
Production and Role of Free Radicals and Reactive Oxygen Species After Facial Nerve Injury



Abstract
1. Introduction
1.1. Facial Nerve
1.2. Reactive Oxygen Species
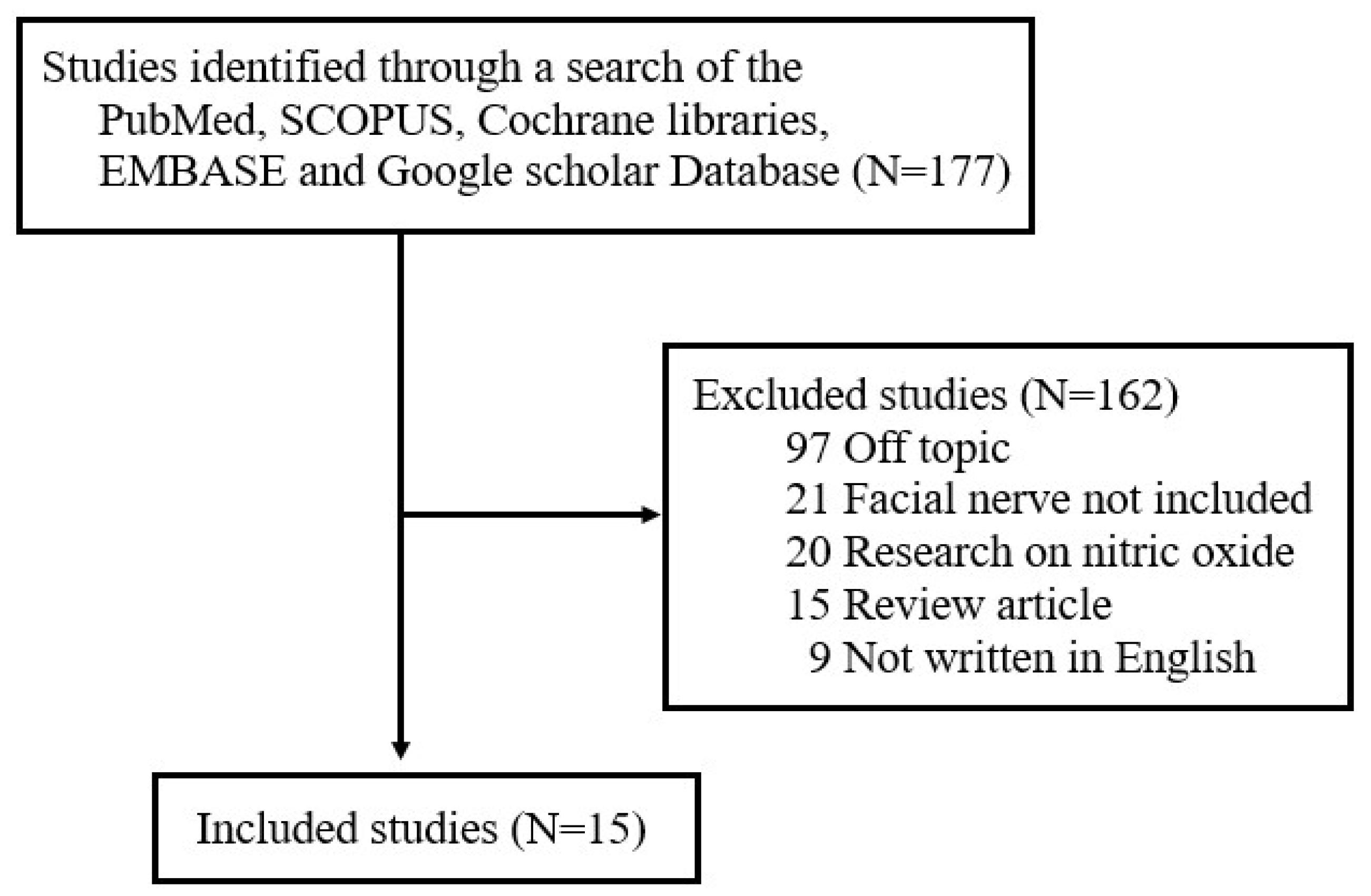
2. Research Methods
3. Results
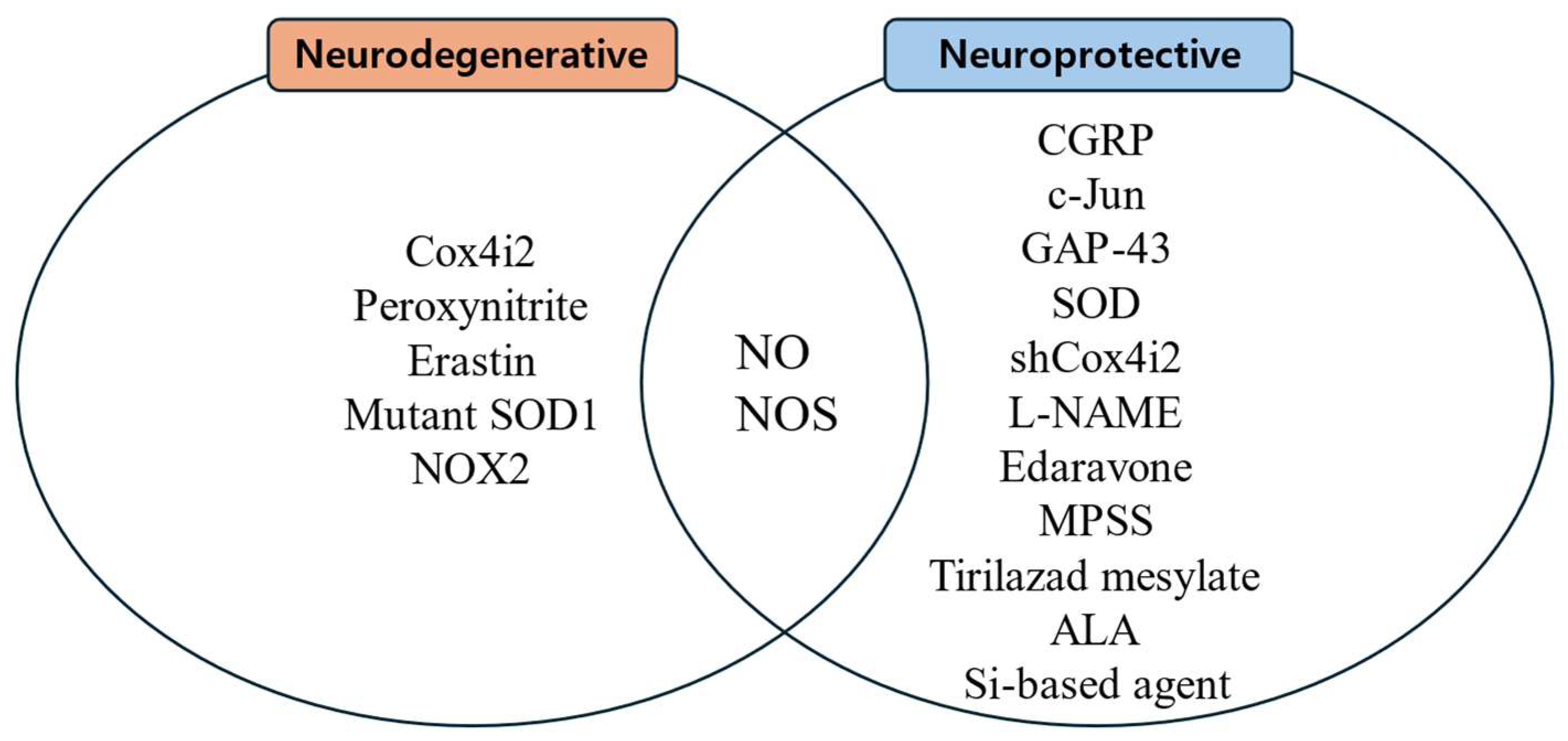
3.1. Facial Nerve Degeneration Through Increased ROS and FR After Facial Nerve Injury
3.1.1. Peroxynitrite, NO, and NOS
3.1.2. Erastin-Induced Ferroptosis
3.1.3. Unscheduled DNA Synthesis and Mitochondrial DNA Synthesis
3.2. Facial Nerve Regeneration by Antioxidants After Facial Nerve Injury
3.2.1. Superoxide Dismutase
3.2.2. SOD and GSH
3.2.3. L-NAME
3.2.4. Methylprednisolone Sodium Succinate
3.2.5. Edaravone
3.2.6. Tirilazad Mesylate
3.2.7. Alpha-Lipoic Acid
3.2.8. Si-Based Agent and MeCbl Combination Therapy
3.3. Age-Dependent Role of ROS and FR in Facial Nerve Injury
3.4. Molecular Mechanisms of ROS and Free Radicals in Facial Nerve Injury
4. Limitations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author/ Year/ Reference | Study Design | Species and/or Sample | Nerve/Injury Method | Detection Method | Target Substance(s) Associated with Free Radicals | Results/Conclusions |
|---|---|---|---|---|---|---|
| Mariotti, R. et al., 1997 [43] | Animal study | 31 Wistar male rats | Unilateral facial nerve transection (left side) | NADPH-d histochemistry, TUNEL assay | NOS, NADPH-d | Facial motoneurons in the non-operated side showed no evidence of NADPH-d positivity at any examined age, whereas most experiments showed positive NADPH-d staining in axotomized facial motoneurons. In cases where the facial nerve was cut on postnatal day 0, NADPH-d staining was undetectable in facial nuclei on either side after 1 or 2 days. For facial nerves transected 1 week postnatally, NADPH-d induction was absent in ipsilateral facial nucleus after 2 days, but was marked in injured facial motoneurons 4 days after the axotomy. NADPH-d activity was observed in facial motoneurons following axotomy, beginning from the end of the first postnatal week and continuing through adulthood, a period during which neuronal loss was less pronounced compared to that in newborns. The time course of NADPH-d induction in facial motoneurons following postnatal axotomy exhibits remarkable age dependence. By contrast, 1 week was sufficient to observe an intense NADPH-d induction in facial motoneurons axotomized in the adult. These data support the view that NOS induction in motoneurons requires a certain amount of time after nerve injury and highlight the age-dependent nature of the temporal gradient. |
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Byun, J.Y. Facial paralysis disorders. Anatomy and evaluation of facial nerve. In Korean Society of Otorhinolaryngology−Head and Neck Surgery; Koonja Publishing: Seoul, Republic of Korea, 2018; Volume 3, pp. 913–932. [Google Scholar]
- Dobie, R.A.; Fredrickson, J.M.; Harker, L.A. Test of facial nerve function. In Otolaryngology Head and Neck Surgery; Mosby: New York, NY, USA, 1998; Volume 3, pp. 2757–2766. [Google Scholar]
- Francis, H.W.; Flint, P.W.; Haughey, B.H.; Lund, V.J. Anatomy of the temporal bone, external ear, and middle ear. In Cummings Otolaryngology-Head and Neck Surgery; Saunders: Philadelphia, PA, USA, 2015; Volume 6, pp. 1984–1985. [Google Scholar]
- Lee, J.D. Facial paralysis disorders. In Korean Society of Otorhinolaryngology−Head and Neck Surgery; Koonja Publishing: Seoul, Republic of Korea, 2018; Volume 3, pp. 933–942. [Google Scholar]
- Montserrat, L.; Benito, M. Facial synkinesis and aberrant regeneration of facial nerve. Adv. Neurol. 1988, 49, 211–244. [Google Scholar] [PubMed]
