
The Janus Face of Oxidative Stress and Hydrogen Sulfide: Insights into Neurodegenerative Disease Pathogenesis

 ,
,  , , and
, , and
Abstract
1. Introduction
2. Oxidative Stress: A Janus-Faced Phenomenon
3. Hydrogen Sulfide Signaling: Another Janus-Faced Regulator
4. The Interplay Between Oxidative Stress and H2S
5. Implications for Neurodegenerative Disease Pathogenesis
6. Current and Prospective Therapeutic Strategies
7. Controversies, Challenges, and Future Directions
8. Conclusions
Funding
Conflicts of Interest
References
- Lamptey, R.N.L.; Chaulagain, B.; Trivedi, R.; Gothwal, A.; Layek, B.; Singh, J. A Review of the Common Neurodegenerative Disorders: Current Therapeutic Approaches and the Potential Role of Nanotherapeutics. Int. J. Mol. Sci. 2022, 23, 1851. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Model. Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Munteanu, C.; Iordan, D.A.; Hoteteu, M.; Popescu, C.; Postoiu, R.; Onu, I.; Onose, G. Mechanistic Intimate Insights into the Role of Hydrogen Sulfide in Alzheimer’s Disease: A Recent Systematic Review. Int. J. Mol. Sci. 2023, 24, 15481. [Google Scholar] [CrossRef]
- Islinger, M.; Voelkl, A.; Fahimi, H.D.; Schrader, M. The peroxisome: An update on mysteries 2.0. Histochem. Cell Biol. 2018, 150, 443–471. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Aitchison, J.D. Peroxisomes take shape. Nat. Rev. Mol. Cell Biol. 2013, 14, 803–817. [Google Scholar] [CrossRef]
- Gadhave, D.G.; Sugandhi, V.V.; Jha, S.K.; Nangare, S.N.; Gupta, G.; Singh, S.K.; Dua, K.; Cho, H.; Hansbro, P.M.; Paudel, K.R. Neurodegenerative disorders: Mechanisms of degeneration and therapeutic approaches with their clinical relevance. Ageing Res. Rev. 2024, 99, 102357. [Google Scholar] [CrossRef]
- Jellinger, K.A. Basic mechanisms of neurodegeneration: A critical update. J. Cell. Mol. Med. 2010, 14, 457–487. [Google Scholar] [CrossRef]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef]
- Ayeni, E.A.; Aldossary, A.M.; Ayejoto, D.A.; Gbadegesin, L.A.; Alshehri, A.A.; Alfassam, H.A.; Afewerky, H.K.; Almughem, F.A.; Bello, S.M.; Tawfik, E.A. Neurodegenerative Diseases: Implications of Environmental and Climatic Influences on Neurotransmitters and Neuronal Hormones Activities. Int. J. Environ. Res. Public Health 2022, 19, 12495. [Google Scholar] [CrossRef]
- Siokas, V.; Dardiotis, E. Advances in Neurodegenerative Diseases. Neurol. Int. 2022, 14, 336. [Google Scholar] [CrossRef]
- Agnello, L.; Gambino, C.M.; Ciaccio, A.M.; Masucci, A.; Vassallo, R.; Tamburello, M.; Scazzone, C.; Sasso, B.L.; Ciaccio, M. Molecular Biomarkers of Neurodegenerative Disorders: A Practical Guide to Their Appropriate Use and Interpretation in Clinical Practice. Int. J. Mol. Sci. 2024, 25, 4323. [Google Scholar] [CrossRef] [PubMed]
- Klemmensen, M.M.; Borrowman, S.H.; Pearce, C.; Pyles, B.; Chandra, B. Mitochondrial dysfunction in neurodegenerative disorders. Neurotherapeutics 2023, 21, e00292. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Li, J.; Wuliji, O.; Li, W.; Jiang, Z.-G.; Ghanbari, H.A. Oxidative Stress and Neurodegenerative Disorders. Int. J. Mol. Sci. 2013, 14, 24438–24475. [Google Scholar] [CrossRef]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Doser, R.L.; Hoerndli, F.J. Regulation of neuronal excitability by reactive oxygen species and calcium signaling: Insights into brain aging. Curr. Res. Neurobiol. 2021, 2, 100012. [Google Scholar] [CrossRef] [PubMed]
- Patel, M. Targeting Oxidative Stress in Central Nervous System Disorders. Trends Pharmacol. Sci. 2016, 37, 768–778. [Google Scholar] [CrossRef]
- Liu, Y.-Z.; Wang, Y.-X.; Jiang, C.-L. Inflammation: The Common Pathway of Stress-Related Diseases. Front. Hum. Neurosci. 2017, 11, 316. [Google Scholar] [CrossRef]
- Hawiger, J.; Zienkiewicz, J. Decoding inflammation, its causes, genomic responses, and emerging countermeasures. Scand. J. Immunol. 2019, 90, e12812. [Google Scholar] [CrossRef]
- Abdolmaleky, H.M.; Zhou, J.-R. Gut Microbiota Dysbiosis, Oxidative Stress, Inflammation, and Epigenetic Alterations in Metabolic Diseases. Antioxidants 2024, 13, 985. [Google Scholar] [CrossRef]
- Shandilya, S.; Kumar, S.; Jha, N.K.; Kesari, K.K.; Ruokolainen, J. Interplay of gut microbiota and oxidative stress: Perspective on neurodegeneration and neuroprotection. J. Adv. Res. 2021, 38, 223–244. [Google Scholar] [CrossRef] [PubMed]
- Alam, R.; Abdolmaleky, H.M.; Zhou, J. Microbiome, inflammation, epigenetic alterations, and mental diseases. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2017, 174, 651–660. [Google Scholar] [CrossRef]
- Munteanu, C.; Onose, G.; Rotariu, M.; Poștaru, M.; Turnea, M.; Galaction, A.I. Role of Microbiota-Derived Hydrogen Sulfide (H2S) in Modulating the Gut–Brain Axis: Implications for Alzheimer’s and Parkinson’s Disease Pathogenesis. Biomedicines 2024, 12, 2670. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Hong, Y.; Boiti, A.; Vallone, D.; Foulkes, N.S. Reactive Oxygen Species Signaling and Oxidative Stress: Transcriptional Regulation and Evolution. Antioxidants 2024, 13, 312. [Google Scholar] [CrossRef]
- Jomova, K.; Raptova, R.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Reactive oxygen species, toxicity, oxidative stress, and antioxidants: Chronic diseases and aging. Arch. Toxicol. 2023, 97, 2499–2574. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Gambini, J.; Stromsnes, K. Oxidative Stress and Inflammation: From Mechanisms to Therapeutic Approaches. Biomedicines 2022, 10, 753. [Google Scholar] [CrossRef]
- Ndhlala, A.R.; Moyo, M.; Van Staden, J. Natural Antioxidants: Fascinating or Mythical Biomolecules? Molecules 2010, 15, 6905–6930. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, S.; Li, Y.; Liu, Z.; Mi, L.; Cai, Y.; Wang, X.; Chen, L.; Ran, H.; Xiao, D.; et al. Suppression of mitochondrial ROS by prohibitin drives glioblastoma progression and therapeutic resistance. Nat. Commun. 2021, 12, 3720. [Google Scholar] [CrossRef]
