

The Interplay Between Autophagy and Apoptosis in the Mechanisms of Action of Stilbenes in Cancer Cells

Abstract
1. Introduction
2. Autophagy
3. Apoptosis
4. The Role of Autophagy and Apoptosis in Cancer Cells
5. The Interplay Between Autophagy and Apoptosis
6. Autophagy and Apoptosis in the Mechanisms of Action of Stilbenes in Cancer Cells
6.1. RES
6.1.1. Lung Cancer
6.1.2. Oral Cancer
6.1.3. Esophageal Cancer
6.1.4. Colon Cancer
6.1.5. Breast Cancer
6.1.6. Ovarian Cancer
6.1.7. Cervical Cancer
6.1.8. Endometrial Cancer
6.1.9. Renal Cancer
6.1.10. Glioblastoma
6.1.11. Leukemia
6.1.12. Multiple Myeloma
6.2. PTER
6.2.1. Lung Cancer
6.2.2. Oral Cancer
6.2.3. Colon Cancer
6.2.4. Pancreatic Cancer
6.2.5. Breast Cancer
6.2.6. Bladder Cancer
6.2.7. Leukemia
6.3. PIC
6.3.1. Neuroblastoma
6.3.2. Leukemia
6.4. OXYRES
Neuroblastoma
6.5. PIN
Leukemia
6.6. CA-4
Osteosarcoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cell Line | Effects | Compound Concentration/ Treatment Time | Ref. | |
|---|---|---|---|---|---|
| RES | A549 | Autophagy: ↑ LC3-II/LC3-I (WB) ↑ Beclin-1 (WB) ↓ p62/SQSTM1 (WB) LC3-GFP fluorescent puncta (FM) | Apoptosis: phosphatidylserine externalization (FC) | 25, 100, 200 µM | [165] |
| ↓ p-Akt/Akt (WB), ↓ p-mTOR/mTOR (WB) ↑ p-p38/p38 (WB), ↓ p-p70S6K/p70S6K (WB) | |||||
| A549 | Autophagy: ↑ LC3-II/LC3-I (WB) ↑ Beclin 1 (WB) ↓ p62/SQSTM1 (WB) MDC fluorescent puncta (FM) accumulation of autophagic vacuoles (TEM) | Apoptosis: phosphatidylserine externalization (FC) DNA fragmentation (FC) caspase-3 cleavage (WB) ↑ Bax (WB) ↓ Bcl-2 (WB) | 25, 50, 100 µM/48 h | [166] | |
| ↓ p-Akt/Akt (WB), ↑ p53 (WB), ↓ p-MDM2 (WB) | |||||
| A549 | Autophagy: ↑ LC3-II (WB) LC3 fluorescent puncta (CM) | Apoptosis: phosphatidylserine externalization (FC) ↓ mitochondrial membrane potential (CM) PARP-1 cleavage (WB) caspase-3 cleavage (WB) caspase-3 activity (C) ↑ Bax (WB), ↓ Bcl-2 (WB) cytochrome c release (WB) | 100, 200, 300 µM/6, 12, 24 h | [167] | |
| A549 | Autophagy: ↑ LC3-II/LC3-I (WB) ↑ Beclin 1 (WB) RFP-GFP-LC3 fluorescent puncta (CM) MDC fluorescent puncta (FM) ↑ AVOs (FC) accumulation of autophagic vacuoles (TEM) | Apoptosis: phosphatidylserine externalization (FC) DNA fragmentation (FC) ↑ Bax (WB) ↓ Bcl-2 (WB) | 25, 30, 50, 75, 80 µM/48 h | [168] | |
| ↑ NGFR (WB), ↑ p-AMPK/AMPK (WB), ↓ p-mTOR/mTOR (WB) | |||||
| PC9/G /gefitinib resistant | Autophagy: ↑ LC3-II (WB) MDC fluorescent puncta (FM, FC) | Apoptosis: phosphatidylserine externalization (FC) condensation and fragmentation of nuclei (FM) caspase-3 cleavage (WB) | 40 µM/72 h | [169] | |
| ↑ p53 (WB), ↑ p21 (WB) | |||||
| CAR/ cisplatin resistant | Autophagy: ↑ LC3-II (WB) ↑ Beclin 1 (WB) ↑ ATG5 (WB), ↑ ATG7 (WB) ↑ ATG12 (WB) ↑ ATG14 (WB) ↑ ATG16L1 (WB) AVOs (FM) LC3-GFP fluorescent puncta (FM) MDC fluorescent puncta (FM) LysoTracker Red DND-99 fluorescent puncta (FM) | Apoptosis: DNA condensation and fragmentation (FM) caspase-3 cleavage (WB) caspase-9 cleavage (WB) ↑ cytochrome c (WB) ↑ Apaf-1 (WB), ↑ AIF (WB) ↑ Endo G (WB) ↑ Bax (WB), ↑ Bad (WB) ↓ Bcl-2 (WB), ↓ p-Bad (WB) | 25, 50, 100 µM/24, 48 h | [170] | |
| ↑ p-AMPK (WB), ↓ p-Akt (WB), ↓ p-mTOR (WB) ↑ PI3K class III (WB), ↓ Rubicon (WB) | |||||
| EC109 | Autophagy: ↑ LC3-II (WB), ↑ Beclin 1 (WB) ↑ ATG5 (WB) LC3 fluorescent puncta (FM) MDC fluorescent puncta (FM) accumulation of autophagic vacuoles (TEM) | Apoptosis: phosphatidylserine externalization (FC) ↑ sub-G0/G1 (FC) ↓ mitochondrial membrane potential (FC) caspase-3 cleavage (WB) caspase-3 activation (C) ↑ Bax (WB), ↓ Bcl-2 (WB) chromatin condensation (FM) | 10, 50, 100, 150 µM/6, 12, 24, 48 h | [171] | |
| ↑ p-AMPK (WB) | |||||
| EC9706 | Autophagy: ↑ LC3-II (WB), ↑ Beclin-1 (WB) ↑ ATG5 (WB) LC3 fluorescent puncta (FM) MDC fluorescent puncta (FM) | Apoptosis: phosphatidylserine externalization (FC) ↑ sub-G0/G1 (FC) caspase-3 cleavage (WB) ↑ Bax (WB), ↓ Bcl-2 (WB) chromatin condensation (FM) | 10, 50, 100, 150 µM/6, 12, 24, 48 h | [171] | |
| ↑ p-AMPK (WB) | |||||
| HT-29 | Autophagy: ↑ LC3-II (WB) accumulation of autophagic vacuoles (TEM) LC3 fluorescent puncta (CM) | Apoptosis: phosphatidylserine externalization (FC) caspase-3 cleavage (WB) caspase-8 cleavage (WB) PARP cleavage (WB) | 150 µM/24, 48, 72 h | [172] | |
| DLD1 | Autophagy: ↑ LC3-II (WB), ↑ Beclin 1(WB) GFP–LC3 fluorescent puncta (FM) LC3-FITC fluorescent puncta (FM) Beclin 1–GFP fluorescent puncta (FM) Lamp-1 fluorescent puncta (FM) MDC fluorescent puncta (FM) | Apoptosis: phosphatidylserine externalization (FC) DNA fragmentation (FM) | 100 µM/15 min, 1, 2, 4, 24, 48 h | [173] | |
| MDA-MB231 | Autophagy: ↑ LC3-II (WB), ↑ Beclin 1 (WB) | Apoptosis: caspase-3 cleavage (WB) caspase-3 activation (SF) | 50, 60, 120 µM/12, 24, 36, 48 h | [174] | |
| A2780 | Autophagy: accumulation of autophagic vacuoles (TEM) MDC fluorescent puncta (FM) | Apoptosis: caspase-9 cleavage (WB) cytochrome c release (WB) | 25, 50, 100 µM/6, 12, 24, 36, 48 h | [175] | |
| OVCAR-3 | Autophagy: ↑ ATG5 (WB), ↑ LC3-II (WB) ↑ p62/SQSTM1 (WB) | Apoptosis: phosphatidylserine externalization (FC) ↓ mitochondrial membrane potential (FC, FM) caspase-3 activation (C, CM) caspase-3 cleavage (WB) ↑ PARP activity (C) | 30, 100 µM /12, 24, 48, 72 h | [176] | |
| HeLa | Autophagy: ↑ LC3-II (WB) LC3-GFP fluorescent puncta (FM) MDC fluorescent puncta (FM) accumulation of autophagic vacuoles (TEM) | Apoptosis: phosphatidylserine externalization (FC) caspase-3 cleavage (WB) cytochrome c release (WB) | 100 µM/6, 12, 24, 48, 72 h | [177] | |
| Ishikawa | Autophagy: ↑ LC3-II (WB) LC3 fluorescent puncta (CM) | Apoptosis: phosphatidylserine externalization (FC) ↑ sub-G1 fraction (FC) PARP cleavage (WB) | 25, 100 µM/24, 48, 72 h | [178] | |
| ↑ p-AMPKα (WB), ↑ p-ERK (WB), ↓ p-AKT (WB) | |||||
| 786-O | Autophagy: ↑ LC3-II (WB) accumulation of autophagic vacuoles (FC, FM) | Apoptosis: phosphatidylserine externalization (FC) ↓ mitochondrial membrane potential (FC) caspase-3 activation (FC) PARP cleavage (WB) | 20, 40 µM/24, 48 h | [179] | |
| ↓ p-ERK/ERK (WB), ↑ p-JNK/JNK (WB), ↑ p-p38/p38 (WB) | |||||
| U251 | Autophagy: ↑ LC3-II (WB) ↑ Beclin 1 (WB) LC3 fluorescent puncta (CM) MDC fluorescent puncta (FM) | Apoptosis: ↑ sub-G1 (FC) ↓ mitochondrial membrane potential (FC, FM), chromatin condensation and nuclei fragmentation (FM) | 150 µM/3, 6, 12, 24, 48, 72 h | [180] | |
| K562 | Autophagy: ↑ LC3-II (WB) accumulation of autophagic vacuoles (TEM) | Apoptosis: phosphatidylserine externalization (FC) caspase-3 activation (C) fragmentation of nuclei (FM) | 12.5, 25, 50, 100 µM/4 days | [181] | |
| ↑ glycophorin A (FC), ↑ CD71 (FC), ↑ Band3 (FC) | |||||
| K562 /imatinib-sensitive | Autophagy: ↑ LC3-II (WB) ↑ p62/SQSTM1 (WB) accumulation of autophagic vacuoles (TEM) | Apoptosis: caspase-3 activation (SF) | 2, 5, 10, 25, 50 µM/2, 4, 8, 16, 24, 48 h | [182] | |
| ↑ p-AMPKα(T172) (WB), ↓ p-mTOR(S2448) (WB) ↓ p-p70/85-S6K (T389) (WB), ↓ p-S6 ribosomal (S235/236) (WB) ↓ p-4EBP1 (T37/46) (WB), ↑ p-JNK 2/3 (T183/Y185) (WB) ↑ p-JNK 1 (T183/Y185) (WB), ↑ p-c-Jun (S63) (WB) | |||||
| K562 /imatinib-resistant | Autophagy: ↑ LC3-II (WB), ↑ p62/SQSTM1 (WB) accumulation of autophagic vacuoles (TEM) | Apoptosis: caspase-3 activation (SF) | 2, 5, 10, 25, 50 µM/2, 4, 8, 16, 24, 48 h | [182] | |
| ↑ p-AMPKα(T172) (WB), ↓ p-mTOR(S2448) (WB) ↓ p-p70/85-S6K (T389) (WB), ↓ p-S6 ribosomal (S235/236) (WB) ↓ p-4EBP1 (T37/46) (WB), ↑ p-JNK 2/3 (T183/Y185) (WB) ↑ p-JNK 1 (T183/Y185) (WB), ↑ p-c-Jun (S63) (WB) | |||||
| K562/ adriamycin resistant | Autophagy: ↑ LC3-II (WB), ↑ Beclin 1 (WB) ↓ p62/SQSTM1 (WB) ↑ cathepsin D (WB) accumulation of autophagic vacuoles (TEM) MDC fluorescent puncta (FM) | Apoptosis: phosphatidylserine externalization (FC) caspase-3 cleavage (WB) ↓ Bcl-2 (WB), ↑ Bax (WB) | 40, 80 µM/24, 48, 72 h | [183] | |
| MOLT-4 | Autophagy: ↑ LC3-II (WB) ↑ p62/SQSTM1 (WB) LC3 fluorescent puncta (FM) | Apoptosis: caspase-3 activation (FC) ↓ mitochondrial membrane potential (FC) phosphatidylserine externalization (FC) ↑ sub-G1 (FC) PARP1 cleavage (WB) DNA fragmentation (AGE) condensation and fragmentation of nuclei (FM) | 41 µM/45 min, 2, 4, 6, 12, 24, 48 h | [184] | |
| HL-60 | Autophagy: ↑ LC3-II (WB) ↑ p62/SQSTM1 (24 h) (WB) ↓ p62/SQSTM1 (48 h, 72 h) (WB) LC3 fluorescent puncta (FM) | Apoptosis: ↑ caspase-3 activation (FC) ↓ mitochondrial membrane potential (FC) phosphatidylserine externalization (FC) ↑ sub-G1 (FC) PARP1 cleavage (WB) DNA fragmentation (AGE) condensation and fragmentation of nuclei (FM) | 43 µM/45 min, 2, 4, 6, 12, 24, 48, 72 h | [184] | |
| HL-60 | Autophagy: ↑ LC3-II (WB) ↑ p62/SQSTM1(WB) ↑ Beclin-1 (WB) ↑ ATG5 (WB) LC3 fluorescent puncta (FM) | Apoptosis: ↓ mitochondrial membrane potential (SF) phosphatidylserine externalization (FC) caspase-3 cleavage (WB) ↑ caspase-3 activation (C) caspase-8 cleavage (WB) Bid cleavage (WB) ↑ Bax/Bcl-2 (WB), ↑ Fas (WB) ↑ FasL (WB) | 12,5, 25, 50, 100 µM/3, 6, 12, 24, 48 h | [185] | |
| ↑ p-AMPK/AMPK (WB), ↑ pLKB1/LKB1 (WB) ↓ p-AKT/AKT(WB), ↓ p-p70S6K/p70S6K (WB) | |||||
| U266 | Autophagy: ↑ LC3-II (WB), ↑ Beclin 1 (WB) | Apoptosis: phosphatidylserine externalization (FC) caspase-3 cleavage (WB) PARP cleavage (WB) ↓ Survivin (WB) | 50, 100 µM/48 h | [186] | |
| ↑ p-AMPKα (WB), ↓ p-mTOR (WB) ↓ p-p70S6K (WB), ↓ p-4EBP1 (WB) | |||||
| RPMI-8226 | Autophagy: ↑ LC3-II (WB), ↑ Beclin 1 (WB) | Apoptosis: phosphatidylserine externalization (FC) caspase-3 cleavage (WB) PARP cleavage (WB) ↓ Survivin (WB) | 50, 100 µM/48 h | [186] | |
| ↑ p-AMPKα (WB), ↓ p-mTOR (WB) ↓ p-p70S6K (WB), ↓ p-4EBP1 (WB) | |||||
| NCI-H929 | Autophagy: ↑ LC3-II (WB), ↑ Beclin 1 (WB) | Apoptosis: phosphatidylserine externalization (FC) caspase-3 cleavage (WB) PARP cleavage (WB) ↓ Survivin (WB) | 50, 100 µM/48 h | [186] | |
| ↑ p-AMPKα (WB), ↓ p-mTOR (WB) ↓ p-p70S6K (WB), ↓ p-4EBP1 (WB) | |||||
| PTER | A549 | Autophagy: ↑ LC3-II (WB), ↑ Beclin 1 (WB) ↓ p62/SQSTM1 (WB) GFP-LC3 fluorescent puncta (FM) ↑ AVOs (FC) | Apoptosis: phosphatidylserine externalization (FC) ↑ sub-G0/G1 (FC) condensed and fragmented nuclei (FM) | 50, 75, 100 µM/ 24, 48, 72 h | [221] |
| ↓ p-PI3K (WB), ↓ p-AKT (WB) ↓ p-JNK (WB), ↑ p-ERK (WB) | |||||
| A549/D16 /docetaxel resistant | Autophagy: ↑ LC3-II (WB), ↑ Beclin 1 (WB) ↓ p62/SQSTM1 (WB) GFP-LC3 fluorescent puncta (FM) ↑ AVOs (FC) | Apoptosis: phosphatidylserine externalization (FC) ↑ sub-G0/G1 (FC) condensed and fragmented nuclei (FM) | 50, 75, 100 µM/24, 48, 72 h | [221] | |
| ↓ p-PI3K (WB), ↓ p-AKT (WB) ↓ p-JNK (WB), ↑ p-ERK (WB) | |||||
| SAS | Autophagy: ↑ LC3-II (WB), ↑ Beclin 1 (WB) GFP-LC3 fluorescent puncta (FM) ↑ AVOs (FC, FM) | Apoptosis: phosphatidylserine externalization (FC) caspase-3 cleavage (WB) caspase-8 cleavage (WB) caspase-9 cleavage (WB) PARP cleavage (WB) condensed and fragmented nuclei (FM) | 10, 20, 40 µM/6, 12, 18, 24, 48 h | [222] | |
| ↓ pp-38 (WB), ↓ p-AKT (WB), ↓ p-ERK1/2(WB) ↓ p-mTOR (WB), ↓ p-ULK (Ser757) (WB), ↑ p-ULK (Ser555) (WB) ↑ p-JNK1/2 (WB), ↑ p-AMPK (WB), ↑ p-Raptor (WB) | |||||