- Moran, C.J.; Neely, J.G. Patterns of facial nerve synkinesis. Laryngoscope 1996, 106, 1491–1496. [Google Scholar] [CrossRef] [PubMed]
- Gantz, B.J.; Rubinstein, J.T.; Gidley, P.; Woodworth, G.G. Surgical management of Bell’s palsy. Laryngoscope 1999, 109, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- McCabe, B.F. Injuries to the facial nerve. Laryngoscope 1972, 182, 1981–1986. [Google Scholar]
- Jeon, B.J.; Kim, J. Surgical treatment and rehabilitation of facial paralysis. In Korean Society of Otorhinolaryngology−Head and Neck Surgery; Koonja Publishing: Seoul, Republic of Korea, 2018; Volume 3, pp. 943–959. [Google Scholar]
- Lam, A.Q.; Tran, T.P.C.; Tran, D.V.; Tran, H.X.; Fox, A.J.; Tran, L.V. Rehabilitation Surgery for Peripheral Facial Nerve Injury after Facial Trauma. Int. Arch. Otorhinolaryngol. 2024, 28, e509–e516. [Google Scholar] [CrossRef]
- Novak, C.B. Rehabilitation strategies for facial nerve injuries. Semin. Plast. Surg. 2004, 18, 47–52. [Google Scholar]
- Gordin, E.; Lee, T.S.; Ducic, Y.; Arnaoutakis, D. Facial nerve trauma: Evaluation and considerations in management. Craniomaxillofac. Trauma Reconstr. 2015, 8, 1–13. [Google Scholar] [CrossRef]
- Van Haesendonck, G.; Jorissen, C.; Lammers, M. Guidelines for the initial management of acute facial nerve palsy. B-ENT 2022, 18, 67–72. [Google Scholar] [CrossRef]
- Yang, H.S. The Reality of Reactive Oxygen Species. Acute and Critical Care. J. Korean Soc. Emerg. Med. 1990, 5, 87–97. [Google Scholar]
- Hong, Y.; Boiti, A.; Vallone, D.; Foulkes, N.S. Reactive Oxygen Species Signaling and Oxidative Stress: Transcriptional Regulation and Evolution. Antioxidants 2024, 13, 312. [Google Scholar] [CrossRef]
- Turpaev, K.T. Reactive oxygen species and regulation of gene expression. Biochemistry 2002, 67, 281–292. [Google Scholar] [PubMed]
- Sies, H.; Jones, P.D. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Review Mol. Cell Biol. 2020, 21, 362–383. [Google Scholar]
- Hyong, I.H.; Moon, S.E.; Bae, S.S. Review of Reactive Oxygen. J. Korean Soc. Phys. Med. 2006, 1, 139–146. [Google Scholar]
- Hong, J.K. A Study on Skin Aging Caused by Free-Radical and on Efficacy of Antioxidant Vitamins. Asian J. Beauty Cosmetol. 2009, 7, 51–62. [Google Scholar]
- Kim, K.S. The Role of Antioxidants in Anti-Aging Treatments; The Korean Academy of Geriatric and Gerontology: Seoul, Republic of Korea, 2004; pp. 379–392. [Google Scholar]
- Cary, E.J.; McCarty, M.F. Potential Clinical Implications for High Dose Nutritional Antioxidants. Med. Hypotheses 1984, 13, 77–98. [Google Scholar]
- Keane, J.A.; Ealy, A.D. An Overview of Reactive Oxygen Species Damage Occurring during In Vitro Bovine Oocyte and Embryo Development and the Efficacy of Antioxidant Use to Limit These Adverse Effects. Animals 2024, 14, 330. [Google Scholar] [CrossRef]
- Dumanovic, J.; Nepovimova, E.; Natic, M.; Kuca, K.; Jacevic, V. The Significance of Reactive Oxygen Species and Antioxidant Defense System in Plants: A Concise Overview. Front. Plant Sci. 2021, 11, 552969. [Google Scholar]
- Yeung, A.W.K.; Tzvetkov, N.T.; Georgieva, M.G.; Ognyanov, I.V.; Kordos, K.; Jóźwik, A.; Kühl, T.; Perry, G.; Petralia, M.C.; Mazzon, E.; et al. Reactive Oxygen Species and Their Impact in Neurodegenerative Diseases: Literature Landscape Analysis. Antioxid. Redox Signal. 2021, 34, 402–420. [Google Scholar]
- Nicol, M. Vitamins and Immunity. Allerg. Immunol. 1993, 25, 70–73. [Google Scholar]
- Oh, Y.J.; Yon, D.K.; Choi, Y.S.; Lee, J.; Yeo, J.H.; Kim, S.S.; Lee, J.M.; Yeo, S.G. Induction of Nitric Oxide and Its Role in Facial Nerve Regeneration According to the Method of Facial Nerve Injury. Antioxidants 2024, 13, 741. [Google Scholar] [CrossRef]