- Gomes, A.P.; Blenis, J. A nexus for cellular homeostasis: The interplay between metabolic and signal transduction pathways. Curr. Opin. Biotechnol. 2015, 34, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Dogaru, B.G.; Munteanu, C. The Role of Hydrogen Sulfide (H2S) in Epigenetic Regulation of Neurodegenerative Diseases: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 12555. [Google Scholar] [CrossRef]
- Olas, B. Hydrogen sulfide in signaling pathways. Clin. Chim. Acta 2015, 439, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Munteanu, C.; Galaction, A.I.; Poștaru, M.; Rotariu, M.; Turnea, M.; Blendea, C.D. Hydrogen Sulfide Modulation of Matrix Metalloproteinases and CD147/EMMPRIN: Mechanistic Pathways and Impact on Atherosclerosis Progression. Biomedicines 2024, 12, 1951. [Google Scholar] [CrossRef]
- Andrés, C.M.C.; de la Lastra, J.M.P.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. Chemistry of Hydrogen Sulfide—Pathological and Physiological Functions in Mammalian Cells. Cells 2023, 12, 2684. [Google Scholar] [CrossRef]
- Munteanu, C.; Popescu, C.; Vlădulescu-Trandafir, A.-I.; Onose, G. Signaling Paradigms of H2S-Induced Vasodilation: A Comprehensive Review. Antioxidants 2024, 13, 1158. [Google Scholar] [CrossRef]
- Munteanu, C.; Turnea, M.A.; Rotariu, M. Hydrogen Sulfide: An Emerging Regulator of Oxidative Stress and Cellular Homeostasis—A Comprehensive One-Year Review. Antioxidants 2023, 12, 1737. [Google Scholar] [CrossRef]
- Kimura, H. Production and Physiological Effects of Hydrogen Sulfide. Antioxid. Redox Signal. 2014, 20, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Lin, H.C. Hydrogen Sulfide in Physiology and Diseases of the Digestive Tract. Microorganisms 2015, 3, 866–889. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Qian, L.-L.; Wang, R.-X. Hydrogen Sulfide-Induced Vasodilation: The Involvement of Vascular Potassium Channels. Front. Pharmacol. 2022, 13, 911704. [Google Scholar] [CrossRef] [PubMed]
- Aroca, A.; Gotor, C.; Bassham, D.C.; Romero, L.C. Hydrogen Sulfide: From a Toxic Molecule to a Key Molecule of Cell Life. Antioxidants 2020, 9, 621. [Google Scholar] [CrossRef]
- Kabil, O.; Vitvitsky, V.; Banerjee, R. Sulfur as a Signaling Nutrient Through Hydrogen Sulfide. Annu. Rev. Nutr. 2014, 34, 171–205. [Google Scholar] [CrossRef] [PubMed]
- Munteanu, C.; Dogaru, G.; Rotariu, M.; Onose, G. Therapeutic gases used in balneotherapy and rehabilitation medicine-scientific relevance in the last ten years (2011–2020)—Synthetic literature review. Balneo PRM Res. J. 2021, 12, 111–122. [Google Scholar] [CrossRef]
- Muro, P.; Zhang, L.; Li, S.; Zhao, Z.; Jin, T.; Mao, F.; Mao, Z. The emerging role of oxidative stress in inflammatory bowel disease. Front. Endocrinol. 2024, 15, 1390351. [Google Scholar] [CrossRef]
- Moghadam, Z.M.; Henneke, P.; Kolter, J. From Flies to Men: ROS and the NADPH Oxidase in Phagocytes. Front. Cell Dev. Biol. 2021, 9, 628991. [Google Scholar] [CrossRef]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide Dismutases: Role in Redox Signaling, Vascular Function, and Diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef]
- Porras, C.A.; Bai, Y. Respiratory supercomplexes: Plasticity and implications. Front. Biosci. 2015, 20, 621. [Google Scholar]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef]
- Stoica, S.I.; Bleotu, C.; Ciobanu, V.; Ionescu, A.M.; Albadi, I.; Onose, G.; Munteanu, C. Considerations about Hypoxic Changes in Neuraxis Tissue Injuries and Recovery. Biomedicines 2022, 10, 481. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Cross-Talk between NADPH Oxidase and Mitochondria: Role in ROS Signaling and Angiogenesis. Cells 2020, 9, 1849. [Google Scholar] [CrossRef]
- Ganguly, U.; Kaur, U.; Chakrabarti, S.S.; Sharma, P.; Agrawal, B.K.; Saso, L. Oxidative Stress, Neuroinflammation, and NADPH Oxidase: Implications in the Pathogenesis and Treatment of Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2021, 2021, 7086512. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Lushchak, V.; Storey, K.B. Oxidative stress concept updated: Definitions, classifications and regulatory pathways implicated. EXCLI J. 2021, 20, 956–967. [Google Scholar] [CrossRef]
- Villalpando-Rodriguez, G.E.; Gibson, S.B. Reactive Oxygen Species (ROS) Regulates Different Types of Cell Death by Acting as a Rheostat. Oxid. Med. Cell. Longev. 2021, 2021, 9912436. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Bresgen, N.; Kovacs, M.; Lahnsteiner, A.; Felder, T.K.; Rinnerthaler, M. The Janus-Faced Role of Lipid Droplets in Aging: Insights from the Cellular Perspective. Biomolecules 2023, 13, 912. [Google Scholar] [CrossRef]
- Ngo, V.; Duennwald, M.L. Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants 2022, 11, 2345. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Kong, A. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol. Carcinog. 2008, 48, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Canning, P. Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch. Biochem. Biophys. 2017, 617, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Naidu, S.D.; Dinkova-Kostova, A.T. KEAP1, a cysteine-based sensor and a drug target for the prevention and treatment of chronic disease. Open Biol. 2020, 10, 200105. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Huang, Y.; Li, W.; Su, Z.-Y.; Kong, A.-N.T. The complexity of the Nrf2 pathway: Beyond the antioxidant response. J. Nutr. Biochem. 2015, 26, 1401–1413. [Google Scholar] [CrossRef]
- Moubarak, M.M.; Zottola, A.C.P.; Larrieu, C.M.; Cuvellier, S.; Daubon, T.; Martin, O.C.B. Exploring the multifaceted role of NRF2 in brain physiology and cancer: A comprehensive review. Neuro-Oncol. Adv. 2023, 6, vdad160. [Google Scholar] [CrossRef]
- Giuliani, C.; Bucci, I.; Napolitano, G. The Role of the Transcription Factor Nuclear Factor-kappa B in Thyroid Autoimmunity and Cancer. Front. Endocrinol. 2018, 9, 471. [Google Scholar] [CrossRef]
