
Cell-Permeable Microprotein from Panax Ginseng Protects Against Doxorubicin-Induced Oxidative Stress and Cardiotoxicity

 ,
,  ,
,  ,
,  , ,
, ,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
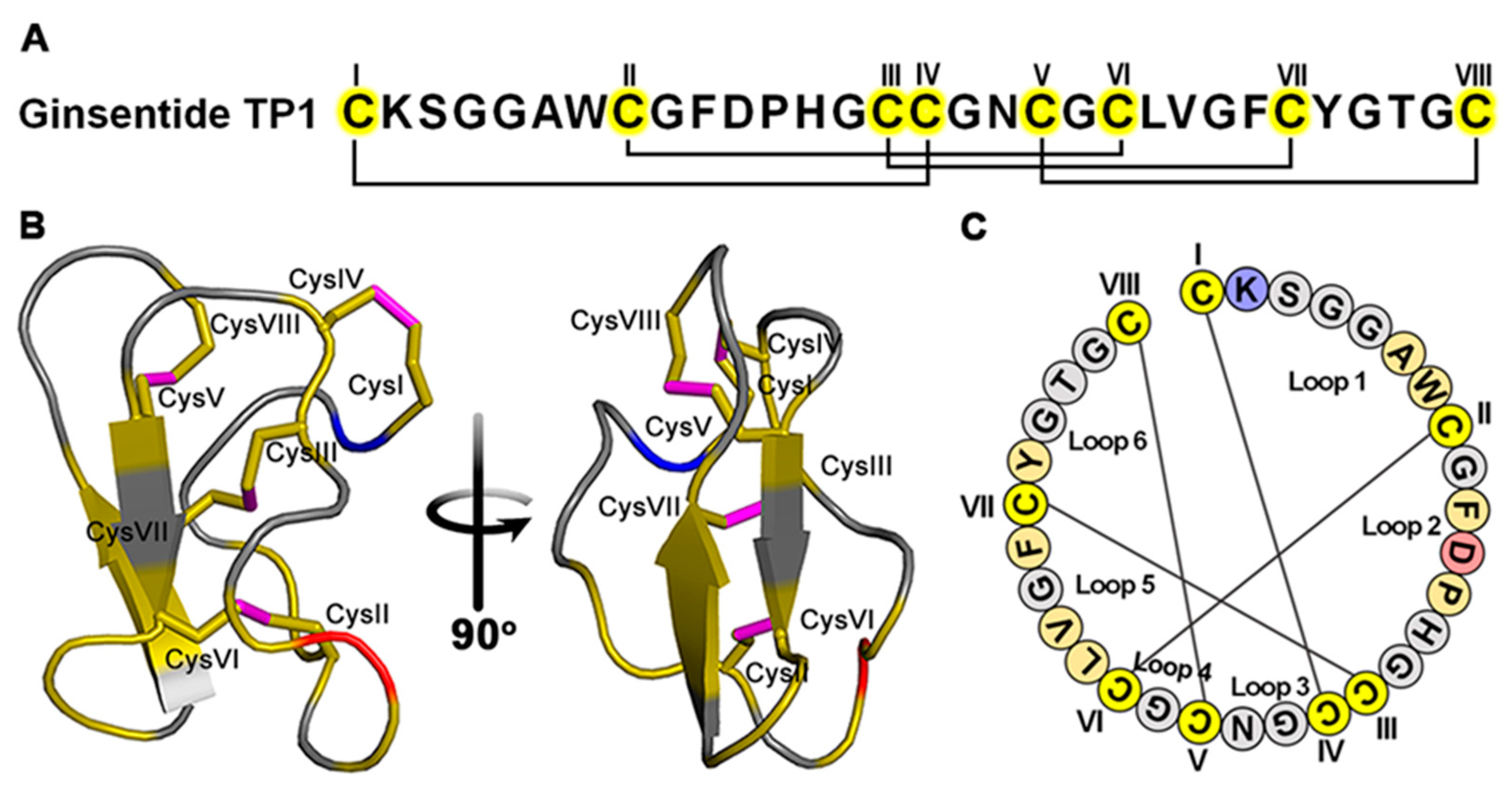
2.2. Isolation and Purification of Ginsentide TP1
2.3. Culture of Cells and Subsequent Treatment
2.4. LDH Assay
2.5. MTT Assay
2.6. Western Blotting
2.7. Immunoassay Using ELISA
2.8. Apoptosis Assay
2.9. ROS Detection
2.10. Nucleus Staining with Hoechst 33342
2.11. Mitochondrial Membrane Potential (MMP) Detection
2.12. Intracellular Calcium Measurement
2.13. Ethics Declarations
2.14. Zebrafish Maintenance and Chemical Treatment
2.15. Morphological Abnormalities Assessment
2.16. Cardiac Functionality Assessment
2.17. Doxorubicin-Induced Acute Myocardial Injury in Mice and Cardiac Functions Assessment
Grouping
2.18. Statistical Analyses
3. Results
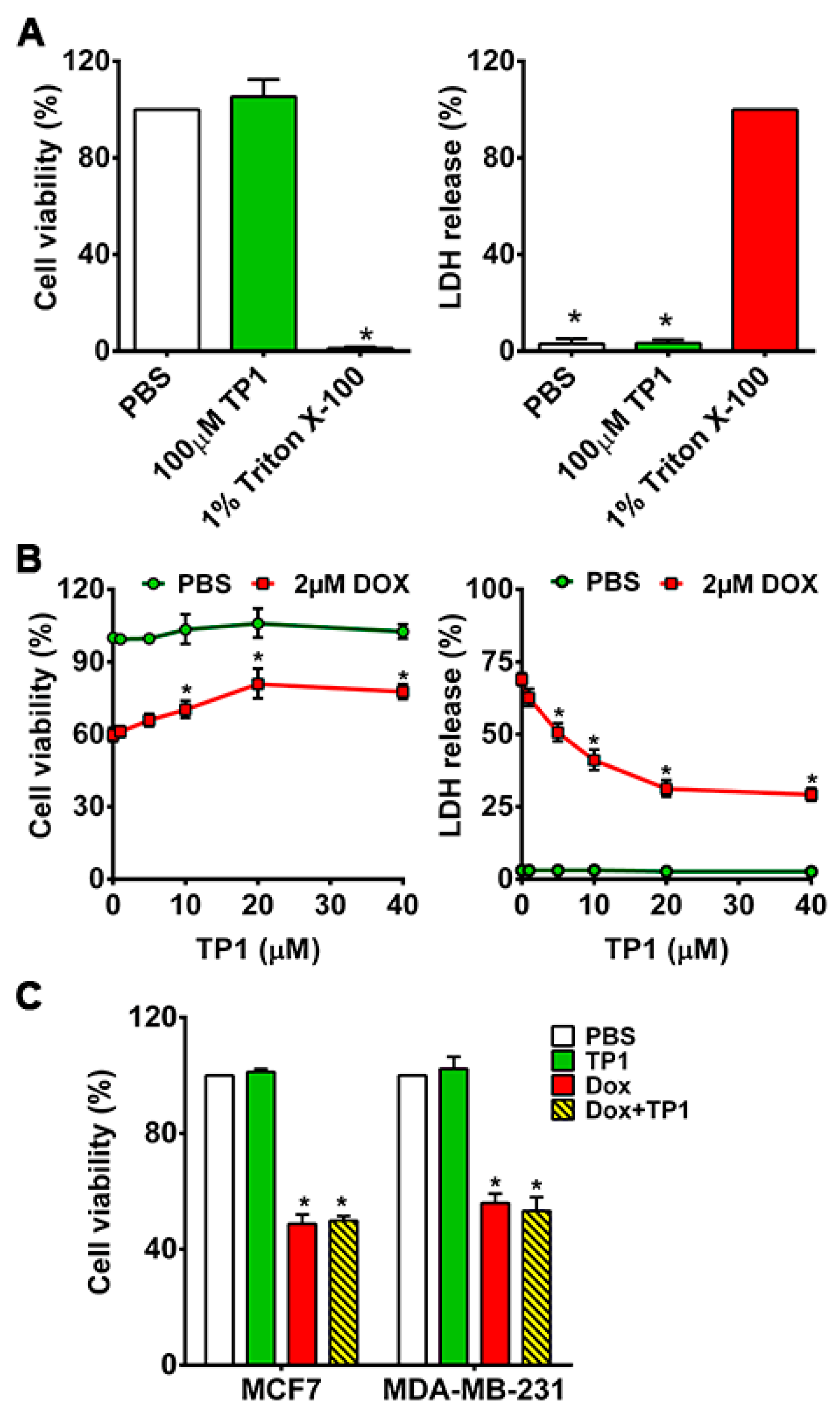
3.1. TP1 Prevents DOX-Induced Cytotoxicity and Increases the Survival of Cardiomyocytes
3.2. TP1 Co-Treatment Does Not Compromise DOX Anticancer Efficacy
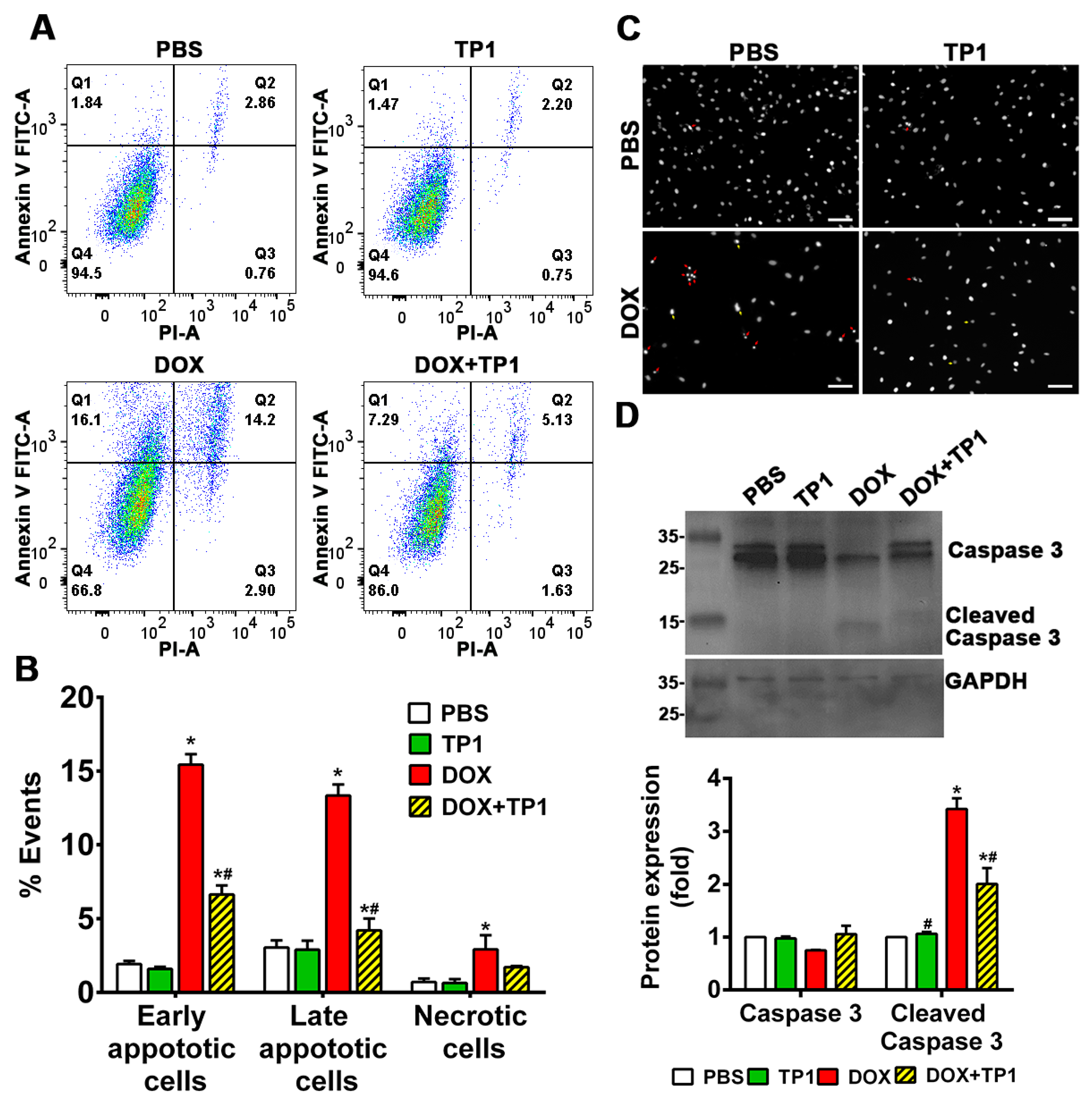
3.3. TP1 Inhibits DOX-Induced Apoptosis of H9c2 Cells
3.4. TP1 Modulates Intracellular Calcium (Ca2+) Homeostasis in DOX-Treated H9c2 Cells
3.5. TP1 Restores MMP in DOX-Treated H9c2 Cells
3.6. TP1 Reduces DOX-Mediated ROS Production in H9c2 Cells
3.7. TP1 Pre-Empts DOX-Induced Inflammatory Responses in H9c2 Cells
3.8. TP1 Mitigates DOX-Induced Cardiotoxicity in Zebrafish
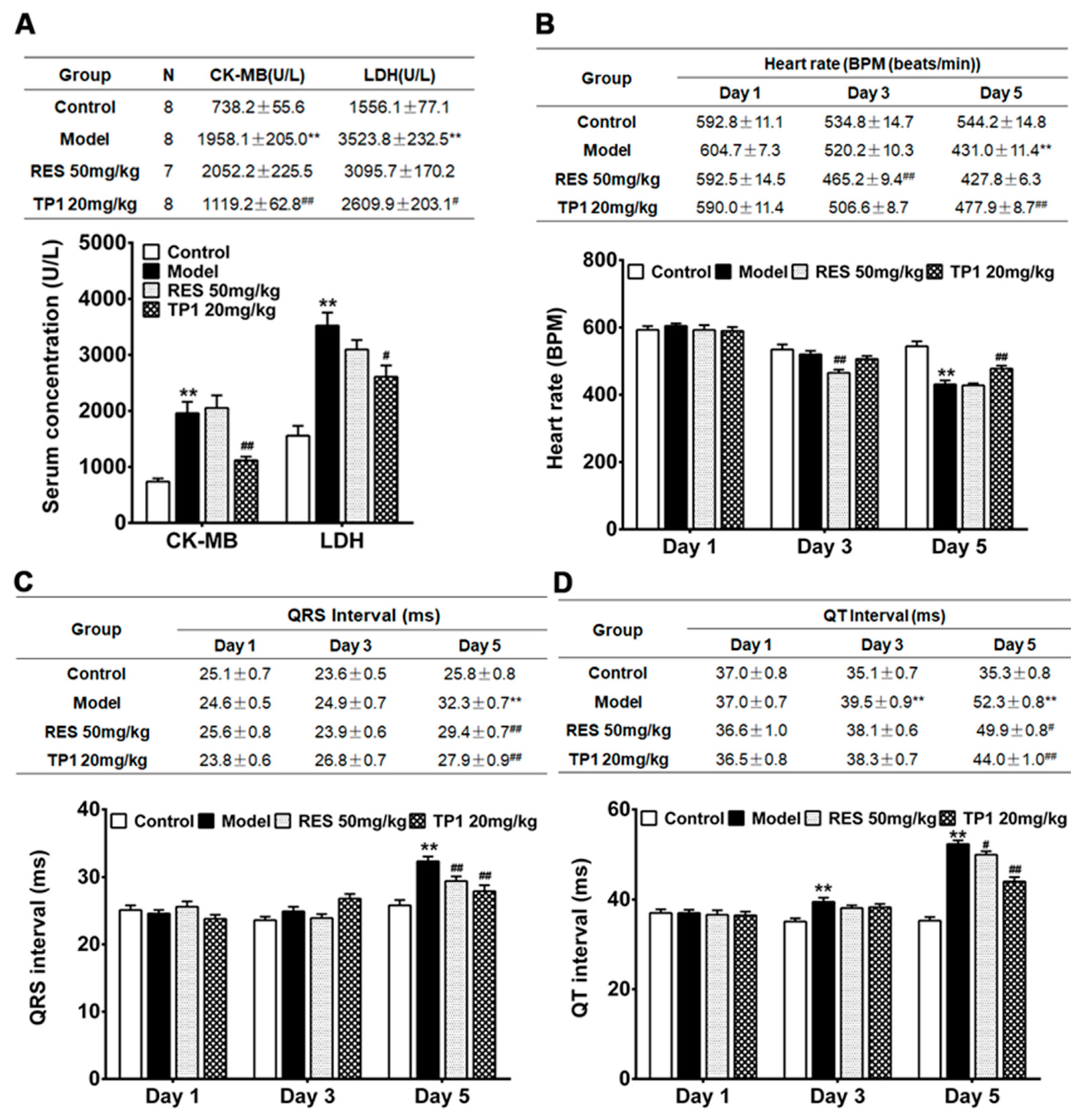
3.9. Cardioprotective Property of TP1 in Mitigating DOX-Induced Cardiotoxicity in a Murine Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACN | Acetonitrile |
| ANOVA | Analysis of variance |
| Ca2+ | Calcium ion |
| CRPs | Cysteine-rich peptides |
| DOX | Doxorubicin |
| EF | Ejection fraction |
| ECG | Electrocardiograms |
| ER | Endoplasmic reticulum |
| eNOs | Endothelial nitric oxide synthase |
| ELISA | Enzyme-linked immunosorbent assay |
| EPO | Erythropoietin |
| GFP | Green fluorescent protein |
| IL-6 | Interleukin 6 |
| LDH | Lactate dehydrogenase |
| MMP | Mitochondrial membrane potential |
| NO | Nitric oxide |
| ROS | Reactive oxygen species |
| GSH | Reduced glutathione |
| RP-HPLC | Reversed-phase high-performance liquid chromatography |
| Rh123 | Rhodamine 123 |
| CK-MB | Serum concentration of creatine kinase isoenzyme |
| SER | Smooth endoplasmic reticulum |
| SD | Standard deviation |
| SE | Standard error |
| SEM | Standard error of the mean |
| TFA | Trifluoroacetic acid |
| TNF-α | Tumour necrosis factor-α |
References
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef]