- Huang, L.; Bian, M.; Zhang, J.; Jiang, L. Iron Metabolism and Ferroptosis in Peripheral Injury. Oxid. Med. Cell Longev. 2022, 2022, 5918218. [Google Scholar] [PubMed]
- Rochette, L.; Dogona, G.; Rigal, E.; Zeller, M.; Cottin, Y.; Vergely, C. Lipid Peroxidation and Iron Metabolism: Two Corner Stones in the Homeostasis Control of Ferroptosis. Int. J. Mol. Sci. 2023, 24, 449. [Google Scholar] [CrossRef] [PubMed]
- Yeh, T.Y.; Liu, P.H. Inhibition of nitric oxide production enhances the activity of facial nerve tubulin polymerization and the ability of tau to promote microtubule assembly after neurorrhaphy. Neurochem. Int. 2021, 150, 105183. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.H.; Yang, L.H.; Wang, T.Y.; Wang, Y.J.; Tseng, G.F. Proximity of Lesioning Determines Response of Facial Motoneurons to Peripheral Axotomy. J. Neurotrauma 2006, 23, 1857–1873. [Google Scholar] [CrossRef]
- Gao, D.; Huang, Y.; Sun, X.; Yang, J.; Chen, J.; He, J. Overexpression of c-Jun inhibits erastin-induced ferroptosis in Schwann cells and promotes repair of facial nerve function. J. Cell Mol. Med. 2002, 26, 2191–2204. [Google Scholar] [CrossRef]
- Korr, H.; Philippi, C.; Schiefer, H.J.; Graeber, M.B.; Kreuztberg, G.W. Unscheduled DNA synthesis and mitochondrial DNA synthetic rate following injury of the facial nerve. Acta Neuropathol. 1997, 94, 557–566. [Google Scholar] [CrossRef]
- Mohri, D.; Satomi, F.; Kondo, E.; Fukuoka, T.; Sakagami, M.; Noguchi, K. Change in gene expression in facial nerve nuclei and the effect of superoxide dismutase in a rat model of ischemic facial paralysis. Brain Res. 2001, 893, 227–236. [Google Scholar]
- Mariotti, R.; Cristino, L.; Bressan, C.; Boscolo, S.; Bentivoglio, M. Altered reation of facial motoneurons to axonal damage in the presymptomatic phase of a murine model familial amyotrophic lateral sclerosis. Neuroscience 2002, 115, 331–335. [Google Scholar] [CrossRef]
- Chang, B.; Guan, H.; Wang, X.; Chen, Z.; Zhu, W.; Wei, X.; Li, S. Cox4i2 Triggers and Increase in Reactive Oxygen Species, Leading to Ferroptosis and Apoptosis in HHV7 Infected Schwann Cells. Front. Mol. Biosci. 2021, 8, 660072. [Google Scholar] [CrossRef]
- Ruan, R.S.; Leong, S.K.; Yeoh, K.H. The role of nitric oxide in facial motoneuronal death. Brain Res. 1995, 698, 163–168. [Google Scholar] [CrossRef]
- Chen, Y.S.; Tseng, F.Y.; Tan, C.T.; Lin-Shiau, S.Y.; Hsu, C.J. Effects of methylprednisolone on nitric oxide formation and survival of facial motor neurons after axotomy. Brain Res. 2008, 1197, 23–31. [Google Scholar] [PubMed]
- Hato, N.; Kohno, H.; Yamada, H.; Takahashi, H.; Gyo, K. Role of Nitric Oxide in the Onset of Facial Nerve Palsy by HSV-1 Infection. JAMA Otolaryngol.–Head Neck Surg. 2013, 139, 1339–1342. [Google Scholar] [PubMed]
- Takeda, T.; Takeda, S.; Takumida, M.; Okada, T.; Kakigi, A.; Nakatani, H.; Hamda, M.; Yamakawa, K. Protective effects of edaravone against ischemia-induced facial paralysis. Auris Nasus Larynx 2008, 35, 321–327. [Google Scholar] [PubMed]
- Hall, E.D.; Smith, S.L.; Oostveen, J.A. Inhibition of Lipid Peroxidation Attenuates Axotomy-Induced Apoptotic Degeneration of Facial Motor Neurons in Neonatal Rats. J. Neurosci. Res. 1996, 44, 293–299. [Google Scholar]
- Yoo, M.C.; Ryu, I.Y.; Choi, J.W.; Lee, J.M.; Byun, J.Y.; Yeo, S.G. Nicotinamide Adenine Dinucleotide Phosphate Oxidase 2 Expression and Effects of Alpha Lipoic Acid on Recovery in a Rat Model of Facial Nerve Injury. Biomedicines 2022, 10, 291. [Google Scholar] [CrossRef]