- Schmidlin, C.J.; Dodson, M.B.; Madhavan, L.; Zhang, D.D. Redox regulation by NRF2 in aging and disease. Free. Radic. Biol. Med. 2019, 134, 702–707. [Google Scholar] [CrossRef]
- Averill-Bates, D. Reactive oxygen species and cell signaling. Review. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2023, 1871, 119573. [Google Scholar] [CrossRef]
- Checa, J.; Aran, J.M. Reactive Oxygen Species: Drivers of Physiological and Pathological Processes. J. Inflamm. Res. 2020, 13, 1057–1073. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Ghosh, K.K.; Chakrabortty, S.; Gulyás, B.; Padmanabhan, P.; Ball, W.B. Mitochondrial Reactive Oxygen Species in Infection and Immunity. Biomolecules 2024, 14, 670. [Google Scholar] [CrossRef]
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2017, 19, 92. [Google Scholar] [CrossRef] [PubMed]
- Vignais, P.V. The superoxide-generating NADPH oxidase: Structural aspects and activation mechanism. Cell. Mol. Life Sci. CMLS 2002, 59, 1428–1459. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xie, S.; Ahmed, S.; Wang, F.; Gu, Y.; Zhang, C.; Chai, X.; Wu, Y.; Cai, J.; Cheng, G. Antimicrobial Activity and Resistance: Influencing Factors. Front. Pharmacol. 2017, 8, 364. [Google Scholar] [CrossRef]
- Ye, Z.-W.; Zhang, J.; Townsend, D.M.; Tew, K.D. Oxidative stress, redox regulation and diseases of cellular differentiation. Biochim. Et Biophys. Acta (BBA)-Gen. Subj. 2014, 1850, 1607–1621. [Google Scholar] [CrossRef]
- Su, L.-J.; Zhang, J.-H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.-Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxidative Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef]
- Melis, J.P.; van Steeg, H.; Luijten, M. Oxidative DNA Damage and Nucleotide Excision Repair. Antioxidants Redox Signal. 2013, 18, 2409–2419. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Houldsworth, A. Role of oxidative stress in neurodegenerative disorders: A review of reactive oxygen species and prevention by antioxidants. Brain Commun. 2023, 6, fcad356. [Google Scholar] [CrossRef]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef]
- Cirino, G.; Szabo, C.; Papapetropoulos, A. Physiological roles of hydrogen sulfide in mammalian cells, tissues, and organs. Physiol. Rev. 2023, 103, 31–276. [Google Scholar] [CrossRef] [PubMed]
- Zuhra, K.; Augsburger, F.; Majtan, T.; Szabo, C. Cystathionine-β-synthase: Molecular Regulation and Pharmacological Inhibition. Biomolecules 2020, 10, 697. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-H.; Wang, F.; You, S.-J.; Cao, Y.-J.; Cao, L.-D.; Han, Q.; Liu, C.-F.; Hu, L.-F. Dysregulation of cystathionine γ-lyase (CSE)/hydrogen sulfide pathway contributes to ox-LDL-induced inflammation in macrophage. Cell. Signal. 2013, 25, 2255–2262. [Google Scholar] [CrossRef]
- Sen, U.; Sathnur, P.B.; Kundu, S.; Givvimani, S.; Coley, D.M.; Mishra, P.K.; Qipshidze, N.; Tyagi, N.; Metreveli, N.; Tyagi, S.C. Increased endogenous H2S generation by CBS, CSE, and 3MST gene therapy improves ex vivo renovascular relaxation in hyperhomocysteinemia. Am. J. Physiol. Physiol. 2012, 303, C41–C51. [Google Scholar] [CrossRef]
- Kabil, O.; Motl, N.; Banerjee, R. H2S and its role in redox. Signaling 2014, 1844, 1355–1366. [Google Scholar] [CrossRef]
- Bindu Paul, C.D.; Solomon Snyder, T.H.; Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Regulators of the transsulfuration pathway. Br. J. Pharmacol. 2019, 176, 583–593. [Google Scholar] [CrossRef]
- Zheng, H.; Chen, H.; Cai, Y.; Shen, M.; Li, X.; Han, Y.; Deng, X.; Cao, H.; Liu, J.; Li, H.; et al. Hydrogen sulfide-mediated persulfidation regulates homocysteine metabolism and enhances ferroptosis in non-small cell lung cancer. Mol. Cell 2024, 84, 4016–4030.e6. [Google Scholar] [CrossRef]
- Casadevall, A.; Fang, F.C.; Imperiale, M.J. The Epistemic Value of Gain of Function Experiments. mSphere 2024, 9, e0071423. [Google Scholar] [CrossRef]
- Dupont, C.; Armant, D.R.; Brenner, C.A. Epigenetics: Definition, Mechanisms and Clinical Perspective. Semin. Reprod. Med. 2009, 27, 351–357. [Google Scholar] [CrossRef]
- Sun, H.-J.; Wu, Z.-Y.; Nie, X.-W.; Bian, J.-S. Role of Hydrogen Sulfide and Polysulfides in Neurological Diseases: Focus on Protein S-Persulfidation. Curr. Neuropharmacol. 2021, 19, 868–884. [Google Scholar] [CrossRef]
- Islam, K.N.; Nguyen, I.D.; Islam, R.; Pirzadah, H.; Malik, H. Roles of Hydrogen Sulfide (H2S) as a Potential Therapeutic Agent in Cardiovascular Diseases: A Narrative Review. Cureus 2024, 16, e64913. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Li, Y.; Li, L.; Xu, S.; Feng, X.; Liu, S. Hydrogen Sulfide (H2S)-Releasing Compounds: Therapeutic Potential in Cardiovascular Diseases. Front. Pharmacol. 2018, 9, 1066. [Google Scholar] [CrossRef] [PubMed]
- Shahid, A.; Bhatia, M. Hydrogen Sulfide: A Versatile Molecule and Therapeutic Target in Health and Diseases. Biomolecules 2024, 14, 1145. [Google Scholar] [CrossRef]
- Lv, B.; Chen, S.; Tang, C.; Jin, H.; Du, J.; Huang, Y. Hydrogen sulfide and vascular regulation—An update. J. Adv. Res. 2021, 27, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Bian, J.-S.; Olson, K.R.; Zhu, Y.-C. Hydrogen Sulfide: Biogenesis, Physiology, and Pathology. Oxidative Med. Cell. Longev. 2016, 2016, 6549625. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Lian, X.; Bhatia, M. Hydrogen Sulfide: A Gaseous Mediator and Its Key Role in Programmed Cell Death, Oxidative Stress, Inflammation and Pulmonary Disease. Antioxidants 2022, 11, 2162. [Google Scholar] [CrossRef]
- Bian, J.-S. Editorial: Hydrogen sulfide: Physiology, Pharmacology and Toxicology, Volume II. Front. Pharmacol. 2022, 13, 943101. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. Gasotransmitter hydrogen sulfide signaling in neuronal health and disease. Biochem. Pharmacol. 2018, 149, 101–109. [Google Scholar] [CrossRef]