| OECM-1 | Autophagy: ↑ LC3-II (WB), ↑ Beclin 1 (WB) GFP-LC3 fluorescent puncta (FM) ↑ AVOs (FC, FM) | Apoptosis: phosphatidylserine externalization (FC) caspase-3 cleavage (WB) caspase-8 cleavage (WB) caspase-9 cleavage (WB) PARP cleavage (WB) condensed and fragmented nuclei (FM) | 10, 20, 40 µM/6, 12, 18, 24, 48 h | [222] | |
| ↓ pp-38 (WB), ↓ p-AKT (WB), ↓ p-ERK1/2 (WB), ↓ p-mTOR (WB) ↓ p-ULK (Ser757) (WB), ↑ p-ULK (Ser555) (WB) ↑ p-JNK1/2 (WB), ↑ p-AMPK (WB), ↑ p-Raptor (WB) | |||||
| CAR/ cisplatin resistant | Autophagy: ↑ LC3-II (WB), ↑ Beclin 1 (WB) ↑ ATG5 (WB), ↑ ATG7 (WB) ↑ ATG12 (WB) ↑ AVOs (FM) MDC fluorescent puncta (FM) | Apoptosis: caspase-3 activation (C) caspase-9 activation (C) caspase-3 cleavage (WB) caspase-7 cleavage (WB) caspase-9 cleavage (WB) PARP cleavage (WB) ↓ Bcl-2 (WB), ↑ Bax (WB) ↑ cytochrome c (WB) DNA fragmentation (FC) chromatin condensation (FM) | 25, 50, 75, 100 µM/24, 48 h | [223] | |
| ↓ p-AKT (WB), ↓ MDR1 (WB) | |||||
| HT-29 | Autophagy: ↑ ULK1 (qRT-PCR) ↑ AMBRA1 (qRT-PCR) ↑ MAP1LC3A (qRT-PCR) | Apoptosis: caspase-3 activation (C) ↑ sub-G1 (FC) DNA fragmentation (C) ↑ BAX (qRT-PCR) | 10, 40, 60 µM/6, 12, 24, 48, 72 h | [224] | |
| ↓ p-AKT/total AKT (C), ↓ p-STAT/total STAT (C) | |||||
| BxPC-3 | Autophagy: ↑ LC3-II (WB), ↑ Beclin 1 (WB) ↑ p62/SQSTM1 (WB) ↑ AVOs (FM, FC) | Apoptosis: phosphatidylserine externalization (FC) ↑ sub-G0/G1 (FC) caspase-3 cleavage (WB) ↓ Bcl-XL (WB), ↓ Bcl-2 (WB) | 75, 100, 125µM/12, 24, 36, 48 h | [225] | |
| ↓ p-AKT (WB), ↓ p-mTOR (WB), ↑ p-ERK (WB) ↓ p-p38 (WB), ↓ p-JNK (WB) | |||||
| MIA PaCa-2 | Autophagy: ↑ LC3-II (WB), ↑ Beclin 1 (WB) ↑ p62/SQSTM1 (WB) ↑ AVOs (FM, FC) accumulation of autophagic vacuoles (TEM) | Apoptosis: phosphatidylserine externalization (FC) ↑ sub-G0/G1 (FC) caspase-3 cleavage (WB) caspase-8 cleavage (WB) caspase-9 cleavage (WB) ↓ Bcl-XL (WB), ↓ Bcl-2 (WB) ↑ Bax (WB) | 75, 100, 125 µM/12, 24, 36, 48 h | [225] | |
| ↓ p-AKT (WB), ↓ p-mTOR (WB), ↑ p-ERK (WB) | |||||
| MCF-7 | Autophagy: ↑ LC3-II (WB) | Apoptosis: phosphatidylserine externalization (FC) PARP cleavage (WB) chromatin condensation (FM) | 50, 100 µM/24 h | [226] | |
| Bcap-37 | Autophagy: ↑ LC3-II (WB) | Apoptosis: phosphatidylserine externalization (FC) PARP cleavage (WB) chromatin condensation (FM) | 50, 100 µM/24 h | [226] | |
| T24 | Autophagy: ↑ LC3-II (WB) ↑ AVOs (FC) accumulation of autophagic vacuoles (TEM) | Apoptosis: phosphatidylserine externalization (FC) ↑ sub-G0/G1 (FC) caspase-3 activation (SF) chromatin condensation and nuclei fragmentation (FM) ↓ Bcl-2 (WB), ↓ Bcl-XL (WB) | 50, 75, 100 µM/24, 48, 72 h | [227] | |
| ↓ p-AKT (WB), ↓ pp70S6K (WB), ↑ p-ERK1/2 (WB) | |||||
| T24R/ chemoresistant | Autophagy: ↑ LC3-II (WB) ↑ AVOs (FC) accumulation of autophagic vacuoles (TEM) | Apoptosis: phosphatidylserine externalization (FC) ↑ sub-G0/G1 (FC) caspase-3 activation (SF) chromatin condensation and nuclei fragmentation (FM) ↓ Bcl-2 (WB), ↓ Bcl-XL (WB) | 50, 75, 100 µM/24, 48, 72 h | [227] | |
| HL-60 | Autophagy: ↑ LC3-II (WB) LC3 fluorescent puncta (CM) accumulation of autophagic vacuoles (TEM) | Apoptosis: phosphatidylserine externalization (FC) ↑ sub-G1 (FC) DNA fragmentation (AGE) ↓ mitochondrial membrane potential (FC) caspase-3 activation (FC) chromatin condensation and nuclei fragmentation (CM) | 1, 10, 43, 100 µM/2, 4, 6, 12, 24, 48, 72 h | [228] | |
| PIC | SH-SY5Y | Autophagy: accumulation of autophagic vacuoles (FM) ↑ BECN1 (qRT-PCR) ↑ ATG5 (qRT-PCR) ↑ ATG7 (qRT-PCR) ↑ ATG12 (qRT-PCR) ↑ MAP 1LC3A (qRT-PCR) ↑ MAP 1LC3B (qRT-PCR) | Apoptosis: phosphatidylserine externalization (FC) DNA fragmentation (C) ↓ mitochondrial membrane potential (FM) ↑ CASP3 (qRT-PCR) ↑ CASP8 (qRT-PCR) ↑ CASP9 (qRT-PCR) ↑ BAX (qRT-PCR) ↑ FADD (qRT-PCR) | 50, 100 µM/72 h | [257] |
| MOLT-4 | Autophagy: ↑ LC3-II (WB) ↓ p62/SQSTM1 (WB) LC3 fluorescent puncta (FM) | Apoptosis: phosphatidylserine externalization (FC) ↑ sub-G1 (FC) DNA fragmentation (AGE) PARP1 cleavage (WB) chromatin condensation and nuclei fragmentation (FM) ↓ mitochondrial membrane potential (FC) caspase-3 activation (FC) | 45.5 µM/ 45 min, 2, 4, 6, 12, 24, 48, 72, 96 h, 3 x 96 h | [258] | |
| ↑ P-gp (FC), ↓ BCRP (FC) | |||||
| OXYRES | SH-SY5Y | Autophagy: ↑ LC3-II (WB), ↑ Beclin 1 (WB) ↑ ATG5 (WB), ↑ ATG7 (WB) ↑ AVOs (FC, FM) LC3 fluorescent puncta (FM) | Apoptosis: phosphatidylserine externalization (FC) ↓ mitochondrial membrane potential (FC) ↓ Bcl-2 (WB), ↑ Bax (WB) caspase-3 cleavage (WB) caspase-9 cleavage (WB) fragmented nuclei (FM) | 40, 80, 120, 160 µM/2, 4, 6, 8, 10, 12 h | [274] |
| ↓ p-AKT (WB), ↓ p-mTOR (WB), ↓ p-S6 (WB) ↑ p-p38 (WB), ↓ p-ERK1/2 (WB) | |||||
| PIN | THP-1 | Autophagy: ↑ LC3-II (WB) ↓ p62/SQSTM1, 4, 6 h (WB) ↑ p62/SQSTM1, 12, 24 h (WB) LC3 fluorescent puncta (FM) | Apoptosis: phosphatidylserine externalization (FC) caspase-3 cleavage (WB) caspase-3 activation (SF) | 0.1, 1, 10, 50, 100 µM/4, 6, 12, 24 h | [281] |
| ↑ p-AMPKα (WB), ↓ AMPKα (WB) | |||||
| U937 | Autophagy: ↑ LC3-II (WB) ↓ p62/SQSTM1, 4, 6 h (WB) ↑ p62/SQSTM1, 12, 24 h (WB) LC3 fluorescent puncta (FM) | Apoptosis: phosphatidylserine externalization (FC) caspase-3 cleavage (WB) caspase-3 activation (SF) | 0.1, 1, 10, 50, 100 µM/4, 6, 12, 24 h | [281] | |
| ↓ AMPKα (WB) | |||||
| CA-4 | SJSA | Autophagy: ↑ LC3-II (WB) ↓ p62/SQSTM1 (WB) GFP-LC3 fluorescent puncta (CM) | Apoptosis: ↑ sub-G0/G1 (FC) caspase-3 cleavage (WB) caspase-8 cleavage (WB) caspase-9 cleavage (WB) PARP cleavage (WB) | 1, 5, 10 nM/24, 48, 72 h | [284] |
| MG63.2 | Autophagy: ↑ LC3-II (WB) ↓ p62/SQSTM1 (WB) GFP-LC3 fluorescent puncta (CM) | Apoptosis: ↑ sub-G0/G1 (FC) caspase-3 cleavage (WB) caspase-8 cleavage (WB) caspase-9 cleavage (WB) PARP cleavage (WB) | 1, 5, 10 nM/24, 48, 72 h | [284] | |
7. Clinical Perspectives of Stilbenes in Cancer Therapy
8. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Rivière, C.; Pawlus, A.D.; Mérillon, J.M. Natural stilbenoids: Distribution in the plant kingdom and chemotaxonomic interest in Vitaceae. Nat. Prod. Rep. 2012, 29, 1317–1333. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Xie, C.-F.; Wang, X.-N.; Lou, H.-X. Stilbenoids. In Natural Produccts:Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes; Ramawat, K., Mérillon, J.-M., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2013; pp. 1901–1949. [Google Scholar]
- Soleas, G.J.; Diamandis, E.P.; Goldberg, D.M. Resveratrol: A molecule whose time has come? And gone? Clin. Biochem. 1997, 30, 91–113. [Google Scholar] [CrossRef] [PubMed]
- Gorham, J. Biochemistry of the Stilbenoids, 1st ed.; Springer: Dordrecht, The Netherlands, 1995. [Google Scholar]
- Niesen, D.B.; Hessler, C.; Seeram, N.P. Beyond resveratrol: A review of natural stilbenoids identified from 2009–2013. J. Berry Res. 2013, 3, 181–196. [Google Scholar] [CrossRef]
- Su, X.; Zhou, D.; Li, N. Bioactive stilbenes from plants. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier: Amsterdam, The Netherlands, 2022; Volume 73, pp. 265–403. [Google Scholar]
- Cassidy, A.; Hanley, B. Isoflavones, lignans and stilbenes—Origins, metabolism and potential importance to human health. J Sci. Food Agric. 2000, 80, 1044–1062. [Google Scholar] [CrossRef]
- Rimando, A.M.; Kalt, W.; Magee, J.B.; Dewey, J.; Ballington, J.R. Resveratrol, pterostilbene, and piceatannol in vaccinium berries. J. Agric. Food Chem. 2004, 52, 4713–4719. [Google Scholar] [CrossRef]
- Viñas, P.; Campillo, N.; Martínez-Castillo, N.; Hernández-Córdoba, M. Solid-phase microextraction on-fiber derivatization for the analysis of some polyphenols in wine and grapes using gas chromatography-mass spectrometry. J. Chromatogr. A 2009, 1216, 1279–1284. [Google Scholar] [CrossRef]
- Viñas, P.; Martínez-Castillo, N.; Campillo, N.; Hernández-Córdoba, M. Directly suspended droplet microextraction with in injection-port derivatization coupled to gas chromatography-mass spectrometry for the analysis of polyphenols in herbal infusions, fruits and functional foods. J. Chromatogr. A 2011, 1218, 639–646. [Google Scholar] [CrossRef]
- Guerrero, R.F.; Cantos-Villar, E.; Puertas, B.; Richard, T. Daily Preharvest UV-C Light Maintains the High Stilbenoid Concentration in Grapes. J. Agric. Food Chem. 2016, 64, 5139–5147. [Google Scholar] [CrossRef] [PubMed]
- Somkuwar, R.G.; Bhange, M.A.; Oulkar, D.P.; Sharma, A.K.; Ahammed Shabeer, T.P. Estimation of polyphenols by using HPLC-DAD in red and white wine grape varieties grown under tropical conditions of India. J. Food Sci. Technol. 2018, 55, 4994–5002. [Google Scholar] [CrossRef]
- Sáez, V.; Pastene, E.; Vergara, C.; Mardones, C.; Hermosín-Gutiérrez, I.; Gómez-Alonso, S.; Gómez, M.V.; Theoduloz, C.; Riquelme, S.; von Baer, D. Oligostilbenoids in Vitis vinifera L. Pinot Noir grape cane extract: Isolation, characterization, in vitro antioxidant capacity and anti-proliferative effect on cancer cells. Food Chem. 2018, 265, 101–110. [Google Scholar] [CrossRef]
- Aliaño-González, M.J.; Richard, T.; Cantos-Villar, E. Grapevine Cane Extracts: Raw Plant Material, Extraction Methods, Quantification, and Applications. Biomolecules 2020, 10, 1195. [Google Scholar] [CrossRef]
- Romero-Pérez, A.I.; Ibern-Gómez, M.; Lamuela-Raventós, R.M.; de La Torre-Boronat, M.C. Piceid, the major resveratrol derivative in grape juices. J. Agric. Food Chem. 1999, 47, 1533–1536. [Google Scholar] [CrossRef]
- Wang, Y.; Catana, F.; Yang, Y.; Roderick, R.; van Breemen, R.B. An LC-MS method for analyzing total resveratrol in grape juice, cranberry juice, and in wine. J. Agric. Food Chem. 2002, 50, 431–435. [Google Scholar] [CrossRef]
- Vitrac, X.; Bornet, A.; Vanderlinde, R.; Valls, J.; Richard, T.; Delaunay, J.C.; Mérillon, J.M.; Teissédre, P.L. Determination of stilbenes (delta-viniferin, trans-astringin, trans-piceid, cis- and trans-resveratrol, epsilon-viniferin) in Brazilian wines. J. Agric. Food Chem. 2005, 53, 5664–5669. [Google Scholar] [CrossRef]
- Gurbuz, O.; Goçmen, D.; Dagdelen, F.; Gursoy, M.; Aydin, S.; Sahin, I.; Buyukuysal, L.; Usta, M. Determination of flavan-3-ols and trans-resveratrol in grapes and wine using HPLC with fluorescence detection. Food Chem. 2007, 100, 518–525. [Google Scholar] [CrossRef]
- Presta, M.A.; Bruyneel, B.; Zanella, R.; Kool, J.; Krabbe, J.G.; Lingeman, H. Determination of Flavonoids and Resveratrol in Wine by Turbulent-Flow Chromatography-LC-MS. Chromatographia 2009, 69, 167–173. [Google Scholar] [CrossRef]
- Montes, R.; García-López, M.; Rodríguez, I.; Cela, R. Mixed-mode solid-phase extraction followed by acetylation and gas chromatography mass spectrometry for the reliable determination of trans-resveratrol in wine samples. Anal. Chim. Acta 2010, 673, 47–53. [Google Scholar] [CrossRef]
- Cvejic, J.M.; Djekic, S.V.; Petrovic, A.V.; Atanackovic, M.T.; Jovic, S.M.; Brceski, I.D.; Gojkovic-Bukarica, L.C. Determination of trans- and cis-resveratrol in Serbian commercial wines. J. Chromatogr. Sci. 2010, 48, 229–234. [Google Scholar] [CrossRef]