- Sritharan, S.; Sivalingam, N. A comprehensive review on time-tested anticancer drug doxorubicin. Life Sci. 2021, 278, 119527. [Google Scholar] [CrossRef] [PubMed]
- Force, T.; Kolaja, K.L. Cardiotoxicity of kinase inhibitors: The prediction and translation of preclinical models to clinical outcomes. Nat. Rev. Drug Discov. 2011, 10, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Yeh, E.T.; Bickford, C.L. Cardiovascular complications of cancer therapy: Incidence, pathogenesis, diagnosis, and management. J. Am. Coll. Cardiol. 2009, 53, 2231–2247. [Google Scholar] [CrossRef]
- Christidi, E.; Brunham, L.R. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2021, 12, 339. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharmacogenet. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef]
- Chong, E.G.; Lee, E.H.; Sail, R.; Denham, L.; Nagaraj, G.; Hsueh, C.T. Anthracycline-induced cardiotoxicity: A case report and review of literature. World J. Cardiol. 2021, 13, 28–37. [Google Scholar] [CrossRef]
- Lipshultz, S.E.; Colan, S.D.; Gelber, R.D.; Perez-Atayde, A.R.; Sallan, S.E.; Sanders, S.P. Late cardiac effects of doxorubicin therapy for acute lymphoblastic leukemia in childhood. N. Engl. J. Med. 1991, 324, 808–815. [Google Scholar] [CrossRef]
- Legha, S.S.; Benjamin, R.S.; Mackay, B.; Ewer, M.; Wallace, S.; Valdivieso, M.; Rasmussen, S.L.; Blumenschein, G.R.; Freireich, E.J. Reduction of doxorubicin cardiotoxicity by prolonged continuous intravenous infusion. Ann. Intern. Med. 1982, 96, 133–139. [Google Scholar] [CrossRef]
- Casper, E.S.; Gaynor, J.J.; Hajdu, S.I.; Magill, G.B.; Tan, C.; Friedrich, C.; Brennan, M.F. A prospective randomized trial of adjuvant chemotherapy with bolus versus continuous infusion of doxorubicin in patients with high-grade extremity soft tissue sarcoma and an analysis of prognostic factors. Cancer 1991, 68, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Ludke, A.R.; Al-Shudiefat, A.A.; Dhingra, S.; Jassal, D.S.; Singal, P.K. A concise description of cardioprotective strategies in doxorubicin-induced cardiotoxicity. Can. J. Physiol. Pharmacol. 2009, 87, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Mirhadi, E.; Mashreghi, M.; Askarizadeh, A.; Mehrabian, A.; Alavizadeh, S.H.; Arabi, L.; Badiee, A.; Jaafari, M.R. Redox-sensitive doxorubicin liposome: A formulation approach for targeted tumor therapy. Sci. Rep. 2022, 12, 11310. [Google Scholar] [CrossRef] [PubMed]
- Di Francesco, M.; Celia, C.; Cristiano, M.C.; d’Avanzo, N.; Ruozi, B.; Mircioiu, C.; Cosco, D.; Di Marzio, L.; Fresta, M. Doxorubicin Hydrochloride-Loaded Nonionic Surfactant Vesicles to Treat Metastatic and Non-Metastatic Breast Cancer. ACS Omega 2021, 6, 2973–2989. [Google Scholar] [CrossRef]