- Giuffrè, A.; Vicente, J.B. Hydrogen Sulfide Biochemistry and Interplay with Other Gaseous Mediators in Mammalian Physiology. Oxidative Med. Cell. Longev. 2018, 2018, 6290931. [Google Scholar] [CrossRef]
- Flannigan, K.L.; Ferraz, J.G.P.; Wang, R.; Wallace, J.L. Enhanced Synthesis and Diminished Degradation of Hydrogen Sulfide in Experimental Colitis: A Site-Specific, Pro-Resolution Mechanism. PLoS ONE 2013, 8, e71962. [Google Scholar] [CrossRef]
- Wu, D.; Hu, Q.; Zhu, D. An Update on Hydrogen Sulfide and Nitric Oxide Interactions in the Cardiovascular System. Oxidative Med. Cell. Longev. 2018, 2018, 4579140. [Google Scholar] [CrossRef] [PubMed]
- Pandey, T.; Kaundal, R.S.; Pandey, V. Biophysical characterization of hydrogen sulfide: A fundamental exploration in understanding significance in cell signaling. Biophys. Chem. 2024, 314, 107317. [Google Scholar] [CrossRef] [PubMed]
- Corsello, T.; Komaravelli, N.; Casola, A. Role of Hydrogen Sulfide in NRF2- and Sirtuin-Dependent Maintenance of Cellular Redox Balance. Antioxidants 2018, 7, 129. [Google Scholar] [CrossRef]
- Predmore, B.L.; Lefer, D.J.; Gojon, G. Hydrogen Sulfide in Biochemistry and Medicine. Antioxid. Redox Signal. 2012, 17, 119–140. [Google Scholar] [CrossRef] [PubMed]
- Raevsky, R.I.; Katrukha, V.A.; Khramova, Y.V.; Bilan, D.S. Biochemistry of Redox-Active Sulfur Compounds in Mammalian Cells and Approaches to Their Detection (A Review). Russ. J. Bioorganic Chem. 2024, 50, 1237–1262. [Google Scholar] [CrossRef]
- Mishanina, T.V.; Libiad, M.; Banerjee, R. Biogenesis of reactive sulfur species for signaling by hydrogen sulfide oxidation pathways. Nat. Chem. Biol. 2015, 11, 457–464. [Google Scholar] [CrossRef]
- Myszkowska, J.; Derevenkov, I.; Makarov, S.V.; Spiekerkoetter, U.; Hannibal, L. Biosynthesis, Quantification and Genetic Diseases of the Smallest Signaling Thiol Metabolite: Hydrogen Sulfide. Antioxidants 2021, 10, 1065. [Google Scholar] [CrossRef]
- Kimura, Y.; Goto, Y.; Kimura, H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid Redox Signal. 2010, 12, 1–13. [Google Scholar] [CrossRef]
- Pietri, R.; Román-Morales, E.; López-Garriga, J. Hydrogen Sulfide and Hemeproteins: Knowledge and Mysteries. Antioxid. Redox Signal. 2011, 15, 393–404. [Google Scholar] [CrossRef]
- Viegas, J.; Esteves, A.F.; Cardoso, E.M.; Arosa, F.A.; Vitale, M.; Taborda-Barata, L. Biological Effects of Thermal Water-Associated Hydrogen Sulfide on Human Airways and Associated Immune Cells: Implications for Respiratory Diseases. Front. Public Health 2019, 7, 128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tang, Z.-H.; Ren, Z.; Qu, S.-L.; Liu, M.-H.; Liu, L.-S.; Jiang, Z.-S. Hydrogen Sulfide, the Next Potent Preventive and Therapeutic Agent in Aging and Age-Associated Diseases. Mol. Cell. Biol. 2013, 33, 1104–1113. [Google Scholar] [CrossRef]
- Stein, A.; Bailey, S.M. Redox biology of hydrogen sulfide: Implications for physiology, pathophysiology, and pharmacology. Redox Biol. 2013, 1, 32–39. [Google Scholar] [CrossRef]
- Cao, X.; Ding, L.; Xie, Z.-Z.; Yang, Y.; Whiteman, M.; Moore, P.K.; Bian, J.S. A Review of Hydrogen Sulfide Synthesis, Metabolism, and Measurement: Is Modulation of Hydrogen Sulfide a Novel Therapeutic for Cancer? Antioxid. Redox Signal. 2019, 31, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Liu, H.; Shi, X.; Prokosch, V. Hydrogen Sulfide: Novel Endogenous and Exogenous Modulator of Oxidative Stress in Retinal Degeneration Diseases. Molecules 2021, 26, 2411. [Google Scholar] [CrossRef]
- Sommer, O.; Aug, R.L.; Schmidt, A.J.; Heiser, P.; Schulz, E.; Vedder, H.; Clement, H.-W. Hydrogen Sulfide Affects Radical Formation in the Hippocampus of LPS Treated Rats and the Effect of Antipsychotics on Hydrogen Sulfide Forming Enzymes in Human Cell Lines. Front. Psychiatry 2018, 9, 501. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Yuan, Z.; Chen, X.; Chen, Y.; Yao, M.; Chen, Y.; Li, X.; Chen, Y.; Ding, W.; Xia, C.; et al. Hydrogen sulfide responsive nanoplatforms: Novel gas responsive drug delivery carriers for biomedical applications. Asian J. Pharm. Sci. 2023, 19, 100858. [Google Scholar] [CrossRef]
- Perridon, B.W.; Leuvenink, H.G.; Hillebrands, J.L.; van Goor, H.; Bos, E.M. The role of hydrogen sulfide in aging and age-related pathologies. Aging 2016, 8, 2264–2289. [Google Scholar] [CrossRef]
- Cornwell, A.; Badiei, A. From Gasotransmitter to Immunomodulator: The Emerging Role of Hydrogen Sulfide in Macrophage Biology. Antioxidants 2023, 12, 935. [Google Scholar] [CrossRef]
- Wang, Y.-Z.; Ngowi, E.E.; Wang, D.; Qi, H.-W.; Jing, M.-R.; Zhang, Y.-X.; Cai, C.-B.; He, Q.-L.; Khattak, S.; Khan, N.H.; et al. The Potential of Hydrogen Sulfide Donors in Treating Cardiovascular Diseases. Int. J. Mol. Sci. 2021, 22, 2194. [Google Scholar] [CrossRef]
- Fatima, G.; Mahdi, A.A.; Alhmadi, H.B.; Medvedev, O. Unveiling Hydrogen Sulfide: A New Frontier in Neuroprotection and Neuromodulation. Indian J. Clin. Biochem. 2024, 1–11. [Google Scholar] [CrossRef]
- Zhao, Y.; Biggs, T.D.; Xian, M. Hydrogen sulfide (H2S) releasing agents: Chemistry and biological applications. Chem. Commun. 2014, 50, 11788–11805. [Google Scholar] [CrossRef] [PubMed]
- Shefa, U.; Kim, M.-S.; Jeong, N.Y.; Jung, J. Antioxidant and Cell-Signaling Functions of Hydrogen Sulfide in the Central Nervous System. Oxidative Med. Cell. Longev. 2018, 2018, 1873962. [Google Scholar] [CrossRef] [PubMed]
- Jeong, N.Y.; Jung, J.; Tabassum, R. Therapeutic importance of hydrogen sulfide in age-associated neurodegenerative diseases. Neural Regen. Res. 2020, 15, 653–662. [Google Scholar] [CrossRef]
- Bithi, N.; Link, C.; Henderson, Y.O.; Kim, S.; Yang, J.; Li, L.; Wang, R.; Willard, B.; Hine, C. Dietary restriction transforms the mammalian protein persulfidome in a tissue-specific and cystathionine γ-lyase-dependent manner. Nat. Commun. 2021, 12, 1745. [Google Scholar] [CrossRef]