- Paulo, L.; Domingues, F.; Queiroz, J.A.; Gallardo, E. Development and validation of an analytical method for the determination of trans- and cis-resveratrol in wine: Analysis of its contents in 186 Portuguese red wines. J. Agric. Food Chem. 2011, 59, 2157–2168. [Google Scholar] [CrossRef] [PubMed]
- Cacho, J.I.; Campillo, N.; Viñas, P.; Hernández-Córdoba, M. Stir bar sorptive extraction with gas chromatography-mass spectrometry for the determination of resveratrol, piceatannol and oxyresveratrol isomers in wines. J. Chromatogr. A 2013, 1315, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Di Fabio, E.; Incocciati, A.; Palombarini, F.; Boffi, A.; Bonamore, A.; Macone, A. Ethylchloroformate Derivatization for GC-MS Analysis of Resveratrol Isomers in Red Wine. Molecules 2020, 25, 4603. [Google Scholar] [CrossRef]
- Suprun, A.R.; Dubrovina, A.S.; Tyunin, A.P.; Kiselev, K.V. Profile of Stilbenes and Other Phenolics in Fanagoria White and Red Russian Wines. Metabolites 2021, 11, 231. [Google Scholar] [CrossRef]
- Chiva-Blanch, G.; Urpi-Sarda, M.; Rotchés-Ribalta, M.; Zamora-Ros, R.; Llorach, R.; Lamuela-Raventós, R.M.; Estruch, R.; Andrés-Lacueva, C. Determination of resveratrol and piceid in beer matrices by solid-phase extraction and liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2011, 1218, 698–705. [Google Scholar] [CrossRef]
- Ehala, S.; Vaher, M.; Kaljurand, M. Characterization of phenolic profiles of Northern European berries by capillary electrophoresis and determination of their antioxidant activity. J. Agric. Food Chem. 2005, 53, 6484–6490. [Google Scholar] [CrossRef]
- Song, W.; Wang, H.J.; Bucheli, P.; Zhang, P.F.; Wei, D.Z.; Lu, Y.H. Phytochemical profiles of different mulberry (Morus sp.) species from China. J. Agric. Food Chem. 2009, 57, 9133–9140. [Google Scholar] [CrossRef]
- Sobolev, V.S.; Cole, R.J. trans-resveratrol content in commercial peanuts and peanut products. J. Agric. Food Chem. 1999, 47, 1435–1439. [Google Scholar] [CrossRef]
- Tokusoglu, O.; Unal, M.K.; Yemis, F. Determination of the phytoalexin resveratrol (3,5,4’-trihydroxystilbene) in peanuts and pistachios by high performance liquid chromatographic diode array (HPLC-DAD) and gas chromatography-mass spectrometry (GC-MS). J. Agric. Food Chem. 2005, 53, 5003–5009. [Google Scholar] [CrossRef]
- Hurst, W.J.; Glinski, J.A.; Miller, K.B.; Apgar, J.; Davey, M.H.; Stuart, D.A. Survey of the trans-resveratrol and trans-piceid content of cocoa-containing and chocolate products. J. Agric. Food Chem. 2008, 56, 8374–8378. [Google Scholar] [CrossRef]
- Ragab, A.S.; Van Fleet, J.; Jankowski, B.; Park, J.H.; Bobzin, S.C. Detection and quantitation of resveratrol in tomato fruit (Lycopersicon esculentum Mill.). J. Agric. Food Chem. 2006, 54, 7175–7179. [Google Scholar] [CrossRef] [PubMed]
- Kageura, T.; Matsuda, H.; Morikawa, T.; Toguchida, I.; Harima, S.; Oda, M.; Yoshikawa, M. Inhibitors from rhubarb on lipopolysaccharide-induced nitric oxide production in macrophages: Structural requirements of stilbenes for the activity. Bioorg Med. Chem. 2001, 9, 1887–1893. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Yang, Z.D.; Shi, D.F.; Yao, X.J.; Wang, M.G. Inhibition of Monoamine Oxidase by Stilbenes from Rheum palmatum. Iran. J. Pharm. Res. 2016, 15, 885–892. [Google Scholar]
- Hathway, D.E.; Seakins, J.W. Hydroxystilbenes of Eucalyptus wandoo. Biochem. J. 1959, 72, 369–374. [Google Scholar] [CrossRef]
- Tian, L.W.; Xu, M.; Li, Y.; Li, X.Y.; Wang, D.; Zhu, H.T.; Yang, C.R.; Zhang, Y.J. Phenolic compounds from the branches of Eucalyptus maideni. Chem. Biodivers. 2012, 9, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Suprun, A.R.; Dubrovina, A.S.; Aleynova, O.A.; Kiselev, K.V. The Bark of the Spruce Picea jezoensis Is a Rich Source of Stilbenes. Metabolites 2021, 11, 714. [Google Scholar] [CrossRef]
- Kiselev, K.V.; Grigorchuk, V.P.; Ogneva, Z.V.; Suprun, A.R.; Dubrovina, A.S. Stilbene biosynthesis in the needles of spruce Picea jezoensis. Phytochemistry 2016, 131, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Latva-Mäenpää, H.; Wufu, R.; Mulat, D.; Sarjala, T.; Saranpää, P.; Wähälä, K. Stability and Photoisomerization of Stilbenes Isolated from the Bark of Norway Spruce Roots. Molecules 2021, 26, 1036. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Xuan, L.; Xu, Y.; Bai, D.; Zhong, D. Constituents from Polygonum cuspidatum. Chem. Pharm. Bull. 2002, 50, 605–608. [Google Scholar] [CrossRef]
- Lin, L.L.; Lien, C.Y.; Cheng, Y.C.; Ku, K.L. An effective sample preparation approach for screening the anticancer compound piceatannol using HPLC coupled with UV and fluorescence detection. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 853, 175–182. [Google Scholar] [CrossRef]
- Havidz, K.A.; Puspitasari, N.; Azminah; Yanuar, A.; Artha, Y.; Mun’im, A. HMG-CoA Reductase Inhibitory Activity of Gnetum gnemon Seed Extract and Identification of Potential Inhibitors for Lowering Cholesterol Level. J. Young Pharm. 2017, 9, 559–565. [Google Scholar] [CrossRef]
- Ali, Z.; Tanaka, T.; Iliya, I.; Iinuma, M.; Furusawa, M.; Ito, T.; Nakaya, K.; Murata, J.; Darnaedi, D. Phenolic constituents of Gnetum klossii. J. Nat. Prod. 2003, 66, 558–560. [Google Scholar] [CrossRef]
- Liu, Y.; Harinantenaina, L.; Brodie, P.J.; Bowman, J.D.; Cassera, M.B.; Slebodnick, C.; Callmander, M.W.; Randrianaivo, R.; Rakotobe, E.; Rasamison, V.E.; et al. Bioactive compounds from Stuhlmannia moavi from the Madagascar dry forest. Bioorganic Med. Chem. 2013, 21, 7591–7594. [Google Scholar] [CrossRef]
- Adrian, M.; Jeandet, P.; Douillet-Breuil, A.C.; Tesson, L.; Bessis, R. Stilbene content of mature Vitis vinifera berries in response to UV-C elicitation. J. Agric. Food Chem. 2000, 48, 6103–6105. [Google Scholar] [CrossRef] [PubMed]
- Douillet-Breuil, A.C.; Jeandet, P.; Adrian, M.; Bessis, R. Changes in the phytoalexin content of various Vitis spp. in response to ultraviolet C elicitation. J. Agric. Food Chem. 1999, 47, 4456–4461. [Google Scholar] [CrossRef] [PubMed]
- Sobolev, V.S.; Khan, S.I.; Tabanca, N.; Wedge, D.E.; Manly, S.P.; Cutler, S.J.; Coy, M.R.; Becnel, J.J.; Neff, S.A.; Gloer, J.B. Biological activity of peanut (Arachis hypogaea) phytoalexins and selected natural and synthetic Stilbenoids. J. Agric. Food Chem. 2011, 59, 1673–1682. [Google Scholar] [CrossRef]
- Devgun, M.; Nanda, A.; Ansari, S.H. Comparison of conventional and non conventional methods of extraction of heartwood of Pterocarpus marsupium Roxb. Acta Pol. Pharm. 2012, 69, 475–485. [Google Scholar] [PubMed]
- Zhang, M.; Zhao, G.; Guo, J.; Liu, B.; Jiang, X.; Yin, Y. A GC-MS Protocol for Separating Endangered and Non-endangered. Molecules 2019, 24, 799. [Google Scholar] [CrossRef]
- Fuendjiep, V.; Wandji, J.; Tillequin, F.; Mulholland, D.A.; Budzikiewicz, H.; Fomum, Z.T.; Nyemba, A.M.; Koch, M. Chalconoid and stilbenoid glycosides from Guibourtia tessmanii. Phytochemistry 2002, 60, 803–806. [Google Scholar] [CrossRef]
- Jin, Q.; Yang, J.; Ma, L.; Cai, J.; Li, J. Comparison of Polyphenol Profile and Inhibitory Activities Against Oxidation and α-Glucosidase in Mulberry (Genus Morus) Cultivars from China. J. Food Sci. 2015, 80, C2440–C2451. [Google Scholar] [CrossRef]
- Matsui, Y.; Sugiyama, K.; Kamei, M.; Takahashi, T.; Suzuki, T.; Katagata, Y.; Ito, T. Extract of passion fruit (Passiflora edulis) seed containing high amounts of piceatannol inhibits melanogenesis and promotes collagen synthesis. J. Agric. Food Chem. 2010, 58, 11112–11118. [Google Scholar] [CrossRef]
- Liudvytska, O.; Ponczek, M.B.; Ciesielski, O.; Krzyżanowska-Kowalczyk, J.; Kowalczyk, M.; Balcerczyk, A.; Kolodziejczyk-Czepas, J. Rheum rhaponticum and Rheum rhabarbarum Extracts as Modulators of Endothelial Cell Inflammatory Response. Nutrients 2023, 15, 949. [Google Scholar] [CrossRef]
- Brinker, A.M.; Seigler, D.S. Isolation and identification of piceatannol as a phytoalexin from sugarcane. Phytochemistry 1991, 30, 3229–3232. [Google Scholar] [CrossRef]
- Ma, X.; Li, Y.; Lv, C.; Liu, B.; Yuan, C.; Huang, W.; Luo, Q.; Xiao, Y.; Sun, C.; Li, T.; et al. Modulation of Keap1-Nrf2-ARE signaling pathway by oxyresveratrol, a derivative of resveratrol from grape skin. Food Biosci. 2022, 50, 102162. [Google Scholar] [CrossRef]
- Ayinampudi, S.; Wang, Y.H.; Avula, B.; Smillie, T.J.; Khan, I.A. Quantitative Analysis of Oxyresveratrol in Different Plant Parts of Morus Species and Related Genera by HPTLC and HPLC. JPC-J. Planar Chromat.-Modern TLC 2011, 24, 125–129. [Google Scholar] [CrossRef]
- Maneechai, S.; Likhitwitayawuid, K.; Sritularak, B.; Palanuvej, C.; Ruangrungsi, N.; Sirisa-Ard, P. Quantitative analysis of oxyresveratrol content in Artocarpus lakoocha and ‘Puag-Haad’. Med. Princ. Pract. 2009, 18, 223–227. [Google Scholar] [CrossRef]
- Deguchi, T.; Tamai, A.; Asahara, K.; Miyamoto, K.; Miyamoto, A.; Nomura, M.; Kawata-Tominaga, T.; Yoshioka, Y.; Murata, K. Anti-tyrosinase and Anti-oxidative Activities by Asana: The Heartwood of Pterocarpus marsupium. Nat. Prod. Commun. 2019, 14, 1934578X19883727. [Google Scholar] [CrossRef]
- Ohguchi, K.; Tanaka, T.; Iliya, I.; Ito, T.; Iinuma, M.; Matsumoto, K.; Akao, Y.; Nozawa, Y. Gnetol as a potent tyrosinase inhibitor from genus Gnetum. Biosci. Biotechnol. Biochem. 2003, 67, 663–665. [Google Scholar] [CrossRef]
- Xiang, W.; Jiang, B.; Li, X.M.; Zhang, H.J.; Zhao, Q.S.; Li, S.H.; Sun, H.D. Constituents of Gnetum montanum. Fitoterapia 2002, 73, 40–42. [Google Scholar] [CrossRef]
- Kloypan, C.; Jeenapongsa, R.; Sri-in, P.; Chanta, S.; Dokpuang, D.; Tip-pyang, S.; Surapinit, N. Stilbenoids from Gnetum macrostachyum attenuate human platelet aggregation and adhesion. Phytother Res. 2012, 26, 1564–1568. [Google Scholar] [CrossRef]
- Nik Azmina, N.F.; Norizan, A.; Syah, Y.M.; Khairunissa, N.; Zawawi, N.A.; Yusof, M.I. A new stilbenoid compound from the lianas of Gnetum microcarpum. Nat. Prod. Commun. 2014, 9, 1743–1744. [Google Scholar]
- Huang, K.S.; Wang, Y.H.; Li, R.L.; Lin, M. Stilbene dimers from the lianas of Gnetum hainanense. Phytochemistry 2000, 54, 875–881. [Google Scholar] [CrossRef]
- Laavola, M.; Nieminen, R.; Leppänen, T.; Eckerman, C.; Holmbom, B.; Moilanen, E. Pinosylvin and monomethylpinosylvin, constituents of an extract from the knot of Pinus sylvestris, reduce inflammatory gene expression and inflammatory responses in vivo. J. Agric. Food Chem. 2015, 63, 3445–3453. [Google Scholar] [CrossRef] [PubMed]
- Verkasalo, E.; Mottonen, V.; Roitto, M.; Vepsalainen, J.; Kumar, A.; Ilvesniemi, H.; Siwale, W.; Julkunen-Tiitto, R.; Raatikainen, O.; Sikanen, L. Extractives of Stemwood and Sawmill Residues of Scots Pine (Pinus sylvestris L.) for Biorefining in Four Climatic Regions in Finland-Phenolic and Resin Acid Compounds. Forests 2021, 12, 192. [Google Scholar] [CrossRef]
- Dumas, M.T.; Hubbes, M.; Strunz, G.M. Identification of Some Compounds Associated with Resistance of Pinus densiflora to Fomes annosus. Eur. J. For Pathol. 1983, 13, 151–160. [Google Scholar] [CrossRef]
