficiency, cardiovascular anomalies, and metabolic dysfunction, all linked to heightened oxidative stress. KS is associated with hypogonadism, metabolic syndrome, and neurocognitive challenges, further exacerbated by oxidative damage. The aneuploid condition predisposes to increased oxidative stress in other SCAs, including 47,XXX and 47,XYY, as well as in high-grade aneuploidies. Emerging evidence highlights the therapeutic potential of antioxidants, including vitamin C, vitamin E, glutathione precursors, polyphenols, and melatonin. These interventions, when combined with hormonal therapies such as estrogen replacement in TS or testosterone replacement in KS, demonstrate synergistic effects in restoring redox balance and mitigating systemic complications. Conclusions: Oxidative stress significantly impacts the progression of SCAs in pediatric populations, amplifying risks across metabolic, cardiovascular, and neurocognitive domains. Early, tailored antioxidant strategies, integrated with syndrome-specific hormonal therapies, could reduce long-term complications and improve patient outcomes. Future research should focus on standardizing protocols to optimize these interventions for pediatric patients with SCAs.
1. Introduction
Sex chromosome aneuploidies (SCAs) are among the most common chromosomal abnormalities, affecting approximately 1 in 500 live births, with the prevalence varying based on the specific condition [1,2,3]. The most frequent SCAs include Klinefelter syndrome (KS), Turner syndrome (TS), 47,XXX syndrome, and 47,XYY syndrome, along with less common variants and mosaicism forms [4,5].
SCAs are associated with a wide spectrum of clinical manifestations, including developmental delay, cognitive impairment, dysmorphic features, gonadal dysfunction, cardiovascular anomalies, and metabolic disturbances [6,7]. The variability in phenotypic presentation is influenced by the specific aneuploidy, mosaicism degree, and dosage effects of sex chromosome-linked genes [8]. Advances in prenatal diagnostic techniques and improved clinical management have contributed to earlier detection and better outcomes, emphasizing the need for a deeper understanding of underlying pathophysiological mechanisms [9,10,11].
Oxidative stress, characterized by an imbalance between reactive oxygen species (ROS) production and antioxidant defense mechanisms, has been implicated in the pathogenesis of numerous pediatric disorders [12,13]. Emerging evidence suggests that SCAs may also involve oxidative stress, contributing to the development of associated comorbidities such as cardiovascular disease, diabetes, and neurodevelopmental disorders [14,15,16,17]. Notably, oxidative stress has been extensively studied in autosomal aneuploidies such as trisomy 21, where altered redox homeostasis plays a critical role in phenotypic manifestations [18,19]. The role of oxidative stress in SCAs, however, remains underexplored. Specific genes located on sex chromosomes, such as those regulating oxidative balance and mitochondrial function, as well as autosomal genes, may contribute to increased ROS production and decreased antioxidant capacity [20,21,22,23,24,25]. Understanding these mechanisms could illuminate the etiology of SCA-related complications and inform therapeutic strategies targeting oxidative stress.
This review aims to provide an overview of oxidative stress in SCAs in pediatric populations, emphasizing its potential implications for metabolic, cardiovascular, and neurodevelopmental health. Given the limited research specifically addressing oxidative stress in SCAs, we also incorporate relevant findings from autosomal aneuploidies to provide a broader context. Rather than presenting an exhaustive systematic analysis, our goal is to synthesize current knowledge on oxidative stress-related mechanisms in SCAs and discuss potential antioxidant strategies that may contribute to mitigating disease progression. While this review does not claim to exhaustively cover all aspects of SCAs and oxidative stress, it underscores the importance of this research avenue for advancing pediatric and reproductive medicine.
2. Materials and Methods
For this narrative review, we conducted a comprehensive literature search using MEDLINE/PubMed, Scopus, and Web of Science to identify studies related to oxidative stress and antioxidant strategies in SCAs, especially in pediatric populations. The search was performed with no restrictions on publication year to ensure the inclusion of all relevant literature. We used the following keywords and Medical Subject Headings (MeSH) terms in various combinations: “Oxidative stress” (unique ID: D015444), “Antioxidants” (unique ID: D000975), “Sex chromosome aneuploidies” (no MeSH term, free text search), “Klinefelter syndrome” (unique ID: D007661), “Turner syndrome” (unique ID: D014424), and “Pediatrics” (unique ID: D010372).
Eligible studies included original research articles (randomized and non-randomized clinical trials, prospective observational studies, retrospective cohort studies, and case–control studies) that investigated oxidative stress and/or antioxidant interventions in SCAs. Additionally, we included review articles discussing oxidative stress mechanisms, antioxidant therapies, or metabolic, cardiovascular, and neurodevelopmental implications in SCAs. Given the limited research on oxidative stress in SCAs, we incorporated case reports and studies on oxidative stress also in autosomal aneuploidies (e.g., trisomy 21) when they provided insights relevant to SCAs. Exclusion criteria were as follows: non-English language manuscripts; articles that did not provide specific data on oxidative stress mechanisms or antioxidant interventions, in particular for pediatric age; and abstract, books, conference proceedings, letters to the editor, and editorials. The selection process was conducted independently by two authors (R.P. and F.Pa.) who screened and reviewed all the studies meeting the inclusion criteria. A total of 161 articles were included, covering oxidative stress mechanisms, antioxidant strategies, and potential clinical implications in pediatric SCAs.
3. Overview of ROS
ROS refer to oxygen-containing molecules that are more reactive than molecular oxygen (O2) [26]. They arise due to the partial reduction of oxygen during metabolic processes, such as mitochondrial respiration [27]. The term encompasses both free radicals (molecules with one or more unpaired electrons) and non-radical derivatives of oxygen that exhibit high reactivity [28]. Key examples of ROS include the following:
- Superoxide anion (O2•−): a free radical formed by the one-electron reduction of oxygen [29].
- Hydrogen peroxide (H2O2): a non-radical ROS that can diffuse through membranes and act as a signaling molecule [30].
- Hydroxyl radical (•OH): an extremely reactive free radical generated from H2O2 via the Fenton reaction [31].
- Singlet oxygen (1O2): a highly reactive non-radical form of oxygen produced by energy transfer to molecular oxygen [32].
- Peroxynitrite (ONOO−): formed by the reaction of nitric oxide (NO) with superoxide; it is classified as a reactive nitrogen species but often grouped with ROS [33].
ROS can arise from internal sources such as mitochondrial electron transport, peroxisomal oxidases, and nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX), as well as from external sources, including ultraviolet (UV) radiation, pollution, and certain drugs (Figure 1). They serve dual roles in biological systems, acting as critical signaling molecules under physiological conditions and as damaging agents under pathological circumstances [34].
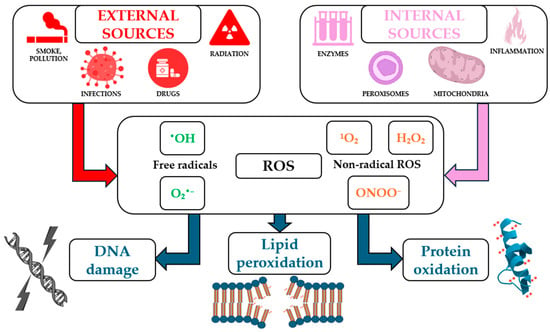
Figure 1.
Main internal and external sources of reactive oxygen species (ROS) and their biological effects (not all the mechanisms illustrated in the figure have been detailed or discussed in the text of the article).
In controlled amounts, ROS are essential for various cellular processes:
- Signal transduction: ROS, particularly H2O2, function as secondary messengers in signaling pathways [35]. For instance, H2O2 can reversibly oxidize thiol groups in proteins, regulating kinases and phosphatases involved in growth, differentiation, and immune responses [36].
- Host defense: ROS generated by phagocytes (via NOX enzymes) are crucial for destroying pathogens. This process, known as the respiratory burst, produces O2•−, which is converted into bactericidal species such as hypochlorous acid (HOCl) [37].
- Redox homeostasis: they regulate the expression of antioxidant genes through redox-sensitive transcription factors like nuclear factor erythroid 2-related factor 2 [38].
- Cellular adaptation to stress: moderate ROS levels activate stress response pathways, such as heat shock proteins and autophagy, promoting cell survival [39].
Excessive ROS production or impaired antioxidant defenses lead to oxidative stress, contributing to numerous diseases, including neurodegenerative disorders, cancer, cardiovascular diseases, and chronic inflammation [34]. Moreover, the free radical theory of aging suggests that accumulated oxidative damage over time contributes to cellular senescence and organismal aging [40].
The chemical reactions involving ROS are diverse and depend on the type of ROS and their specific biological or chemical context. The key reactions can be categorized as generation, conversion, and interaction with biological molecules.
3.1. Generation Reactions
ROS are produced through various physiological and environmental processes, in both enzymatic and non-enzymatic reactions.
Enzymatic production
- NOX: these enzymes catalyze the transfer of electrons from NADPH to molecular oxygen, forming O2•− [41].
- Xanthine oxidase: produces O2•− and H2O2 during purine metabolism [42].
- Cytochrome P450 enzymes: generate O2•− as a byproduct during detoxification reactions [43].
Non-enzymatic production
- Electron leakage from the mitochondrial electron transport chain during oxidative phosphorylation can reduce oxygen to O2•− [44].
- UV radiation and ionizing radiation split water molecules, producing •OH [45,46].
3.2. Conversion Reactions
Once generated, ROS are often converted into other species via chemical or enzymatic reactions:
- Superoxide dismutase (SOD) converts O2•− into H2O2: 2O2•− + 2H+ → H2O2 + O2 [47].
- Fenton reaction: H2O2 reacts with transition metals (e.g., Fe2+), producing •OH: H2O2 + Fe2+ → •OH + OH− + Fe3+ [48].
- Haber-Weiss reaction: the reaction between O2•− and H2O2 to produce •OH, which occurs in the presence of metal ions, particularly iron (Fe2+ or Fe3+): O2•− + H2O2 → •OH + OH− + O2 [49].
3.3. Interactions with Biological Molecules
ROS react with lipids, proteins, and DNA, leading to significant biological effects [50]:
- Lipid peroxidation: lipid radicals and malondialdehyde (MDA) are generated, disrupting membrane integrity [51].
- Protein oxidation: oxidation of amino acids, such as cysteine and methionine, can modify enzyme activity and signaling pathways [52].
- DNA damage: ROS can induce strand breaks and base modifications, such as the conversion of guanine to 8-oxoguanine, contributing to mutagenesis [53].
4. Parental Meiotic Errors and Offspring Susceptibility to Oxidative Stress in SCAs
SCAs arise from meiotic nondisjunction, which can occur in both maternal and paternal meiosis, although maternal nondisjunction is more common. Errors in the first and second meiotic divisions, influenced by factors such as recombination failures and maternal age, are primary contributors to these aneuploidies. KS (47,XXY) can arise from nondisjunction in either parent, with approximately equal contributions from maternal or paternal origins [54,55]. 47,XXX syndrome predominantly results from maternal nondisjunction, with a small percentage arising from paternal errors [54,55]. 47,XYY syndrome usually results from nondisjunction during paternal meiosis II [55,56]. TS, in contrast, often results from postzygotic mitotic errors or nondisjunction during paternal meiosis, leading to different karyotypes, all of which lack X-chromosomal material [57]. More complex SCAs include the high-grade sex chromosome aneuploidies (HGAs) with more than 47 chromosomes that arise from a maternal or paternal non-disjunction event during meiosis I or II [58]. The distinct mechanisms underlying these errors include improper chromosomal pairing, cohesion defects, and spindle assembly checkpoint failures [59]. These disruptions lead to aneuploid gametes, which, when involved in fertilization, result in offspring with SCAs.
Once SCAs are established in the zygote, the aneuploid condition, as well as the number of X chromosomes, may predispose the offspring to increased oxidative stress (Table 1). It is hypothesized that the chromosomal imbalance alters gene expression profiles, particularly in genes involved in redox regulation, further increasing the susceptibility of cells to oxidative damage. SCAs impact cellular and tissue function through both cis and trans gene expression effects [60]. Cis effects, particularly on X-Y gametologs (homologous genes on X and Y chromosomes that evolved from a single ancestral gene and retained related functions [61]), are consistent across tissue types, influencing critical processes like gene regulation and chromatin dynamics. In contrast, trans effects exhibit tissue specificity, potentially driving varied clinical outcomes in neurodevelopment, reproduction, and metabolism [60]. Therefore, the potential oxidative imbalance likely exacerbates the various comorbidities associated with SCAs [62].
While parental meiotic errors are the primary cause of SCAs, the resultant chromosomal abnormalities set the stage for oxidative stress-mediated pathogenesis in the offspring. The supernumerary sex chromosomes carry imprinted genes that influence cellular metabolism and ROS production, linking the meiotic origin of SCAs with the downstream oxidative stress observed in affected individuals.
The degree of oxidative stress susceptibility appears to vary depending on the specific aneuploidy and the genes involved [60]. For example, the presence of an extra X chromosome in 47,XXY, or 47,XXX may increase the gene dosage of X-linked pro-oxidant genes that escape X-inactivation. In 47,XYY individuals, for instance, the Y-linked gene involvement in redox regulation may have a role. However, the evidence is not uniformly consistent. Thus, while gene dosage and meiotic origin clearly contribute to oxidative vulnerability, the precise degree and mechanisms remain incompletely defined and are likely modulated by epigenetic, environmental, and sex-specific factors.