- Akbari, M.; Sogutdelen, E.; Juriasingani, S.; Sener, A. Hydrogen Sulfide: Emerging Role in Bladder, Kidney, and Prostate Malignancies. Oxidative Med. Cell. Longev. 2019, 2019, 2360945. [Google Scholar] [CrossRef]
- Liu, J.; Mesfin, F.M.; Hunter, C.E.; Olson, K.R.; Shelley, W.C.; Brokaw, J.P.; Manohar, K.; Markel, T.A. Recent Development of the Molecular and Cellular Mechanisms of Hydrogen Sulfide Gasotransmitter. Antioxidants 2022, 11, 1788. [Google Scholar] [CrossRef]
- Munteanu, C.; Hoteteu, M.; Munteanu, D.; Onose, G. The effects of Mineral Waters from Slănic Moldova’s Spring 1 and Spring 1 bis on Fibroblast activity: An In Vitro Study. Balneo PRM Res. J. 2023, 14, 591. [Google Scholar] [CrossRef]
- Munteanu, C.; Hoteteu, M.; Munteanu, D.; Onose, G. Mineral waters from Spring 1 and Spring 1 bis from Slănic Moldova—Molecular mechanisms responsible for triggering the prophylactic and therapeutic effects. Balneo PRM Res. J. 2023, 14, 592. [Google Scholar] [CrossRef]
- Munteanu, C.; Popescu, C.; Munteanu, D.; Hoteteu, M.; Iliescu, M.G.; Ionescu, E.V.; Stanciu, L.; Oprea, D.; Minea, M.; Oprea, C.; et al. Biological Evaluation of Balneotherapeutic Mud and Sulfurous Mineral Waters: Insights from In Vivo and In Vitro Studies. Balneo PRM Res. J. 2024, 15, 702. [Google Scholar] [CrossRef]
- Hoteteu, M.; Munteanu, C.; Ionescu, E.V.; Almășan, R.E.; Balnear, T.; Sanatorium, R. Bioactive substances of the Techirghiol therapeutic mud. Balneo Res. J. 2018, 9, 5–10. [Google Scholar] [CrossRef]
- Martelli, A.; Testai, L.; Citi, V.; Marino, A.; Bellagambi, F.G.; Ghimenti, S.; Breschi, M.C.; Calderone, V. Pharmacological characterization of the vascular effects of aryl isothiocyanates: Is hydrogen sulfide the real player? Vasc. Pharmacol. 2014, 60, 32–41. [Google Scholar] [CrossRef]
- Meng, G.; Ma, Y.; Xie, L.; Ferro, A.; Ji, Y. Emerging role of hydrogen sulfide in hypertension and related cardiovascular diseases. Br. J. Pharmacol. 2014, 172, 5501–5511. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.J.; Chakraborty, S.; Miller, E.; Pieper, A.A.; Paul, B.D. Hydrogen sulfide signalling in neurodegenerative diseases. Br. J. Pharmacol. 2023, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kolluru, G.K.; Shen, X.; Bir, S.C.; Kevil, C.G. Hydrogen sulfide chemical biology: Pathophysiological roles and detection. Nitric Oxide 2013, 35, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71. [Google Scholar] [CrossRef]
- Li, Q.; Lancaster, J.R. Chemical foundations of hydrogen sulfide biology. Nitric Oxide 2013, 35, 21–34. [Google Scholar] [CrossRef]
- Zhang, J.; Corpas, F.J.; Li, J.; Xie, Y. Hydrogen Sulfide and Reactive Oxygen Species, Antioxidant Defense, Abiotic Stress Tolerance Mechanisms in Plants. Int. J. Mol. Sci. 2022, 23, 9463. [Google Scholar] [CrossRef]
- Munteanu, C.; Onose, G.; Poștaru, M.; Turnea, M.; Rotariu, M.; Galaction, A.I. Hydrogen Sulfide and Gut Microbiota: Their Synergistic Role in Modulating Sirtuin Activity and Potential Therapeutic Implications for Neurodegenerative Diseases. Pharmaceuticals 2024, 17, 1480. [Google Scholar] [CrossRef]
- Olson, K.R.; Gao, Y.; Arif, F.; Arora, K.; Patel, S.; DeLeon, E.R.; Sutton, T.R.; Feelisch, M.; Cortese-Krott, M.M.; Straub, K.D. Metabolism of hydrogen sulfide (H2S) and Production of Reactive Sulfur Species (RSS) by superoxide dismutase. Redox Biol. 2018, 15, 74–85. [Google Scholar] [CrossRef]
- Wang, M.; Tang, W.; Zhu, Y.Z. An Update on AMPK in Hydrogen Sulfide Pharmacology. Front. Pharmacol. 2017, 8, 810. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Wang, J.; Xu, M.; Chen, S.; Li, X.; Sun, A.; Lou, N.; Ni, Y. Hydrogen sulfide protects against high glucose-induced lipid metabolic disturbances in 3T3-L1 adipocytes via the AMPK signaling pathway. Mol. Med. Rep. 2019, 20, 4119–4124. [Google Scholar] [CrossRef]
- Jeong, N.Y.; Jung, J.; Tabassum, R. Protective effect of hydrogen sulfide on oxidative stress-induced neurodegenerative diseases. Neural Regen. Res. 2020, 15, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.P. Oxidative Stress in Health and Disease. Biomedicines 2023, 11, 2925. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 6. [Google Scholar] [CrossRef]
- Dai, D.-F.; Chiao, Y.A.; Marcinek, D.J.; Szeto, H.H.; Rabinovitch, P.S. Mitochondrial oxidative stress in aging and healthspan. Longev. Heal. 2014, 3, 6. [Google Scholar] [CrossRef]
- Rumpf, S.; Sanal, N.; Marzano, M. Energy metabolic pathways in neuronal development and function. Oxf. Open Neurosci. 2023, 2, kvad004. [Google Scholar] [CrossRef]
- Disabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef]
- Vikram, A.; Anish, R.; Kumar, A.; Tripathi, D.N.; Kaundal, R.K. Oxidative Stress and Autophagy in Metabolism and Longevity. Oxidative Med. Cell. Longev. 2017, 2017, 3451528. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Ferraro, E.; Giammarioli, A.M.; Chiandotto, S.; Spoletini, I.; Rosano, G. Exercise-Induced Skeletal Muscle Remodeling and Metabolic Adaptation: Redox Signaling and Role of Autophagy. Antioxid. Redox Signal. 2014, 21, 154–176. [Google Scholar] [CrossRef]
- Watts, M.E.; Pocock, R.; Claudianos, C.; Chowen, J.A.; Arevalo, M.A.; Rosenberger, T.A. Brain Energy and Oxygen Metabolism: Emerging Role in Normal Function and Disease. Front. Mol. Neurosci. 2018, 11, 216. [Google Scholar] [CrossRef]
- Calabrese, E.J. Hormesis: A fundamental concept in biology. Microb. Cell 2014, 1, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Venkat, P.; Chopp, M.; Chen, J. New insights into coupling and uncoupling of cerebral blood flow and metabolism in the brain. Croat. Med. J. 2016, 57, 223–228. [Google Scholar] [CrossRef]
- Mosley, R.L.; Benner, E.J.; Kadiu, I.; Thomas, M.; Boska, M.D.; Hasan, K.; Laurie, C.; Gendelman, H.E. Neuroinflammation, Oxidative Stress and the Pathogenesis of Parkinson’s Disease. Clin. Neurosci. Res. 2006, 6, 261–281. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Cao, L.; Ding, L.; Bian, J.-S. A New Hope for a Devastating Disease: Hydrogen Sulfide in Parkinson’s Disease. Mol. Neurobiol. 2017, 55, 3789–3799. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Cui, G.; Peng, S.-Y.; Wu, X.; Chen, G. Research progress of hydrogen sulfide in Alzheimer’s disease from laboratory to hospital: A narrative review. Med. Gas Res. 2020, 10, 125–129. [Google Scholar] [CrossRef]