- Celimene, C.C.; Micales, J.A.; Ferge, L.; Young, R.A. Efficacy of Pinosylvins against White-Rot and Brown-Rot Fungi. Holzforschung 1999, 53, 491–497. [Google Scholar] [CrossRef]
- Pietarinen, S.P.; Willfor, S.M.; Ahotupa, M.O.; Hemming, J.E.; Holmbom, B.R. Knotwood and Bark Extracts: Strong Antioxidants From Waste Materials. J. Wood Sci. 2006, 52, 436–444. [Google Scholar] [CrossRef]
- Yao, C.-S.; Lin, M.; Liu, X.; Wang, Y. Stilbenes from Gnetum cleistostachyum. Acta Chim. Sin. 2003, 61, 1331–1334. [Google Scholar]
- Suresh Babu, K.; Tiwari, A.K.; Srinivas, P.V.; Ali, A.Z.; China Raju, B.; Rao, J.M. Yeast and mammalian alpha-glucosidase inhibitory constituents from Himalayan rhubarb Rheum emodi Wall.ex Meisson. Bioorganic Med. Chem. Lett. 2004, 14, 3841–3845. [Google Scholar] [CrossRef] [PubMed]
- Bahtiar, A.; Setyowati, H.T.; Mahanani, R.R.; Wati, A.; Arsianti, A.; Fadilah, F. Rhaponticin contained Rheum officinale root extract improved Postmenopause symptom of Ovariectomized Rat. J. Adv. Pharm. Technol. Res. 2021, 12, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Leng, L.F.; Lai, Q.; Lv, D.; Yin, J.L.; Zeng, G.Z. Study on the chemical composition of Caesalpinia sinensis. Nat. Prod. Res. 2022, 36, 5559–5566. [Google Scholar] [CrossRef]
- Lui, A.C.W.; Pow, K.C.; Lin, N.; Lam, L.P.Y.; Liu, G.; Godwin, I.D.; Fan, Z.; Khoo, C.J.; Tobimatsu, Y.; Wang, L.; et al. Regioselective stilbene O-methylations in Saccharinae grasses. Nat. Commun. 2023, 14, 3462. [Google Scholar] [CrossRef]
- Pettit, G.R.; Singh, S.B.; Hamel, E.; Lin, C.M.; Alberts, D.S.; Garcia-Kendall, D. Isolation and structure of the strong cell growth and tubulin inhibitor combretastatin A-4. Experientia 1989, 45, 209–211. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, S.C.N.; Assalin, M.R.; Nobre, S.; Melo, I.S.; Moraes, R.M.; Ferracini, V.L.; Cerdeira, A.L. Determination of Combretastatin A-4 in Combretum leprosum. Planta Med. 2010, 76, 53. [Google Scholar] [CrossRef]
- Paul, B.; Masih, I.; Deopujari, J.; Charpentier, C. Occurrence of resveratrol and pterostilbene in age-old darakchasava, an ayurvedic medicine from India. J. Ethnopharmacol. 1999, 68, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Signorelli, P.; Ghidoni, R. Resveratrol as an anticancer nutrient: Molecular basis, open questions and promises. J. Nutr. Biochem. 2005, 16, 449–466. [Google Scholar] [CrossRef] [PubMed]
- Nonomura, S.; Kanagawa, H.; Makimot, A. Chemical constituents of polygonaceous plants.I.Studies on the components of Ko-J O-Kon. (Polygonum cuspidatum Sieb Et Zucc.). Yakugaku Zasshi 1963, 83, 988–990. [Google Scholar] [CrossRef]
- Langcake, P.; Pryce, R.J. A new class of phytoalexins from grapevines. Experientia 1977, 33, 151–152. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Daim, M.M.; Zakhary, N.I.; Aleya, L.; Bungǎu, S.G.; Bohara, R.A.; Siddiqi, N.J. Aging, Metabolic, and Degenerative Disorders: Biomedical Value of Antioxidants. Oxidative Med. Cell Longev. 2018, 2018, 2098123. [Google Scholar] [CrossRef]
- Abdel-Daim, M.M.; Abo-El-Sooud, K.; Aleya, L.; Bungǎu, S.G.; Najda, A.; Saluja, R. Alleviation of Drugs and Chemicals Toxicity: Biomedical Value of Antioxidants. Oxidative Med. Cell Longev. 2018, 2018, 6276438. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, T.; Mihis, A.G. Resveratrol as a privileged molecule with antioxidant activity. Food Chem. Adv. 2023, 3, 100539. [Google Scholar] [CrossRef]
- Gulcin, I. Antioxidant properties of resveratrol: A structure-activity insight. Innov. Food Sci. Emerg. Technol. 2010, 11, 2010–2218. [Google Scholar] [CrossRef]
- Rimando, A.M.; Cuendet, M.; Desmarchelier, C.; Mehta, R.G.; Pezzuto, J.M.; Duke, S.O. Cancer chemopreventive and antioxidant activities of pterostilbene, a naturally occurring analogue of resveratrol. J. Agric. Food Chem. 2002, 50, 3453–3457. [Google Scholar] [CrossRef]
- Murias, M.; Jäger, W.; Handler, N.; Erker, T.; Horvath, Z.; Szekeres, T.; Nohl, H.; Gille, L. Antioxidant, prooxidant and cytotoxic activity of hydroxylated resveratrol analogues: Structure-activity relationship. Biochem. Pharmacol. 2005, 69, 903–912. [Google Scholar] [CrossRef]
- Kavas, G.O.; Ayral, P.A.; Elhan, A.H. The effects of resveratrol on oxidant/antioxidant systems and their cofactors in rats. Adv. Clin. Exp. Med. 2013, 22, 151–155. [Google Scholar]
- Stivala, L.A.; Savio, M.; Carafoli, F.; Perucca, P.; Bianchi, L.; Maga, G.; Forti, L.; Pagnoni, U.M.; Albini, A.; Prosperi, E.; et al. Specific structural determinants are responsible for the antioxidant activity and the cell cycle effects of resveratrol. J. Biol. Chem. 2001, 276, 22586–22594. [Google Scholar] [CrossRef]
- Waffo Teguo, P.; Fauconneau, B.; Deffieux, G.; Huguet, F.; Vercauteren, J.; Merillon, J.M. Isolation, identification, and antioxidant activity of three stilbene glucosides newly extracted from vitis vinifera cell cultures. J. Nat. Prod. 1998, 61, 655–657. [Google Scholar] [CrossRef]
- Spanier, G.; Xu, H.; Xia, N.; Tobias, S.; Deng, S.; Wojnowski, L.; Forstermann, U.; Li, H. Resveratrol reduces endothelial oxidative stress by modulating the gene expression of superoxide dismutase 1 (SOD1), glutathione peroxidase 1 (GPx1) and NADPH oxidase subunit (Nox4). J. Physiol. Pharmacol. 2009, 60 (Suppl. S4), 111–116. [Google Scholar]
- Eskelinen, E.L. Autophagy: Supporting cellular and organismal homeostasis by self-eating. Int. J. Biochem. Cell Biol. 2019, 111, 1–10. [Google Scholar] [CrossRef]
- Peng, H.; Lavker, R.M. Nucleophagy: A New Look at Past Observations. J. Invest. Dermatol. 2016, 136, 1316–1318. [Google Scholar] [CrossRef]
- Eskelinen, E.L. Maturation of autophagic vacuoles in Mammalian cells. Autophagy 2005, 1, 1–10. [Google Scholar] [CrossRef]
- Eskelinen, E.L. Novel insights into autophagosome biogenesis revealed by cryo-electron tomography. FEBS Lett 2024, 598, 9–16. [Google Scholar] [CrossRef]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Mammalian Autophagy: How Does It Work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef]
- Zhen, Y.; Stenmark, H. Autophagosome Biogenesis. Cells 2023, 12, 668. [Google Scholar] [CrossRef]
- Melia, T.J. Growing thin-How bulk lipid transport drives expansion of the autophagosome membrane but not of its lumen. Curr. Opin. Cell Biol. 2023, 83, 102190. [Google Scholar] [CrossRef]
- Wirawan, E.; Vanden Berghe, T.; Lippens, S.; Agostinis, P.; Vandenabeele, P. Autophagy: For better or for worse. Cell Res. 2012, 22, 43–61. [Google Scholar] [CrossRef]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef]
- Yim, W.W.; Mizushima, N. Lysosome biology in autophagy. Cell Discov. 2020, 6, 6. [Google Scholar] [CrossRef]
- Yuan, J.; Ofengeim, D. A guide to cell death pathways. Nat. Rev. Mol. Cell Biol. 2024, 25, 379–395. [Google Scholar] [CrossRef]
- Xu, G.; Shi, Y. Apoptosis signaling pathways and lymphocyte homeostasis. Cell Res. 2007, 17, 759–771. [Google Scholar] [CrossRef]
- Moyer, A.; Tanaka, K.; Cheng, E.H. Apoptosis in Cancer Biology and Therapy. Annu. Rev. Pathol. 2025, 20, 303–328. [Google Scholar] [CrossRef]
- Kroemer, G. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat. Med. 1997, 3, 614–620. [Google Scholar] [CrossRef]
- Nagata, S. Apoptosis by growth factors. Cell 1997, 88, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Shakeri, R.; Kheirollahi, A.; Davoodi, J. Apaf-1: Regulation and function in cell death. Biochimie 2017, 135, 111–125. [Google Scholar] [CrossRef] [PubMed]
- van Gurp, M.; Festjens, N.; van Loo, G.; Saelens, X.; Vandenabeele, P. Mitochondrial intermembrane proteins in cell death. Biochem Biophys Res Commun 2003, 304, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef]
- Suzuki, Y.; Imai, Y.; Nakayama, H.; Takahashi, K.; Takio, K.; Takahashi, R. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell 2001, 8, 613–621. [Google Scholar] [CrossRef]
- Eskelinen, E.L. The dual role of autophagy in cancer. Curr. Opin. Pharmacol. 2011, 11, 294–300. [Google Scholar] [CrossRef]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Vicencio, J.M.; Criollo, A.; Maiuri, M.C.; Kroemer, G. Anti- and pro-tumor functions of autophagy. Biochim. Biophys Acta. 2009, 1793, 1524–1532. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Gammoh, N.; Ryan, K.M. Autophagy and autophagy-related pathways in cancer. Nat. Rev. Mol. Cell Biol. 2023, 24, 560–575. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Liang, C.; Feng, P.; Ku, B.; Oh, B.H.; Jung, J.U. UVRAG: A new player in autophagy and tumor cell growth. Autophagy 2007, 3, 69–71. [Google Scholar] [CrossRef]
- Song, Y.J.; Zhang, S.S.; Guo, X.L.; Sun, K.; Han, Z.P.; Li, R.; Zhao, Q.D.; Deng, W.J.; Xie, X.Q.; Zhang, J.W.; et al. Autophagy contributes to the survival of CD133+ liver cancer stem cells in the hypoxic and nutrient-deprived tumor microenvironment. Cancer Lett. 2013, 339, 70–81. [Google Scholar] [CrossRef]
- Peng, Q.; Qin, J.; Zhang, Y.; Cheng, X.; Wang, X.; Lu, W.; Xie, X.; Zhang, S. Autophagy maintains the stemness of ovarian cancer stem cells by FOXA2. J. Exp. Clin. Cancer Res. 2017, 36, 171. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Vitale, I.; Pietrocola, F.; Guilbaud, E.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostini, M.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; et al. Apoptotic cell death in disease-Current understanding of the NCCD 2023. Cell Death Differ. 2023, 30, 1097–1154. [Google Scholar] [CrossRef]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef] [PubMed]
- Nigam, M.; Mishra, A.P.; Deb, V.K.; Dimri, D.B.; Tiwari, V.; Bungau, S.G.; Bungau, A.F.; Radu, A.F. Evaluation of the association of chronic inflammation and cancer: Insights and implications. Biomed. Pharmacother. 2023, 164, 115015. [Google Scholar] [CrossRef]
- Wang, M.; Chen, S.; He, X.; Yuan, Y.; Wei, X. Targeting inflammation as cancer therapy. J. Hematol. Oncol. 2024, 17, 13. [Google Scholar] [CrossRef]
- Monkkonen, T.; Debnath, J. Inflammatory signaling cascades and autophagy in cancer. Autophagy 2018, 14, 190–198. [Google Scholar] [CrossRef]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Hong, J.T. Roles of NF-κB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, A.D.; Eisenstein, M.; Ber, Y.; Bialik, S.; Kimchi, A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol. Cell 2011, 44, 698–709. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Xi, H.; Wang, S.; Wang, B.; Hong, X.; Liu, X.; Li, M.; Shen, R.; Dong, Q. The role of interaction between autophagy and apoptosis in tumorigenesis (Review). Oncol. Rep. 2022, 48, 208. [Google Scholar] [CrossRef]
- Norman, J.M.; Cohen, G.M.; Bampton, E.T. The in vitro cleavage of the hAtg proteins by cell death proteases. Autophagy 2010, 6, 1042–1056. [Google Scholar] [CrossRef]