Table 1.
Main oxidative stress findings across sex chromosome aneuploidies.
Table 1.
Main oxidative stress findings across sex chromosome aneuploidies.
| Karyotypes | Key Findings on Oxidative Stress | References | |
|---|---|---|---|
| Single X Chromosome | 45,X (Turner syndrome, TS) | Reduced antioxidant capacity, increased oxidative stress markers (e.g., lipid peroxidation, reduced glutathione levels)—see Table 2 | [63,64] |
| Mosaic TS (e.g., 45,X/46,XX) | Oxidative stress may vary depending on the degree of mosaicism; similar trends to 45,X are observed | ||
| Mosaic TS with Y material (e.g., 45,X/46,XY) | Similar to 45,X, but limited data on specific oxidative stress markers in this subgroup | ||
| 47,XYY | Limited evidence: oxidative stress not well characterized in this aneuploidy | [65], “Limited data” | |
| Multiple X Chromosomes | 47,XXY (Klinefelter syndrome) | Increased levels of oxidative stress biomarkers; associated with metabolic syndrome, which exacerbates oxidative stress—see Table 2 | [65,66,67] |
| 47,XXX | Limited studies: oxidative imbalance likely less pronounced than in other sex chromosome aneuploidies | [68], “Limited data” | |
| Tetrasomies and pentasomies with supernumerary X (and/or Y) chromosomes | Insufficient data; presumed oxidative stress due to multiple X (and/or Y) chromosomes and severe aneuploidy effects | [69], “Limited data” |
Table 2.
Oxidative stress mechanisms and impact in Turner syndrome and Klinefelter syndrome. Abbreviations: ROS, reactive oxygen species; XIAP, X-chromosome-linked inhibitor of apoptosis protein; SOD, superoxide dismutase; NADPH, nicotinamide adenine dinucleotide phosphate; NOX, NADPH oxidase; GPx, glutathione peroxidase.
Table 2.
Oxidative stress mechanisms and impact in Turner syndrome and Klinefelter syndrome. Abbreviations: ROS, reactive oxygen species; XIAP, X-chromosome-linked inhibitor of apoptosis protein; SOD, superoxide dismutase; NADPH, nicotinamide adenine dinucleotide phosphate; NOX, NADPH oxidase; GPx, glutathione peroxidase.
| Turner Syndrome | Klinefelter Syndrome | |
|---|---|---|
| Genetic Alteration | Lack of X chromosome material (45,X or different karyotypes) increases susceptibility to oxidative stress. | Extra X chromosomes (most common karyotype: 47,XXY) lead to increased ROS production and reduced antioxidant capacity. |
| Oxidative Stress Response | Altered stress pathways, with increased oxidative burden due to estrogen deficiency. | Elevated ROS and impaired antioxidant defense, especially in spermatozoa. |
| Molecular Mechanisms | XIAP regulates mitochondrial antioxidants (SOD-2), reducing oxidative stress. | Increased NADPH production enhances ROS generation via sperm NOX. |
| Antioxidant Defenses | Estrogen upregulates SOD, catalase, and scavenges free radicals; estrogen replacement may help restore defenses. | Testosterone modulates antioxidant enzymes (SOD, GPx), with testosterone replacement improving oxidative stress. |
| Cardiovascular Implications | Oxidative stress leads to endothelial dysfunction, hypertension, and aortic issues. | ROS in endothelial cells cause dysfunction, contributing to cardiovascular risk. |
| Metabolic Issues | Oxidative stress impairs insulin signaling and promotes metabolic syndrome. | Excess adipose ROS exacerbates metabolic dysfunction and systemic inflammation. |
| Neurodevelopmental Challenges | Oxidative stress in the brain may contribute to cognitive and developmental deficits. | Mitochondrial dysfunction and ROS accumulation affect cognitive development. |
5. SCAs and Oxidative Stress in Pediatric Populations
Oxidative stress results from an imbalance between the production of ROS and the body’s ability to neutralize them through antioxidants. Excessive ROS can damage lipids, proteins, and DNA, leading to cellular dysfunction and contributing to the development of chronic diseases [50]. Recent studies have suggested that the altered gene dosage associated with SCAs may result in disrupted metabolic and oxidative processes, offering insight into the pathogenic mechanisms of these syndromes [66,67].
Research indicates that oxidative stress markers differ between sexes, with boys exhibiting higher levels of oxidative stress markers and lower levels of antioxidant defenses compared to girls during the neonatal period. This discrepancy is attributed to the more active estrogen metabolism in females, which enhances glutathione metabolism, a critical antioxidant pathway [70].
These findings suggest that antioxidant strategies should be personalized, considering the sex of the individual to effectively restore redox homeostasis. Investigating oxidative stress markers in SCAs has been proposed as a promising avenue to elucidate these processes.
5.1. Turner Syndrome
TS is a genetic condition caused by the complete or partial absence of one X chromosome, which affects approximately 1 in 2500 live female births [71]. It encompasses a range of karyotypes, with 40–50% of cases presenting as 45,X (complete loss of one X chromosome). Mosaic forms, such as 45,X/46,XX or 45,X/47,XXX, occur in 15–25% of cases. Isochromosomes are found in 20% of cases, and ring chromosomes are relatively rare. Additionally, 10–12% of individuals possess varying amounts of Y chromosome material, with about 3% showing a 45,X/46,XY mosaic karyotype [57]. While its clinical presentation is diverse—ranging from short stature and ovarian insufficiency to cardiovascular anomalies and metabolic dysfunction [57]—there is growing evidence that oxidative stress plays a central role in the pathogenesis and progression of many complications associated with TS [63] (Table 2). This underscores the importance of understanding oxidative stress mechanisms and exploring antioxidant-based strategies, particularly in pediatric and adolescent populations.
TS exhibits a distinct oxidative stress response compared to normal 46,XX cells. Transcriptomic analysis reveals that over 350 transcripts are differentially expressed in 45,X cells under mild oxidative stress conditions. These cells show increased susceptibility to oxidative stress and altered regulation of stress-related molecular pathways. The differential expression of transcription factors in 45,X cells suggests that the altered stress response may contribute to the various phenotypes and comorbidities observed in TS, such as short stature, osteoporosis, and ovarian malfunction [63]. Furthermore, the X-chromosome plays a significant role in managing oxidative stress through mechanisms involving the X-chromosome-linked inhibitor of apoptosis protein (XIAP), encoded by the XIAP gene, and its regulation of mitochondrial antioxidants. XIAP has been shown to play a crucial role in reducing oxidative stress in brain injury models, such as hypoxia–ischemia and cerebral irradiation. Its overexpression leads to a significant decrease in oxidative stress markers by upregulating certain mitochondrial antioxidants, including SOD-2. This upregulation helps in reducing cytochrome c release from mitochondria, thereby mitigating oxidative damage [72]. An additional factor potentially influencing oxidative stress in TS is the embryonic origin of the condition. TS cases derived from 46,XY embryos may differ in their oxidative stress response compared to those derived from 46,XX embryos, given differences in Y-linked gene expression during early development. However, current evidence addressing this distinction is scarce, and further research is needed to determine whether embryonic origin impacts oxidative stress susceptibility in TS.
Estrogen deficiency, a hallmark of TS due to ovarian dysgenesis, has been implicated in increased oxidative stress [73]. Estrogens exert antioxidant effects by upregulating the expression of antioxidant enzymes like SOD and catalase, as well as by directly scavenging free radicals [74]. The absence of these protective effects in TS may lead to a higher oxidative burden, particularly during adolescence, a critical period for hormonal regulation. Based on the results of studies on estrogen replacement therapy (ERT) in postmenopausal women [75], it is plausible to state that it could partially restore antioxidant defenses, particularly when initiated early in adolescence; however, its role in oxidative stress management warrants further investigation [76].
The synergy between ERT and antioxidant therapies likely involves estrogen-mediated activation of redox-sensitive transcription factors (e.g., Nrf2), which upregulate SOD, glutathione peroxidase (GPx), and catalase [77]. Estrogens also stabilize mitochondrial membranes, reducing ROS leakage [78]. ERT may thus potentiate the effects of dietary antioxidants by enhancing endogenous antioxidant gene expression [79].
Girls with TS are predisposed to congenital heart defects (e.g., bicuspid aortic valve, coarctation of the aorta) and acquired cardiovascular complications, such as hypertension and aortic dilation or dissection [71]. These conditions are closely associated with oxidative stress, which promotes endothelial dysfunction, inflammation, and vascular remodeling. Elevated ROS in vascular tissues induce endothelial dysfunction by impairing NO bioavailability and promoting inflammation, thrombosis, and arterial stiffness [80]. Soto and colleagues demonstrated significant alterations in antioxidant enzyme activities, particularly in SOD isoforms, in the aortic tissue of patients with TS, suggesting a compensatory response to oxidative stress. Moreover, an impaired antioxidant defense system was depicted in the presence of heightened oxidative damage with increased levels of lipid peroxidation markers such as MDA. A decreased expression of endothelial NO synthase, which could contribute to endothelial dysfunction and aortic complications, was also found [64].
Patients with TS are at increased risk for metabolic syndrome, including insulin resistance, dyslipidemia, and central adiposity, even in pediatric age [81]. Oxidative stress has a well-established role in impairing insulin signaling pathways and contributing to β-cell dysfunction [82]. Moreover, adipose tissue is a significant source of ROS, perpetuating oxidative damage in metabolic pathways and contributing to chronic low-grade inflammation [83]. This is particularly concerning during adolescence, as the onset of metabolic complications often coincides with puberty. The interplay between oxidative stress and chronic low-grade inflammation in TS exacerbates metabolic risks, making early intervention essential.
Cognitive challenges and neurodevelopmental differences, particularly in visuospatial processing and executive function, are frequent in TS [71]. Emerging research suggests that oxidative stress in the brain, through mitochondrial dysfunction and neuroinflammation, in the form of ROS accumulation in neural tissues, may contribute to these deficits [84]. Pediatric populations may be particularly vulnerable, as the developing brain is highly sensitive to oxidative damage [85,86,87].
5.2. Klinefelter Syndrome
KS is characterized by the presence of one or more extra X chromosomes in males (47,XXY being the most common karyotype) and affects approximately 1 in 500–660 live male births [88]. While KS is often recognized for its impact on fertility and hypogonadism, it also involves a broad range of systemic complications, including metabolic syndrome, cardiovascular diseases, and neurodevelopmental challenges, with numerous biomarkers potentially predictive or prognostic of alterations strictly connected to the syndrome [89,90,91,92]. Oxidative stress has emerged as a key contributor to these complications (Table 2). The additional genetic material can lead to increased ROS production and reduced antioxidant capacity, contributing to defining the typical phenotype of KS [66]. Additionally, oxidative stress is linked to male infertility through the generation of excess free radicals by spermatozoa, which may result from defective Sertoli cell function, a characteristic shared with KS. This defect leads to inadequate removal of residual cytoplasm during sperm maturation, leaving excess cytoplasmic components that enhance free radical production. One potential mechanism involves elevated glucose-6-phosphate dehydrogenase levels, which increase NADPH production, fueling free radical generation via sperm NOX [65]. However, further research is needed to clarify the exact mechanisms involved.
The oxidative stress in patients with KS can further complicate their clinical management and overall health outcomes [80]. Understanding its role in KS, particularly during the pediatric and adolescent years, is critical for developing targeted antioxidant strategies that may mitigate long-term health risks [93].
Testosterone plays an important role in maintaining redox balance through its influence on antioxidant enzyme activity, with both pro-oxidant and antioxidant effects [94]. Research indicates that testosterone modulates the endogenous antioxidant systems, impacting the activity and expression of cellular antioxidants. For instance, testosterone signaling via the androgen receptor has been shown to have both pro- and antioxidant effects on the heart, influencing the natural antioxidant system glutathione [95]. Furthermore, androgen deprivation in the rat prostate has been shown to induce oxidative stress through the elevation of ROS and the reduction of key ROS-detoxifying enzymes such as SOD and GPx. Testosterone replacement therapy (TRT) in these cases partially restored the expression of these antioxidant enzymes, indicating its potential therapeutic role in reducing oxidative stress [96]. In cases of male secondary hypogonadism, TRT has been observed to significantly enhance levels of coenzyme Q10 and total antioxidant capacity, suggesting an interrelationship between different antioxidants and a potential reduction in oxidative stress [97]. Additionally, the management of oxidative stress in male hypogonadism, particularly in the context of age-related non-communicable chronic diseases, may benefit from conventional hormone replacement therapy, dietary antioxidant supplementation, and lifestyle changes [98]. In summary, testosterone plays a significant role in regulating oxidative stress and antioxidant defenses. Hypogonadism, as seen in KS, leads to reduced antioxidant capacity. The hypogonadism observed in KS leads to reduced antioxidant defenses, such as decreased levels of SOD and GPx, contributing to heightened oxidative stress, but TRT may offer a partial restoration of these defenses, highlighting its potential therapeutic benefits. Testosterone replacement appears to exert its antioxidant effects by also modulating NOX activity and enhancing the expression of mitochondrial antioxidant enzymes. This hormonal–oxidative stress interaction may restore redox homeostasis in KS, particularly in tissues such as testis, adipose, and neural tissue [96]. Co-administration of antioxidants with TRT may optimize therapeutic outcomes.