- Lee, M.-K. Clinical usefulness of liposomal formulations in cancer therapy: Lessons from the experiences of doxorubicin. J. Pharm. Investig. 2018, 49, 203–214. [Google Scholar] [CrossRef]
- Abraham, S.A.; Waterhouse, D.N.; Mayer, L.D.; Cullis, P.R.; Madden, T.D.; Bally, M.B. The liposomal formulation of doxorubicin. Methods Enzymol. 2005, 391, 71–97. [Google Scholar] [CrossRef]
- Hosseini, A.; Sahebkar, A. Reversal of Doxorubicin-induced Cardiotoxicity by Using Phytotherapy: A Review. J. Pharmacopunct. 2017, 20, 243–256. [Google Scholar] [CrossRef]
- Lipshultz, S.E.; Rifai, N.; Dalton, V.M.; Levy, D.E.; Silverman, L.B.; Lipsitz, S.R.; Colan, S.D.; Asselin, B.L.; Barr, R.D.; Clavell, L.A.; et al. The effect of dexrazoxane on myocardial injury in doxorubicin-treated children with acute lymphoblastic leukemia. N. Engl. J. Med. 2004, 351, 145–153. [Google Scholar] [CrossRef]
- Tebbi, C.K.; London, W.B.; Friedman, D.; Villaluna, D.; De Alarcon, P.A.; Constine, L.S.; Mendenhall, N.P.; Sposto, R.; Chauvenet, A.; Schwartz, C.L. Dexrazoxane-associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric Hodgkin’s disease. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007, 25, 493–500. [Google Scholar] [CrossRef]
- Swain, S.M.; Whaley, F.S.; Gerber, M.C.; Weisberg, S.; York, M.; Spicer, D.; Jones, S.E.; Wadler, S.; Desai, A.; Vogel, C.; et al. Cardioprotection with dexrazoxane for doxorubicin-containing therapy in advanced breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1997, 15, 1318–1332. [Google Scholar] [CrossRef]
- Legha, S.S.; Wang, Y.M.; Mackay, B.; Ewer, M.; Hortobagyi, G.N.; Benjamin, R.S.; Ali, M.K. Clinical and pharmacologic investigation of the effects of alpha-tocopherol on adriamycin cardiotoxicity. Ann. N. Y. Acad. Sci. 1982, 393, 411–418. [Google Scholar] [CrossRef]
- Myers, C.; Bonow, R.; Palmeri, S.; Jenkins, J.; Corden, B.; Locker, G.; Doroshow, J.; Epstein, S. A randomized controlled trial assessing the prevention of doxorubicin cardiomyopathy by N-acetylcysteine. Semin. Oncol. 1983, 10, 53–55. [Google Scholar] [PubMed]
- Choi, S.H.; Jung, S.W.; Lee, B.H.; Kim, H.J.; Hwang, S.H.; Kim, H.K.; Nah, S.Y. Ginseng pharmacology: A new paradigm based on gintonin-lysophosphatidic acid receptor interactions. Front. Pharmacol. 2015, 6, 245. [Google Scholar] [CrossRef] [PubMed]
- Buettner, C.; Yeh, G.Y.; Phillips, R.S.; Mittleman, M.A.; Kaptchuk, T.J. Systematic review of the effects of ginseng on cardiovascular risk factors. Ann. Pharmacother. 2006, 40, 83–95. [Google Scholar] [CrossRef]