- Pagliarini, V.; Wirawan, E.; Romagnoli, A.; Ciccosanti, F.; Lisi, G.; Lippens, S.; Cecconi, F.; Fimia, G.M.; Vandenabeele, P.; Corazzari, M.; et al. Proteolysis of Ambra1 during apoptosis has a role in the inhibition of the autophagic pro-survival response. Cell Death Differ. 2012, 19, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- Wirawan, E.; Vande Walle, L.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; De Rycke, R.; Verspurten, J.; Declercq, W.; et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010, 1, e18. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef]
- Hou, W.; Han, J.; Lu, C.; Goldstein, L.A.; Rabinowich, H. Autophagic degradation of active caspase-8: A crosstalk mechanism between autophagy and apoptosis. Autophagy 2010, 6, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.M. p53 and autophagy in cancer: Guardian of the genome meets guardian of the proteome. Eur. J. Cancer 2011, 47, 44–50. [Google Scholar] [CrossRef]
- Livesey, K.M.; Kang, R.; Vernon, P.; Buchser, W.; Loughran, P.; Watkins, S.C.; Zhang, L.; Manfredi, J.J.; Zeh, H.J.; Li, L.; et al. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res. 2012, 72, 1996–2005. [Google Scholar] [CrossRef]
- Pu, T.; Zhang, X.P.; Liu, F.; Wang, W. Coordination of the nuclear and cytoplasmic activities of p53 in response to DNA damage. Biophys J. 2010, 99, 1696–1705. [Google Scholar] [CrossRef]
- Szkudelska, K.; Okulicz, M.; Hertig, I.; Szkudelski, T. Resveratrol ameliorates inflammatory and oxidative stress in type 2 diabetic Goto-Kakizaki rats. Biomed. Pharmacother. 2020, 125, 110026. [Google Scholar] [CrossRef]
- Zhao, X.; Cui, Q.; Fu, Q.; Song, X.; Jia, R.; Yang, Y.; Zou, Y.; Li, L.; He, C.; Liang, X.; et al. Antiviral properties of resveratrol against pseudorabies virus are associated with the inhibition of IκB kinase activation. Sci. Rep. 2017, 7, 8782. [Google Scholar] [CrossRef] [PubMed]
- Miyashiro, C.A.; Bernegossi, J.; Bonifácio, B.V.; de Toledo, L.G.; Ramos, M.A.S.; Bauab, T.M.; Chorilli, M. Development and characterization of a novel liquid crystalline system containing sodium alginate for incorporation of trans-resveratrol intended for treatment of buccal candidiasis. Pharmazie 2020, 75, 179–185. [Google Scholar] [CrossRef]
- Spósito, L.; Fonseca, D.; Gonçalves Carvalho, S.; Sábio, R.M.; Marena, G.D.; Bauab, T.M.; Bagliotti Meneguin, A.; Parreira, P.; L Martins, M.C.; Chorilli, M. Engineering resveratrol-loaded chitosan nanoparticles for potential use against Helicobacter pylori infection. Eur. J. Pharm. Biopharm. 2024, 199, 114280. [Google Scholar] [CrossRef]
- El-Sayed, S.A.M.; Fouad, G.I.; Rizk, M.Z.; Beherei, H.H.; Mabrouk, M. Comparative Neuroprotective Potential of Nanoformulated and Free Resveratrol Against Cuprizone-Induced Demyelination in Rats. Mol. Neurobiol. 2024, 62, 2710–2725. [Google Scholar] [CrossRef] [PubMed]
- Ozpak, L.; Bağca, B.G. Neuroprotective effects of resveratrol through modulation of PI3K/Akt/GSK-3β pathway and metalloproteases. IUBMB Life 2024, 76, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Gimblet, C.J.; Kruse, N.T.; Geasland, K.; Michelson, J.; Sun, M.; Mandukhail, S.R.; Wendt, L.H.; Eyck, P.T.; Pierce, G.L.; Jalal, D.I. Effect of Resveratrol on Endothelial Function in Patients with CKD and Diabetes: A Randomized Controlled Trial. Clin. J. Am. Soc. Nephrol. 2024, 19, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Yang, Y.; Guo, J.; Ma, T.; Hu, Y.; Huang, L.; He, Y.; Xi, J. Resveratrol Inhibits Zinc Deficiency-Induced Mitophagy and Exerts Cardiac Cytoprotective Effects. Biol. Trace Elem. Res. 2024, 202, 1669–1682. [Google Scholar] [CrossRef]
- Chakraborty, S.; Levenson, A.S.; Biswas, P.K. Structural insights into Resveratrol’s antagonist and partial agonist actions on estrogen receptor alpha. BMC Struct. Biol. 2013, 13, 27. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Feng, F.; Deng, Y. Resveratrol Regulates Glucose and Lipid Metabolism in Diabetic Rats by Inhibition of PDK1/AKT Phosphorylation and HIF-1α Expression. Diabetes Metab Syndr Obes. 2023, 16, 1063–1074. [Google Scholar] [CrossRef]
- Li, W.; Li, C.; Ma, L.; Jin, F. Resveratrol inhibits viability and induces apoptosis in the small-cell lung cancer H446 cell line via the PI3K/Akt/c-Myc pathway. Oncol. Rep. 2020, 44, 1821–1830. [Google Scholar] [CrossRef]
- Xiao, Y.; Duan, Y.; Wang, Y.; Yin, X. Resveratrol suppresses malignant progression of oral squamous cell carcinoma cells by inducing the ZNF750/RAC1 signaling pathway. Bioengineered 2021, 12, 2863–2873. [Google Scholar] [CrossRef]
- Song, M.; Qu, Y.; Jia, H.; Zhang, Y.; Liu, S.; Laster, K.V.; Choi, B.Y.; Tian, J.; Gu, T.; Chen, H.; et al. Targeting TAOK1 with resveratrol inhibits esophageal squamous cell carcinoma growth in vitro and in vivo. Mol. Carcinog. 2024, 63, 991–1008. [Google Scholar] [CrossRef]
- Qin, X.; Luo, H.; Deng, Y.; Yao, X.; Zhang, J.; He, B. Resveratrol inhibits proliferation and induces apoptosis via the Hippo/YAP pathway in human colon cancer cells. Biochem. Biophys. Res. Commun. 2022, 636, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Zheng, A.; Zhai, H.; Zhang, T. Resveratrol mediated the proliferation and apoptosis of gastric cancer cells by modulating the PI3K/Akt/P53 signaling pathway. Biochem. Biophys. Res. Commun. 2024, 723, 150186. [Google Scholar] [CrossRef]
- Dai, W.; Wang, F.; Lu, J.; Xia, Y.; He, L.; Chen, K.; Li, J.; Li, S.; Liu, T.; Zheng, Y.; et al. By reducing hexokinase 2, resveratrol induces apoptosis in HCC cells addicted to aerobic glycolysis and inhibits tumor growth in mice. Oncotarget 2015, 6, 13703–13717. [Google Scholar] [CrossRef]
- Qin, Y.; Ma, Z.; Dang, X.; Li, W.; Ma, Q. Effect of resveratrol on proliferation and apoptosis of human pancreatic cancer MIA PaCa-2 cells may involve inhibition of the Hedgehog signaling pathway. Mol. Med. Rep. 2014, 10, 2563–2567. [Google Scholar] [CrossRef] [PubMed]
- Venkatadri, R.; Muni, T.; Iyer, A.K.; Yakisich, J.S.; Azad, N. Role of apoptosis-related miRNAs in resveratrol-induced breast cancer cell death. Cell Death Dis. 2016, 7, e2104. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tong, L.; Luo, Y.; Li, X.; Chen, G.; Wang, Y. Resveratrol inhibits the proliferation and induces the apoptosis in ovarian cancer cells via inhibiting glycolysis and targeting AMPK/mTOR signaling pathway. J. Cell Biochem. 2018, 119, 6162–6172. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Tian, H.; Lin, S.; Mo, J.; Li, Z.; Chen, X.; Liu, J. Resveratrol inhibits proliferation and promotes apoptosis via the androgen receptor splicing variant 7 and PI3K/AKT signaling pathway in LNCaP prostate cancer cells. Oncol. Lett. 2020, 20, 169. [Google Scholar] [CrossRef]
- Zhao, S.; Tang, L.; Chen, W.; Su, J.; Li, F.; Chen, X.; Wu, L. Resveratrol-induced apoptosis is associated with regulating the miR-492/CD147 pathway in malignant melanoma cells. Naunyn. Schmiedebergs Arch. Pharmacol. 2021, 394, 797–807. [Google Scholar] [CrossRef]
- Wang, B.; Liu, J.; Gong, Z. Resveratrol induces apoptosis in K562 cells via the regulation of mitochondrial signaling pathways. Int. J. Clin. Exp. Med. 2015, 8, 16926–16933. [Google Scholar]
- Jara, P.; Spies, J.; Cárcamo, C.; Arancibia, Y.; Vargas, G.; Martin, C.; Salas, M.; Otth, C.; Zambrano, A. The Effect of Resveratrol on Cell Viability in the Burkitt’s Lymphoma Cell Line Ramos. Molecules 2017, 23, 14. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A.; Sethi, G.; Vadhan-Raj, S.; Bueso-Ramos, C.; Takada, Y.; Gaur, U.; Nair, A.S.; Shishodia, S.; Aggarwal, B.B. Resveratrol inhibits proliferation, induces apoptosis, and overcomes chemoresistance through down-regulation of STAT3 and nuclear factor-kappaB-regulated antiapoptotic and cell survival gene products in human multiple myeloma cells. Blood 2007, 109, 2293–2302. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Cao, N.; Li, Z.; Han, J.; Li, L. Resveratrol, an activator of SIRT1, induces protective autophagy in non-small-cell lung cancer via inhibiting Akt/mTOR and activating p38-MAPK. Onco Targets Ther. 2018, 11, 7777–7786. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Li, J.; Yang, Y.; Zhao, X.; Liu, Y.; Jiang, Y.; Zhou, L.; Feng, Y.; Yu, Y.; Cheng, Y. Resveratrol modulates the apoptosis and autophagic death of human lung adenocarcinoma A549 cells via a p53-dependent pathway: Integrated bioinformatics analysis and experimental validation. Int. J. Oncol. 2020, 57, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Wei, S.; Xiao, T.; Li, G. LC3B/p62-mediated mitophagy protects A549 cells from resveratrol-induced apoptosis. Life Sci. 2021, 271, 119139. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Fan, Y.; Zhang, Y.; Liu, Y.; Yu, Y.; Ma, M. Resveratrol Induces Autophagy and Apoptosis in Non-Small-Cell Lung Cancer Cells by Activating the NGFR-AMPK-mTOR Pathway. Nutrients 2022, 14, 2413. [Google Scholar] [CrossRef]
- Zhu, Y.; He, W.; Gao, X.; Li, B.; Mei, C.; Xu, R.; Chen, H. Resveratrol overcomes gefitinib resistance by increasing the intracellular gefitinib concentration and triggering apoptosis, autophagy and senescence in PC9/G NSCLC cells. Sci. Rep. 2015, 5, 17730. [Google Scholar] [CrossRef]
- Chang, C.H.; Lee, C.Y.; Lu, C.C.; Tsai, F.J.; Hsu, Y.M.; Tsao, J.W.; Juan, Y.N.; Chiu, H.Y.; Yang, J.S.; Wang, C.C. Resveratrol-induced autophagy and apoptosis in cisplatin-resistant human oral cancer CAR cells: A key role of AMPK and Akt/mTOR signaling. Int. J. Oncol. 2017, 50, 873–882. [Google Scholar] [CrossRef]
- Tang, Q.; Li, G.; Wei, X.; Zhang, J.; Chiu, J.F.; Hasenmayer, D.; Zhang, D.; Zhang, H. Resveratrol-induced apoptosis is enhanced by inhibition of autophagy in esophageal squamous cell carcinoma. Cancer Lett. 2013, 336, 325–337. [Google Scholar] [CrossRef]
- Miki, H.; Uehara, N.; Kimura, A.; Sasaki, T.; Yuri, T.; Yoshizawa, K.; Tsubura, A. Resveratrol induces apoptosis via ROS-triggered autophagy in human colon cancer cells. Int. J. Oncol. 2012, 40, 1020–1028. [Google Scholar] [CrossRef]
- Trincheri, N.F.; Follo, C.; Nicotra, G.; Peracchio, C.; Castino, R.; Isidoro, C. Resveratrol-induced apoptosis depends on the lipid kinase activity of Vps34 and on the formation of autophagolysosomes. Carcinogenesis 2008, 29, 381–389. [Google Scholar] [CrossRef]
- Prabhu, V.; Srivastava, P.; Yadav, N.; Amadori, M.; Schneider, A.; Seshadri, A.; Pitarresi, J.; Scott, R.; Zhang, H.; Koochekpour, S.; et al. Resveratrol depletes mitochondrial DNA and inhibition of autophagy enhances resveratrol-induced caspase activation. Mitochondrion 2013, 13, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Opipari, A.W.; Tan, L.; Boitano, A.E.; Sorenson, D.R.; Aurora, A.; Liu, J.R. Resveratrol-induced autophagocytosis in ovarian cancer cells. Cancer Res. 2004, 64, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Qin, Z.; Li, F.; Zhang, H.; Fang, Z.; Hao, E. Apoptotic Cell Death Induced by Resveratrol Is Partially Mediated by the Autophagy Pathway in Human Ovarian Cancer Cells. PLoS ONE 2015, 10, e0129196. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.F.; Wu, C.L.; Huang, S.C.; Wu, C.M.; Hsiao, J.R.; Yo, Y.T.; Chen, Y.H.; Shiau, A.L.; Chou, C.Y. Cathepsin L mediates resveratrol-induced autophagy and apoptotic cell death in cervical cancer cells. Autophagy 2009, 5, 451–460. [Google Scholar] [CrossRef]
