
Discovery of Novel Tofacitinib–ADTOH Molecular Hybridization Derivatives for the Treatment of Ulcerative Colitis

 ,
,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
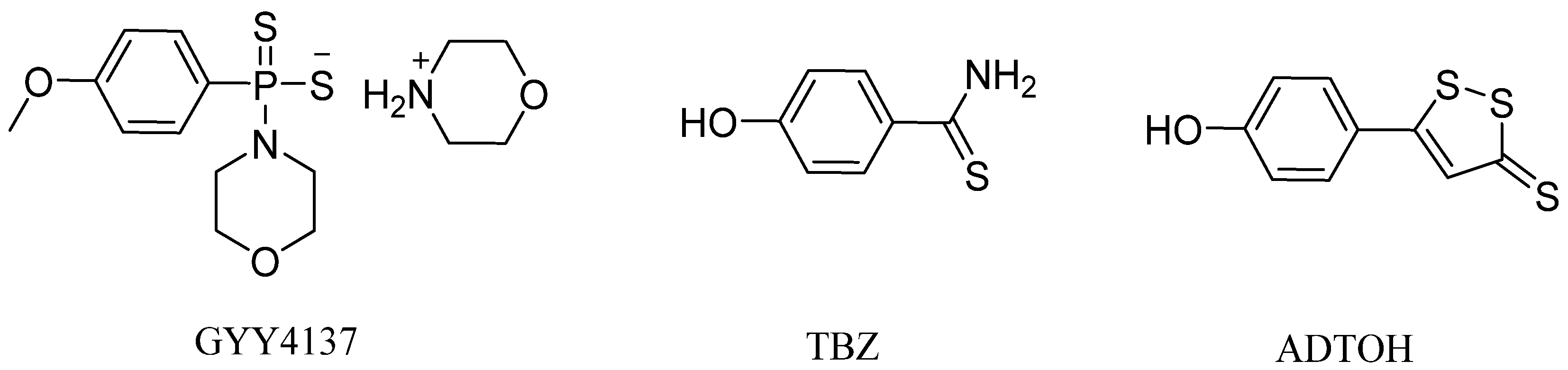
1. Introduction
2. Experimental Section
2.1. Chemistry Section
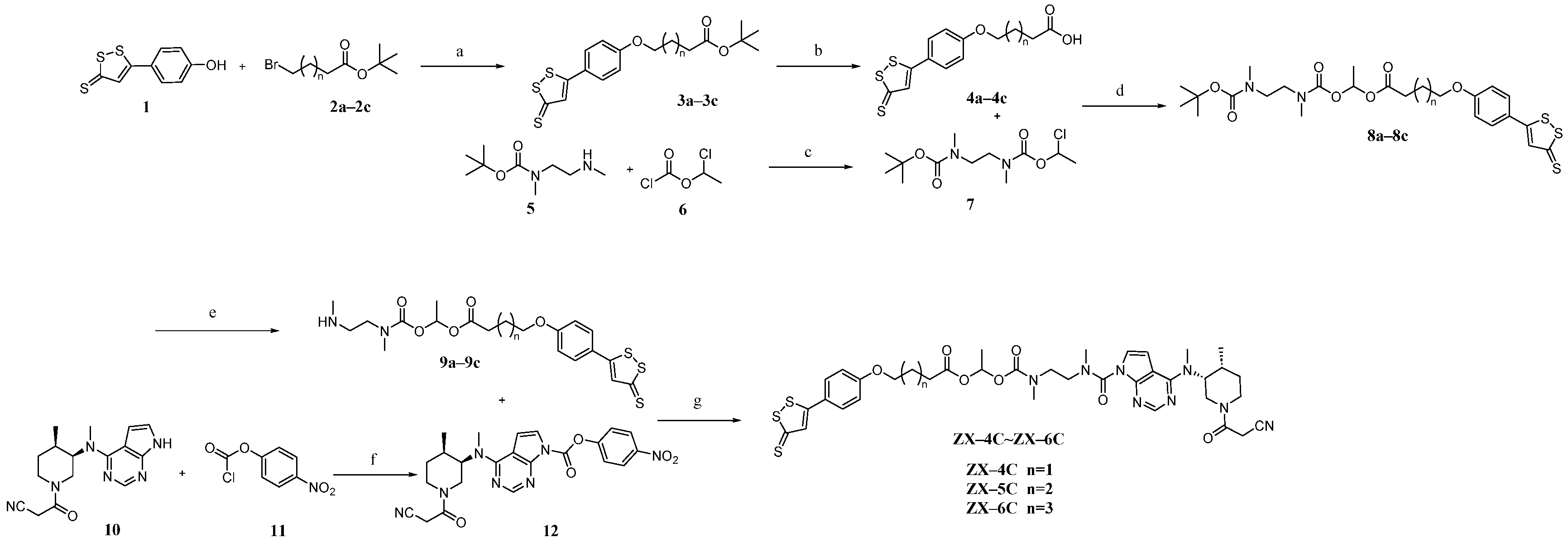
2.1.1. General Synthetic Procedure A for Intermediates 3a–c
2.1.2. General Synthetic Procedure B for Intermediates 4a–c
2.1.3. Synthetic Procedure for Intermediate 7