- Wee, J.J.; Mee Park, K.; Chung, A.S. Biological Activities of Ginseng and Its Application to Human Health. In Herbal Medicine: Biomolecular and Clinical Aspects, 2nd ed.; Benzie, I.F.F., Wachtel-Galor, S., Eds.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2011. [Google Scholar]
- Wang, H.; Peng, D.; Xie, J. Ginseng leaf-stem: Bioactive constituents and pharmacological functions. Chin. Med. 2009, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Ratan, Z.A.; Haidere, M.F.; Hong, Y.H.; Park, S.H.; Lee, J.O.; Lee, J.; Cho, J.Y. Pharmacological potential of ginseng and its major component ginsenosides. J. Ginseng Res. 2021, 45, 199–210. [Google Scholar] [CrossRef]
- Nah, S.Y.; Kim, D.H.; Rhim, H. Ginsenosides: Are any of them candidates for drugs acting on the central nervous system? CNS Drug Rev. 2007, 13, 381–404. [Google Scholar] [CrossRef]
- Chi, N.C.; Shaw, R.M.; Jungblut, B.; Huisken, J.; Ferrer, T.; Arnaout, R.; Scott, I.; Beis, D.; Xiao, T.; Baier, H.; et al. Genetic and physiologic dissection of the vertebrate cardiac conduction system. PLoS Biol. 2008, 6, e109. [Google Scholar] [CrossRef]
- Tam, J.P.; Nguyen, G.K.T.; Loo, S.; Wang, S.; Yang, D.; Kam, A. Ginsentides: Cysteine and Glycine-rich Peptides from the Ginseng Family with Unusual Disulfide Connectivity. Sci. Rep. 2018, 8, 16201. [Google Scholar] [CrossRef]
- Dutta, B.; Loo, S.; Kam, A.; Sze, S.K.; Tam, J.P. Ginsentide TP1 Protects Hypoxia-Induced Dysfunction and ER Stress-Linked Apoptosis. Cells 2023, 12, 1401. [Google Scholar] [CrossRef]
- Loo, S.; Kam, A.; Dutta, B.; Zhang, X.; Feng, N.; Sze, S.K.; Liu, C.-F.; Wang, X.; Tam, J.P. Broad-spectrum ginsentides are principal bioactives in unraveling the cure-all effects of ginseng. Acta Pharm. Sin. B 2023, 14, 653–666. [Google Scholar] [CrossRef]
- Tam, J.P.; Huang, J.; Loo, S.; Li, Y.; Kam, A. Ginsentide-like Coffeetides Isolated from Coffee Waste Are Cell-Penetrating and Metal-Binding Microproteins. Molecules 2023, 28, 6556. [Google Scholar] [CrossRef]
- Wallace, K.B.; Sardao, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef]
- Shi, S.; Chen, Y.; Luo, Z.; Nie, G.; Dai, Y. Role of oxidative stress and inflammation-related signaling pathways in doxorubicin-induced cardiomyopathy. Cell Commun. Signal 2023, 21, 61. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, S.J.; Kim, B.J.; Rah, S.Y.; Chung, S.M.; Im, M.J.; Kim, U.H. Doxorubicin-induced reactive oxygen species generation and intracellular Ca2+ increase are reciprocally modulated in rat cardiomyocytes. Exp. Mol. Med. 2006, 38, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Gille, L.; Nohl, H. Analyses of the molecular mechanism of adriamycin-induced cardiotoxicity. Free Radic. Biol. Med. 1997, 23, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Povea-Cabello, S.; Oropesa-Avila, M.; de la Cruz-Ojeda, P.; Villanueva-Paz, M.; de la Mata, M.; Suarez-Rivero, J.M.