KS is strongly associated with obesity and dyslipidemia, which are linked to oxidative stress. As abovementioned, excess adipose tissue generates ROS, which exacerbates systemic inflammation and metabolic dysfunction [83]. Pediatric patients with KS often display early signs of metabolic syndrome, highlighting the need for timely interventions [99]. Endothelial dysfunction and insulin resistance are also common in KS, even in very young subjects, and contribute to the elevated risk of cardiovascular disease [100]. Oxidative damage to endothelial cells promotes inflammation, thrombosis, and vascular remodeling, amplifying cardiovascular risks even in children and adolescents [80].
Neurodevelopmental challenges, including deficits in executive function, language processing, and social cognition, are frequently seen in pediatric patients with KS [90,101,102]. Mitochondrial dysfunction and oxidative stress in neural tissues may underlie these deficits, particularly during critical periods of brain development in childhood and adolescence. The aforementioned neurological alterations resulting from oxidative stress in TS also apply to KS [84,85,86,87].
5.3. 47,XXX, 47,XYY, and HGAs
47,XXX (or triple X or trisomy X) syndrome affects approximately 1 in 1000 female births. While many individuals with this condition are asymptomatic, children with 47,XXX syndrome commonly present with language and motor delays, hypotonia, cognitive deficits, learning disabilities, and psychological disorders including depression, anxiety, and attention deficits [68]. 47,XYY (or Jacob) syndrome, which occurs in approximately 1 in 1000 male births, is also often underdiagnosed [103]. Common clinical features include tall stature, learning disabilities, mild developmental delays, and behavioral problems, including a higher prevalence of attention-deficit/hyperactivity disorder and autism spectrum disorders [104]. Overexpression of the genes escaping X inactivation might account for the phenotypic differences observed in 47,XXX syndrome, just as the 47,XYY phenotype could primarily be attributed to the abnormal gene dosage resulting from the extra Y chromosome [105]. Tetrasomies and pentasomies with supernumerary X and/or Y chromosomes are extremely rare and tend to present with more severe clinical features, including intellectual disability, growth abnormalities, and significant neuropsychiatric disturbances [55,69,106,107].
While clinical descriptions of 47,XXX and 47,XYY syndromes are relatively well established, evidence regarding oxidative stress in these and HGAs remains sparse and largely speculative. Due to the rarity of these conditions and the paucity of dedicated studies, the following considerations are primarily hypothetical and should be interpreted with caution.
Though oxidative stress has not represented a primary focus in studies of HGAs, 47,XXX, and 47,XYY syndromes, emerging research indicates that the relationship between oxidative stress and SCAs can be described as bidirectional, where oxidative stress both contributes to and is exacerbated by aneuploidies Some proposed mechanisms, based primarily on experimental and in vitro models, suggest that oxidative stress induces ploidy changes through disruption of spindle assembly checkpoint function, energy shortages, centrosome over-replication, and microtubule-kinetochore disorganization [108].
A recent hypothesis suggests that oxidative modifications of proteins crucial for mitosis and the cytoskeleton might lead to disruptions in the cell cycle and interfere with cell division [109]. ROS have been shown to alter structural proteins such as vimentin, actin, and tubulin, which can disrupt the formation of the spindle apparatus, cause chromosome misalignment, and result in failure of cytokinesis [110,111]. On the flip side, the presence of aneuploidy, including SCAs, creates cellular imbalances that might contribute to further oxidative stress. Cells with abnormal chromosome numbers may experience increased metabolic stress and mitochondrial dysfunction; for example, the extra genetic material in individuals with trisomies (or tetrasomies or pentasomies) may lead to cellular stress, increasing the production of ROS and further aggravating oxidative damage [112]. This may suggest a potentially vicious cycle in which oxidative stress contributes to chromosomal instability, and chromosomal abnormalities in turn promote oxidative damage. This reciprocal relationship suggests that both oxidative stress and SCAs can influence each other, potentially amplifying the effects of each and contributing to the cellular dysfunction observed in these conditions.
While these mechanisms are biologically plausible, the limited number of dedicated studies and the small sample sizes available prevent definitive conclusions. Phenotypic variability, mosaicism, and environmental modifiers further complicate interpretation. Given the speculative nature of current hypotheses, further research using well-powered, phenotype-stratified studies is essential to determine whether oxidative stress plays a causative or merely correlative role in HGAs.
5.4. Sex Chromosome-Linked and Autosomal Gene Contributors to Oxidative Stress in SCAs
Understanding the genetic contributors to oxidative stress in SCAs is crucial for developing targeted therapeutic strategies. Among the mechanisms discussed, X-linked genes, such as XIAP, are key players in modulating oxidative stress. XIAP plays an essential role in inhibiting apoptosis by directly binding to and inhibiting caspases, thereby protecting cells from oxidative damage. Reduced expression of XIAP has been associated with increased vulnerability to oxidative stress in TS, as the protective effect of this gene is diminished due to the lack of a second X chromosome [22,72]. In addition to caspase inhibition, XIAP also promotes the expression of antioxidant enzymes such as SOD-2, with its dysfunction being particularly relevant in pediatric tissues, especially the brain and cardiovascular system [63] (Table 3).
Table 3.
Oxidative stress-related genes differentially expressed in SCAs: function, redox involvement, and pathological relevance. Abbreviations: ADHD, attention deficit hyperactivity disorder; ADP, adenosine diphosphate; ATP, adenosine triphosphate; BMP, bone morphogenetic protein; DUOX, dual oxidase; H2O2, hydrogen peroxide; KS, Klinefelter syndrome; MAPK, mitogen-activated protein kinase; NADPH, nicotinamide adenine dinucleotide phosphate; ROS, reactive oxygen species; SOD-2, superoxide dismutase-2; TF, transcription factor; TS, Turner syndrome.
In addition to XIAP, several other X-linked genes are implicated in oxidative stress regulation. SLC25A5, which encodes a mitochondrial adenine nucleotide translocator, is critical for ATP/ADP exchange across the mitochondrial membrane. Dysregulation of SLC25A5 leads to an imbalance in mitochondrial function and excessive production of ROS, potentially contributing to oxidative stress in SCAs [23,113]. This pattern has been confirmed in fibroblast models from Turner syndrome patients [63]. Moreover, mitochondrial dysfunction is frequently reported in SCAs [20], and the interplay between mitochondrial and sex chromosome-linked genetic factors remains poorly understood. Emerging evidence suggests that sex chromosome-linked genes, such as SLC25A5, may mediate mitochondrial dysfunction in SCAs [23,113].
Transcription factors encoded by EGR1, KLF4, and SOX11 are also differentially expressed in X monosomy and participate in multiple oxidative stress-related pathways. EGR1 is associated with cell proliferation and apoptosis regulation and acts as a central hub in Turner-associated gene networks. KLF4 and KLF2, which encode two mechanosensitive transcription factors, regulate endothelial homeostasis and antioxidant responses; their downregulation may contribute to the increased vascular risk observed in SCAs. Meanwhile, SOX11 is involved in osteogenic and neurodevelopmental processes and is overexpressed in TS, where it may underlie characteristic skeletal anomalies [63].
The interplay among the Y chromosome, its genes, and oxidative stress is complex and multifaceted. While significant progress has been made in understanding their roles in male infertility and other health issues, further research is essential to unravel the underlying mechanisms and develop effective interventions. The Y chromosome, through genes such as sex-determining region Y (SRY), regulates the expression of X-linked genes like monoamine oxidase A (MAO-A), which plays a key role in brain function and development. This SRY-mediated regulation of MAO A highlights the broader influence of the Y chromosome beyond its role in reproduction, potentially affecting cellular responses to oxidative stress and contributing to sex-specific differences in redox homeostasis [24]. MAO-A generates H2O2 during the catabolism of catecholamines [114], and chronic accumulation of ROS may contribute to neurodevelopmental disorders such as attention-deficit/hyperactivity disorder and autism spectrum disorders in 47,XYY and 47,XXY individuals.
Notably, oxidative stress in SCAs is not solely influenced by genes on the sex chromosomes. Mitochondrial function plays a pivotal role in ROS production, and mitochondrial dysfunction is frequently reported in SCAs [20]. Genes not located on the sex chromosomes also contribute significantly to oxidative homeostasis. One such gene is GPX4, which encodes GPx-4, an antioxidant enzyme that prevents ferroptosis by reducing phospholipid peroxides. Although not SCA-specific, its activity is crucial in protecting tissues such as the brain and testes, and its dysfunction may be particularly relevant in KS, where oxidative stress may exacerbate testicular and cognitive impairment [115].
Moreover, the upregulation of NOX4, a member of the NADPH oxidase family responsible for H2O2 production, has been documented in both 45,X and 46,XX cells under oxidative stress, suggesting a role in metabolic dysregulation and thyroid dysfunction [63]. Other autosomal stress-responsive genes of interest include GADD45B, which modulates mitogen-activated protein kinase (MAPK) signaling and is implicated in inflammation and cancer progression, and DUOXA1, a maturation factor for dual oxidase (DUOX) enzymes involved in thyroid ROS metabolism. Downregulation of DUOXA1 may contribute to the thyroid abnormalities frequently reported in Turner syndrome [63].
Furthermore, oxidative stress has been identified as a potential underlying cause of idiopathic premature ovarian failure (POF). Women with POF—where X-chromosome anomalies play a significant role in the pathogenesis—exhibit significantly higher levels of ROS compared to controls, suggesting that oxidative stress may contribute to ovarian dysfunction [25]. It is also worth noting that even a slight increase in ROS levels during early development may elevate the risk of SCAs. In mouse models, increased oxidative stress during the first mitotic divisions has been shown to promote chromosomal missegregation, highlighting the importance of centromeric proteins and genomic stability in SCA pathogenesis [21].
The connection between chromosomal aneuploidy and oxidative stress is increasingly supported by transcriptomic and functional studies showing that gene dosage alterations directly impact redox-sensitive pathways [116]. In SCAs, this relationship is not merely associative but may be causal, as several X- and Y-linked genes implicated in oxidative homeostasis escape X-inactivation or are overexpressed due to chromosomal gain [22,23,24]. A hypothetical model integrating genetic, mitochondrial, and hormonal factors suggests a direct mechanistic link between SCAs and oxidative stress imbalance. The following pathway is proposed (Figure 2):
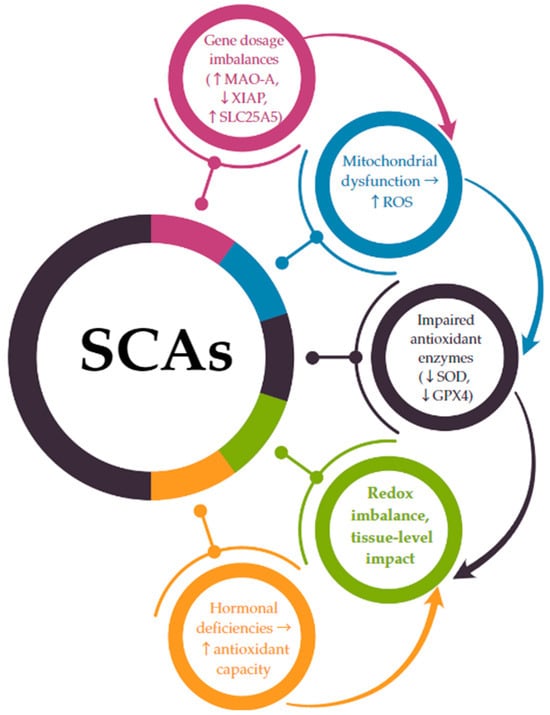
Figure 2.
A mechanistic link between SCA-related gene dosage and oxidative stress imbalance. ↑ = increase; ↓ = decrease; → = leads to.
- -
- SCAs lead to gene dosage imbalances due to monosomy (e.g., 45,X) or the presence of supernumerary chromosomes (e.g., 47,XXY; 47,XYY).
- -
- Genes that escape X-inactivation or are overexpressed on the Y chromosome (such as MAO-A, SLC25A5, and XIAP) are dysregulated, disrupting mitochondrial function and increasing ROS production.
- -
- These changes are compounded by a reduced antioxidant enzymatic defense (e.g., decreased expression of SOD, GPX4).
- -
- The resulting redox imbalance contributes to cellular and systemic dysfunction, particularly in cardiovascular, metabolic, and neurodevelopmental systems.
Hormonal deficiencies, notably estrogen in TS and testosterone in KS, exacerbate the impaired redox balance by failing to adequately support antioxidant pathways.
While some aspects of this model remain hypothetical, the accumulation of gene expression, molecular, and clinical data offers a compelling basis for future translational research. Future research should aim to clarify the contributions of these sex chromosome-linked and autosomal genes to oxidative stress and explore potential therapeutic interventions. Antioxidant therapies, such as N-acetylcysteine and vitamin E, may provide a means to mitigate oxidative damage in individuals with SCAs. Additionally, personalized approaches targeting specific genetic vulnerabilities, such as XIAP or SLC25A5, could improve outcomes in these patients.
5.5. Comparative Insights: Oxidative Stress in Other Pediatric Genetic Syndromes
While oxidative stress appears to be a hallmark of SCAs due to gene dosage imbalance and hormonal deficiencies, it is also a common pathogenic feature in various autosomal genetic syndromes, often contributing to the clinical phenotype through mitochondrial dysfunction, inflammation, and impaired antioxidant responses.