- Koyama, Y.; Harada, S.; Sato, T.; Kobayashi, Y.; Yanagawa, H.; Iwahashi, T.; Tanaka, H.; Ohata, K.; Imai, T.; Ohta, Y.; et al. Therapeutic strategy for facial paralysis based on the combined application of Si-based agent and methylcobalamin. Biochem. Biophys. Rep. 2022, 32, 101388. [Google Scholar]
- Mariotti, R.; Peng, Z.C.; Kristensson, K.; Bentivoglio, M. Age Dependent Induction of Nitric Oxide Synthase Activity in Facial Motoneurons after Axotomy. Exp. Neurol. 1997, 145, 361–370. [Google Scholar]
- Christman, J.W.; Blackwell, T.S.; Juurlink, B.H. Redox regulation of nuclear factor kappa B: Therapeutic potential for attenuating inflammatory responses. Brain Pathol. 2000, 10, 153–162. [Google Scholar]
- Shiau, J.P.; Chuang, Y.T.; Cheng, Y.B.; Tang, J.Y.; Hou, M.F.; Yen, C.Y.; Chang, H.W. Impacts of Oxidative Stress and PI3K/AKT/mTOR on Metabolism and the Future Direction of Investigating Fucoidan-Modulated Metabolism. Antioxidants 2022, 11, 911. [Google Scholar] [CrossRef]
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro. 2020, 12, 1759091419899782. [Google Scholar]
- Yang, D.; Elner, S.G.; Bian, Z.M.; Till, G.O.; Petty, H.R.; Elner, V.M. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp. Eye Res. 2007, 85, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Terenghi, G. Peripheral nerve regeneration and neurotrophic factors. J. Anat. 1999, 194, 1–14. [Google Scholar] [CrossRef] [PubMed]
- McGregor, C.E.; English, A.W. The Role of BDNF in Peripheral Nerve Regeneration: Activity-Dependent Treatments and Val66Met. Front. Cell. Neurosci. 2019, 12, 522. [Google Scholar] [CrossRef] [PubMed]
- Averill-Bates, D. Reactive oxygen species and cell signaling. Review. Biochim. Biophys. Acta Mol. Cell Res. 2024, 1871, 119573. [Google Scholar] [CrossRef]
- Wright, C.J.; Dennery, P.A. Manipulation of gene expression by oxygen: A primer from bedside to bench. Pediatr. Res. 2009, 66, 3–10. [Google Scholar] [CrossRef]
- Fu, H.; Hardy, J.; Duff, K.E. Selective vulnerability in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1350–1358. [Google Scholar] [CrossRef]
- Johri, A.; Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef]
- Jha, M.K.; Jeon, S.; Suk, K. Glia as a Link between Neuroinflammation and Neuropathic Pain. Immune Netw. 2012, 12, 41–47. [Google Scholar] [CrossRef]
- Lee, K.H.; Cha, M.; Lee, B.H. Neuroprotective Effect of Antioxidants in the Brain. Int. J. Mol. Sci. 2020, 21, 7152. [Google Scholar] [CrossRef]

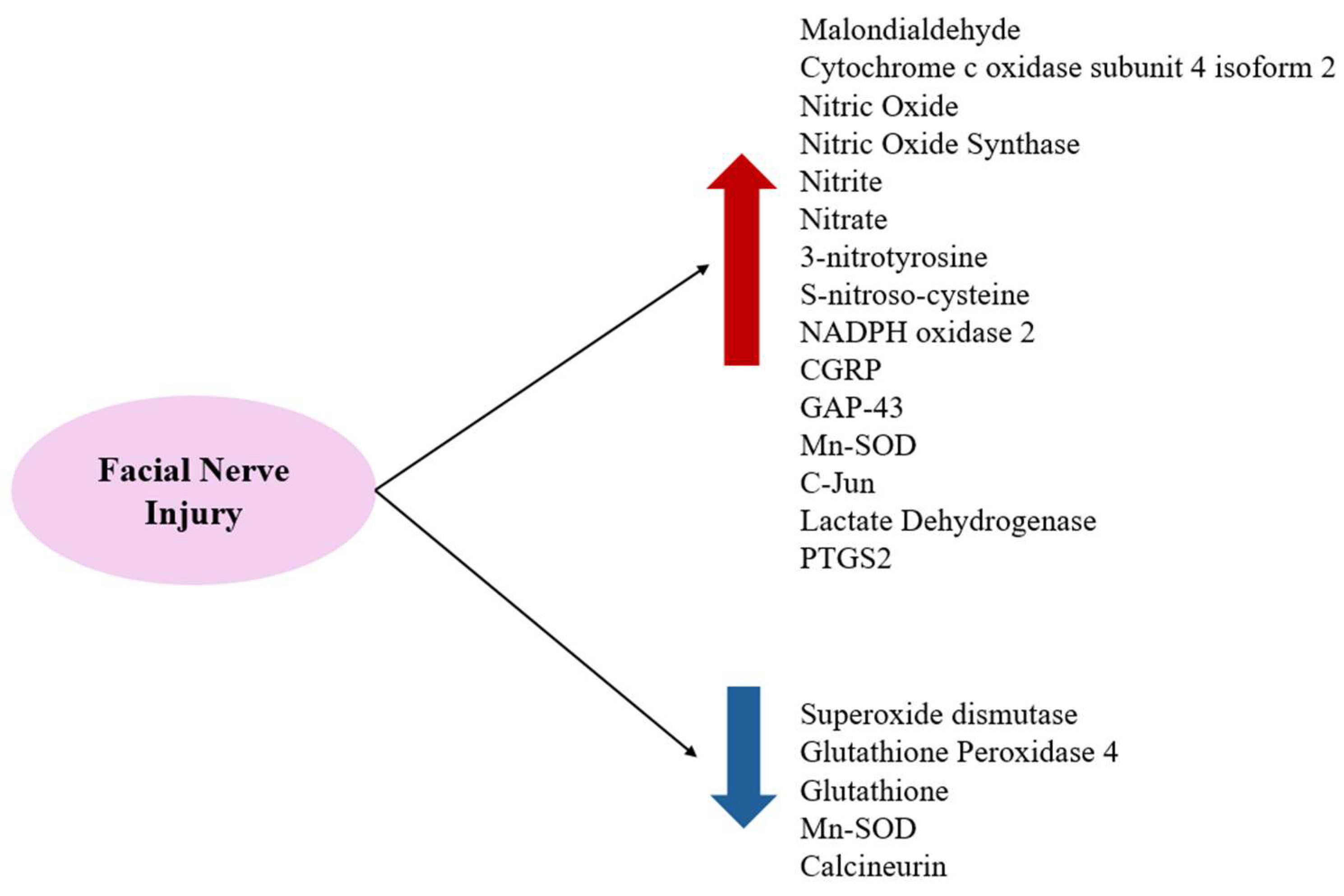
| Type of Injury | Increased | Decreased |
|---|---|---|
| Viral infection | c-Jun COX4I2 LDH MDA NO NO2− (nitrite), NO3− (nitrate) NOS PTGS2 | GSH GPX4 SOD |
| Axotomy | c-Jun CGRP GAP-43 Mn-SOD (after distal axotomy) NO NOS NOX2 3-NT, SNO-Cys | Calcineurin Mn-SOD (after proximal axotomy) |
| Author/ Year/ Reference | Study Design | Species and/or Sample | Nerve/Injury Method | Detection Method | Target Substance(s) Associated with Free Radicals | Results/Conclusions |
|---|---|---|---|---|---|---|
| Yeh, T.Y. et al., 2021 [29] | Animal study | Male Sprague Dawley rats | Left facial nerve transection | L-NAME treatment, immunoprecipitation, microtubule assembly assay, immunohistochemistry | NO, peroxynitrite, 3-NT, SNO-Cys | 3-NT and SNO-Cys (nitrosative stress biomarkers) were highly upregulated in proximal and distal stumps of reconnected facial nerves 3 days and 1 week after neurorrhaphy. 3-NT expression was substantially reduced at 2 weeks, whereas that of SNO-Cys was maintained. The inhibition of NO production with L-NAME prevented the upregulation of SNO-Cys and enhanced the polymerization activity of tubulins and the ability of tau to promote microtubule assembly. NO or peroxynitrite reduces the polymerization of normal rat facial nerve tubulins and the ability of tau to promote microtubule assembly. Preventing NO- or peroxynitrite-induced alterations in tubulin or tau may be clinically useful for facilitating microtubule assembly and the consequent recovery of axon regeneration. |