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Cotan, D.; Ybot-Gonzalez, P.; et al. Dynamic Reorganization of the Cytoskeleton during Apoptosis: The Two Coffins Hypothesis. Int. J. Mol. Sci. 2017, 18, 2393. [Google Scholar] [CrossRef]
- Shinlapawittayatorn, K.; Chattipakorn, S.C.; Chattipakorn, N. The effects of doxorubicin on cardiac calcium homeostasis and contractile function. J. Cardiol. 2022, 80, 125–132. [Google Scholar] [CrossRef]
- Fernandez-Chas, M.; Curtis, M.J.; Niederer, S.A. Mechanism of doxorubicin cardiotoxicity evaluated by integrating multiple molecular effects into a biophysical model. Br. J. Pharmacol. 2018, 175, 763–781. [Google Scholar] [CrossRef]
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-induced cardiotoxicity: An update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed. Pharmacother. 2021, 139, 111708. [Google Scholar] [CrossRef]
- Huigsloot, M.; Tijdens, I.B.; Mulder, G.J.; van de Water, B. Differential regulation of doxorubicin-induced mitochondrial dysfunction and apoptosis by Bcl-2 in mammary adenocarcinoma (MTLn3) cells. J. Biol. Chem. 2002, 277, 35869–35879. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Wang, L.; Qiao, Y.; Zhou, Q.; Li, H.; Chen, S.; Yin, D.; Huang, Q.; He, M. Doxorubicin Induces Endotheliotoxicity and Mitochondrial Dysfunction via ROS/eNOS/NO Pathway. Front. Pharmacol. 2019, 10, 1531. [Google Scholar] [CrossRef] [PubMed]
- Kalivendi, S.V.; Konorev, E.A.; Cunningham, S.; Vanamala, S.K.; Kaji, E.H.; Joseph, J.; Kalyanaraman, B. Doxorubicin activates nuclear factor of activated T-lymphocytes and Fas ligand transcription: Role of mitochondrial reactive oxygen species and calcium. Biochem. J. 2005, 389, 527–539. [Google Scholar] [CrossRef]
- Zepeda-Quiróz, I.; Sánchez-Barrera, H.; Colín-Val, Z.; Robledo-Cadena, D.X.; Rodríguez-Enríquez, S.; López-Marure, R. Curcumin promotes oxidative stress, apoptosis and autophagy in H9c2 rat cardiomyoblasts. Mol. Cell. Toxicol. 2020, 16, 441–453. [Google Scholar] [CrossRef]
- Ahmed, A.Z.; Satyam, S.M.; Shetty, P.; D’Souza, M.R. Methyl Gallate Attenuates Doxorubicin-Induced Cardiotoxicity in Rats by Suppressing Oxidative Stress. Scientifica 2021, 2021, 6694340. [Google Scholar] [CrossRef]