A well-characterized example is Down syndrome, in which the triplication of chromosome 21 leads to overexpression of genes involved in redox regulation. Notably, the SOD1 gene, encoding superoxide dismutase 1, is located on chromosome 21 and is overexpressed in these individuals. This results in an increased dismutation of O2•− into H2O2, which, if not adequately neutralized by catalase or GPx, leads to the accumulation of ROS and oxidative damage to cellular components such as DNA, proteins, and lipids [18,19]. Mitochondrial dysfunction is also a major contributor to oxidative stress in Down syndrome and has been implicated in the early onset of Alzheimer’s disease and neurodegeneration observed in this population [18].
Another illustrative condition is Williams–Beuren syndrome, caused by a microdeletion at 7q11.23 and characterized by cardiovascular abnormalities, connective tissue disorders, and cognitive deficits. Recent evidence suggests that elastin haploinsufficiency contributes to vascular oxidative stress through increased NOX activity and reduced endothelial NO availability, resulting in endothelial dysfunction and inflammation [117,118]. Elevated markers of lipid peroxidation and reduced glutathione levels have been reported in both serum and vascular tissues of these patients, highlighting a systemic oxidative imbalance [119].
These comparisons underscore that oxidative stress is not exclusive to SCAs but rather a shared pathogenic mechanism across various genetic syndromes. However, the molecular underpinnings differ considerably. In Down syndrome, the imbalance stems from excess antioxidant enzyme activity and mitochondrial vulnerability, while in Williams–Beuren syndrome, the loss of vascular structural integrity and redox regulation is central. In SCAs, oxidative stress arises from altered expression of sex chromosome-linked genes and hormonal dysregulation. Understanding these differences is essential for developing syndrome-specific antioxidant strategies.
6. Supplementing Dietary Antioxidants as a Therapeutic Strategy
While the underlying mechanisms and clinical presentations differ between syndromes, antioxidant dietary strategies can provide a common therapeutic approach to mitigate oxidative damage and improve health outcomes in pediatric and adolescent patients [120]. However, it is important to recognize that nutritional needs, antioxidant metabolism, and optimal dosages may vary by age and sex (and karyotype) (Table 4). To enhance clinical applicability, dietary reference values (DRVs) for pediatric populations are included wherever available, particularly highlighting differences between males and females. While precise recommendations tailored to each specific SCA remain under-researched, these general guidelines can provide a foundation for appropriate supplementation in clinical practice.
Table 4.
Suggested antioxidant approaches by SCA type and age group. Abbreviations: HGAs, high-grade sex chromosome aneuploidies.
Vitamin C, a potent water-soluble antioxidant, neutralizes ROS and regenerates other antioxidants like vitamin E [121]. It supports vascular health by enhancing endothelial function and reducing inflammation [122], which is crucial for both TS, where cardiovascular anomalies such as aortic coarctation are prevalent [123], and KS, which is associated with endothelial dysfunction and increased cardiovascular risks [124]. Supplementation with vitamin C may also help address metabolic complications, including insulin resistance seen in both syndromes [125,126]. The recommended dietary allowance (RDA) for vitamin C varies by age and sex: 1–3 years: 15 mg/day; 4–8 years: 25 mg/day; 9–13 years: 45 mg/day; 14–18 years: 75 mg/day (males), 65 mg/day (females) [127,128]. Studies in other populations with similar oxidative stress profiles suggest that vitamin C supplementation improves vascular health and reduces oxidative biomarkers [129].
As a lipid-soluble antioxidant, vitamin E protects cell membranes from lipid peroxidation [130]. It is particularly relevant for reducing atherosclerosis risks in TS, where lipid metabolism is often disrupted [131], and improving insulin sensitivity in KS [100]. Combining vitamins C and E could enhance their benefits [132], offering a synergistic approach to managing oxidative stress-related cardiovascular and metabolic complications in both aneuploidies. The RDA for vitamin E is as follows: 1–3 years: 6 mg/day; 4–8 years: 7 mg/day; 9–13 years: 11 mg/day; 14–18 years: 15 mg/day (both sexes) [128]. In addition, increased intake of antioxidants, including vitamins C and E, beyond the usual dietary and supplemental levels has been linked to higher sperm count and improved motility in a sample of healthy non-smoking men, attenuating the impact of age on sperm motility, to some extent [133]. This evidence shows that antioxidant intake may be associated with semen quality in healthy males, a relevant consideration for KS.
Glutathione, a key intracellular antioxidant, plays a central role in maintaining redox balance [134]. Reduced glutathione levels have been implicated in oxidative damage [135]. Therefore, supplementation with N-acetylcysteine, a precursor of glutathione, may boost endogenous antioxidant defenses, helping to mitigate oxidative stress linked to neurocognitive deficits, vascular damage, and metabolic dysfunction [136,137]. While there are no specific DRVs for N-acetylcysteine, its pediatric use has been studied in conditions such as cystic fibrosis and respiratory disorders, suggesting a potential role in SCAs where oxidative stress is heightened.
Dietary polyphenols, such as flavonoids and catechins found in fruits, vegetables, and green tea, offer strong anti-inflammatory and antioxidant effects [138,139,140,141,142]. Their role in reducing adipose-related inflammation, improving endothelial function, and protecting against neurodegeneration makes them a promising dietary intervention for SCAs [143,144,145]. Encouraging polyphenol-rich diets in pediatric and adolescent patients may serve as a practical and non-invasive antioxidant strategy [146]. However, optimal intake levels remain undefined for children, and more research is needed to establish specific guidelines.
Carotenoids, a class of lipid-soluble antioxidants found in colorful fruits and vegetables, provide significant protection against oxidative damage by scavenging free radicals and quenching singlet oxygen [147]. Among them, beta-carotene, lutein, and zeaxanthin are particularly well-studied for their roles in maintaining cellular health and preventing oxidative damage to lipids, proteins, and DNA [148]. Beta-carotene serves as a precursor to vitamin A, contributing to immune function and vision health, while also reducing oxidative stress markers [149]. Lutein and zeaxanthin are crucial for protecting neural tissues, particularly the retina, against oxidative injury [150]. These carotenoids may offer added benefits for individuals with SCAs by targeting oxidative stress-related complications, such as neurodevelopmental deficits or cardiovascular risks [151]. The RDA for vitamin A (beta-carotene equivalent) in pediatrics is as follows: 1–3 years: 300 µg/day; 4–8 years: 400 µg/day; 9–13 years: 600 µg/day; 14–18 years: 900 µg/day (males), 700 µg/day (females).
Melatonin, beyond its role in regulating circadian rhythms, acts as a powerful antioxidant that scavenges ROS and enhances endogenous antioxidant enzyme activity [152]. Its neuroprotective effects could be particularly beneficial for addressing cognitive and psychological challenges in SCAs in pediatric populations [153,154]. For instance, melatonin supplementation may help mitigate oxidative damage in neural tissues [155,156] implicated in the neurodevelopmental and cognitive difficulties in SCAs. Moreover, studies have indicated distinct alterations in melatonin secretion in both KS, characterized by potential sleep disorders with elevated baseline melatonin levels that decrease with testosterone therapy, and TS, which shows abnormal day–night rhythms and desynchronized melatonin patterns that are unaffected by estrogen treatment [157,158,159,160,161,162]. While melatonin use in pediatrics remains an area of ongoing research, its safety profile in children has been well-documented in sleep disorders, suggesting potential applicability for SCA-related neurodevelopmental issues.
In general, encouraging balanced diets rich in natural antioxidants, including vitamins, minerals, and polyphenols, can provide a sustainable and holistic approach to reducing oxidative stress [163]. A Mediterranean-style diet, emphasizing fruits, vegetables, whole grains, and healthy fats, may be particularly effective in pediatric populations with SCAs [164]. Additionally, pairing antioxidants with syndrome-specific therapies can enhance outcomes. For instance, combining antioxidant supplementation with growth hormone and ERT in TS or TRT in KS may address both oxidative stress and the hormonal imbalances characteristic of each syndrome [165,166]. Early intervention may therefore mitigate neurocognitive issues, as well as reduce the risk of long-term cardiovascular and metabolic complications [167,168]. Among the various antioxidant strategies explored, we propose the following prioritization—summarized in Table 5—based on current evidence of efficacy, safety in pediatric populations, and relevance to SCA pathophysiology.
Table 5.
Prioritization of antioxidant interventions in pediatric SCAs. Abbreviations: ROS, reactive oxygen species.
7. Conclusions