| Liu, P.H. et al., 2006 [30] | Animal study | Adult female Wistar rats | Distal and proximal facial axotomy | Light microscopy, electron microscopy, immunohistochemistry, TUNEL assay, cresyl-violet staining | nNOS, calcineurin, Mn-SOD | Distal lesions resulted in the upregulation of NOS expression in neurons and the persistent downregulation of neuronal expression of the NOS-activating enzyme calcineurin. They also led to the transient downregulation of neuronal expression of Mn-SOD and modest neuronal loss. Proximal axotomy led to the upregulation of NOS but transient downregulation in the expression of calcineurin and Mn-SOD at 4 weeks after injury. nNOS expression in proximally axotomized FMNs was more likely to produce NO, beginning 1 week after lesioning, than nNOS in distantly axotomized FMNs if other intracellular factors that affected nNOS function were not greatly altered in the two cases. The mitochondrial Mn-SOD expression pattern in proximally axotomized FMNs further suggests that these mitochondria are more likely to face serious superoxide toxicity than those in distally axotomized cells. |
| Gao, D. et al., 2022 [31] | Animal study | Sprague Dawley rats | Right facial nerve injury with AAV-mediated expression of c-Jun or c-Jun siRNA | Immunofluorescence, cell viability, PI staining, ROS, ELISA, lentivirus transfection, Western blotting, RT-PCR | c-Jun, GPX4, GSH, PTSG2, MDA, Fe2+, ROS | The ferroptosis activator erastin decreased GPX4 and c-Jun protein expression and GSH content, while increasing PTSG2, NRF2, and HO-1 protein, and MDA, Fe2+, and ROS content. These effects were inhibited by c-Jun overexpression but were reversed by siRNA-mediated c-Jun knockdown. c-Jun can inhibit ferroptosis in the facial nerve and promote facial nerve recovery and S-100 expression, allowing the nerve to form a more morphologically normal myelin sheath. |
| Korr, H. et al., 1997 [32] | Animal study | Adult male Wistar rats | Right facial nerve transection | Light microscopic autoradiography, Northern blotting, electron microscopic autoradiography | FRs | In addition to the increased rate of mtDNA synthesis in facial motoneurons 12 h after axotomy, UDS and mtDNA synthesis significantly increased in the regenerating facial nucleus 4 days after axotomy. FRs produced by mitochondria in injured nerve cells can cause nonspecific DNA damage, followed by immediate repair. |
| Author/ Year/ Reference | Study Design | Species and/or Sample | Nerve/Injury Method | Detection Method | Target Substance(s) Associated with Free Radicals | Results/Conclusions |
|---|---|---|---|---|---|---|
| Mohri, D. et al., 2001 [33] | Animal study | Male Sprague Dawley rats | The induction of the transient paralysis of the facial nerve by the embolization of vessels supplying the temporal bone | Confocal laser-scanning microscopy, in situ hybridization, histochemistry | SOD | This study examining the effect of systemic administration of SOD on facial paralysis and mRNA expression in facial nerve nuclei following vascular embolization showed that CGRP, c-Jun, and GAP-43 mRNA exhibited distinct patterns of induction and a time course of increase after ischemic nerve injury (p < 0.05). SOD treatment clearly alleviated behavioral impairments and decreased CGRP mRNA expression on day 3 after injury (p < 0.01). Ischemic facial nerve injury can be attenuated by pre-treatment with SOD, providing indirect evidence that FRs may be partially responsible for the development of peripheral nerve injury after ischemia. |