- Fukuda, T.; Oda, K.; Wada-Hiraike, O.; Sone, K.; Inaba, K.; Ikeda, Y.; Makii, C.; Miyasaka, A.; Kashiyama, T.; Tanikawa, M.; et al. Autophagy inhibition augments resveratrol-induced apoptosis in Ishikawa endometrial cancer cells. Oncol Lett. 2016, 12, 2560–2566. [Google Scholar] [CrossRef]
- Yao, H.; Fan, M.; He, X. Autophagy suppresses resveratrol-induced apoptosis in renal cell carcinoma 786-O cells. Oncol Lett. 2020, 19, 3269–3277. [Google Scholar] [CrossRef]
- Li, J.; Qin, Z.; Liang, Z. The prosurvival role of autophagy in Resveratrol-induced cytotoxicity in human U251 glioma cells. BMC Cancer 2009, 9, 215. [Google Scholar] [CrossRef]
- Yan, H.W.; Hu, W.X.; Zhang, J.Y.; Wang, Y.; Xia, K.; Peng, M.Y.; Liu, J. Resveratrol induces human K562 cell apoptosis, erythroid differentiation, and autophagy. Tumor Biol. 2014, 35, 5381–5388. [Google Scholar] [CrossRef]
- Puissant, A.; Robert, G.; Fenouille, N.; Luciano, F.; Cassuto, J.P.; Raynaud, S.; Auberger, P. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res. 2010, 70, 1042–1052. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, Z.; Chen, J.; Yi, J.; Cheng, J.; Dun, W.; Wei, H. Resveratrol induces autophagic apoptosis via the lysosomal cathepsin D pathway in human drug-resistant K562/ADM leukemia cells. Exp. Ther. Med. 2018, 15, 3012–3019. [Google Scholar] [CrossRef] [PubMed]
- Siedlecka-Kroplewska, K.; Wozniak, M.; Kmiec, Z. The wine polyphenol resveratrol modulates autophagy and induces apoptosis in MOLT-4 and HL-60 human leukemia cells. J. Physiol. Pharmacol. 2019, 70, 825–838. [Google Scholar] [CrossRef]
- Fan, Y.; Chiu, J.F.; Liu, J.; Deng, Y.; Xu, C.; Zhang, J.; Li, G. Resveratrol induces autophagy-dependent apoptosis in HL-60 cells. BMC Cancer 2018, 18, 581. [Google Scholar] [CrossRef]
- Ma, R.; Yu, D.; Peng, Y.; Yi, H.; Wang, Y.; Cheng, T.; Shi, B.; Yang, G.; Lai, W.; Wu, X.; et al. Resveratrol induces AMPK and mTOR signaling inhibition-mediated autophagy and apoptosis in multiple myeloma cells. Acta Biochim. Biophys. Sin. 2021, 53, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Savio, M.; Ferraresi, A.; Corpina, C.; Vandenberghe, S.; Scarlata, C.; Sottile, V.; Morini, L.; Garavaglia, B.; Isidoro, C.; Stivala, L.A. Resveratrol and Its Analogue 4,4’-Dihydroxy-trans-stilbene Inhibit Lewis Lung Carcinoma Growth In Vivo through Apoptosis, Autophagy and Modulation of the Tumour Microenvironment in a Murine Model. Biomedicines 2022, 10, 1784. [Google Scholar] [CrossRef]
- Davoodvandi, A.; Darvish, M.; Borran, S.; Nejati, M.; Mazaheri, S.; Reza Tamtaji, O.; Hamblin, M.R.; Masoudian, N.; Mirzaei, H. The therapeutic potential of resveratrol in a mouse model of melanoma lung metastasis. Int. Immunopharmacol. 2020, 88, 106905. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Niu, H.; Cui, D.; Huang, G.; Li, J.; Tian, H.; Xu, X.; Liang, F.; Chen, R. Resveratrol triggers autophagy-related apoptosis to inhibit the progression of colorectal cancer via inhibition of FOXQ1. Phytother. Res. 2024, 38, 3218–3239. [Google Scholar] [CrossRef]
- Dias, S.J.; Li, K.; Rimando, A.M.; Dhar, S.; Mizuno, C.S.; Penman, A.D.; Levenson, A.S. Trimethoxy-resveratrol and piceatannol administered orally suppress and inhibit tumor formation and growth in prostate cancer xenografts. Prostate 2013, 73, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.J.; Qi, M.; Li, N.; Lei, Y.H.; Zhang, D.M.; Chen, J.X. Natural products and their derivatives: Promising modulators of tumor immunotherapy. J. Leukoc. Biol. 2020, 108, 493–508. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Shin, H.; Kim, J. In vivo Anti-Cancer Effects of Resveratrol Mediated by NK Cell Activation. J. Innate Immun. 2021, 13, 94–106. [Google Scholar] [CrossRef]
- Cheuk, I.W.; Chen, J.; Siu, M.; Ho, J.C.; Lam, S.S.; Shin, V.Y.; Kwong, A. Resveratrol enhanced chemosensitivity by reversing macrophage polarization in breast cancer. Clin. Transl. Oncol. 2022, 24, 854–863. [Google Scholar] [CrossRef]
- Morvan, M.G.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat Rev Cancer 2016, 16, 7–19. [Google Scholar] [CrossRef]
- Khan, S.U.; Khan, M.U.; Azhar Ud Din, M.; Khan, I.M.; Khan, M.I.; Bungau, S.; Hassan, S.S.U. Reprogramming tumor-associated macrophages as a unique approach to target tumor immunotherapy. Front. Immunol. 2023, 14, 1166487. [Google Scholar] [CrossRef]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef]
- Dasari, S.K.; Bialik, S.; Levin-Zaidman, S.; Levin-Salomon, V.; Merrill, A.H.; Futerman, A.H.; Kimchi, A. Signalome-wide RNAi screen identifies GBA1 as a positive mediator of autophagic cell death. Cell Death Differ. 2017, 24, 1288–1302. [Google Scholar] [CrossRef]
- Zhang, J.; Chiu, J.; Zhang, H.; Qi, T.; Tang, Q.; Ma, K.; Lu, H.; Li, G. Autophagic cell death induced by resveratrol depends on the Ca(2+)/AMPK/mTOR pathway in A549 cells. Biochem. Pharmacol. 2013, 86, 317–328. [Google Scholar] [CrossRef]
- Rasheduzzaman, M.; Jeong, J.K.; Park, S.Y. Resveratrol sensitizes lung cancer cell to TRAIL by p53 independent and suppression of Akt/NF-κB signaling. Life Sci. 2018, 208, 208–220. [Google Scholar] [CrossRef]
- Wu, Q.J.; Zhang, T.N.; Chen, H.H.; Yu, X.F.; Lv, J.L.; Liu, Y.Y.; Liu, Y.S.; Zheng, G.; Zhao, J.Q.; Wei, Y.F.; et al. The sirtuin family in health and disease. Signal. Transduct Target. Ther. 2022, 7, 402. [Google Scholar] [CrossRef]
- Yang, Y.P.; Hu, L.F.; Zheng, H.F.; Mao, C.J.; Hu, W.D.; Xiong, K.P.; Wang, F.; Liu, C.F. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol. Sin. 2013, 34, 625–635. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Lin, C.J.; Lee, C.C.; Shih, Y.L.; Lin, T.Y.; Wang, S.H.; Lin, Y.F.; Shih, C.M. Resveratrol enhances the therapeutic effect of temozolomide against malignant glioma in vitro and in vivo by inhibiting autophagy. Free Radic. Biol. Med. 2012, 52, 377–391. [Google Scholar] [CrossRef]
- Sipos, D.; Raposa, B.L.; Freihat, O.; Simon, M.; Mekis, N.; Cornacchione, P.; Kovács, Á. Glioblastoma: Clinical Presentation, Multidisciplinary Management, and Long-Term Outcomes. Cancers 2025, 17, 146. [Google Scholar] [CrossRef]
- Fan, Y.; Chen, M.; Meng, J.; Yu, L.; Tu, Y.; Wan, L.; Fang, K.; Zhu, W. Arsenic trioxide and resveratrol show synergistic anti-leukemia activity and neutralized cardiotoxicity. PLoS ONE 2014, 9, e105890. [Google Scholar] [CrossRef]
- Rosiak, N.; Tykarska, E.; Cielecka-Piontek, J. Enhanced Antioxidant and Neuroprotective Properties of Pterostilbene (Resveratrol Derivative) in Amorphous Solid Dispersions. Int. J. Mol. Sci. 2024, 25, 2774. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, H.; Ji, S.; Jia, P.; Li, Y.; Wang, T. Resveratrol and its derivative pterostilbene attenuate oxidative stress-induced intestinal injury by improving mitochondrial redox homeostasis and function via SIRT1 signaling. Free Radic. Biol. Med. 2021, 177, 1–14. [Google Scholar] [CrossRef]
- Shih, Y.H.; Tsai, P.J.; Chen, Y.L.; Pranata, R.; Chen, R.J. Assessment of the Antibacterial Mechanism of Pterostilbene against Bacillus cereus through Apoptosis-like Cell Death and Evaluation of Its Beneficial Effects on the Gut Microbiota. J. Agric. Food Chem. 2021, 69, 12219–12229. [Google Scholar] [CrossRef]
- Wasilewicz, A.; Zwirchmayr, J.; Kirchweger, B.; Bojkova, D.; Cinatl, J.; Rabenau, H.F.; Rollinger, J.M.; Beniddir, M.A.; Grienke, U. Discovery of anti-SARS-CoV-2 secondary metabolites from the heartwood of Pterocarpus santalinus using multi-informative molecular networking. Front. Mol. Biosci. 2023, 10, 1202394. [Google Scholar] [CrossRef]
- Patil, R.; Telang, G.; Aswar, U.; Vyas, N. Comparative analyses of anti-inflammatory effects of Resveratrol, Pterostilbene and Curcumin: In-silico and in-vitro evidences. Silico Pharmacol. 2024, 12, 38. [Google Scholar] [CrossRef]
- Li, S.; Wang, H.; Zhou, Y. JAK2/STAT3 pathway mediates beneficial effects of pterostilbene on cardiac contractile and electrical function in the setting of myocardial reperfusion injury. Physiol. Res. 2022, 71, 489–499. [Google Scholar] [CrossRef]
- Zhao, X.; Shi, A.; Ma, Q.; Yan, X.; Bian, L.; Zhang, P.; Wu, J. Nanoparticles prepared from pterostilbene reduce blood glucose and improve diabetes complications. J. Nanobiotechnology 2021, 19, 191. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, T.; Chen, X.; Cheng, J.; Wang, L. Pterostilbene regulates cell proliferation and apoptosis in non-small-cell lung cancer via targeting COX-2. Biotechnol. Appl. Biochem. 2023, 70, 106–119. [Google Scholar] [CrossRef]
- Pan, M.H.; Chang, Y.H.; Badmaev, V.; Nagabhushanam, K.; Ho, C.T. Pterostilbene induces apoptosis and cell cycle arrest in human gastric carcinoma cells. J. Agric. Food. Chem. 2007, 55, 7777–7785. [Google Scholar] [CrossRef]
- Khalil, M.I.; Agamy, A.F.; Elshewemi, S.S.; Sultan, A.S.; Abdelmeguid, N.E. Pterostilbene induces apoptosis in hepatocellular carcinoma cells: Biochemical, pathological, and molecular markers. Saudi. J. Biol. Sci. 2023, 30, 103717. [Google Scholar] [CrossRef]
- Mannal, P.W.; Alosi, J.A.; Schneider, J.G.; McDonald, D.E.; McFadden, D.W. Pterostilbene inhibits pancreatic cancer in vitro. J. Gastrointest. Surg. 2010, 14, 873–879. [Google Scholar] [CrossRef]
- Nutakul, W.; Sobers, H.S.; Qiu, P.; Dong, P.; Decker, E.A.; McClements, D.J.; Xiao, H. Inhibitory effects of resveratrol and pterostilbene on human colon cancer cells: A side-by-side comparison. J. Agric. Food Chem. 2011, 59, 10964–10970. [Google Scholar] [CrossRef]
- Chakraborty, A.; Gupta, N.; Ghosh, K.; Roy, P. In vitro evaluation of the cytotoxic, anti-proliferative and anti-oxidant properties of pterostilbene isolated from Pterocarpus marsupium. Toxicol. Vitr. 2010, 24, 1215–1228. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Wu, Y.; Lv, C.; Li, X.; Cao, X.; Yang, M.; Feng, D.; Luo, Z. Pterostilbene exerts antitumor activity against human osteosarcoma cells by inhibiting the JAK2/STAT3 signaling pathway. Toxicology 2013, 304, 120–131. [Google Scholar] [CrossRef]
- Siedlecka-Kroplewska, K.; Jozwik, A.; Kaszubowska, L.; Kowalczyk, A.; Boguslawski, W. Pterostilbene induces cell cycle arrest and apoptosis in MOLT4 human leukemia cells. Folia. Histochem. Cytobiol. 2012, 50, 574–580. [Google Scholar] [CrossRef]
- Hsieh, M.J.; Lin, C.W.; Yang, S.F.; Sheu, G.T.; Yu, Y.Y.; Chen, M.K.; Chiou, H.L. A combination of pterostilbene with autophagy inhibitors exerts efficient apoptotic characteristics in both chemosensitive and chemoresistant lung cancer cells. Toxicol. Sci. 2014, 137, 65–75. [Google Scholar] [CrossRef]
- Ko, C.P.; Lin, C.W.; Chen, M.K.; Yang, S.F.; Chiou, H.L.; Hsieh, M.J. Pterostilbene induce autophagy on human oral cancer cells through modulation of Akt and mitogen-activated protein kinase pathway. Oral. Oncol. 2015, 51, 593–601. [Google Scholar] [CrossRef]