2.1.4. General Synthetic Procedure C for Intermediates 8a–c
2.1.5. General Synthetic Procedure D for Intermediates 9a–c
2.1.6. Synthetic Procedure for Intermediate 12
2.1.7. General Synthetic Procedure E for Target Compounds ZX-4C~ZX-6C
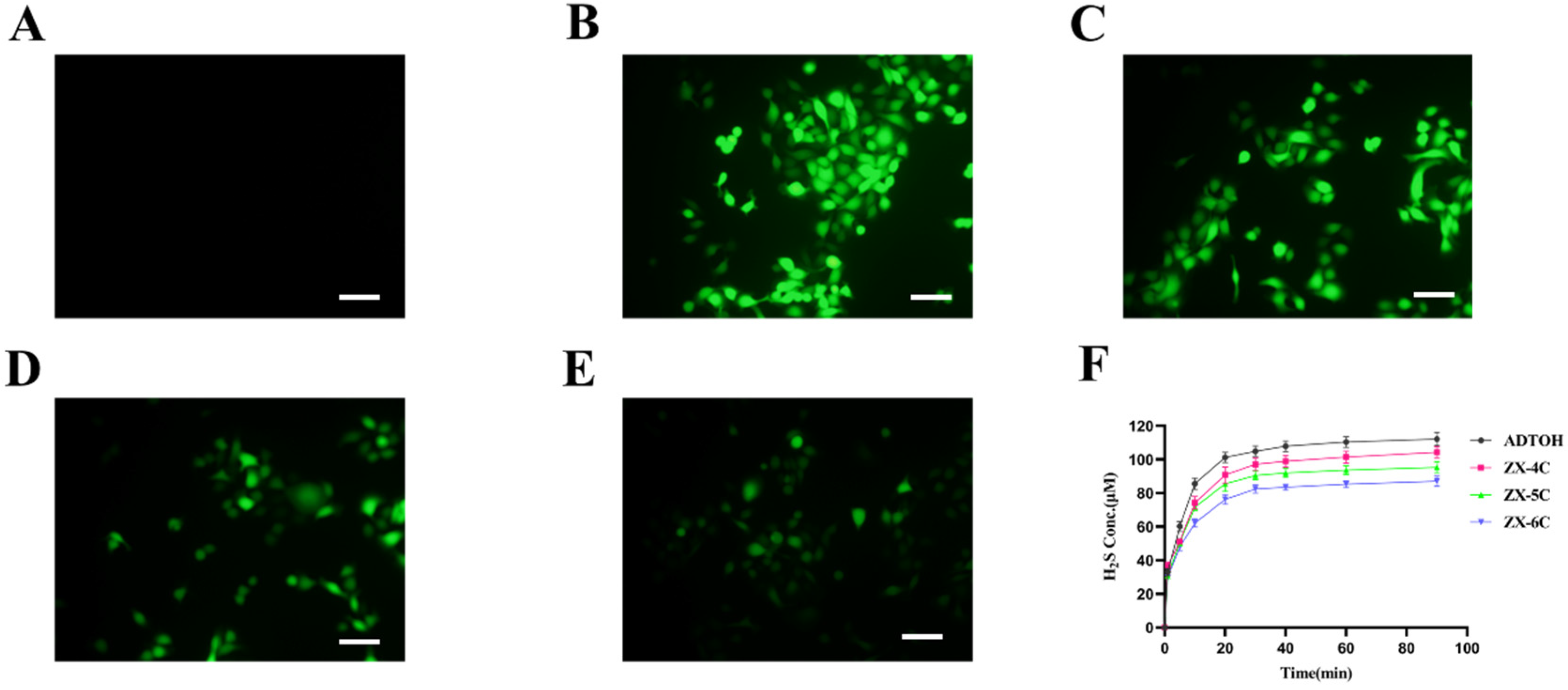
2.2. Evaluation of Hydrogen Sulfide Release
2.3. Fluorescent Probe to Study H2S Release in Cultured Cells
2.4. Esterase-Responsive Drug Release as Monitored by HPLC
2.5. Cell Culture