| Mariotti, R. et al., 2002 [34] | Animal study | Mice carrying a mutated SOD1 gene and wild-type mice (B6SJL) | Axotomy of FALS facial motoneurons | NADPH-d histochemistry, immunohistochemistry, avidin-biotin peroxidase protocol, cresyl violet staining | SOD1, NOS | Axotomy elicited high NOS expression in facial motoneurons of wild-type mice after 2–3 weeks, whereas induction was very weak or absent in Sod1-mutant transgenic mice. At 1 month post-axotomy, the loss of facial motoneurons was significantly higher in mutant mice than in wild-type littermates. SOD1 mutation interferes with the oxidative cascade elicited by axonal injury in cranial motoneurons. Moreover, the adverse gain of function of mutant SOD1 enhances the vulnerability of motoneurons to peripheral stressful conditions. |
| Chang, B. et al., 2021 [35] | Animal study | 23-week-old female Wistar rats | Induction of facial nerve injury by HHV7 inoculation | Cell culture and HHV7 infection, immunofluorescence staining, qRT-PCR, Western blotting, lipid peroxidation assay, flow cytometry, Phen green staining, TUNEL assay | SOD, MDA, GSH, COX4I2 | HHV7-induced facial nerve injury increased levels of MDA and decreased levels of SOD and GSH, reflecting lipid peroxidation and weakening of antioxidant capacity. Transfection of shCox4i2 into HHV7-treated SCs relieved oxidative stress and decreased Cox4i2 expression. The same trend was observed for ROS concentrations (p < 0.01). The increased expression of Cox4i2 in HHV7-infected SCs promoted the production of ROS, whereas the knockdown of Cox4i2 expression in these cells caused a relative decrease in ROS levels. |
| Ruan, R.S. et al., 1995 [36] | Animal study | 28 male Wistar albino rats | Left facial nerve avulsion | Immunocytochemistry, NADPH-d histochemistry | NOS, NADPH-d | In unoperated FMNs, NADPH-d staining in neurons was weak and NOS immunoreactivity was absent. After nerve injury, NADPH-d reactivity increased as early as 2 days, and NOS-positive neurons emerged by day 5. The intensity and number of both markers increased progressively, peaking at 20 days post-injury, after which NOS expression declined. By 30 days, >60% of neurons were lost in the operated FMNs, but neuronal loss was reduced to 43% by the daily administration of L-NAME. NADPH-d and NOS play roles in the neuronal response to injury, and NOS inhibition may protect against neuronal loss. |
| Chen, Y.S. et al., 2007 [37] | Animal study | 83 Hartley albino guinea pigs | Right facial nerve transection | Immunohistochemistry, cresyl violet staining, NO/ozone chemiluminescence | nNOS, NO | Facial nerve transection induced a significant increase in NO formation in the brainstem by 1 week in both MPSS- and saline-treated groups that lasted to the end of the study (4 weeks). MPSS administration appeared to delay the increase in NOS expression and NO formation during the first 1–2 weeks after facial nerve transection compared with saline-treated controls (p < 0.001). The survival rate of FMNs was significantly higher in the MPSS-treated group than in the saline-treated group at 3 and 4 weeks after facial nerve transection (p < 0.05). The administration of a large dose of MPSS produces a delayed increase in NO formation in the brainstem that may attenuate the loss of FMNs. |