- Chang, H.P.; Lu, C.C.; Chiang, J.H.; Tsai, F.J.; Juan, Y.N.; Tsao, J.W.; Chiu, H.Y.; Yang, J.S. Pterostilbene modulates the suppression of multidrug resistance protein1 and triggers autophagic and apoptotic mechanisms in cisplatin-resistant human oral cancer CAR cells via AKT signaling. Int. J. Oncol. 2018, 52, 1504–1514. [Google Scholar] [CrossRef]
- Wawszczyk, J.; Jesse, K.; Smolik, S.; Kapral, M. Mechanism of Pterostilbene-Induced Cell Death in HT-29 Colon Cancer Cells. Molecules 2022, 27, 369. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.J.; Lyu, Y.J.; Chen, Y.Y.; Lee, Y.C.; Pan, M.H.; Ho, Y.S.; Wang, Y.J. Chloroquine Potentiates the Anticancer Effect of Pterostilbene on Pancreatic Cancer by Inhibiting Autophagy and Downregulating the RAGE/STAT3 Pathway. Molecules 2021, 26, 6741. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, L.; Wang, X.; Zhang, J.; Han, W.; Feng, L.; Sun, J.; Jin, H.; Wang, X.J. Pterostilbene simultaneously induces apoptosis, cell cycle arrest and cyto-protective autophagy in breast cancer cells. Am. J. Transl. Res. 2012, 4, 44–51. [Google Scholar] [PubMed]
- Chen, R.J.; Ho, C.T.; Wang, Y.J. Pterostilbene induces autophagy and apoptosis in sensitive and chemoresistant human bladder cancer cells. Mol. Nutr. Food Res. 2010, 54, 1819–1832. [Google Scholar] [CrossRef]
- Siedlecka-Kroplewska, K.; Jozwik, A.; Boguslawski, W.; Wozniak, M.; Zauszkiewicz-Pawlak, A.; Spodnik, J.H.; Rychlowski, M.; Kmiec, Z. Pterostilbene induces accumulation of autophagic vacuoles followed by cell death in HL60 human leukemia cells. J. Physiol. Pharmacol. 2013, 64, 545–556. [Google Scholar]
- He, P.; Li, Y.; Hu, J.; Deng, B.; Tan, Z.; Chen, Y.; Yu, B.; Dong, W. Pterostilbene suppresses gastric cancer proliferation and metastasis by inhibiting oncogenic JAK2/STAT3 signaling: In vitro and in vivo therapeutic intervention. Phytomedicine 2024, 128, 155316. [Google Scholar] [CrossRef]
- Wang, D.; Guo, H.; Yang, H.; Gao, P.; Wei, W. Pterostilbene, An Active Constituent of Blueberries, Suppresses Proliferation Potential of Human Cholangiocarcinoma via Enhancing the Autophagic Flux. Front. Pharmacol. 2019, 10, 1238. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.; Kumar, A.; Rimando, A.M.; Zhang, X.; Levenson, A.S. Resveratrol and pterostilbene epigenetically restore PTEN expression by targeting oncomiRs of the miR-17 family in prostate cancer. Oncotarget 2015, 6, 27214–27226. [Google Scholar] [CrossRef]
- Feng, Y.; Yang, Y.; Fan, C.; Di, S.; Hu, W.; Jiang, S.; Li, T.; Ma, Z.; Chao, D.; Feng, X.; et al. Pterostilbene Inhibits the Growth of Human Esophageal Cancer Cells by Regulating Endoplasmic Reticulum Stress. Cell Physiol. Biochem. 2016, 38, 1226–1244. [Google Scholar] [CrossRef]
- Kapetanovic, I.M.; Muzzio, M.; Huang, Z.; Thompson, T.N.; McCormick, D.L. Pharmacokinetics, oral bioavailability, and metabolic profile of resveratrol and its dimethylether analog, pterostilbene, in rats. Cancer Chemother Pharmacol 2011, 68, 593–601. [Google Scholar] [CrossRef]
- Bastianetto, S.; Dumont, Y.; Han, Y.; Quirion, R. Comparative neuroprotective properties of stilbene and catechin analogs: Action via a plasma membrane receptor site? CNS Neurosci. Ther. 2009, 15, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Docherty, J.J.; McEwen, H.A.; Sweet, T.J.; Bailey, E.; Booth, T.D. Resveratrol inhibition of Propionibacterium acnes. J. Antimicrob. Chemother. 2007, 59, 1182–1184. [Google Scholar] [CrossRef]
- Hung, L.M.; Chen, J.K.; Lee, R.S.; Liang, H.C.; Su, M.J. Beneficial effects of astringinin, a resveratrol analogue, on the ischemia and reperfusion damage in rat heart. Free Radic. Biol. Med. 2001, 30, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, J.; Kundu, J.K.; Surh, Y.J. Piceatannol inhibits phorbol ester-induced expression of COX-2 and iNOS in HR-1 hairless mouse skin by blocking the activation of NF-κB and AP-1. Inflamm. Res. 2014, 63, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Maggiolini, M.; Recchia, A.G.; Bonofiglio, D.; Catalano, S.; Vivacqua, A.; Carpino, A.; Rago, V.; Rossi, R.; Andò, S. The red wine phenolics piceatannol and myricetin act as agonists for estrogen receptor alpha in human breast cancer cells. J. Mol. Endocrinol. 2005, 35, 269–281. [Google Scholar] [CrossRef]
- Minakawa, M.; Miura, Y.; Yagasaki, K. Piceatannol, a resveratrol derivative, promotes glucose uptake through glucose transporter 4 translocation to plasma membrane in L6 myocytes and suppresses blood glucose levels in type 2 diabetic model db/db mice. Biochem. Biophys. Res. Commun. 2012, 422, 469–475. [Google Scholar] [CrossRef]
- Khan, I.; Preeti, K.; Kumar, R.; Kumar Khatri, D.; Bala Singh, S. Piceatannol promotes neuroprotection by inducing mitophagy and mitobiogenesis in the experimental diabetic peripheral neuropathy and hyperglycemia-induced neurotoxicity. Int. Immunopharmacol. 2023, 116, 109793. [Google Scholar] [CrossRef]
- Çınar Ayan, İ.; Güçlü, E.; Vural, H.; Dursun, H.G. Piceatannol induces apoptotic cell death through activation of caspase-dependent pathway and upregulation of ROS-mediated mitochondrial dysfunction in pancreatic cancer cells. Mol. Biol. Rep. 2022, 49, 11947–11957. [Google Scholar] [CrossRef]
- Chowdhury, S.A.; Kishino, K.; Satoh, R.; Hashimoto, K.; Kikuchi, H.; Nishikawa, H.; Shirataki, Y.; Sakagami, H. Tumor-specificity and apoptosis-inducing activity of stilbenes and flavonoids. Anticancer. Res. 2005, 25, 2055–2063. [Google Scholar]
- Hsieh, T.C.; Lin, C.Y.; Lin, H.Y.; Wu, J.M. AKT/mTOR as Novel Targets of Polyphenol Piceatannol Possibly Contributing to Inhibition of Proliferation of Cultured Prostate Cancer Cells. ISRN Urol. 2012, 2012, 272697. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.H.; Moon, D.O.; Choi, Y.H.; Choi, I.W.; Moon, S.K.; Kim, W.J.; Kim, G.Y. Piceatannol enhances TRAIL-induced apoptosis in human leukemia THP-1 cells through Sp1- and ERK-dependent DR5 up-regulation. Toxicol. Vitro 2011, 25, 605–612. [Google Scholar] [CrossRef]
- Kim, Y.H.; Park, C.; Lee, J.O.; Kim, G.Y.; Lee, W.H.; Choi, Y.H.; Ryu, C.H. Induction of apoptosis by piceatannol in human leukemic U937 cells through down-regulation of Bcl-2 and activation of caspases. Oncol. Rep. 2008, 19, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Tolomeo, M.; Grimaudo, S.; Di Cristina, A.; Roberti, M.; Pizzirani, D.; Meli, M.; Dusonchet, L.; Gebbia, N.; Abbadessa, V.; Crosta, L.; et al. Pterostilbene and 3’-hydroxypterostilbene are effective apoptosis-inducing agents in MDR and BCR-ABL-expressing leukemia cells. Int. J. Biochem. Cell Biol. 2005, 37, 1709–1726. [Google Scholar] [CrossRef]
- Kuo, P.L.; Hsu, Y.L. The grape and wine constituent piceatannol inhibits proliferation of human bladder cancer cells via blocking cell cycle progression and inducing Fas/membrane bound Fas ligand-mediated apoptotic pathway. Mol. Nutr. Food Res. 2008, 52, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Larrosa, M.; Tomás-Barberán, F.A.; Espín, J.C. The grape and wine polyphenol piceatannol is a potent inducer of apoptosis in human SK-Mel-28 melanoma cells. Eur. J. Nutr. 2004, 43, 275–284. [Google Scholar] [CrossRef]
- Lee, Y.M.; Lim, D.Y.; Cho, H.J.; Seon, M.R.; Kim, J.K.; Lee, B.Y.; Park, J.H. Piceatannol, a natural stilbene from grapes, induces G1 cell cycle arrest in androgen-insensitive DU145 human prostate cancer cells via the inhibition of CDK activity. Cancer Lett. 2009, 285, 166–173. [Google Scholar] [CrossRef]
- Liu, W.H.; Chang, L.S. Piceatannol induces Fas and FasL up-regulation in human leukemia U937 cells via Ca2+/p38alpha MAPK-mediated activation of c-Jun and ATF-2 pathways. Int. J. Biochem. Cell Biol. 2010, 42, 1498–1506. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.; Haza, A.I. Selective apoptotic effects of piceatannol and myricetin in human cancer cells. J. Appl. Toxicol. 2012, 32, 986–993. [Google Scholar] [CrossRef]
- Vo, N.T.; Madlener, S.; Bago-Horvath, Z.; Herbacek, I.; Stark, N.; Gridling, M.; Probst, P.; Giessrigl, B.; Bauer, S.; Vonach, C.; et al. Pro- and anticarcinogenic mechanisms of piceatannol are activated dose dependently in MCF-7 breast cancer cells. Carcinogenesis 2010, 31, 2074–2081. [Google Scholar] [CrossRef]
- Wieder, T.; Prokop, A.; Bagci, B.; Essmann, F.; Bernicke, D.; Schulze-Osthoff, K.; Dörken, B.; Schmalz, H.G.; Daniel, P.T.; Henze, G. Piceatannol, a hydroxylated analog of the chemopreventive agent resveratrol, is a potent inducer of apoptosis in the lymphoma cell line BJAB and in primary, leukemic lymphoblasts. Leukemia 2001, 15, 1735–1742. [Google Scholar] [CrossRef]
- Wolter, F.; Clausnitzer, A.; Akoglu, B.; Stein, J. Piceatannol, a natural analog of resveratrol, inhibits progression through the S phase of the cell cycle in colorectal cancer cell lines. J. Nutr. 2002, 132, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jia, R.; Wang, C.; Hu, T.; Wang, F. Piceatannol promotes apoptosis via up-regulation of microRNA-129 expression in colorectal cancer cell lines. Biochem. Biophys. Res. Commun. 2014, 452, 775–781. [Google Scholar] [CrossRef]
- Zheng, M.; Wu, Y. Piceatannol suppresses proliferation and induces apoptosis by regulation of the microRNA-21/phosphatase and tensin homolog/protein kinase B signaling pathway in osteosarcoma cells. Mol. Med. Rep. 2020, 22, 3985–3993. [Google Scholar] [CrossRef] [PubMed]
- Güçlü, E.; Ayan, İ.; Çetinkaya, S.; Dursun, H.G.; Vural, H. Piceatannol induces caspase-dependent apoptosis by modulating intracellular reactive oxygen species/mitochondrial membrane potential and enhances autophagy in neuroblastoma cells. J. Appl. Toxicol. 2024, 44, 1714–1724. [Google Scholar] [CrossRef]
- Siedlecka-Kroplewska, K.; Ślebioda, T.; Kmieć, Z. Induction of autophagy, apoptosis and aquisition of resistance in response to piceatannol toxicity in MOLT-4 human leukemia cells. Toxicol. Vitr. 2019, 59, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Jiu, X.; Li, W.; Liu, Y.; Liu, L.; Lu, H. TREM2, a critical activator of pyroptosis, mediates the anti-tumor effects of piceatannol in uveal melanoma cells via caspase 3/GSDME pathway. Int. J. Mol. Med. 2024, 54, 96. [Google Scholar] [CrossRef]
- Khamis, T.; Diab, A.A.A.; Zahra, M.H.; El-Dahmy, S.E.; Abd Al-Hameed, B.A.; Abdelkhalek, A.; Said, M.A.; Abdellatif, H.; Fericean, L.M.; Banatean-Dunea, I.; et al. The Antiproliferative Activity of Adiantum pedatum Extract and/or Piceatannol in Phenylhydrazine-Induced Colon Cancer in Male Albino Rats: The miR-145 Expression of the PI-3K/Akt/p53 and Oct/Sox2/Nanog Pathway. Molecules 2023, 28, 5543. [Google Scholar] [CrossRef]
- Yoshizawa, K.; Tabuchi, M.; Ukai, S.; Suzuki, H.; Kawano, Y.; Takasawa, R. The Putative Glyoxalase 1 Inhibitor Piceatannol Exhibits Both Anxiolytic-like and Antitumor Effects in Mice. Anticancer. Res. 2020, 40, 3271–3276. [Google Scholar] [CrossRef]
- Potter, G.A.; Patterson, L.H.; Wanogho, E.; Perry, P.J.; Butler, P.C.; Ijaz, T.; Ruparelia, K.C.; Lamb, J.H.; Farmer, P.B.; Stanley, L.A.; et al. The cancer preventative agent resveratrol is converted to the anticancer agent piceatannol by the cytochrome P450 enzyme CYP1B1. Br. J. Cancer 2002, 86, 774–778. [Google Scholar] [CrossRef]