- Gheibi, S.; Samsonov, A.P.; Gheibi, S.; Vazquez, A.B.; Kashfi, K. Regulation of carbohydrate metabolism by nitric oxide and hydrogen sulfide: Implications in diabetes. Biochem. Pharmacol. 2020, 176, 113819. [Google Scholar] [CrossRef]
- Kar, S.; Shahshahan, H.R.; Hackfort, B.T.; Yadav, S.K.; Yadav, R.; Kambis, T.N.; Lefer, D.J.; Mishra, P.K. Exercise Training Promotes Cardiac Hydrogen Sulfide Biosynthesis and Mitigates Pyroptosis to Prevent High-Fat Diet-Induced Diabetic Cardiomyopathy. Antioxidants 2019, 8, 638. [Google Scholar] [CrossRef]
- Zuo, J.; Zhang, Z.; Luo, M.; Zhou, L.; Nice, E.C.; Zhang, W.; Wang, C.; Huang, C. Redox signaling at the crossroads of human health and disease. Medcomm 2022, 3, e127. [Google Scholar] [CrossRef]
- Valgimigli, L. Lipid Peroxidation and Antioxidant Protection. Biomolecules 2023, 13, 1291. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.; Kumar, N.; Sahun, M.; Smits, E.; Bogaerts, A.; Privat-Maldonado, A. Modulating the Antioxidant Response for Better Oxidative Stress-Inducing Therapies: How to Take Advantage of Two Sides of the Same Medal? Biomedicines 2022, 10, 823. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, A.J.P.O.; de Oliveira, J.C.P.L.; da Silva Pontes, L.V.; de Souza Júnior, J.F.; Gonçalves, T.A.F.; Dantas, S.H.; de Almeida Feitosa, M.S.; Silva, A.O.; de Medeiros, I.A. ROS: Basic Concepts, Sources, Cellular Signaling, and its Implications in Aging Pathways. Oxid. Med. Cell Longev. 2022, 2022, 1225578. [Google Scholar] [CrossRef]
- Schwedhelm, E.; Maas, R.; Troost, R.; Böger, R.H. Clinical Pharmacokinetics of Antioxidants and Their Impact on Systemic Oxidative Stress. Clin. Pharmacokinet. 2003, 42, 437–459. [Google Scholar] [CrossRef]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative Stress and Neurodegenerative Diseases: A Review of Upstream and Downstream Antioxidant Therapeutic Options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef]
- Lee, K.H.; Cha, M.; Lee, B.H. Neuroprotective Effect of Antioxidants in the Brain. Int. J. Mol. Sci. 2020, 21, 7152. [Google Scholar] [CrossRef]
- Halliwell, B. Understanding mechanisms of antioxidant action in health and disease. Nat. Rev. Mol. Cell Biol. 2023, 25, 13–33. [Google Scholar] [CrossRef] [PubMed]
- Ashok, A.; Andrabi, S.S.; Mansoor, S.; Kuang, Y.; Kwon, B.K.; Labhasetwar, V. Antioxidant Therapy in Oxidative Stress-Induced Neurodegenerative Diseases: Role of Nanoparticle-Based Drug Delivery Systems in Clinical Translation. Antioxidants 2022, 11, 408. [Google Scholar] [CrossRef]
- Manzari, M.T.; Shamay, Y.; Kiguchi, H.; Rosen, N.; Scaltriti, M.; Heller, D.A. Targeted drug delivery strategies for precision medicines. Nat. Rev. Mater. 2021, 6, 351–370. [Google Scholar] [CrossRef]
- Bourgonje, A.R.; Kloska, D.; Grochot-Przęczek, A.; Feelisch, M.; Cuadrado, A.; van Goor, H. Personalized redox medicine in inflammatory bowel diseases: An emerging role for HIF-1α and NRF2 as therapeutic targets. Redox Biol. 2023, 60, 102603. [Google Scholar] [CrossRef]
- Perez-Leal, O.; Barrero, C.A.; Merali, S. Pharmacological stimulation of nuclear factor (erythroid-derived 2)-like 2 translation activates antioxidant responses. J. Biol. Chem. 2017, 292, 14108–14121. [Google Scholar] [CrossRef]
- Liu, S.; Huang, B.; Cao, J.; Wang, Y.; Xiao, H.; Zhu, Y.; Zhang, H. ROS fine-tunes the function and fate of immune cells. Int. Immunopharmacol. 2023, 119, 110069. [Google Scholar] [CrossRef] [PubMed]
- Harder, B.; Jiang, T.; Wu, T.; Tao, S.; de la Vega, M.R.; Tian, W.; Chapman, E.; Zhang, D.D. Molecular mechanisms of Nrf2 regulation and how these influence chemical modulation for disease intervention. Biochem. Soc. Trans. 2015, 43, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Fiorani, M.; Guidarelli, A.; Cantoni, O. Mitochondrial reactive oxygen species: The effects of mitochondrial ascorbic acid vs untargeted and mitochondria-targeted antioxidants. Int. J. Radiat. Biol. 2020, 97, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Lundgren, C.A.K.; Sjöstrand, D.; Biner, O.; Bennett, M.; Rudling, A.; Johansson, A.-L.; Brzezinski, P.; Carlsson, J.; von Ballmoos, C.; Högbom, M. Scavenging of superoxide by a membrane-bound superoxide oxidase. Nat. Chem. Biol. 2018, 14, 788–793. [Google Scholar] [CrossRef]
- Meng, J.; Lv, Z.; Zhang, Y.; Wang, Y.; Qiao, X.; Sun, C.; Chen, Y.; Guo, M.; Han, W.; Ye, A.; et al. Precision Redox: The Key for Antioxidant Pharmacology. Antioxid. Redox Signal. 2021, 34, 1069–1082. [Google Scholar] [CrossRef]
- Drăgoi, C.M.; Diaconu, C.C.; Nicolae, A.C.; Dumitrescu, I.-B. Redox Homeostasis and Molecular Biomarkers in Precision Therapy for Cardiovascular Diseases. Antioxidants 2024, 13, 1163. [Google Scholar] [CrossRef]
- Powell, C.R.; Dillon, K.M.; Matson, J.B. A review of hydrogen sulfide (H2S) donors: Chemistry and potential therapeutic applications. Biochem. Pharmacol. 2018, 149, 110–123. [Google Scholar] [CrossRef]
- Szabo, C. A timeline of hydrogen sulfide (H2S) research: From environmental toxin to biological mediator. Biochem. Pharmacol. 2018, 149, 5–19. [Google Scholar] [CrossRef]
- Wang, W.; Sun, X.; Zhang, H.; Yang, C.; Liu, Y.; Yang, W.; Guo, C.; Wang, C. Controlled release hydrogen sulfide delivery system based on mesoporous silica nanoparticles protects graft endothelium from ischemia–reperfusion injury. Int. J. Nanomed. 2016, 11, 3255–3263. [Google Scholar] [CrossRef]
- Levinn, C.M.; Cerda, M.M.; Pluth, M.D. Activatable Small-Molecule Hydrogen Sulfide Donors. Antioxid. Redox Signal. 2020, 32, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Polhemus, D.J.; Lefer, D.J. Evolution of Hydrogen Sulfide Therapeutics to Treat Cardiovascular Disease. Circ. Res. 2018, 123, 590–600. [Google Scholar] [CrossRef]