| Hato, N. et al., 2013 [38] | Animal study | 62 female Balb/cAjcl mice | Facial paralysis induced by HSV-1 inoculation | Behavioral test, in vivo microdialysis, high-performance liquid chromatography | NO, iNOS, edaravone | In cases where HSV-1 induced facial palsy, which usually occurred 7 days after inoculation, NO levels were significantly higher on the inoculated side than on the control side (p < 0.001). High NO levels in the inoculated side decreased following recovery from palsy. No increase in NO levels was observed in animals without transient facial palsy. The administration of edaravone significantly decreased the incidence of facial palsy. NO produced by iNOS in the facial nerve plays an important role in the onset of facial palsy caused by HSV-1 infection, considered a causative virus of Bell’s palsy. This study elucidated the role of NO in HSV-1-related facial nerve paralysis in mice and evaluated the role of edaravone, an FR scavenger, in preventing paralysis |
| Takeda, T. et al., 2008 [39] | Animal study | Guinea pigs | Ischemic facial palsy induced by the interruption of the petrosal artery | Morphological study, light microscopy, electron microscopy, fluorescence microscopy | ROS, hydroxyl radicals | Edaravone (MCI-186), an extremely potent scavenger of hydroxyl radicals, inhibits not only hydroxyl radicals but also iron-induced peroxidative injuries. Edaravone injection produced signs of facial movement recovery beginning 15 to 19 days after the intervention. It also reduced tissue damage: nerve fibers were relatively intensely stained with Luxol fast blue and, significantly, cell bodies were observed in the geniculate ganglion. The excess generation of ROS likely participates in the development and severity of facial palsy, and the reduction of ROS could attenuate severe nerve damage. |
| Hall, E.D. et al., 1996 [40] | Animal study | 14-day-old male Sprague Dawley rats | Facial nerve transection | Choline acetyltransferase immunocytochemistry, cresyl violet histochemistry | Lipid peroxidation inhibitor tirilazad mesylate (U-74006F) | Following facial nerve axotomy, survival rates of motor neurons in regions A, B, and C of the ipsilateral facial nucleus were 56.2%, 50.6%, and 57.4%, respectively, of those in the non-axotomized contralateral region (p < 0.001). Treatment with U-74006F significantly enhanced motor neuron survival, increasing survival rates in region B and C to 72.8% and 66.7%, respectively, at a dose of 10 mg/kg, and to 64.2% and 67.9% at a dose of 30 mg/kg. When limited to the first 5 days after axotomy, an oral 30 mg/kg dose of U-74006F also significantly blunted retrograde degeneration measured 21 days post-axotomy. The protection of axotomized FMNs by a lipid peroxidation inhibitor strongly suggests that neuronal apoptosis ultimately involves an oxygen radical-induced membrane peroxidative mechanism. |
| Yoo, M.C. et al., 2022 [41] | Animal study | 40 mature Sprague Dawley rats | Left facial nerve crushing injury, axotomy | Behavioral tests, immunohistochemistry | NOX2, ALA | Relative NOX2 expression was higher in the axotomy group than in the crushing group (p < 0.001). Relative NOX expression trended higher in both injury groups that received an injection of ALA, but reached statistical significance only in the axotomy group (p < 0.001). Behavioral tests conducted 4 days after the crushing injury showed better results in the group injected with ALA than in the group without injection of ALA (p = 0.031). Injection with ALA promoted nerve regeneration in a rat model of crushing nerve injury. Moreover, NOX2 expression was significantly higher following facial nerve injury, particularly for a cutting injury. |