- Siedlecka-Kroplewska, K.; Wrońska, A.; Kmieć, Z. Piceatannol, a Structural Analog of Resveratrol, Is an Apoptosis Inducer and a Multidrug Resistance Modulator in HL-60 Human Acute Myeloid Leukemia Cells. Int. J. Mol. Sci. 2021, 22, 10597. [Google Scholar] [CrossRef]
- Likhitwitayawuid, K. Oxyresveratrol: Sources, Productions, Biological Activities, Pharmacokinetics, and Delivery Systems. Molecules 2021, 26, 4212. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Liu, C.; Chen, R. Phenolic constituents from stem bark of Morus wittiorum and their anti-inflammation and cytotoxicity. Zhongguo Zhong Yao Za Zhi 2010, 35, 2700–2703. [Google Scholar] [PubMed]
- Sintuyanon, N.; Phoolcharoen, W.; Pavasant, P.; Sooampon, S. Resveratrol demonstrated higher antiproliferative and antiangiogenic efficacy compared with oxyresveratrol on head and neck squamous cell carcinoma cell lines. Nat. Prod. Commun. 2017, 12, 1781–1784. [Google Scholar] [CrossRef]
- Lv, T.; Jian, Z.; Li, D.; Ao, R.; Zhang, X.; Yu, B. Oxyresveratrol induces apoptosis and inhibits cell viability via inhibition of the STAT3 signaling pathway in Saos-2 cells. Mol. Med. Rep. 2020, 22, 5191–5198. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, G.; Li, C.; Wang, S.; Zhu, M.; Wang, J.; Yue, H.; Ma, X.; Zhen, Y.; Shu, X. Metabolic profile and structure-activity relationship of resveratrol and its analogs in human bladder cancer cells. Cancer Manag. Res. 2019, 11, 4631–4642. [Google Scholar] [CrossRef]
- Radapong, S.; Chan, K.; Sarker, S.D.; Ritchie, K.J. Oxyresveratrol Modulates Genes Associated with Apoptosis, Cell Cycle Control and DNA Repair in MCF-7 Cells. Front. Pharmacol. 2021, 12, 694562. [Google Scholar] [CrossRef]
- Lee, S.G.; Lee, D.G.; Joo, Y.H.; Chung, N. Synergistic inhibitory effects of the oxyresveratrol and dacarbazine combination against melanoma cells. Oncol. Lett. 2021, 22, 667. [Google Scholar] [CrossRef]
- Sunilkumar, D.; Drishya, G.; Chandrasekharan, A.; Shaji, S.K.; Bose, C.; Jossart, J.; Perry, J.J.P.; Mishra, N.; Kumar, G.B.; Nair, B.G. Oxyresveratrol drives caspase-independent apoptosis-like cell death in MDA-MB-231 breast cancer cells through the induction of ROS. Biochem. Pharmacol. 2020, 173, 113724. [Google Scholar] [CrossRef]
- Lin, T.A.; Lin, W.S.; Chou, Y.C.; Nagabhushanam, K.; Ho, C.T.; Pan, M.H. Oxyresveratrol inhibits human colon cancer cell migration through regulating epithelial-mesenchymal transition and microRNA. Food Funct. 2021, 12, 9658–9668. [Google Scholar] [CrossRef]
- Liu, Y.; Ren, W.; Bai, Y.; Wan, L.; Sun, X.; Xiong, W.; Zhang, Y.Y.; Zhou, L. Oxyresveratrol prevents murine H22 hepatocellular carcinoma growth and lymph node metastasis via inhibiting tumor angiogenesis and lymphangiogenesis. J. Nat. Med. 2018, 72, 481–492. [Google Scholar] [CrossRef]
- Rahman, M.A.; Bishayee, K.; Sadra, A.; Huh, S.O. Oxyresveratrol activates parallel apoptotic and autophagic cell death pathways in neuroblastoma cells. Biochim. Biophys. Acta Gen Subj 2017, 1861, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Koskela, A.; Reinisalo, M.; Hyttinen, J.M.; Kaarniranta, K.; Karjalainen, R.O. Pinosylvin-mediated protection against oxidative stress in human retinal pigment epithelial cells. Mol. Vis. 2014, 20, 760–769. [Google Scholar] [PubMed]
- Lee, S.K.; Lee, H.J.; Min, H.Y.; Park, E.J.; Lee, K.M.; Ahn, Y.H.; Cho, Y.J.; Pyee, J.H. Antibacterial and antifungal activity of pinosylvin, a constituent of pine. Fitoterapia 2005, 76, 258–260. [Google Scholar] [CrossRef]
- Chen, M.K.; Liu, Y.T.; Lin, J.T.; Lin, C.C.; Chuang, Y.C.; Lo, Y.S.; Hsi, Y.T.; Hsieh, M.J. Pinosylvin reduced migration and invasion of oral cancer carcinoma by regulating matrix metalloproteinase-2 expression and extracellular signal-regulated kinase pathway. Biomed. Pharmacother. 2019, 117, 109160. [Google Scholar] [CrossRef]
- González-Sarrías, A.; Espín-Aguilar, J.C.; Romero-Reyes, S.; Puigcerver, J.; Alajarín, M.; Berná, J.; Selma, M.V.; Espín, J.C. Main Determinants Affecting the Antiproliferative Activity of Stilbenes and Their Gut Microbiota Metabolites in Colon Cancer Cells: A Structure-Activity Relationship Study. Int. J. Mol. Sci. 2022, 23, 15102. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Chung, H.J.; Park, H.J.; Kim, G.D.; Ahn, Y.H.; Lee, S.K. Suppression of Src/ERK and GSK-3/β-catenin signaling by pinosylvin inhibits the growth of human colorectal cancer cells. Food Chem. Toxicol. 2013, 55, 424–433. [Google Scholar] [CrossRef]
- Yatkin, E.; Polari, L.; Laajala, T.D.; Smeds, A.; Eckerman, C.; Holmbom, B.; Saarinen, N.M.; Aittokallio, T.; Mäkelä, S.I. Novel Lignan and stilbenoid mixture shows anticarcinogenic efficacy in preclinical PC-3M-luc2 prostate cancer model. PLoS ONE 2014, 9, e93764. [Google Scholar] [CrossRef]
- Song, J.; Seo, Y.; Park, H. Pinosylvin enhances leukemia cell death via down-regulation of AMPKα expression. Phytother Res. 2018, 32, 2097–2104. [Google Scholar] [CrossRef]
- Karatoprak, G.; Küpeli Akkol, E.; Genç, Y.; Bardakci, H.; Yücel, Ç.; Sobarzo-Sánchez, E. Combretastatins: An Overview of Structure, Probable Mechanisms of Action and Potential Applications. Molecules 2020, 25, 2560. [Google Scholar] [CrossRef]
- Dark, G.G.; Hill, S.A.; Prise, V.E.; Tozer, G.M.; Pettit, G.R.; Chaplin, D.J. Combretastatin A-4, an agent that displays potent and selective toxicity toward tumor vasculature. Cancer Res. 1997, 57, 1829–1834. [Google Scholar]
- Wang, H.; Li, W.; Xu, J.; Zhang, T.; Zuo, D.; Zhou, Z.; Lin, B.; Wang, G.; Wang, Z.; Sun, W.; et al. NDRG1 inhibition sensitizes osteosarcoma cells to combretastatin A-4 through targeting autophagy. Cell Death Dis. 2017, 8, e3048. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Kwah, M.X.; Liu, C.; Ma, Z.; Shanmugam, M.K.; Ding, L.; Xiang, X.; Ho, P.C.; Wang, L.; Ong, P.S.; et al. Resveratrol for cancer therapy: Challenges and future perspectives. Cancer Lett. 2021, 515, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Howells, L.M.; Berry, D.P.; Elliott, P.J.; Jacobson, E.W.; Hoffmann, E.; Hegarty, B.; Brown, K.; Steward, W.P.; Gescher, A.J. Phase I randomized, double-blind pilot study of micronized resveratrol (SRT501) in patients with hepatic metastases--safety, pharmacokinetics, and pharmacodynamics. Cancer Prev. Res. 2011, 4, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Tian, W.; Ning, J.; Xiao, G.; Zhou, Y.; Wang, Z.; Zhai, Z.; Tanzhu, G.; Yang, J.; Zhou, R. Cancer stem cells: Advances in knowledge and implications for cancer therapy. Signal. Transduct. Target. Ther. 2024, 9, 170. [Google Scholar] [CrossRef]
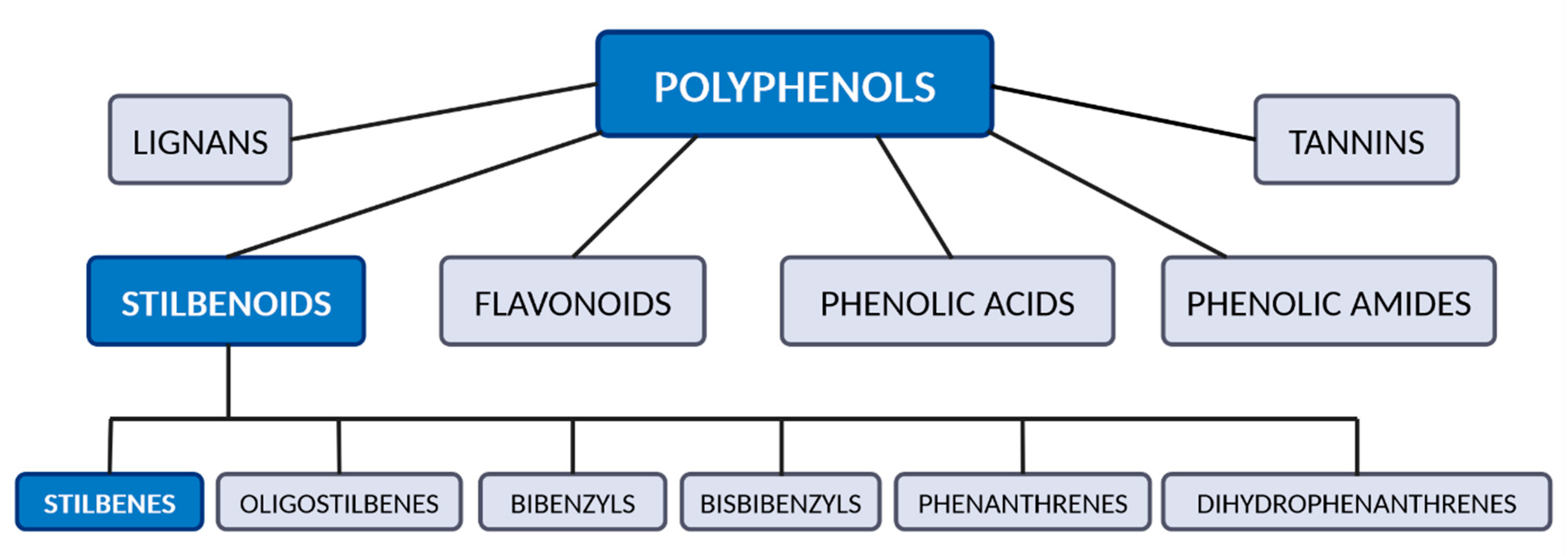
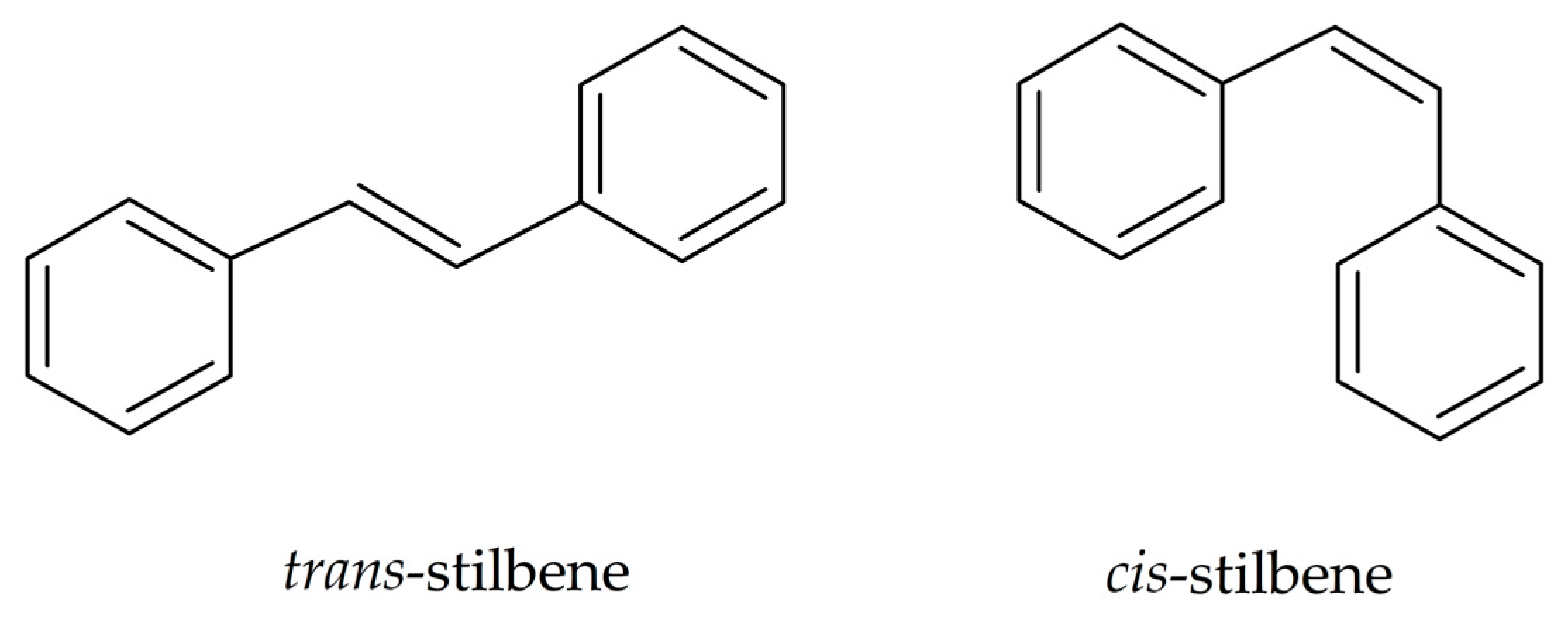

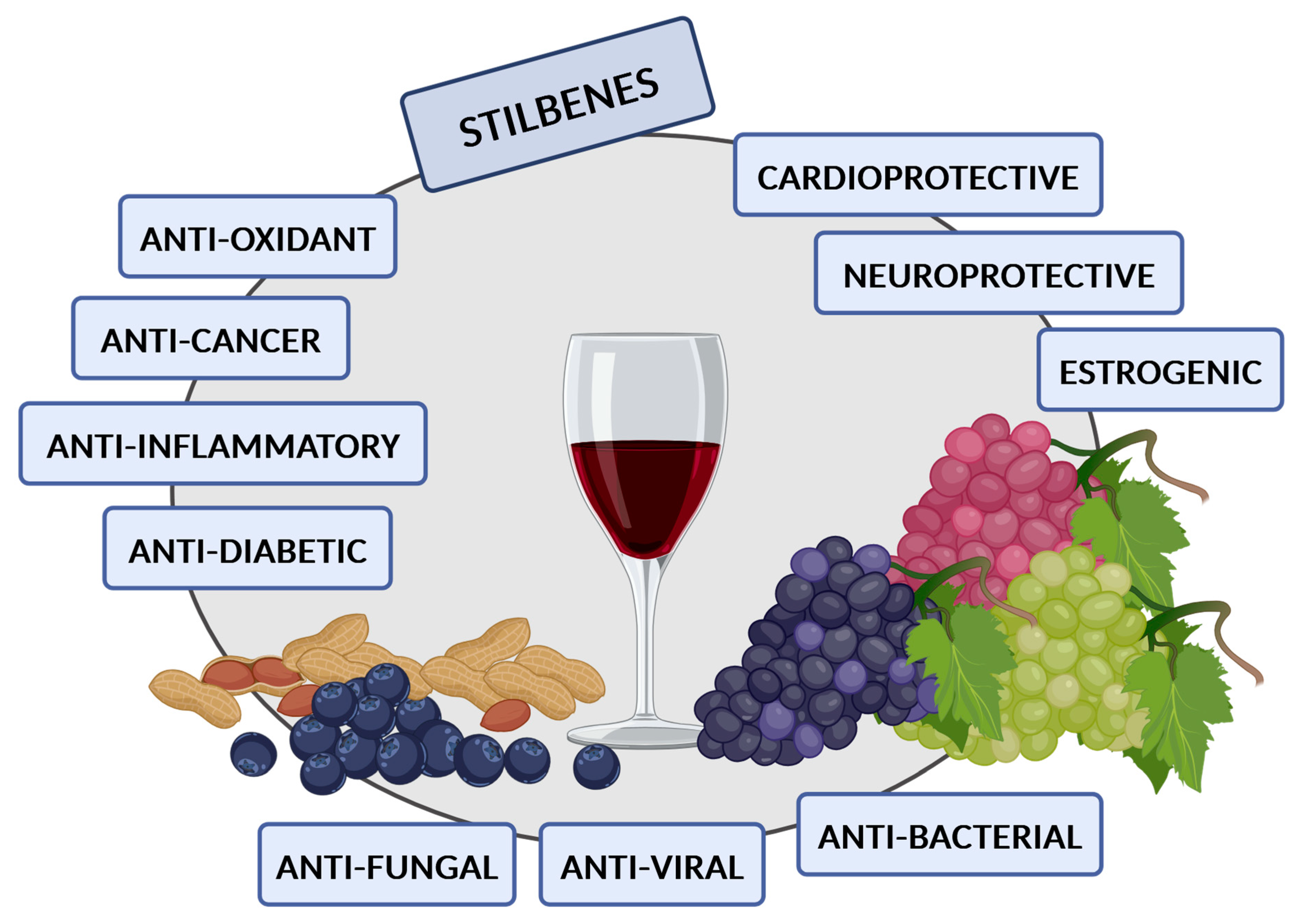
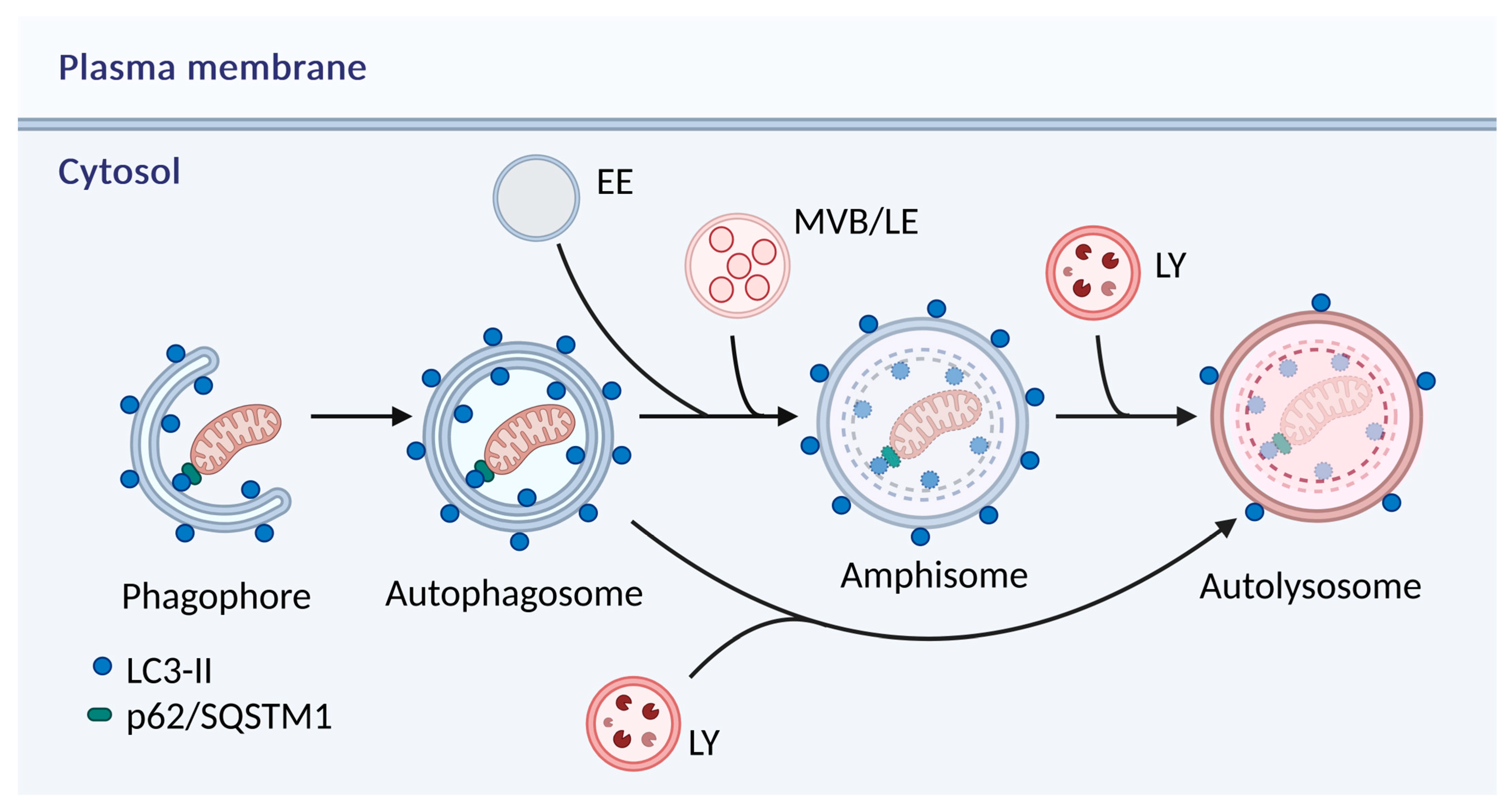
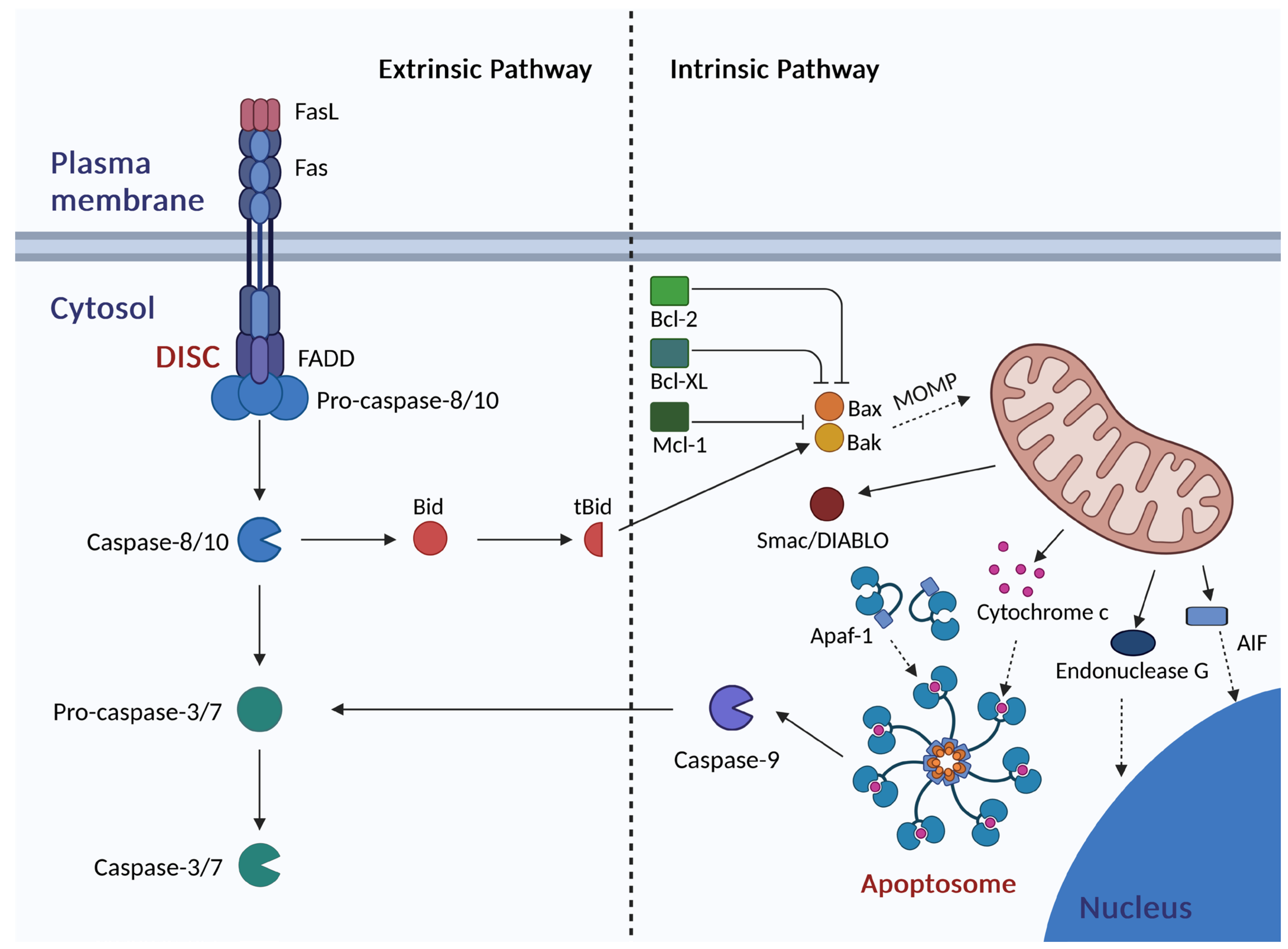
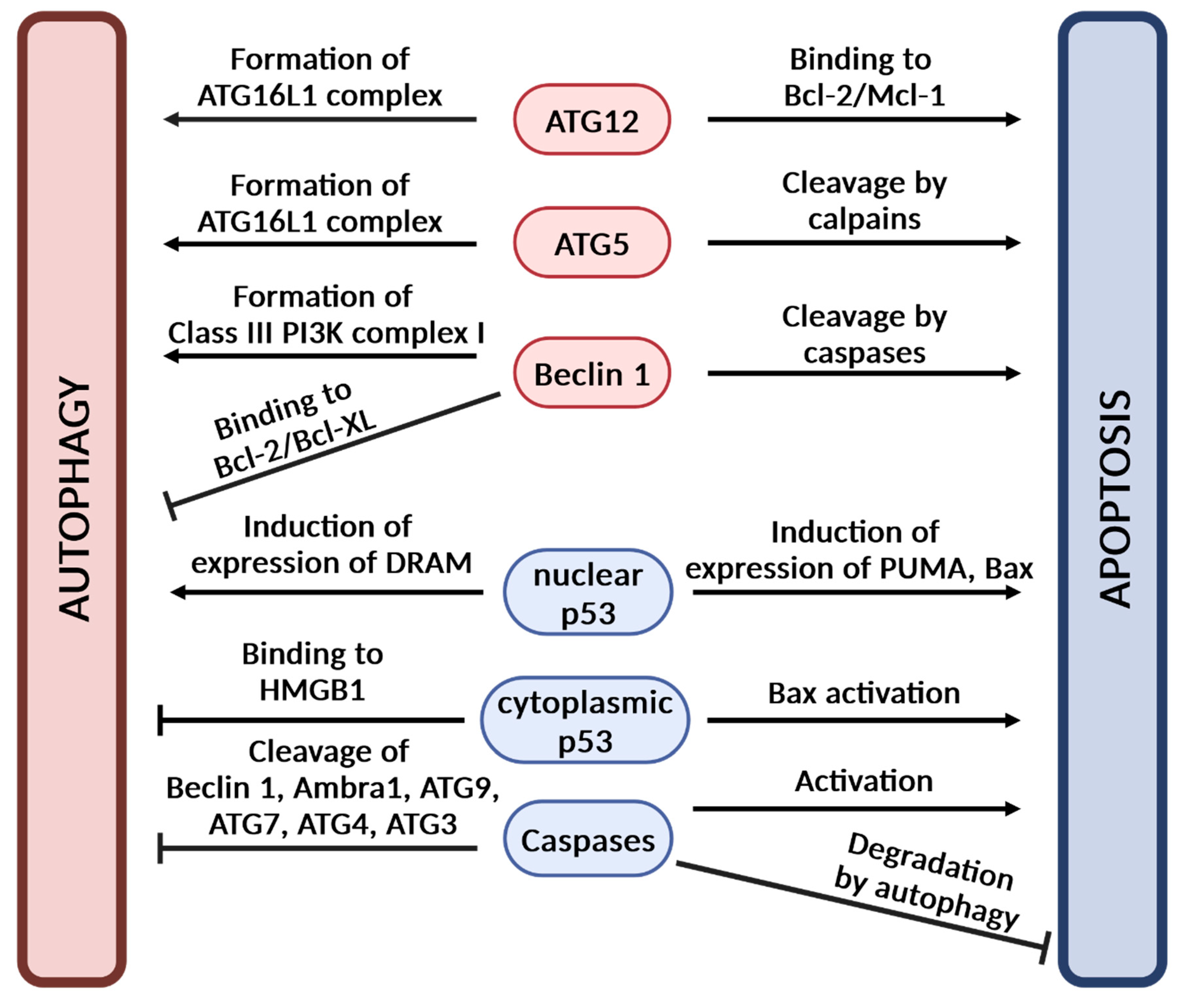
| Stilbenes | Plants/Plant-Derived Dietary Products |
|---|---|
| RES | Grape [8,9,10,11,12], Grape cane [13,14], Grape juice [10,15,16] Red wine [9,16,17,18,19,20,21,22,23,24,25], White wine [9,18,19,20,21,23,25], Rose wine [9,19,21,23] Beer [26] Apple juice [10], Peach juice [10] Lowbush blueberry [8], Sparkleberry [8], Rabbiteye blueberry [8] Highbush blueberry [8], Elliott’s blueberry [8], Cranberry [8,16,27] Bilberry [8,27], Deerberry [8], Lingonberry [8] Partridgeberry [8], Cowberry [27], Mulberry [28] Strawberry [27], Red currant [27] Peanut [29,30], Peanut butter [29], Pistachio [30] Cocoa powder [31], Chocolate [31] Tomato [32] Green tea [10], Black tea [10], Red tea [10], Camomile [10] Rhubarb Rheum undulatum [33], Rhubarb Rheum palmatum [34] Eucalyptus [35,36] Spruce [37], Spruce Picea jezoensis [38], Spruce Picea abies [39] Polygonum cuspidatum [40,41] Gnetum gnemon [42], Gnetum klossii [43] Stuhlmannia moavi [44] |
| PTER | Grape [45,46], Grape cane [14] Rabbiteye blueberry [8], Deerberry [8] Peanut [47] Pterocarpus marsupium [48], Pterocarpus santalinus [49] Pterocarpus tinctorius [49] Guibourtia tessmanii [50] |
| PIC | Grape [9,10,11,12], Grape cane [13,14], Grape juice [10] Red wine [9,23,25], White wine [23,25], Rose wine [23] Apple juice [10], Peach juice [10] Highbush blueberry [8], Deerberry [8], Mulberry [51] Peanut [41] Passion fruit seeds [52] Green tea [10], Black tea [10], Red tea [10], Camomile [10] Rhubarb Rheum undulatum [33], Rhubarb Rheum palmatum [34] Rhubarb Rheum rhaponticum [53], Rhubarb Rheum rhabarbarum [53] Sugar cane [54] Spruce [37], Spruce Picea jezoensis [38], Spruce Picea abies [39] Eucalyptus [36] Polygonum cuspidatum [41] Stuhlmannia moavi [44] |
| OXYRES | Grape [55] Red wine [23], White wine [23], Rose wine [23] Mulberry [28,56] Artocarpus lakoocha [57] Pterocarpus marsupium [58] |
| GNE | Gnetum gnemon [42,59], Gnetum montanum [60], Gnetum klossii [43] Gnetum macrostachyum [61], Gnetum microcarpum [62] Gnetum hainanense [63] |
| PIN | Pinus sylvestris [64,65], Pinus densiflora [66], Pinus resinosa [67,68] Pinus banksiana [67,68], Pinus strobus [68], Picea glauca [67] Gnetum cleistostachyum [69] |
| RHAP | Rhubarb Rheum emodi [70], Rhubarb Rheum palmatum [34] Rhubarb Rheum undulatum [33], Rhubarb Rheum officinale [71] Rhubarb Rheum rhaponticum [53], Rhubarb Rheum rhabarbarum [53] Caesalpinia sinensis [72] Stuhlmannia moavi [44] Gnetum hainanense [63], Gnetum cleistostachyum [69] |
| ISORHAP | Grape [11], Grape cane [13] Spruce [37], Spruce Picea jezoensis [38], Spruce Picea abies [39] Gnetum macrostachyum [61], Gnetum cleistostachyum [69] Gnetum gnemon [42], Gnetum klossii [43], Gnetum hainanense [63] Rhubarb Rheum rhaponticum [53], Rhubarb Rheum rhabarbarum [53] Stuhlmannia moavi [44] Saccharum spontaneum (wild sugar cane) [73] |
| DEOXYRHAP | Rhubarb Rheum emodi [70], Rhubarb Rheum palmatum [34] Rhubarb Rheum undulatum [33], Rhubarb Rheum rhaponticum [53] Rhubarb Rheum rhabarbarum [53] Gnetum cleistostachyum [69] |
| CA-4 | Combretum caffrum [74], Combretum leprosum [75] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siedlecka-Kroplewska, K.; Kmiec, Z.; Zmijewski, M.A. The Interplay Between Autophagy and Apoptosis in the Mechanisms of Action of Stilbenes in Cancer Cells. Antioxidants 2025, 14, 339. https://doi.org/10.3390/antiox14030339
Siedlecka-Kroplewska K, Kmiec Z, Zmijewski MA. The Interplay Between Autophagy and Apoptosis in the Mechanisms of Action of Stilbenes in Cancer Cells. Antioxidants. 2025; 14(3):339. https://doi.org/10.3390/antiox14030339
Chicago/Turabian StyleSiedlecka-Kroplewska, Kamila, Zbigniew Kmiec, and Michal Aleksander Zmijewski. 2025. "The Interplay Between Autophagy and Apoptosis in the Mechanisms of Action of Stilbenes in Cancer Cells" Antioxidants 14, no. 3: 339. https://doi.org/10.3390/antiox14030339
APA StyleSiedlecka-Kroplewska, K., Kmiec, Z., & Zmijewski, M. A. (2025). The Interplay Between Autophagy and Apoptosis in the Mechanisms of Action of Stilbenes in Cancer Cells. Antioxidants, 14(3), 339. https://doi.org/10.3390/antiox14030339