Oxidative stress emerges as a central, yet modifiable, contributor to the progression of SCAs in pediatric populations. This review underscores the importance of early antioxidant strategies, particularly in conjunction with syndrome-specific hormonal therapies, to mitigate long-term neurocognitive, metabolic, and cardiovascular complications. Despite promising evidence, several research gaps remain. Priority areas include longitudinal studies on antioxidant use in pediatric SCA cohorts; exploration of gene–antioxidant interactions, particularly involving XIAP, SLC25A5, and MAO-A; standardization of pediatric antioxidant dosing protocols; and trials assessing combined antioxidant and hormonal therapy in adolescents. A precision medicine approach integrating genetic, metabolic, and clinical factors is warranted to optimize care for children with SCAs.
Author Contributions
Conceptualization, R.P. and F.P.; methodology, R.P., L.T., G.F., S.V. and M.F.; writing—original draft preparation, R.P., F.P., F.T. and B.D.; writing—review and editing, L.L., I.P., G.F., M.F. and L.T.; supervision, I.P., S.V., F.C., M.F. and L.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors thank the IBBC-CNR and the Sapienza University of Rome in Rome, Italy.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Tartaglia, N.R.; Howell, S.; Sutherland, A.; Wilson, R.; Wilson, L. A Review of Trisomy X (47,XXX). Orphanet J. Rare Dis. 2010, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, M.; Rochira, V.; Pasquali, D.; Balercia, G.; Jannini, E.A.; Ferlin, A.; On behalf of the Klinefelter ItaliaN Group (KING). Klinefelter Syndrome (KS): Genetics, Clinical Phenotype and Hypogonadism. J. Endocrinol. Investig. 2017, 40, 123–134. [Google Scholar] [CrossRef]
- Profeta, G.; Micangeli, G.; Tarani, F.; Paparella, R.; Ferraguti, G.; Spaziani, M.; Isidori, A.M.; Menghi, M.; Ceccanti, M.; Fiore, M.; et al. Sexual Developmental Disorders in Pediatrics. Clin. Ter. 2022, 173, 475–488. [Google Scholar] [CrossRef]
- Gravholt, C.H.; Andersen, N.H.; Conway, G.S.; Dekkers, O.M.; Geffner, M.E.; Klein, K.O.; Lin, A.E.; Mauras, N.; Quigley, C.A.; Rubin, K.; et al. Clinical Practice Guidelines for the Care of Girls and Women with Turner Syndrome: Proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur. J. Endocrinol. 2017, 177, G1–G70. [Google Scholar] [CrossRef]
- Visootsak, J.; Graham, J.M. Klinefelter Syndrome and Other Sex Chromosomal Aneuploidies. Orphanet J. Rare Dis. 2006, 1, 42. [Google Scholar] [CrossRef] [PubMed]
- Gravholt, C.H.; Jensen, A.S.; Høst, C.; Bojesen, A. Body Composition, Metabolic Syndrome and Type 2 Diabetes in Klinefelter Syndrome. Acta Paediatr. 2011, 100, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Sybert, V.P.; McCauley, E. Turner’s Syndrome. N. Engl. J. Med. 2004, 351, 1227–1238. [Google Scholar] [CrossRef]
- Ricciardi, G.; Cammisa, L.; Bove, R.; Picchiotti, G.; Spaziani, M.; Isidori, A.M.; Aceti, F.; Giacchetti, N.; Romani, M.; Sogos, C. Clinical, Cognitive and Neurodevelopmental Profile in Tetrasomies and Pentasomies: A Systematic Review. Children 2022, 9, 1719. [Google Scholar] [CrossRef]
- Song, J.-P.; Jiang, Y.-F.; Gao, T.-X.-Z.; Yao, Y.-Y.; Liu, L.-J.; Xu, R.-H.; Yi, M.-Q.; Yu, C.-J.; Wang, W.-P.; Li, H. Performance of Non-Invasive Prenatal Screening for Sex Chromosome Aneuploidies and Parental Decision-Making. Chin. Med. J. Engl. 2020, 133, 1617–1619. [Google Scholar] [CrossRef]
- Micangeli, G.; Paparella, R.; Tarani, F.; Menghi, M.; Ferraguti, G.; Carlomagno, F.; Spaziani, M.; Pucarelli, I.; Greco, A.; Fiore, M.; et al. Clinical Management and Therapy of Precocious Puberty in the Sapienza University Pediatrics Hospital of Rome, Italy. Children 2023, 10, 1672. [Google Scholar] [CrossRef]
- Micangeli, G.; Profeta, G.; Colloridi, F.; Pirro, F.; Tarani, F.; Ferraguti, G.; Spaziani, M.; Isidori, A.M.; Menghi, M.; Fiore, M.; et al. The Role of the Pediatrician in the Management of the Child and Adolescent with Gender Dysphoria. Ital. J. Pediatr. 2023, 49, 71. [Google Scholar] [CrossRef] [PubMed]
- Micangeli, G.; Menghi, M.; Profeta, G.; Tarani, F.; Mariani, A.; Petrella, C.; Barbato, C.; Ferraguti, G.; Ceccanti, M.; Tarani, L.; et al. The Impact of Oxidative Stress on Pediatrics Syndromes. Antioxidants 2022, 11, 1983. [Google Scholar] [CrossRef]
- Terracina, S.; Tarani, L.; Ceccanti, M.; Vitali, M.; Francati, S.; Lucarelli, M.; Venditti, S.; Verdone, L.; Ferraguti, G.; Fiore, M. The Impact of Oxidative Stress on the Epigenetics of Fetal Alcohol Spectrum Disorders. Antioxidants 2024, 13, 410. [Google Scholar] [CrossRef] [PubMed]
- Tremellen, K. Oxidative Stress and Male Infertility—A Clinical Perspective. Hum. Reprod. Update 2008, 14, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Oroian, B.A.; Ciobica, A.; Timofte, D.; Stefanescu, C.; Serban, I.L. New Metabolic, Digestive, and Oxidative Stress-Related Manifestations Associated with Posttraumatic Stress Disorder. Oxid. Med. Cell. Longev. 2021, 2021, 5599265. [Google Scholar] [CrossRef]
- Shkurat, M.A.; Mashkina, E.V.; Milyutina, N.P.; Shkurat, T.P. The Role of Polymorphism of Redox-Sensitive Genes in the Mechanisms of Oxidative Stress in Obesity and Metabolic Diseases. Ecol. Genet. 2023, 21, 261–287. [Google Scholar] [CrossRef]
- Ferraguti, G.; Terracina, S.; Micangeli, G.; Lucarelli, M.; Tarani, L.; Ceccanti, M.; Spaziani, M.; D’Orazi, V.; Petrella, C.; Fiore, M. NGF and BDNF in Pediatrics Syndromes. Neurosci. Biobehav. Rev. 2023, 145, 105015. [Google Scholar] [CrossRef]
- Lott, I.T.; Head, E. Alzheimer Disease and Down Syndrome: Factors in Pathogenesis. Neurobiol. Aging 2005, 26, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Pagano, G.; Castello, G. Oxidative Stress and Mitochondrial Dysfunction in Down Syndrome. In Neurodegenerative Diseases; Ahmad, S.I., Ed.; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2012; Volume 724, pp. 291–299. ISBN 978-1-4614-0652-5. [Google Scholar]
- Pomatto, L.C.D.; Carney, C.; Shen, B.; Wong, S.; Halaszynski, K.; Salomon, M.P.; Davies, K.J.A.; Tower, J. The Mitochondrial Lon Protease Is Required for Age-Specific and Sex-Specific Adaptation to Oxidative Stress. Curr. Biol. 2017, 27, 1–15. [Google Scholar] [CrossRef]
- Huang, Y.; Ha, S.; Li, Z.; Li, J.; Xiao, W. CHK1-CENP B/MAD2 Is Associated with Mild Oxidative Damage-Induced Sex Chromosome Aneuploidy of Male Mouse Embryos during in Vitro Fertilization. Free Radic. Biol. Med. 2019, 137, 181–193. [Google Scholar] [CrossRef]
- Jevalikar, G.S.; Zacharin, M.; White, M.; Yau, S.W.; Li, W.; Ijspeert, C.; Russo, V.C.; Werther, G.A.; Sabin, M.A. Turner Syndrome Patients with Bicuspid Aortic Valves and Renal Malformations Exhibit Abnormal Expression of X-Linked Inhibitor of Apoptosis Protein (XIAP). J. Pediatr. Endocrinol. Metab. 2015, 28, 1203–1208. [Google Scholar] [CrossRef]
- Clémençon, B.; Babot, M.; Trézéguet, V. The Mitochondrial ADP/ATP Carrier (SLC25 Family): Pathological Implications of Its Dysfunction. Mol. Aspects Med. 2013, 34, 485–493. [Google Scholar] [CrossRef]
- Wu, J.B.; Chen, K.; Li, Y.; Lau, Y.-F.C.; Shih, J.C. Regulation of Monoamine Oxidase A by the SRY Gene on the Y Chromosome. FASEB J. 2009, 23, 4029–4038. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Pathak, D.; Venkatesh, S.; Kriplani, A.; Ammini, A.C.; Dada, R. Chromosomal Abnormalities & Oxidative Stress in Women with Premature Ovarian Failure (POF). Indian J. Med. Res. 2012, 135, 92–97. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Demirci-Çekiç, S.; Özkan, G.; Avan, A.N.; Uzunboy, S.; Çapanoğlu, E.; Apak, R. Biomarkers of Oxidative Stress and Antioxidant Defense. J. Pharm. Biomed. Anal. 2022, 209, 114477. [Google Scholar] [CrossRef]
- Andrés, C.M.C.; Pérez De La Lastra, J.M.; Andrés Juan, C.; Plou, F.J.; Pérez-Lebeña, E. Superoxide Anion Chemistry—Its Role at the Core of the Innate Immunity. Int. J. Mol. Sci. 2023, 24, 1841. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen Peroxide as a Central Redox Signaling Molecule in Physiological Oxidative Stress: Oxidative Eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Leser, M.; Chapman, J.R.; Khine, M.; Pegan, J.; Law, M.; Makkaoui, M.E.; Ueberheide, B.M.; Brenowitz, M. Chemical Generation of Hydroxyl Radical for Oxidative ‘Footprinting’. Protein Pept. Lett. 2019, 26, 61–69. [Google Scholar] [CrossRef]
- Di Mascio, P.; Martinez, G.R.; Miyamoto, S.; Ronsein, G.E.; Medeiros, M.H.G.; Cadet, J. Singlet Molecular Oxygen Reactions with Nucleic Acids, Lipids, and Proteins. Chem. Rev. 2019, 119, 2043–2086. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Sueta, G.; Campolo, N.; Trujillo, M.; Bartesaghi, S.; Carballal, S.; Romero, N.; Alvarez, B.; Radi, R. Biochemistry of Peroxynitrite and Protein Tyrosine Nitration. Chem. Rev. 2018, 118, 1338–1408. [Google Scholar] [CrossRef] [PubMed]
- Voziyan, P.A.; Yazlovitskaya, E.M. Reactive Oxygen Species. J. Bioequivalence Bioavailab. 2014, 6. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Reconciling the Chemistry and Biology of Reactive Oxygen Species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef]
- Forman, H.J.; Maiorino, M.; Ursini, F. Signaling Functions of Reactive Oxygen Species. Biochemistry 2010, 49, 835–842. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Kettle, A.J. Redox Reactions and Microbial Killing in the Neutrophil Phagosome. Antioxid. Redox Signal. 2013, 18, 642–660. [Google Scholar] [CrossRef]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules 2020, 10, 320. [Google Scholar] [CrossRef]
- Dimauro, I.; Mercatelli, N.; Caporossi, D. Exercise-Induced ROS in Heat Shock Proteins Response. Free Radic. Biol. Med. 2016, 98, 46–55. [Google Scholar] [CrossRef]
- Wickens, A.P. Ageing and the Free Radical Theory. Respir. Physiol. 2001, 128, 379–391. [Google Scholar] [CrossRef]
- Pecchillo Cimmino, T.; Ammendola, R.; Cattaneo, F.; Esposito, G. NOX Dependent ROS Generation and Cell Metabolism. Int. J. Mol. Sci. 2023, 24, 2086. [Google Scholar] [CrossRef]