- Lee, Z.W.; Zhou, J.; Chen, C.-S.; Zhao, Y.; Tan, C.-H.; Li, L.; Moore, P.K.; Deng, L.-W. The Slow-Releasing Hydrogen Sulfide Donor, GYY4137, Exhibits Novel Anti-Cancer Effects In Vitro and In Vivo. PLoS ONE 2011, 6, e21077. [Google Scholar] [CrossRef]
- Lee, Y.; Thompson, D.H. Stimuli-responsive liposomes for drug delivery. WIREs Nanomed. Nanobiotechnol. 2017, 9, e1450. [Google Scholar] [CrossRef]
- Ali, H.; Opere, C.; Singh, S. In Vitro-Controlled Release Delivery System for Hydrogen Sulfide Donor. AAPS PharmSciTech 2014, 15, 910–919. [Google Scholar] [CrossRef]
- Do, A.-V.; Smith, R.; Tobias, P.; Carlsen, D.; Pham, E.; Bowden, N.B.; Salem, A.K. Sustained Release of Hydrogen Sulfide (H2S) from Poly(Lactic Acid) Functionalized 4-Hydroxythiobenzamide Microparticles to Protect Against Oxidative Damage. Ann. Biomed. Eng. 2019, 47, 1691–1700. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Shieh, M.; Paul, B.D.; Xian, M. Hydrogen sulfide: Recent development of its dual donors and hybrid drugs. Br. J. Pharmacol. 2023, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Distrutti, E. COXIBs, CINODs and H2S-Releasing NSAIDs: Current Perspectives in the Development of Safer Non Steroidal Anti-Inflammatory Drugs. Curr. Med. Chem. 2011, 18, 3494–3505. [Google Scholar] [CrossRef]
- Miljkovic, J.L.; Burger, N.; Gawel, J.M.; Mulvey, J.F.; Norman, A.A.; Nishimura, T.; Tsujihata, Y.; Logan, A.; Sauchanka, O.; Caldwell, S.T.; et al. Rapid and selective generation of H2S within mitochondria protects against cardiac ischemia-reperfusion injury. Redox Biol. 2022, 55, 102429. [Google Scholar] [CrossRef]
- Corvino, A.; Caliendo, G. Hydrogen Sulfide (H2S)-Donor Molecules: Chemical, Biological, and Therapeutical Tools. Int. J. Mol. Sci. 2024, 25, 7932. [Google Scholar] [CrossRef]
- Yang, C.-T.; Chen, L.; Xu, S.; Day, J.J.; Li, X.; Xian, M. Recent Development of Hydrogen Sulfide Releasing/Stimulating Reagents and Their Potential Applications in Cancer and Glycometabolic Disorders. Front. Pharmacol. 2017, 8, 664. [Google Scholar] [CrossRef] [PubMed]
- Opoku-Damoah, Y.; Zhang, R.; Ta, H.T.; Xu, Z.P. Therapeutic gas-releasing nanomedicines with controlled release: Advances and perspectives. Exploration 2022, 2, 20210181. [Google Scholar] [CrossRef] [PubMed]
- Cacciotti, I.; Ciocci, M.; Di Giovanni, E.; Nanni, F.; Melino, S. Hydrogen Sulfide-Releasing Fibrous Membranes: Potential Patches for Stimulating Human Stem Cells Proliferation and Viability under Oxidative Stress. Int. J. Mol. Sci. 2018, 19, 2368. [Google Scholar] [CrossRef]
- Corvino, A.; Scognamiglio, A.; Fiorino, F.; Perissutti, E.; Santagada, V.; Caliendo, G.; Severino, B. Pills of Multi-Target H2S Donating Molecules for Complex Diseases. Int. J. Mol. Sci. 2024, 25, 7014. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.; Mhatre, S.; Chandler, C.; Opere, C.; Singh, S. Application of Quality by Design in the Development of Hydrogen Sulfide Donor Loaded Polymeric Microparticles. Aaps PharmSciTech 2024, 25, 132. [Google Scholar] [CrossRef]
- Morén, C.; Desouza, R.M.; Giraldo, D.M.; Uff, C. Antioxidant Therapeutic Strategies in Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 9328. [Google Scholar] [CrossRef]
- Kerksick, C.; Willoughby, D. The Antioxidant Role of Glutathione and N-Acetyl-Cysteine Supplements and Exercise-Induced Oxidative Stress. J. Int. Soc. Sports Nutr. 2005, 2, 38–44. [Google Scholar] [CrossRef]
- Flieger, J.; Flieger, W.; Baj, J.; Maciejewski, R. Antioxidants: Classification, Natural Sources, Activity/Capacity Measurements, and Usefulness for the Synthesis of Nanoparticles. Materials 2021, 14, 4135. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Gugliandolo, A.; Bramanti, P.; Mazzon, E. Activation of Nrf2 by Natural Bioactive Compounds: A Promising Approach for Stroke? Int. J. Mol. Sci. 2020, 21, 4875. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Q.; Wang, Y.; Li, J.; Bai, Z.; Zhao, Q.; Wang, Z.; He, D.; Zhang, J.; Chen, Y. Toxicities and beneficial protection of H2S donors based on nonsteroidal anti-inflammatory drugs. MedChemComm 2019, 10, 742–756. [Google Scholar] [CrossRef] [PubMed]
- Gunaydin, C.; Bilge, S.S. Effects of Nonsteroidal Anti-Inflammatory Drugs at the Molecular Level. Eurasian J. Med. 2018, 50, 116–121. [Google Scholar] [CrossRef]
- Li, M.; Li, J.; Zhang, T.; Zhao, Q.; Cheng, J.; Liu, B.; Wang, Z.; Zhao, L.; Wang, C. Syntheses, toxicities and anti-inflammation of H 2 S-donors based on non-steroidal anti-inflammatory drugs. Eur. J. Med. Chem. 2017, 138, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Han, Y.; He, Y.; Liu, J.; Wang, Y. Natural compounds targeting mitochondrial dysfunction: Emerging therapeutics for target organ damage in hypertension. Front. Pharmacol. 2023, 14, 1209890. [Google Scholar] [CrossRef]
- Jia, J.; Wang, Z.; Zhang, M.; Huang, C.; Song, Y.; Xu, F.; Zhang, J.; Li, J.; He, M.; Li, Y.; et al. SQR mediates therapeutic effects of H 2 S by targeting mitochondrial electron transport to induce mitochondrial uncoupling. Sci. Adv. 2020, 6, eaaz5752. [Google Scholar] [CrossRef]
- Griendling, K.K.; Touyz, R.M.; Zweier, J.L.; Dikalov, S.; Chilian, W.; Chen, Y.-R.; Harrison, D.G.; Bhatnagar, A. Measurement of Reactive Oxygen Species, Reactive Nitrogen Species, and Redox-Dependent Signaling in the Cardiovascular System: A Scientific Statement from the American Heart Association. Circ. Res. 2016, 119, E39–E75. [Google Scholar] [CrossRef]
- Calin, M.A.; Manea, D.; Parasca, S.V.; Popescu, C.; Ionescu, E.-V.; Munteanu, C. Hyperspectral imaging reveals that sapropelic mud therapy may improve local tissue oxygenation in elderly. Int. J. Biometeorol. 2024, 69, 591–604. [Google Scholar] [CrossRef]