- Elberry, A.A.; Abdel-Naim, A.B.; Abdel-Sattar, E.A.; Nagy, A.A.; Mosli, H.A.; Mohamadin, A.M.; Ashour, O.M. Cranberry (Vaccinium macrocarpon) protects against doxorubicin-induced cardiotoxicity in rats. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2010, 48, 1178–1184. [Google Scholar] [CrossRef] [PubMed]
- Warpe, V.S.; Mali, V.R.; Bodhankar, S.L.; Mahadik, K.R. Cardioprotective effect of ellagic acid on doxorubicin induced cardiotoxicity in wistar rats. J. Acute Med. 2015, 5, 1–8. [Google Scholar] [CrossRef]
- De Angelis, A.; Urbanek, K.; Cappetta, D.; Piegari, E.; Ciuffreda, L.P.; Rivellino, A.; Russo, R.; Esposito, G.; Rossi, F.; Berrino, L. Doxorubicin cardiotoxicity and target cells: A broader perspective. Cardiooncology 2016, 2, 2. [Google Scholar] [CrossRef]
- Wenningmann, N.; Knapp, M.; Ande, A.; Vaidya, T.R.; Ait-Oudhia, S. Insights into Doxorubicin-induced Cardiotoxicity: Molecular Mechanisms, Preventive Strategies, and Early Monitoring. Mol. Pharmacol. 2019, 96, 219–232. [Google Scholar] [CrossRef]
- Kumar, D.; Kirshenbaum, L.A.; Li, T.; Danelisen, I.; Singal, P.K. Apoptosis in adriamycin cardiomyopathy and its modulation by probucol. Antioxid. Redox Signal 2001, 3, 135–145. [Google Scholar] [CrossRef]
- Chacko, S.M.; Nevin, K.G.; Dhanyakrishnan, R.; Kumar, B.P. Protective effect of p-coumaric acid against doxorubicin induced toxicity in H9c2 cardiomyoblast cell lines. Toxicol. Rep. 2015, 2, 1213–1221. [Google Scholar] [CrossRef] [PubMed]
- Rahbardar, M.G.; Eisvand, F.; Rameshrad, M.; Razavi, B.M.; Hosseinzadeh, H. In Vivo and In Vitro Protective Effects of Rosmarinic Acid against Doxorubicin-Induced Cardiotoxicity. Nutr. Cancer 2022, 74, 747–760. [Google Scholar] [CrossRef]
- Hiona, A.; Lee, A.S.; Nagendran, J.; Xie, X.; Connolly, A.J.; Robbins, R.C.; Wu, J.C. Pretreatment with angiotensin-converting enzyme inhibitor improves doxorubicin-induced cardiomyopathy via preservation of mitochondrial function. J. Thorac. Cardiovasc. Surg. 2011, 142, 396–403.e3. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.; Yoguchi, A.; Takizawa, T.; Yokoyama, T.; Kanda, T.; Kurabayashi, M.; Nagai, R. Mechanism of doxorubicin-induced inhibition of sarcoplasmic reticulum Ca2+-ATPase gene transcription. Circ. Res. 2000, 86, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Mokni, M.; Hamlaoui-Guesmi, S.; Amri, M.; Marzouki, L.; Limam, F.; Aouani, E. Grape seed and skin extract protects against acute chemotherapy toxicity induced by doxorubicin in rat heart. Cardiovasc. Toxicol. 2012, 12, 158–165. [Google Scholar] [CrossRef]
- Jayachandra, R.; Zhao, H.; Cheng, Z.; Luo, L.; Sun, T.; Tan, W. Synthesis of Isosteviol analogues as potential protective agents against Doxorubicin-induced cardiomyopathy in zebrafish embryos. Bioorg. Med. Chem. Lett. 2019, 29, 1705–1709. [Google Scholar] [CrossRef]