- Galbusera, C.; Orth, P.; Fedida, D.; Spector, T. Superoxide Radical Production by Allopurinol and Xanthine Oxidase. Biochem. Pharmacol. 2006, 71, 1747–1752. [Google Scholar] [CrossRef] [PubMed]
- Hrycay, E.G.; Bandiera, S.M. Involvement of Cytochrome P450 in Reactive Oxygen Species Formation and Cancer. Adv. Pharmacol. 2015, 74, 35–84. [Google Scholar]
- He, Z.; Li, Q.; Xu, Y.; Zhang, D.; Pan, X. Production of Extracellular Superoxide Radical in Microorganisms and Its Environmental Implications: A Review. Environ. Pollut. 2023, 338, 122563. [Google Scholar] [CrossRef]
- Attri, P.; Kim, Y.H.; Park, D.H.; Park, J.H.; Hong, Y.J.; Uhm, H.S.; Kim, K.-N.; Fridman, A.; Choi, E.H. Generation Mechanism of Hydroxyl Radical Species and Its Lifetime Prediction during the Plasma-Initiated Ultraviolet (UV) Photolysis. Sci. Rep. 2015, 5, 9332. [Google Scholar] [CrossRef]
- Riley, P.A. Free Radicals in Biology: Oxidative Stress and the Effects of Ionizing Radiation. Int. J. Radiat. Biol. 1994, 65, 27–33. [Google Scholar] [CrossRef]
- Miao, L.; St. Clair, D.K. Regulation of Superoxide Dismutase Genes: Implications in Disease. Free Radic. Biol. Med. 2009, 47, 344–356. [Google Scholar] [CrossRef] [PubMed]
- Muranov, K.O. Fenton Reaction in Vivo and in Vitro. Possibilities and Limitations. Biochem. Mosc. 2024, 89, S112–S126. [Google Scholar] [CrossRef] [PubMed]
- Kehrer, J.P. The Haber–Weiss Reaction and Mechanisms of Toxicity. Toxicology 2000, 149, 43–50. [Google Scholar] [CrossRef]
- Juan, C.A.; Pérez De La Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid Peroxidation in Cell Death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Stadtman, E.R.; Berlett, B.S. Reactive Oxygen-Mediated Protein Oxidation in Aging and Disease. Drug Metab. Rev. 1998, 30, 225–243. [Google Scholar] [CrossRef]
- Fleming, A.M.; Burrows, C.J. Chemistry of ROS-Mediated Oxidation to the Guanine Base in DNA and Its Biological Consequences. Int. J. Radiat. Biol. 2022, 98, 452–460. [Google Scholar] [CrossRef]
- MacDonald, M.; Hassold, T.; Harvey, J.; Wang, L.H.; Morton, N.E.; Jacobs, P. The Origin of 47,XXY and 47,XXX Aneuploidy: Heterogeneous Mechanisms and Role of Aberrant Recombination. Hum. Mol. Genet. 1994, 3, 1365–1371. [Google Scholar] [CrossRef]
- Skuse, D.; Printzlau, F.; Wolstencroft, J. Sex Chromosome Aneuploidies. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 147, pp. 355–376. ISBN 978-0-444-63233-3. [Google Scholar]
- Hassold, T.J.; Hunt, P.A. Missed Connections: Recombination and Human Aneuploidy. Prenat. Diagn. 2021, 41, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Gravholt, C.H.; Viuff, M.H.; Brun, S.; Stochholm, K.; Andersen, N.H. Turner Syndrome: Mechanisms and Management. Nat. Rev. Endocrinol. 2019, 15, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Lorda-Sanchez, I.; Binkert, F.; Hinkel, K.G.; Moser, H.; Rosenkranz, W.; Maechler, M.; Schinzel, A. Uniparental Origin of Sex Chromosome Polysomies. Hum. Hered. 1992, 42, 193–197. [Google Scholar] [CrossRef]
- Nagaoka, S.I.; Hassold, T.J.; Hunt, P.A. Human Aneuploidy: Mechanisms and New Insights into an Age-Old Problem. Nat. Rev. Genet. 2012, 13, 493–504. [Google Scholar] [CrossRef]
- Liu, S.; Akula, N.; Reardon, P.K.; Russ, J.; Torres, E.; Clasen, L.S.; Blumenthal, J.; Lalonde, F.; McMahon, F.J.; Szele, F.; et al. Aneuploidy Effects on Human Gene Expression across Three Cell Types. Proc. Natl. Acad. Sci. USA 2023, 120, e2218478120. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.A.; Makova, K.D. Evolution and Survival on Eutherian Sex Chromosomes. PLoS Genet. 2009, 5, e1000568. [Google Scholar] [CrossRef]
- Lanfranco, F.; Kamischke, A.; Zitzmann, M.; Nieschlag, E. Klinefelter’s Syndrome. Lancet 2004, 364, 273–283. [Google Scholar] [CrossRef]
- Biradar, V.S.; Rajpathak, S.N.; Joshi, S.R.; Deobagkar, D.D. Functional and Regulatory Aspects of Oxidative Stress Response in X Monosomy. Vitro Cell. Dev. Biol.-Anim. 2021, 57, 661–675. [Google Scholar] [CrossRef] [PubMed]
- Soto, M.E.; Soria-Castro, E.; Guarner Lans, V.; Muruato Ontiveros, E.; Iván Hernández Mejía, B.; Jorge Martínez Hernandez, H.; Barragán García, R.; Herrera, V.; Pérez-Torres, I. Analysis of Oxidative Stress Enzymes and Structural and Functional Proteins on Human Aortic Tissue from Different Aortopathies. Oxid. Med. Cell. Longev. 2014, 2014, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Aitken, R.; Krausz, C. Oxidative Stress, DNA Damage and the Y Chromosome. Reproduction 2001, 122, 497–506. [Google Scholar] [CrossRef]
- Aitken, R.J.; Baker, M.A. The Role of Genetics and Oxidative Stress in the Etiology of Male Infertility—A Unifying Hypothesis? Front. Endocrinol. 2020, 11, 581838. [Google Scholar] [CrossRef] [PubMed]
- Tarani, L.; Ceci, F.M.; Carito, V.; Ferraguti, G.; Petrella, C.; Greco, A.; Ralli, M.; Minni, A.; Spaziani, M.; Isidori, A.M.; et al. Neuroimmune Dysregulation in Prepubertal and Adolescent Individuals Affected by Klinefelter Syndrome. Endocr. Metab. Immune Disord. Drug Targets 2023, 23, 105–114. [Google Scholar] [CrossRef]
- Otter, M.; Schrander-Stumpel, C.T.R.M.; Curfs, L.M.G. Triple X Syndrome: A Review of the Literature. Eur. J. Hum. Genet. EJHG 2010, 18, 265–271. [Google Scholar] [CrossRef]
- Tartaglia, N.; Ayari, N.; Howell, S.; D’Epagnier, C.; Zeitler, P. 48,XXYY, 48,XXXY and 49,XXXXY Syndromes: Not Just Variants of Klinefelter Syndrome: 48,XXYY, 48,XXXY and 49,XXXXY Syndromes. Acta Paediatr. 2011, 100, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, J.-C.; Tremblay, A. Sex-Specificity of Oxidative Stress in Newborns Leading to a Personalized Antioxidant Nutritive Strategy. Antioxidants 2018, 7, 49. [Google Scholar] [CrossRef]
- Steiner, M.; Saenger, P. Turner Syndrome. Adv. Pediatr. 2022, 69, 177–202. [Google Scholar] [CrossRef]
- Zhu, C.; Xu, F.; Fukuda, A.; Wang, X.; Fukuda, H.; Korhonen, L.; Hagberg, H.; Lannering, B.; Nilsson, M.; Eriksson, P.S.; et al. X Chromosome-Linked Inhibitor of Apoptosis Protein Reduces Oxidative Stress after Cerebral Irradiation or Hypoxia-Ischemia through up-Regulation of Mitochondrial Antioxidants. Eur. J. Neurosci. 2007, 26, 3402–3410. [Google Scholar] [CrossRef]
- Mohamad, N.-V.; Ima-Nirwana, S.; Chin, K.-Y. Are Oxidative Stress and Inflammation Mediators of Bone Loss Due to Estrogen Deficiency? A Review of Current Evidence. Endocr. Metab. Immune Disord. Drug Targets 2020, 20, 1478–1487. [Google Scholar] [CrossRef]
- Elliot, S.J.; Catanuto, P.; Pereira-Simon, S.; Xia, X.; Pastar, I.; Thaller, S.; Head, C.R.; Stojadinovic, O.; Tomic-Canic, M.; Glassberg, M.K. Catalase, a Therapeutic Target in the Reversal of Estrogen-Mediated Aging. Mol. Ther. J. Am. Soc. Gene Ther. 2022, 30, 947–962. [Google Scholar] [CrossRef] [PubMed]
- Borrás, C.; Ferrando, M.; Inglés, M.; Gambini, J.; Lopez-Grueso, R.; Edo, R.; Mas-Bargues, C.; Pellicer, A.; Viña, J. Estrogen Replacement Therapy Induces Antioxidant and Longevity-Related Genes in Women after Medically Induced Menopause. Oxid. Med. Cell. Longev. 2021, 2021, 8101615. [Google Scholar] [CrossRef]
- White, R.E.; Gerrity, R.; Barman, S.A.; Han, G. Estrogen and Oxidative Stress: A Novel Mechanism That May Increase the Risk for Cardiovascular Disease in Women. Steroids 2010, 75, 788–793. [Google Scholar] [CrossRef] [PubMed]
- Borrás, C.; Gambini, J.; Gómez-Cabrera, M.C.; Sastre, J.; Pallardó, F.V.; Mann, G.E.; Viña, J.; Borrás, C.; Gambini, J.; Gómez-Cabrera, M.C.; et al. Genistein, a Soy Isoflavone, Up-regulates Expression of Antioxidant Genes: Involvement of Estrogen Receptors, ERK1/2, and NFκB. FASEB J. 2006, 20, 2136–2138. [Google Scholar] [CrossRef] [PubMed]
- Felty, Q.; Xiong, W.-C.; Sun, D.; Sarkar, S.; Singh, K.P.; Parkash, J.; Roy, D. Estrogen-Induced Mitochondrial Reactive Oxygen Species as Signal-Transducing Messengers. Biochemistry 2005, 44, 6900–6909. [Google Scholar] [CrossRef]
- Chen, J.; Yager, J.D. Estrogen’s Effects on Mitochondrial Gene Expression: Mechanisms and Potential Contributions to Estrogen Carcinogenesis. Ann. N. Y. Acad. Sci. 2004, 1028, 258–272. [Google Scholar] [CrossRef]
- Hertiš Petek, T.; Petek, T.; Močnik, M.; Marčun Varda, N. Systemic Inflammation, Oxidative Stress and Cardiovascular Health in Children and Adolescents: A Systematic Review. Antioxidants 2022, 11, 894. [Google Scholar] [CrossRef]
- Mavinkurve, M.; O’Gorman, C.S. Cardiometabolic and Vascular Risks in Young and Adolescent Girls with Turner Syndrome. BBA Clin. 2015, 3, 304–309. [Google Scholar] [CrossRef]
- Vilas-Boas, E.A.; Almeida, D.C.; Roma, L.P.; Ortis, F.; Carpinelli, A.R. Lipotoxicity and β-Cell Failure in Type 2 Diabetes: Oxidative Stress Linked to NADPH Oxidase and ER Stress. Cells 2021, 10, 3328. [Google Scholar] [CrossRef]
- Fernández-Sánchez, A.; Madrigal-Santillán, E.; Bautista, M.; Esquivel-Soto, J.; Morales-González, Á.; Esquivel-Chirino, C.; Durante-Montiel, I.; Sánchez-Rivera, G.; Valadez-Vega, C.; Morales-González, J.A. Inflammation, Oxidative Stress, and Obesity. Int. J. Mol. Sci. 2011, 12, 3117–3132. [Google Scholar] [CrossRef]
- Teleanu, D.M.; Niculescu, A.-G.; Lungu, I.I.; Radu, C.I.; Vladâcenco, O.; Roza, E.; Costăchescu, B.; Grumezescu, A.M.; Teleanu, R.I. An Overview of Oxidative Stress, Neuroinflammation, and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 5938. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-González, D.; Carreón-Trujillo, S.; Alvarez-Arellano, L.; Abarca-Merlin, D.M.; Domínguez-López, P.; Salazar-García, M.; Corona, J.C. A Potential Role for Neuroinflammation in ADHD. In Neuroinflammation, Gut-Brain Axis and Immunity in Neuropsychiatric Disorders; Kim, Y.-K., Ed.; Advances in Experimental Medicine and Biology; Springer Nature: Singapore, 2023; Volume 1411, pp. 327–356. ISBN 978-981-19737-5-8. [Google Scholar]
- Usui, N.; Kobayashi, H.; Shimada, S. Neuroinflammation and Oxidative Stress in the Pathogenesis of Autism Spectrum Disorder. Int. J. Mol. Sci. 2023, 24, 5487. [Google Scholar] [CrossRef] [PubMed]