- Duanghathaipornsuk, S.; Farrell, E.J.; Alba-Rubio, A.C.; Zelenay, P.; Kim, D.S. Detection Technologies for Re-active Oxygen Species: Fluorescence and Electrochemical Methods and Their Applications. Biosensors 2021, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Debowska, K.; Debski, D.; Hardy, M.; Jakubowska, M.; Kalyanaraman, B.; Marcinek, A.; Michalski, R.; Michalowski, B.; Ouari, O.; Sikora, A.; et al. Toward selective detection of reactive oxygen and nitrogen species with the use of fluorogenic probes—Limitations, progress, and perspectives. Pharmacol. Rep. 2015, 67, 756–764. [Google Scholar] [CrossRef]
- Mhatre, S.; Rai, A.; Ali, H.; Patil, A.; Singh, N.; Verma, R.; Auden, J.; Chandler, C.; Dash, A.; Opere, C.; et al. Comparison of Colorimetric, Spectroscopic and Electrochemical Techniques for Quantification of Hydrogen Sulfide. BioTechniques 2023, 76, 71–80. [Google Scholar] [CrossRef]
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef] [PubMed]
- Iliadis, S.; Papanikolaou, N.A. Reactive Oxygen Species Mechanisms that Regulate Protein–Protein Interactions in Cancer. Int. J. Mol. Sci. 2024, 25, 9255. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Pluth, M.D. Hydrogen Sulfide Donors Activated by Reactive Oxygen Species. Angew. Chem. Int. Ed. Engl. 2016, 55, 14638–14642. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Jing, G.; Zhang, L.; Chen, C.; Zhu, S. Interplay Among Hydrogen Sulfide, Nitric Oxide, Reactive Oxygen Species, and Mitochondrial DNA Oxidative Damage. Front. Plant Sci. 2021, 12, 701681. [Google Scholar] [CrossRef]
- van der Vlies, A.J.; Ghasemi, M.; Adair, B.M.; Adair, J.H.; Gomez, E.D.; Hasegawa, U. Reactive Oxygen Species-Triggered Hydrogen Sulfide Release and Cancer-Selective Antiproliferative Effect of Anethole Dithiolethione-Containing Polymeric Micelles. Adv. Health Mater. 2023, 12, e2201836. [Google Scholar] [CrossRef]
- Krylatov, A.; Maslov, L.; Tsibulnikov, S.Y.; Voronkov, N.; Boshchenko, A.; Downey, J.; Mentzer, R. The Role of Reactive Oxygen Species, Kinases, Hydrogen Sulfide, and Nitric Oxide in the Regulation of Autophagy and Their Impact on Ischemia and Reperfusion Injury in the Heart. Curr. Cardiol. Rev. 2021, 17, e230421186874. [Google Scholar] [CrossRef]
- Královičová, J.; Bartůněk, A.; Hofmann, J.; Křížek, T.; Kozlík, P.; Roušarová, J.; Ryšánek, P.; Šíma, M.; Slanař, O. Pharmacokinetic Variability in Pre-Clinical Studies: Sample Study with Abiraterone in Rats and Implications for Short-Term Comparative Pharmacokinetic Study Designs. Pharmaceutics 2022, 14, 643. [Google Scholar] [CrossRef]

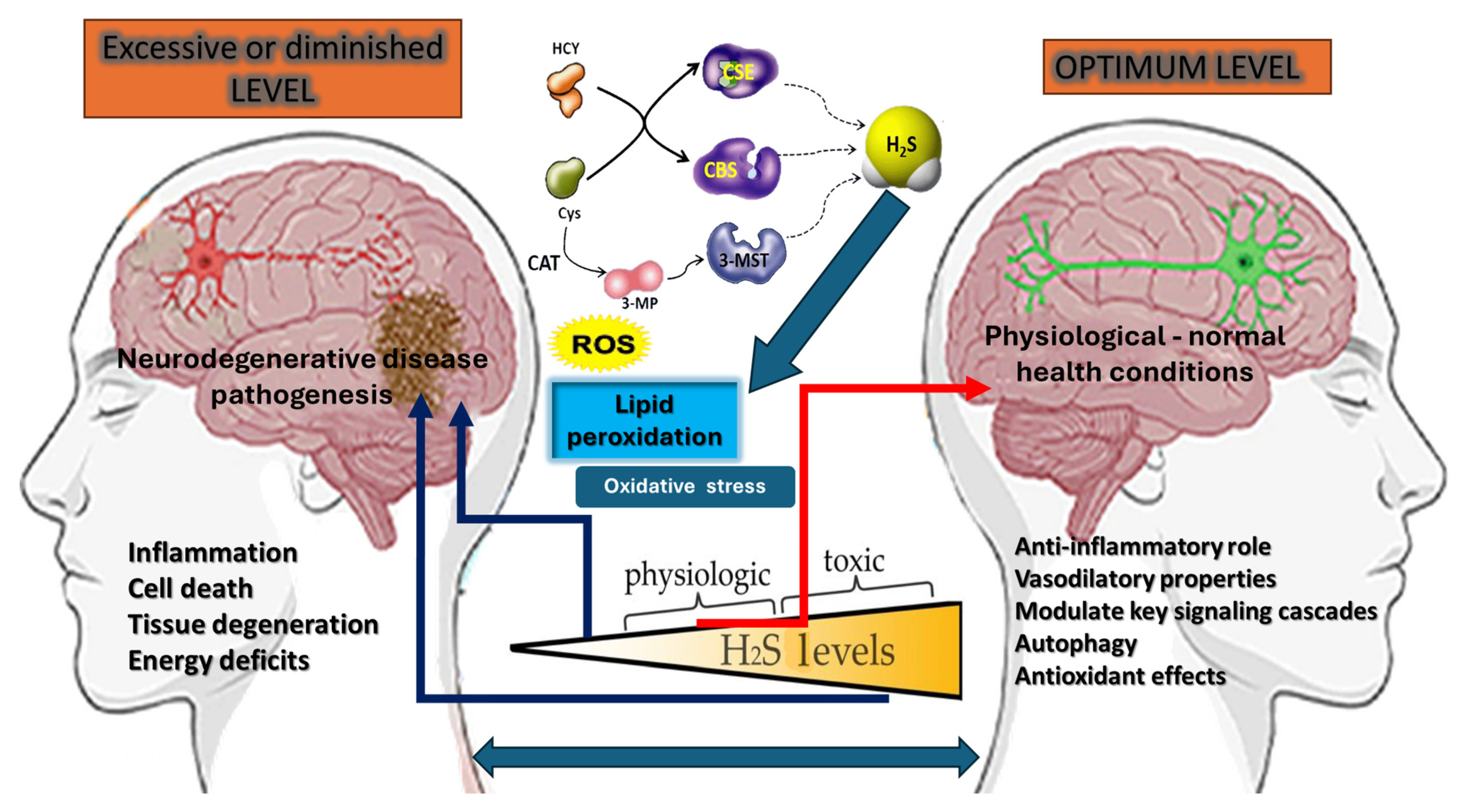

{kind=link}
{kind=link}
| Measurement Method | Sample/Tissue | Reported H2S Level | Observations/Comments | References |
|---|---|---|---|---|
| Methylene blue (strong-acid-liberation) | Rodent brain slices | 50–160 µM | Potential overestimation due to the acid-labile release of sulfane sulfur pools. | [116] |
| Methylene blue + acid reagents | Mammalian plasma or tissue | >30–35 µM | Early measurements. Possibly includes “bound” or polysulfide forms. Generally recognized as artificially high. | [114] |
| Methylene blue | H2S in blood | 2–5 μM | The human colon has the highest luminal concentration of H2S in the body (1.0–3.4 mM). | [117] |
| Gas chromatography (GC) | Rodent tissues | Typically nM to <1 µM | Researchers demonstrated that older, much higher (µM–100 µM) measurements were “dramatically overestimated”. The GC technique helps to avoid artifacts from strong acid steps. | [81,118] |
| GC + chemiluminescence | Blood (mouse) | ~15–50 nM (plasma) | More realistic results for free (unbound) H2S in vivo. | [114,119] |
| Polarographic electrode | Various tissues | 10–300 nM | A polarographic sensor in a sealed system. Helps to avoid volatility losses and acid-liberation artifacts. | [110,114] |
| Polarographic electrode | Blood and tissues (rodents) | 50–200 nM | Researchers concur that free H2S is likely in the tens–hundreds of nM range, at least under baseline physiological conditions. | [114] |
| MBB (monobromobimane) derivatization + HPLC | Human liver or plasma samples | ~0.7–3 µM | Less overestimation than with strong acid, but partial artifactual release can still occur, depending on pH and sample handling. | [116] |
| enzymatic assays, MBB/HPLC, and others | Human plasma | ~0.4–1 µM | Suggests that the submicromolar–micromolar range for free H2S might be physiologically relevant in humans. | [120] |
| GC/polarographic/MBB | Vascular tissues, plasma (rodents) | Typically 100–300 nM | Repeatedly confirms baseline H2S in the ~nM-to-<1 µM bracket in healthy conditions. | [93,120] |
| Various (electrode-based, GC, fluorescent/bimane-based methods) | Cultured cells, animal tissues (brain, kidney, heart, retina) | nM–~1 µM | There is a broader consensus that “physiological” free H2S rarely exceeds a few µM. Levels above 10–50 µM appear toxic (rapid inhibition of cytochrome c oxidase, cellular respiration). | [112,115,121,122] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munteanu, C.; Galaction, A.I.; Onose, G.; Turnea, M.; Rotariu, M. The Janus Face of Oxidative Stress and Hydrogen Sulfide: Insights into Neurodegenerative Disease Pathogenesis. Antioxidants 2025, 14, 360. https://doi.org/10.3390/antiox14030360
Munteanu C, Galaction AI, Onose G, Turnea M, Rotariu M. The Janus Face of Oxidative Stress and Hydrogen Sulfide: Insights into Neurodegenerative Disease Pathogenesis. Antioxidants. 2025; 14(3):360. https://doi.org/10.3390/antiox14030360
Chicago/Turabian StyleMunteanu, Constantin, Anca Irina Galaction, Gelu Onose, Marius Turnea, and Mariana Rotariu. 2025. "The Janus Face of Oxidative Stress and Hydrogen Sulfide: Insights into Neurodegenerative Disease Pathogenesis" Antioxidants 14, no. 3: 360. https://doi.org/10.3390/antiox14030360
APA StyleMunteanu, C., Galaction, A. I., Onose, G., Turnea, M., & Rotariu, M. (2025). The Janus Face of Oxidative Stress and Hydrogen Sulfide: Insights into Neurodegenerative Disease Pathogenesis. Antioxidants, 14(3), 360. https://doi.org/10.3390/antiox14030360