- Maciag, M.; Wnorowski, A.; Mierzejewska, M.; Plazinska, A. Pharmacological assessment of zebrafish-based cardiotoxicity models. Biomed. Pharmacother. 2022, 148, 112695. [Google Scholar] [CrossRef]
- Meng, Y.; Zhong, K.; Xiao, J.; Huang, Y.; Wei, Y.; Tang, L.; Chen, S.; Wu, J.; Ma, J.; Cao, Z.; et al. Exposure to pyrimethanil induces developmental toxicity and cardiotoxicity in zebrafish. Chemosphere 2020, 255, 126889. [Google Scholar] [CrossRef]
- Sarmah, S.; Marrs, J.A. Zebrafish as a Vertebrate Model System to Evaluate Effects of Environmental Toxicants on Cardiac Development and Function. Int. J. Mol. Sci. 2016, 17, 2123. [Google Scholar] [CrossRef]
- Wang, X.; Yang, X.; Wang, J.; Li, L.; Zhang, Y.; Jin, M.; Chen, X.; Sun, C.; Wang, R.; Liu, K. Cardiotoxicity of sanguinarine via regulating apoptosis and MAPK pathways in zebrafish and HL1 cardiomyocytes. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2022, 252, 109228. [Google Scholar] [CrossRef]
- Messerli, F.H.; Bangalore, S.; Bavishi, C.; Rimoldi, S.F. Angiotensin-Converting Enzyme Inhibitors in Hypertension: To Use or Not to Use? J. Am. Coll. Cardiol. 2018, 71, 1474–1482. [Google Scholar] [CrossRef]
- Oliveira, M.S.; Melo, M.B.; Carvalho, J.L.; Melo, I.M.; Lavor, M.S.; Gomes, D.A.; de Goes, A.M.; Melo, M.M. Doxorubicin Cardiotoxicity and Cardiac Function Improvement After Stem Cell Therapy Diagnosed by Strain Echocardiography. J. Cancer Sci. Ther. 2013, 5, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Razmaraii, N.; Babaei, H.; Mohajjel Nayebi, A.; Asadnasab, G.; Ashrafi Helan, J.; Azarmi, Y. Cardioprotective Effect of Phenytoin on Doxorubicin-induced Cardiac Toxicity in a Rat Model. J. Cardiovasc. Pharmacol. 2016, 67, 237–245. [Google Scholar] [CrossRef]
- Li, G.; Li, W.R.; Jin, Y.G.; Jie, Q.Q.; Wang, C.Y.; Wu, L. Tetrandrine Attenuated Doxorubicin-Induced Acute Cardiac Injury in Mice. BioMed Res. Int. 2020, 2020, 2616024. [Google Scholar] [CrossRef] [PubMed]
- Mattila, M.; Soderstrom, M.; Ailanen, L.; Savontaus, E.; Savontaus, M. The Effects of Neuropeptide Y Overexpression on the Mouse Model of Doxorubicin-Induced Cardiotoxicity. Cardiovasc. Toxicol. 2020, 20, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Takemura, G.; Fujiwara, H. Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog. Cardiovasc. Dis. 2007, 49, 330–352. [Google Scholar] [CrossRef]
- Naderi, Y.; Khosraviani, S.; Nasiri, S.; Hajiaghaei, F.; Aali, E.; Jamialahmadi, T.; Banach, M.; Sahebkar, A. Cardioprotective effects of minocycline against doxorubicin-induced cardiotoxicity. Biomed. Pharmacother. 2023, 158, 114055. [Google Scholar] [CrossRef]