- Ildarabadi, A.; Mir Mohammad Ali, S.N.; Rahmani, F.; Mosavari, N.; Pourbakhtyaran, E.; Rezaei, N. Inflammation and Oxidative Stress in Epileptic Children: From Molecular Mechanisms to Clinical Application of Ketogenic Diet. Rev. Neurosci. 2024, 35, 473–488. [Google Scholar] [CrossRef]
- Gravholt, C.H.; Chang, S.; Wallentin, M.; Fedder, J.; Moore, P.; Skakkebæk, A. Klinefelter Syndrome: Integrating Genetics, Neuropsychology, and Endocrinology. Endocr. Rev. 2018, 39, 389–423. [Google Scholar] [CrossRef] [PubMed]
- Pozza, C.; Sesti, F.; Tenuta, M.; Spaziani, M.; Tarantino, C.; Carlomagno, F.; Minnetti, M.; Pofi, R.; Paparella, R.; Lenzi, A.; et al. Testicular Dysfunction in 47,XXY Boys: When It All Begins. A Semilongitudinal Study. J. Clin. Endocrinol. Metab. 2023, 108, 2486–2499. [Google Scholar] [CrossRef]
- Zitzmann, M.; Aksglaede, L.; Corona, G.; Isidori, A.M.; Juul, A.; T’Sjoen, G.; Kliesch, S.; D’Hauwers, K.; Toppari, J.; Słowikowska-Hilczer, J.; et al. European Academy of Andrology Guidelines on Klinefelter Syndrome Endorsing Organization: European Society of Endocrinology. Andrology 2021, 9, 145–167. [Google Scholar] [CrossRef]
- Carlomagno, F.; Minnetti, M.; Angelini, F.; Pofi, R.; Sbardella, E.; Spaziani, M.; Aureli, A.; Anzuini, A.; Paparella, R.; Tarani, L.; et al. Altered Thyroid Feedback Loop in Klinefelter Syndrome: From Infancy Through the Transition to Adulthood. J. Clin. Endocrinol. Metab. 2023, 108, e1329–e1340. [Google Scholar] [CrossRef]
- Paparella, R.; Ferraguti, G.; Fiore, M.; Menghi, M.; Micangeli, G.; Tarani, F.; Ligotino, A.; Messina, M.P.; Ceccanti, M.; Minni, A.; et al. Serum Lipocalin-2 Levels as a Biomarker in Pre- and Post-Pubertal Klinefelter Syndrome Patients: A Pilot Study. Int. J. Mol. Sci. 2024, 25, 2214. [Google Scholar] [CrossRef]
- Nassau, D.E.; Chu, K.Y.; Blachman-Braun, R.; Castellan, M.; Ramasamy, R. The Pediatric Patient and Future Fertility: Optimizing Long-Term Male Reproductive Health Outcomes. Fertil. Steril. 2020, 113, 489–499. [Google Scholar] [CrossRef]
- Tostes, R.C.; Carneiro, F.S.; Carvalho, M.H.C.; Reckelhoff, J.F. Reactive Oxygen Species: Players in the Cardiovascular Effects of Testosterone. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2016, 310, R1–R14. [Google Scholar] [CrossRef]
- Cruz-Topete, D.; Dominic, P.; Stokes, K.Y. Uncovering Sex-Specific Mechanisms of Action of Testosterone and Redox Balance. Redox Biol. 2020, 31, 101490. [Google Scholar] [CrossRef] [PubMed]
- Tam, N.N.C.; Gao, Y.; Leung, Y.-K.; Ho, S.-M. Androgenic Regulation of Oxidative Stress in the Rat Prostate. Am. J. Pathol. 2003, 163, 2513–2522. [Google Scholar] [CrossRef]
- Mancini, A.; Leone, E.; Festa, R.; Grande, G.; Silvestrini, A.; De Marinis, L.; Pontecorvi, A.; Maira, G.; Littarru, G.P.; Meucci, E. Effects of Testosterone on Antioxidant Systems in Male Secondary Hypogonadism. J. Androl. 2008, 29, 622–629. [Google Scholar] [CrossRef]
- Leisegang, K.; Roychoudhury, S.; Slama, P.; Finelli, R. The Mechanisms and Management of Age-Related Oxidative Stress in Male Hypogonadism Associated with Non-Communicable Chronic Disease. Antioxidants 2021, 10, 1834. [Google Scholar] [CrossRef] [PubMed]
- Aksglaede, L.; Molgaard, C.; Skakkebaek, N.E.; Juul, A. Normal Bone Mineral Content but Unfavourable Muscle/Fat Ratio in Klinefelter Syndrome. Arch. Dis. Child. 2008, 93, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Haymana, C.; Aydogdu, A.; Demirci, I.; Dinc, M.; Demir, O.; Torun, D.; Yesildal, F.; Meric, C.; Basaran, Y.; Sonmez, A.; et al. Increased Endothelial Dysfunction and Insulin Resistance in Patients with Klinefelter Syndrome. Endocr. Metab. Immune Disord.-Drug Targets 2018, 18, 401–406. [Google Scholar] [CrossRef]
- Panvino, F.; Paparella, R.; Gambuti, L.; Cerrito, A.; Menghi, M.; Micangeli, G.; Petrella, C.; Fiore, M.; Tarani, L.; Ardizzone, I. Klinefelter Syndrome: A Genetic Disorder Leading to Neuroendocrine Modifications and Psychopathological Vulnerabilities in Children—A Literature Review and Case Report. Children 2024, 11, 509. [Google Scholar] [CrossRef]
- Fiore, M.; Tarani, L.; Radicioni, A.; Spaziani, M.; Ferraguti, G.; Putotto, C.; Gabanella, F.; Maftei, D.; Lattanzi, R.; Minni, A.; et al. Serum Prokineticin-2 in Prepubertal and Adult Klinefelter Individuals. Can. J. Physiol. Pharmacol. 2021, 100, 151–157. [Google Scholar] [CrossRef]
- Berglund, A.; Viuff, M.H.; Skakkebæk, A.; Chang, S.; Stochholm, K.; Gravholt, C.H. Changes in the Cohort Composition of Turner Syndrome and Severe Non-Diagnosis of Klinefelter, 47,XXX and 47,XYY Syndrome: A Nationwide Cohort Study. Orphanet J. Rare Dis. 2019, 14, 16. [Google Scholar] [CrossRef]
- Berglund, A.; Stochholm, K.; Gravholt, C.H. Morbidity in 47,XYY Syndrome: A Nationwide Epidemiological Study of Hospital Diagnoses and Medication Use. Genet. Med. 2020, 22, 1542–1551. [Google Scholar] [CrossRef] [PubMed]
- Rappold, G.A. The Pseudoautosomal Regions of the Human Sex Chromosomes. Hum. Genet. 1993, 92, 315–324. [Google Scholar] [CrossRef]
- Spaziani, M.; Carlomagno, F.; Tarantino, C.; Angelini, F.; Paparella, R.; Tarani, L.; Putotto, C.; Badagliacca, R.; Pozza, C.; Isidori, A.M.; et al. From Klinefelter Syndrome to High Grade Aneuploidies: Expanding the Gene-Dosage Effect of Supernumerary X Chromosomes. J. Clin. Endocrinol. Metab. 2024, 109, dgad730. [Google Scholar] [CrossRef] [PubMed]
- Shimojima Yamamoto, K.; Yamamoto, S.; Imaizumi, T.; Kumada, S.; Yamamoto, T. Uniparental Maternal Tetrasomy X Co-Occurrence with Paternal Nondisjunction: Investigation of the Origin of 48,XXXX. Hum. Genome Var. 2024, 11, 31. [Google Scholar] [CrossRef]
- Andriani, G.A.; Vijg, J.; Montagna, C. Mechanisms and Consequences of Aneuploidy and Chromosome Instability in the Aging Brain. Mech. Ageing Dev. 2017, 161, 19–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-Y.; Liu, L.-N.; Zhao, Z.-B. The Role of ROS Toxicity in Spontaneous Aneuploidy in Cultured Cells. Tissue Cell 2013, 45, 47–53. [Google Scholar] [CrossRef]
- Banan, A.; Fields, J.Z.; Decker, H.; Zhang, Y.; Keshavarzian, A. Nitric Oxide and Its Metabolites Mediate Ethanol-Induced Microtubule Disruption and Intestinal Barrier Dysfunction. J. Pharmacol. Exp. Ther. 2000, 294, 997–1008. [Google Scholar] [CrossRef]
- Baraibar, M.A.; Friguet, B. Changes of the Proteasomal System During the Aging Process. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2012; Volume 109, pp. 249–275. ISBN 978-0-12-397863-9. [Google Scholar]
- Roh, M.; Van Der Meer, R.; Abdulkadir, S.A. Tumorigenic Polyploid Cells Contain Elevated ROS and ARE Selectively Targeted by Antioxidant Treatment. J. Cell. Physiol. 2012, 227, 801–812. [Google Scholar] [CrossRef]
- Palmieri, F. The Mitochondrial Transporter Family SLC25: Identification, Properties and Physiopathology. Mol. Aspects Med. 2013, 34, 465–484. [Google Scholar] [CrossRef]
- Edmondson, D.E.; Binda, C. Monoamine Oxidases. In Membrane Protein Complexes: Structure and Function; Harris, J.R., Boekema, E.J., Eds.; Subcellular Biochemistry; Springer: Singapore, 2018; Volume 87, pp. 117–139. ISBN 978-981-10-7756-2. [Google Scholar]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e21. [Google Scholar] [CrossRef]
- Chen, G.; Li, Z.; Iemura, K.; Tanaka, K. Oxidative Stress Induces Chromosomal Instability through Replication Stress in Fibroblasts from Aged Mice. J. Cell Sci. 2023, 136, jcs260688. [Google Scholar] [CrossRef] [PubMed]
- Kozel, B.A.; Danback, J.R.; Waxler, J.L.; Knutsen, R.H.; de las Fuentes, L.; Reusz, G.S.; Kis, E.; Bhatt, A.B.; Pober, B.R. Williams Syndrome Predisposes to Vascular Stiffness Modified by Antihypertensive Use and Copy Number Changes in NCF1. Hypertens. Dallas Tex 1979 2014, 63, 74–79. [Google Scholar] [CrossRef]
- Kozel, B.A.; Knutsen, R.H.; Ye, L.; Ciliberto, C.H.; Broekelmann, T.J.; Mecham, R.P. Genetic Modifiers of Cardiovascular Phenotype Caused by Elastin Haploinsufficiency Act by Extrinsic Noncomplementation. J. Biol. Chem. 2011, 286, 44926–44936. [Google Scholar] [CrossRef] [PubMed]
- Pober, B.R. Williams-Beuren Syndrome. N. Engl. J. Med. 2010, 362, 239–252. [Google Scholar] [CrossRef]
- Vassalle, C.; Sabatino, L.; Pingitore, A.; Chatzianagnostou, K.; Mastorci, F.; Ceravolo, R. Antioxidants in the Diet and Cognitive Function: Which Role for the Mediterranean Life-Style? J. Prev. Alzheimers Dis. 2016, 4, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Padayatty, S.J.; Katz, A.; Wang, Y.; Eck, P.; Kwon, O.; Lee, J.-H.; Chen, S.; Corpe, C.; Dutta, A.; Dutta, S.K.; et al. Vitamin C as an Antioxidant: Evaluation of Its Role in Disease Prevention. J. Am. Coll. Nutr. 2003, 22, 18–35. [Google Scholar] [CrossRef]
- Aguirre, R.; May, J.M. Inflammation in the Vascular Bed: Importance of Vitamin C. Pharmacol. Ther. 2008, 119, 96–103. [Google Scholar] [CrossRef]
- Dulac, Y.; Pienkowski, C.; Abadir, S.; Tauber, M.; Acar, P. Cardiovascular Abnormalities in Turner’s Syndrome: What Prevention? Arch. Cardiovasc. Dis. 2008, 101, 485–490. [Google Scholar] [CrossRef]
- Spaziani, M.; Radicioni, A.F. Metabolic and Cardiovascular Risk Factors in Klinefelter Syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 334–343. [Google Scholar] [CrossRef]
- Calcaterra, V.; Brambilla, P.; Maffè, G.C.; Klersy, C.; Albertini, R.; Introzzi, F.; Bozzola, E.; Bozzola, M.; Larizza, D. Metabolic Syndrome in Turner Syndrome and Relation Between Body Composition and Clinical, Genetic, and Ultrasonographic Characteristics. Metab. Syndr. Relat. Disord. 2014, 12, 159–164. [Google Scholar] [CrossRef]
- Mameli, C.; Fiore, G.; Sangiorgio, A.; Agostinelli, M.; Zichichi, G.; Zuccotti, G.; Verduci, E. Metabolic and Nutritional Aspects in Paediatric Patients with Klinefelter Syndrome: A Narrative Review. Nutrients 2022, 14, 2107. [Google Scholar] [CrossRef]