| Koyama, Y. et al., 2022 [42] | Animal study | C57BL/6J mice | Left facial nerve compression injury | Behavioral test, electroneuronography, oxidative stress measurement, immunofluorescence staining | Si-based agent, ROS | Combined treatment with a Si-based agent and MeCbl improved clinical scores and neuroregeneration and reduced oxidative stress compared with individual administration. The Si-based agent rapidly rescued the loss of normal facial expressions by promoting myelin sheath formation and alleviating oxidative stress. Combined Si-based agent/MeCbl therapy could be a clinically viable new treatment for facial paralysis. |
| Antioxidant | Efficacy | Potential Advantages | Potential Disadvantages |
|---|---|---|---|
| Alpha-lipoic acid (ALA) | Facilitates functional recovery in nerve injury models; inhibits NOX2 expression | Highly membrane-permeable; provides broad protection against multiple ROS/FR species | Mild adverse effects (e.g., gastrointestinal disturbances) at high doses |
| Superoxide dismutase (SOD) | Promotes recovery from ischemic injury; reduces pro-inflammatory gene expression | Highly effective in directly scavenging ROS, protective against nerve damage | Clinical application limited by administration route and short half-life |
| Glutathione (GSH) | Provides cytoprotection after nerve injury, inhibits ferroptosis | Potent antioxidant effect that promotes cell survival | Rapidly depleted in vivo; continuous replenishment strategies required |
| Edaravone | Prevents and mitigates ischemic and HSV-1-induced nerve palsy | Effectively scavenges various ROS, including hydroxyl radicals; rapid symptom alleviation | Possible side effects including hypersensitivity; cost and limited accessibility |
| Tirilazad mesylate (U-74006F) | Enhances motor neuron survival post-axotomy; inhibits lipid peroxidation | Specialized in protecting cellular membranes by effectively suppressing lipid peroxidation | Limited clinical studies; selective action limited to specific types of FRs |
| Methylprednisolone sodium succinate (MPSS) | Delays NO formation after nerve injury; improves motor neuron survival | Strong anti-inflammatory and antioxidant effects; effective in early-phase treatment of nerve injury | Risk of severe adverse effects such as infection, gastrointestinal bleeding with high-dose or long-term use |
| L-NAME (NOS inhibitor) | Reduces neuronal damage and neuronal loss through inhibition of NO synthesis | Potent neuroprotective effect when administered shortly after injury | Risk of interfering with essential physiological functions mediated by NO (e.g., vasodilation, neurotransmission) |
| Si-based agent and methylcobalamin (MeCbl) | Promotes nerve regeneration and reduces ROS levels after nerve compression injury | Dual beneficial effects through alleviating oxidative stress and enhancing myelin sheath regeneration | Insufficient clinical data; uncertainty regarding long-term efficacy with prolonged administration |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Yeo, J.H.; Kim, S.S.; Lee, J.M.; Yeo, S.G. Production and Role of Free Radicals and Reactive Oxygen Species After Facial Nerve Injury. Antioxidants 2025, 14, 436. https://doi.org/10.3390/antiox14040436
Lee J, Yeo JH, Kim SS, Lee JM, Yeo SG. Production and Role of Free Radicals and Reactive Oxygen Species After Facial Nerve Injury. Antioxidants. 2025; 14(4):436. https://doi.org/10.3390/antiox14040436
Chicago/Turabian StyleLee, Jeongmin, Joon Hyung Yeo, Sung Soo Kim, Jae Min Lee, and Seung Geun Yeo. 2025. "Production and Role of Free Radicals and Reactive Oxygen Species After Facial Nerve Injury" Antioxidants 14, no. 4: 436. https://doi.org/10.3390/antiox14040436
APA StyleLee, J., Yeo, J. H., Kim, S. S., Lee, J. M., & Yeo, S. G. (2025). Production and Role of Free Radicals and Reactive Oxygen Species After Facial Nerve Injury. Antioxidants, 14(4), 436. https://doi.org/10.3390/antiox14040436