- Zilinyi, R.; Czompa, A.; Czegledi, A.; Gajtko, A.; Pituk, D.; Lekli, I.; Tosaki, A. The Cardioprotective Effect of Metformin in Doxorubicin-Induced Cardiotoxicity: The Role of Autophagy. Molecules 2018, 23, 1184. [Google Scholar] [CrossRef]
- van Acker, S.A.; Kramer, K.; Voest, E.E.; Grimbergen, J.A.; Zhang, J.; van der Vijgh, W.J.; Bast, A. Doxorubicin-induced cardiotoxicity monitored by ECG in freely moving mice. A new model to test potential protectors. Cancer Chemother. Pharmacol. 1996, 38, 95–101. [Google Scholar] [CrossRef]
- Wu, B.B.; Leung, K.T.; Poon, E.N. Mitochondrial-Targeted Therapy for Doxorubicin-Induced Cardiotoxicity. Int. J. Mol. Sci. 2022, 23, 1912. [Google Scholar] [CrossRef]
- Dutta, B.; Loo, S.; Kam, A.; Tam, J.P. Plant-derived cell-penetrating microprotein alpha-astratide aM1 targets Akt signaling and alleviates insulin resistance. Cell Mol. Life Sci. 2023, 80, 293. [Google Scholar] [CrossRef] [PubMed]
- Dutta, B.; Huang, J.; To, J.; Tam, J.P. LIR Motif-Containing Hyperdisulfide beta-Ginkgotide is Cytoprotective, Adaptogenic, and Scaffold-Ready. Molecules 2019, 24, 2417. [Google Scholar] [CrossRef] [PubMed]
- Kam, A.; Loo, S.; Dutta, B.; Sze, S.K.; Tam, J.P. Plant-derived mitochondria-targeting cysteine-rich peptide modulates cellular bioenergetics. J. Biol. Chem. 2019, 294, 4000–4011. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dutta, B.; Loo, S.; Kam, A.; Wang, X.; Wei, N.; Luo, K.Q.; Liu, C.-F.; Tam, J.P. Cell-Permeable Microprotein from Panax Ginseng Protects Against Doxorubicin-Induced Oxidative Stress and Cardiotoxicity. Antioxidants 2025, 14, 493. https://doi.org/10.3390/antiox14040493
Dutta B, Loo S, Kam A, Wang X, Wei N, Luo KQ, Liu C-F, Tam JP. Cell-Permeable Microprotein from Panax Ginseng Protects Against Doxorubicin-Induced Oxidative Stress and Cardiotoxicity. Antioxidants. 2025; 14(4):493. https://doi.org/10.3390/antiox14040493
Chicago/Turabian StyleDutta, Bamaprasad, Shining Loo, Antony Kam, Xiaoliang Wang, Na Wei, Kathy Qian Luo, Chuan-Fa Liu, and James P. Tam. 2025. "Cell-Permeable Microprotein from Panax Ginseng Protects Against Doxorubicin-Induced Oxidative Stress and Cardiotoxicity" Antioxidants 14, no. 4: 493. https://doi.org/10.3390/antiox14040493
APA StyleDutta, B., Loo, S., Kam, A., Wang, X., Wei, N., Luo, K. Q., Liu, C.-F., & Tam, J. P. (2025). Cell-Permeable Microprotein from Panax Ginseng Protects Against Doxorubicin-Induced Oxidative Stress and Cardiotoxicity. Antioxidants, 14(4), 493. https://doi.org/10.3390/antiox14040493