- Paparella, R.; Panvino, F.; Leonardi, L.; Pucarelli, I.; Menghi, M.; Micangeli, G.; Tarani, F.; Niceta, M.; Rasio, D.; Pancheva, R.; et al. Water-Soluble Vitamins: Hypo- and Hypervitaminosis in Pediatric Population. Pharmaceutics 2025, 17, 118. [Google Scholar] [CrossRef]
- Institute of Medicine, Food and Nutrition Board. Dietary Reference Intakes: Vitamin C, Vitamin E, Selenium, and Carotenoids; National Academy Press: Washington, DC, USA, 2000. [Google Scholar]
- Carr, A.C.; Maggini, S. Vitamin C and Immune Function. Nutrients 2017, 9, 1211. [Google Scholar] [CrossRef]
- Jiang, Q. Natural Forms of Vitamin E: Metabolism, Antioxidant, and Anti-Inflammatory Activities and Their Role in Disease Prevention and Therapy. Free Radic. Biol. Med. 2014, 72, 76–90. [Google Scholar] [CrossRef]
- Hannah-Shmouni, F.; Schoepp, M.; Abd-Elmoniem, K.Z.; Matta, J.; Ghanem, A.; Hanover, J.A.; Gharib, A.M. Coronary Atherosclerosis in Females with Turner Syndrome. Can. J. Diabetes 2017, 41, S30. [Google Scholar] [CrossRef]
- Strain, J.J.; Mulholland, C.W. Vitamin C and Vitamin E—Synergistic Interactions in Vivo? In Free Radicals and Aging; Emerit, I., Chance, B., Eds.; Birkhäuser: Basel, Switzerland, 1992; pp. 419–422. ISBN 978-3-0348-7462-5. [Google Scholar]
- Eskenazi, B.; Kidd, S.A.; Marks, A.R.; Sloter, E.; Block, G.; Wyrobek, A.J. Antioxidant Intake Is Associated with Semen Quality in Healthy Men. Hum. Reprod. 2005, 20, 1006–1012. [Google Scholar] [CrossRef]
- Couto, N.; Wood, J.; Barber, J. The Role of Glutathione Reductase and Related Enzymes on Cellular Redox Homoeostasis Network. Free Radic. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef]
- Enns, G.M.; Moore, T.; Le, A.; Atkuri, K.; Shah, M.K.; Cusmano-Ozog, K.; Niemi, A.-K.; Cowan, T.M. Degree of Glutathione Deficiency and Redox Imbalance Depend on Subtype of Mitochondrial Disease and Clinical Status. PLoS ONE 2014, 9, e100001. [Google Scholar] [CrossRef]
- Izigov, N.; Farzam, N.; Savion, N. S-Allylmercapto-N-Acetylcysteine up-Regulates Cellular Glutathione and Protects Vascular Endothelial Cells from Oxidative Stress. Free Radic. Biol. Med. 2011, 50, 1131–1139. [Google Scholar] [CrossRef]
- Grinberg, L.; Fibach, E.; Amer, J.; Atlas, D. N-Acetylcysteine Amide, a Novel Cell-Permeating Thiol, Restores Cellular Glutathione and Protects Human Red Blood Cells from Oxidative Stress. Free Radic. Biol. Med. 2005, 38, 136–145. [Google Scholar] [CrossRef]
- Arancibia-Riveros, C.; Domínguez-López, I.; Laveriano-Santos, E.P.; Parilli-Moser, I.; Tresserra-Rimbau, A.; Ruiz-León, A.M.; Sacanella, E.; Casas, R.; Estruch, R.; Bodega, P.; et al. Unlocking the Power of Polyphenols: A Promising Biomarker of Improved Metabolic Health and Anti-Inflammatory Diet in Adolescents. Clin. Nutr. 2024, 43, 1865–1871. [Google Scholar] [CrossRef]
- Tresserra-Rimbau, A. Dietary Polyphenols and Human Health. Nutrients 2020, 12, 2893. [Google Scholar] [CrossRef]
- Carito, V.; Ceccanti, M.; Cestari, V.; Natella, F.; Bello, C.; Coccurello, R.; Mancinelli, R.; Fiore, M. Olive Polyphenol Effects in a Mouse Model of Chronic Ethanol Addiction. Nutrition 2017, 33, 65–69. [Google Scholar] [CrossRef]
- Fiore, M.; Messina, M.P.; Petrella, C.; D’Angelo, A.; Greco, A.; Ralli, M.; Ferraguti, G.; Tarani, L.; Vitali, M.; Ceccanti, M. Antioxidant Properties of Plant Polyphenols in the Counteraction of Alcohol-Abuse Induced Damage: Impact on the Mediterranean Diet. J. Funct. Foods 2020, 71, 104012. [Google Scholar] [CrossRef]
- Petrella, C.; Di Certo, M.G.; Gabanella, F.; Barbato, C.; Ceci, F.M.; Greco, A.; Ralli, M.; Polimeni, A.; Angeloni, A.; Severini1, C.; et al. Mediterranean Diet, Brain and Muscle: Olive Polyphenols and Resveratrol Protection in Neurodegenerative and Neuromuscular Disorders. Curr. Med. Chem. 2021, 28, 7595–7613. [Google Scholar] [CrossRef]
- Ghosh, D.; Scheepens, A. Vascular Action of Polyphenols. Mol. Nutr. Food Res. 2009, 53, 322–331. [Google Scholar] [CrossRef]
- Lau, F.C.; Shukitt-Hale, B.; Joseph, J.A. Nutritional Intervention in Brain Aging: Reducing the Effects of Inflammation and Oxidative Stress. Subcell. Biochem. 2007, 42, 299–318. [Google Scholar]
- De Santis, S.; Clodoveo, M.L.; Cariello, M.; D’Amato, G.; Franchini, C.; Faienza, M.F.; Corbo, F. Polyphenols and Obesity Prevention: Critical Insights on Molecular Regulation, Bioavailability and Dose in Preclinical and Clinical Settings. Crit. Rev. Food Sci. Nutr. 2021, 61, 1804–1826. [Google Scholar] [CrossRef]
- Wisnuwardani, R.W.; De Henauw, S.; Androutsos, O.; Forsner, M.; Gottrand, F.; Huybrechts, I.; Knaze, V.; Kersting, M.; Le Donne, C.; Marcos, A.; et al. Estimated Dietary Intake of Polyphenols in European Adolescents: The HELENA Study. Eur. J. Nutr. 2019, 58, 2345–2363. [Google Scholar] [CrossRef]
- Krinsky, N.I.; Johnson, E.J. Carotenoid Actions and Their Relation to Health and Disease. Mol. Aspects Med. 2005, 26, 459–516. [Google Scholar] [CrossRef]
- Fiedor, J.; Burda, K. Potential Role of Carotenoids as Antioxidants in Human Health and Disease. Nutrients 2014, 6, 466–488. [Google Scholar] [CrossRef]
- Kaulmann, A.; Bohn, T. Carotenoids, Inflammation, and Oxidative Stress--Implications of Cellular Signaling Pathways and Relation to Chronic Disease Prevention. Nutr. Res. 2014, 34, 907–929. [Google Scholar] [CrossRef]
- Landrum, J.T.; Bone, R.A. Lutein, Zeaxanthin, and the Macular Pigment. Arch. Biochem. Biophys. 2001, 385, 28–40. [Google Scholar] [CrossRef]
- Stahl, W.; Sies, H. Bioactivity and Protective Effects of Natural Carotenoids. Biochim. Biophys. Acta 2005, 1740, 101–107. [Google Scholar] [CrossRef]
- Tan, D.-X.; Manchester, L.; Esteban-Zubero, E.; Zhou, Z.; Reiter, R. Melatonin as a Potent and Inducible Endogenous Antioxidant: Synthesis and Metabolism. Molecules 2015, 20, 18886–18906. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Tain, Y.-L.; Sheen, J.-M.; Huang, L.-T. Melatonin Utility in Neonates and Children. J. Formos. Med. Assoc. 2012, 111, 57–66. [Google Scholar] [CrossRef]
- Lee, S.K.M.; Smith, L.; Tan, E.C.K.; Cairns, R.; Grunstein, R.; Cheung, J.M.Y. Melatonin Use in Children and Adolescents: A Scoping Review of Caregiver Perspectives. Sleep Med. Rev. 2023, 70, 101808. [Google Scholar] [CrossRef]
- Chitimus, D.M.; Popescu, M.R.; Voiculescu, S.E.; Panaitescu, A.M.; Pavel, B.; Zagrean, L.; Zagrean, A.-M. Melatonin’s Impact on Antioxidative and Anti-Inflammatory Reprogramming in Homeostasis and Disease. Biomolecules 2020, 10, 1211. [Google Scholar] [CrossRef]
- Garofoli, F.; Franco, V.; Accorsi, P.; Albertini, R.; Angelini, M.; Asteggiano, C.; Aversa, S.; Ballante, E.; Borgatti, R.; Cabini, R.F.; et al. Fate of Melatonin Orally Administered in Preterm Newborns: Antioxidant Performance and Basis for Neuroprotection. J. Pineal Res. 2024, 76, e12932. [Google Scholar] [CrossRef]
- Luboshitzky, R.; Wagner, O.; Lavi, S.; Herer, P.; Lavie, P. Decreased Nocturnal Melatonin Secretion in Patients with Klinefelter’s Syndrome. Clin. Endocrinol. Oxf. 1996, 45, 749–754. [Google Scholar] [CrossRef]
- Caglayan, S.; Ozata, M.; Ozisik, G.; Turan, M.; Bolu, E.; Oktenli, C.; Arslan, N.; Erbil, K.; Gul, D.; Ozdemir, I.C. Plasma Melatonin Concentration before and during Testosterone Replacement in Klinefelter’s Syndrome: Relation to Hepatic Indolamine Metabolism and Sympathoadrenal Activity. J. Clin. Endocrinol. Metab. 2001, 86, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Rouen, A.; Elbaz, M.; Duquesne, E.; Caetano, G.; Léger, D. Multifactorial Sleep Disturbance in Klinefelter Syndrome: A Case Report. Transl. Androl. Urol. 2023, 12, 1204–1210. [Google Scholar] [CrossRef]
- Attanasio, A.; Borrelli, P.; Munding, G.; Gupta, D. Melatonin Day-Night Rhythms in Subjects with Turner Syndrome. Pediatr. Res. 1984, 18, 1227. [Google Scholar] [CrossRef]
- Schober, E.; Waldhauser, F.; Frisch, H.; Schuster, E.; Bieglmayer, C. Melatonin secretion in turner’s syndrome: Lack of effect of oestrogen administration. Clin. Endocrinol. Oxf. 1989, 31, 475–479. [Google Scholar] [CrossRef]
- Paparella, R.; Panvino, F.; Gambuti, L.; Cerrito, A.; Pallante, A.; Micangeli, G.; Menghi, M.; Pisani, F.; Bruni, O.; Ardizzone, I.; et al. Evaluation of Sleep Disorders in Children and Adolescents Affected by Klinefelter Syndrome. Eur. J. Pediatr. 2025, 184, 129. [Google Scholar] [CrossRef] [PubMed]
- Karbasi, S.; Mohamadian, M.; Naseri, M.; Khorasanchi, Z.; Zarban, A.; Bahrami, A.; Ferns, G.A. A Mediterranean Diet Is Associated with Improved Total Antioxidant Content of Human Breast Milk and Infant Urine. Nutr. J. 2023, 22, 11. [Google Scholar] [CrossRef] [PubMed]
- Gantenbein, K.V.; Kanaka-Gantenbein, C. Mediterranean Diet as an Antioxidant: The Impact on Metabolic Health and Overall Wellbeing. Nutrients 2021, 13, 1951. [Google Scholar] [CrossRef]
- Li, P.; Cheng, F.; Xiu, L. Height Outcome of the Recombinant Human Growth Hormone Treatment in Turner Syndrome: A Meta-Analysis. Endocr. Connect. 2018, 7, 573–583. [Google Scholar] [CrossRef]
- Ross, J.L.; Kushner, H.; Kowal, K.; Bardsley, M.; Davis, S.; Reiss, A.L.; Tartaglia, N.; Roeltgen, D. Androgen Treatment Effects on Motor Function, Cognition, and Behavior in Boys with Klinefelter Syndrome. J. Pediatr. 2017, 185, 193–199.e4. [Google Scholar] [CrossRef]
- Chaudhary, P.; Janmeda, P.; Docea, A.O.; Yeskaliyeva, B.; Abdull Razis, A.F.; Modu, B.; Calina, D.; Sharifi-Rad, J. Oxidative Stress, Free Radicals and Antioxidants: Potential Crosstalk in the Pathophysiology of Human Diseases. Front. Chem. 2023, 11, 1158198. [Google Scholar] [CrossRef]
- Poljsak, B. Strategies for Reducing or Preventing the Generation of Oxidative Stress. Oxid. Med. Cell. Longev. 2011, 2011, 194586. [Google Scholar] [CrossRef] [PubMed]
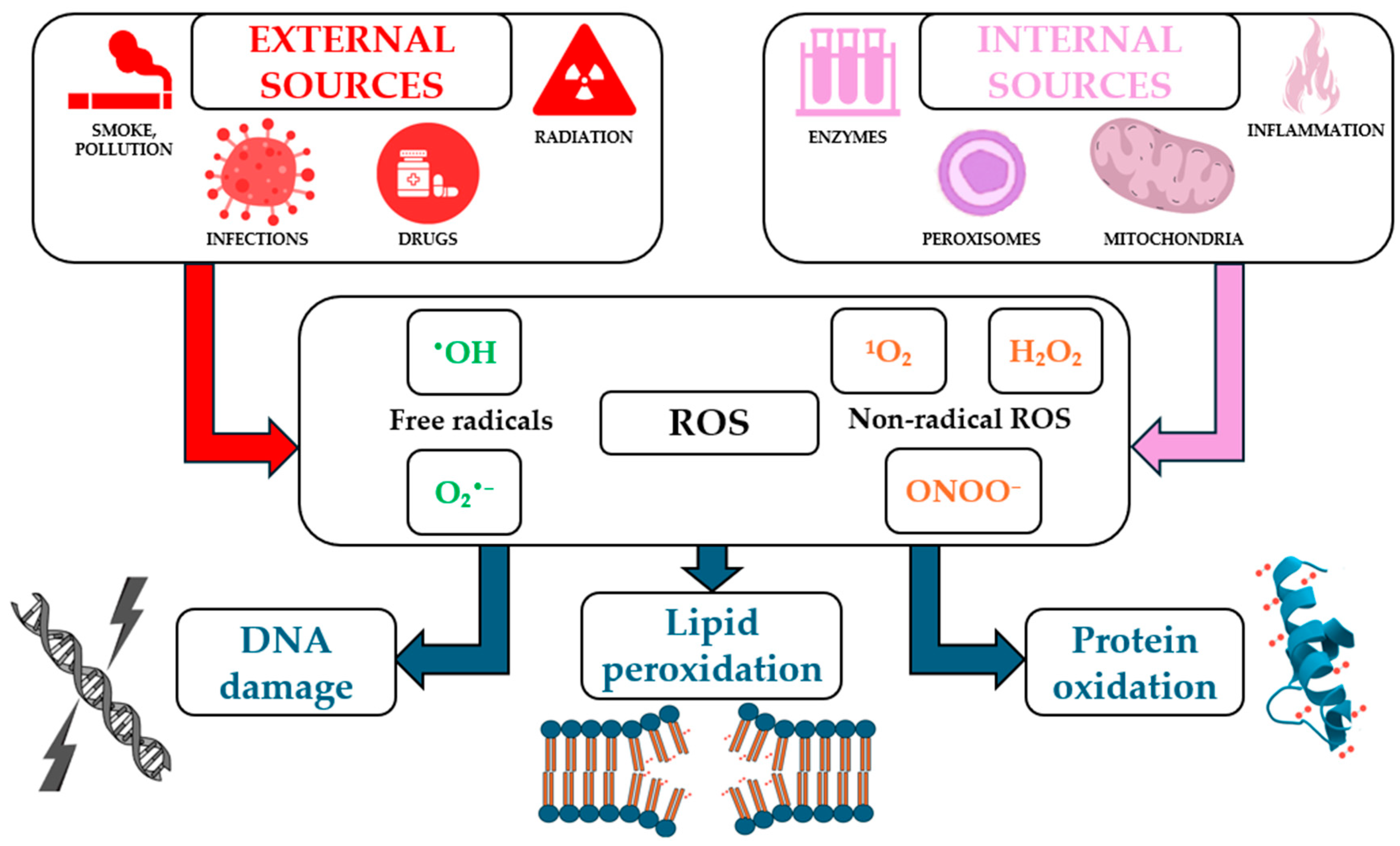
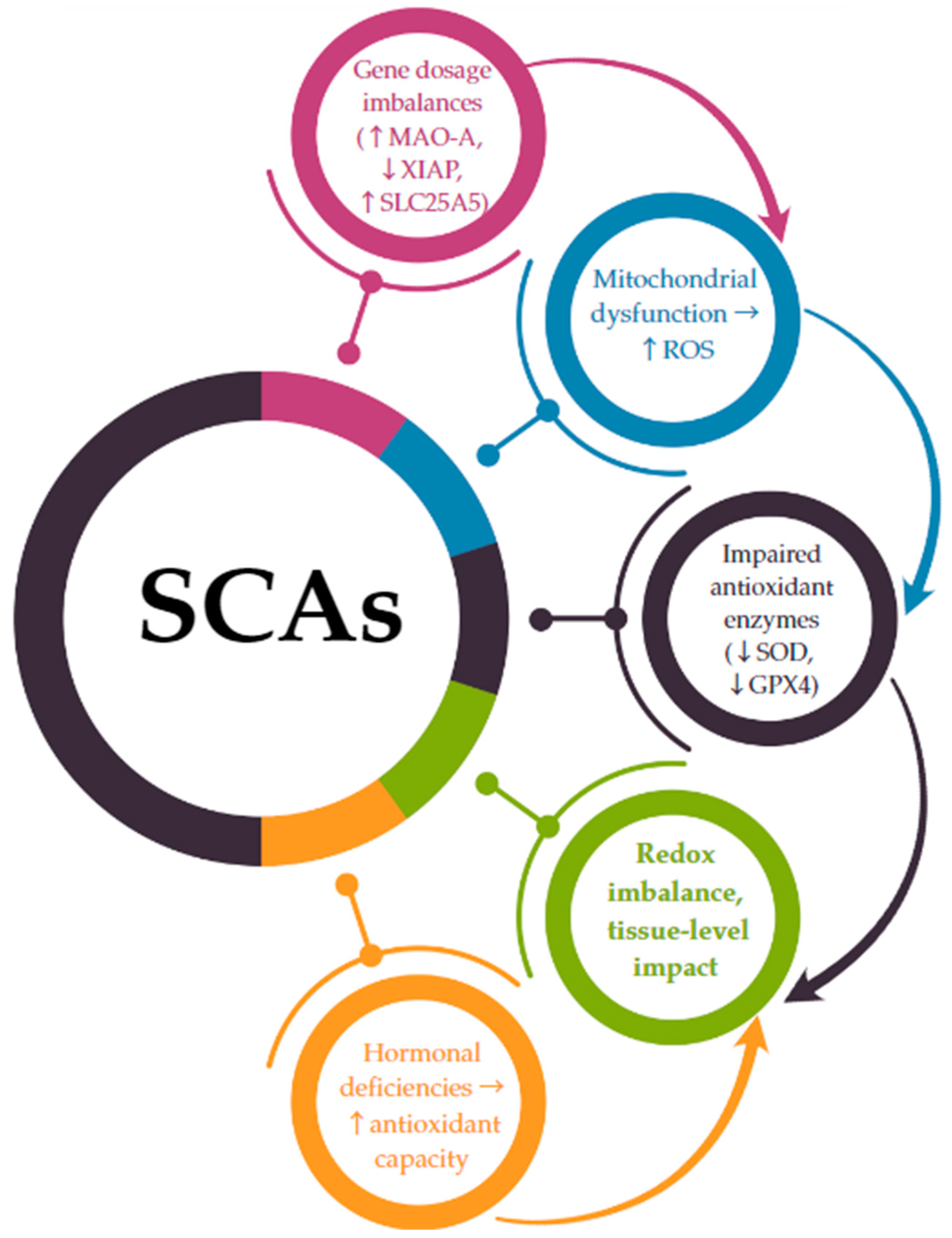
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).