2.6. Cell Viability Assay
2.7. Detection of ROS Levels
2.8. Detection of GPx, SOD Activities, and GSH/GSSG Ratio
2.9. Detection of IL-1β, IL-6, and TNF-α Production
2.10. In Vivo Studies
2.11. Histopathological Assessment
2.12. Statistical Analysis
3. Results and Discussion
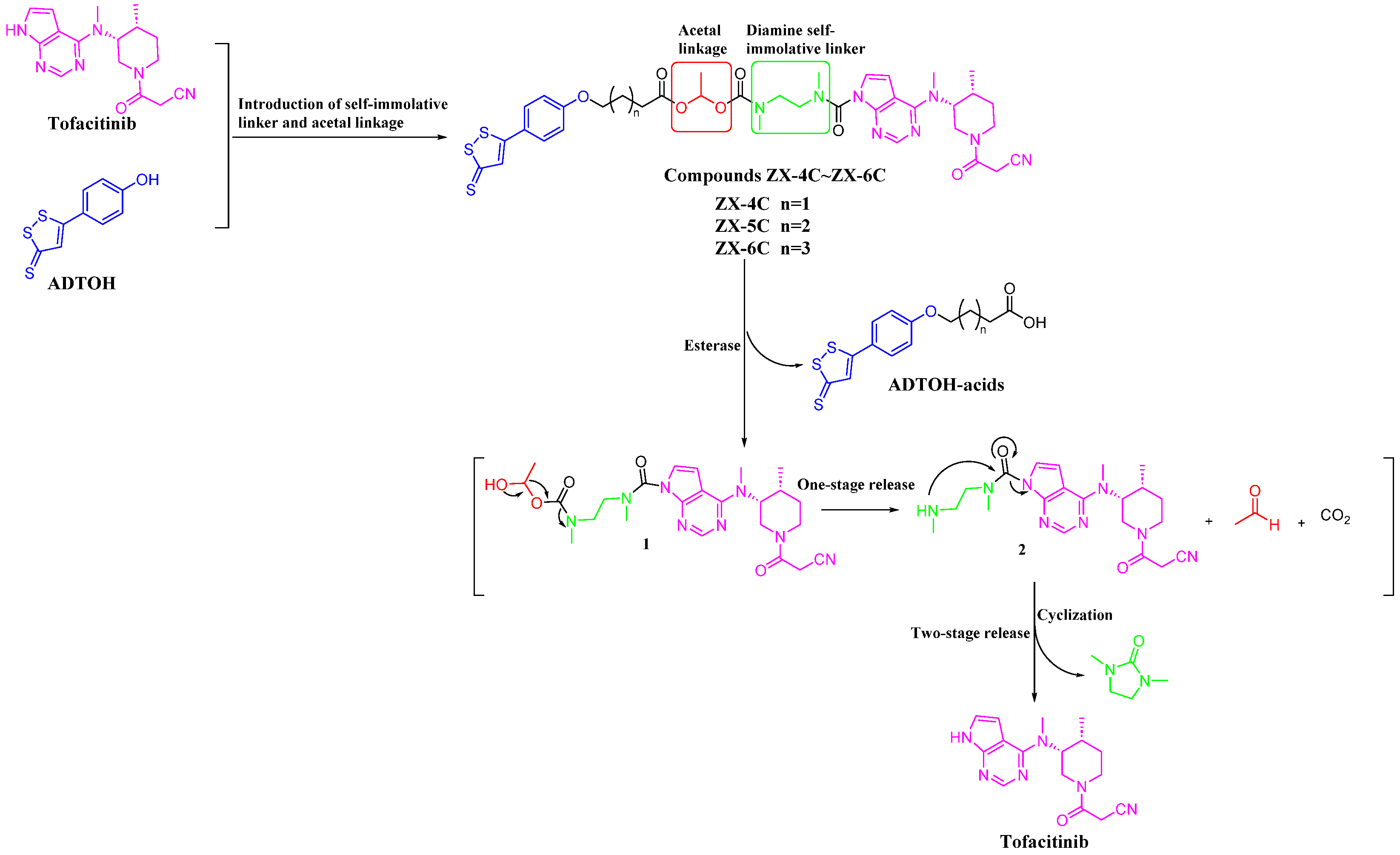
3.1. Molecular Hybridization Strategy to Design Tofacitinib–ADTOH Hybrid Derivatives
3.2. Chemistry
3.3. H2S Release Efficiency of Compounds ZX-4C~ZX-6C
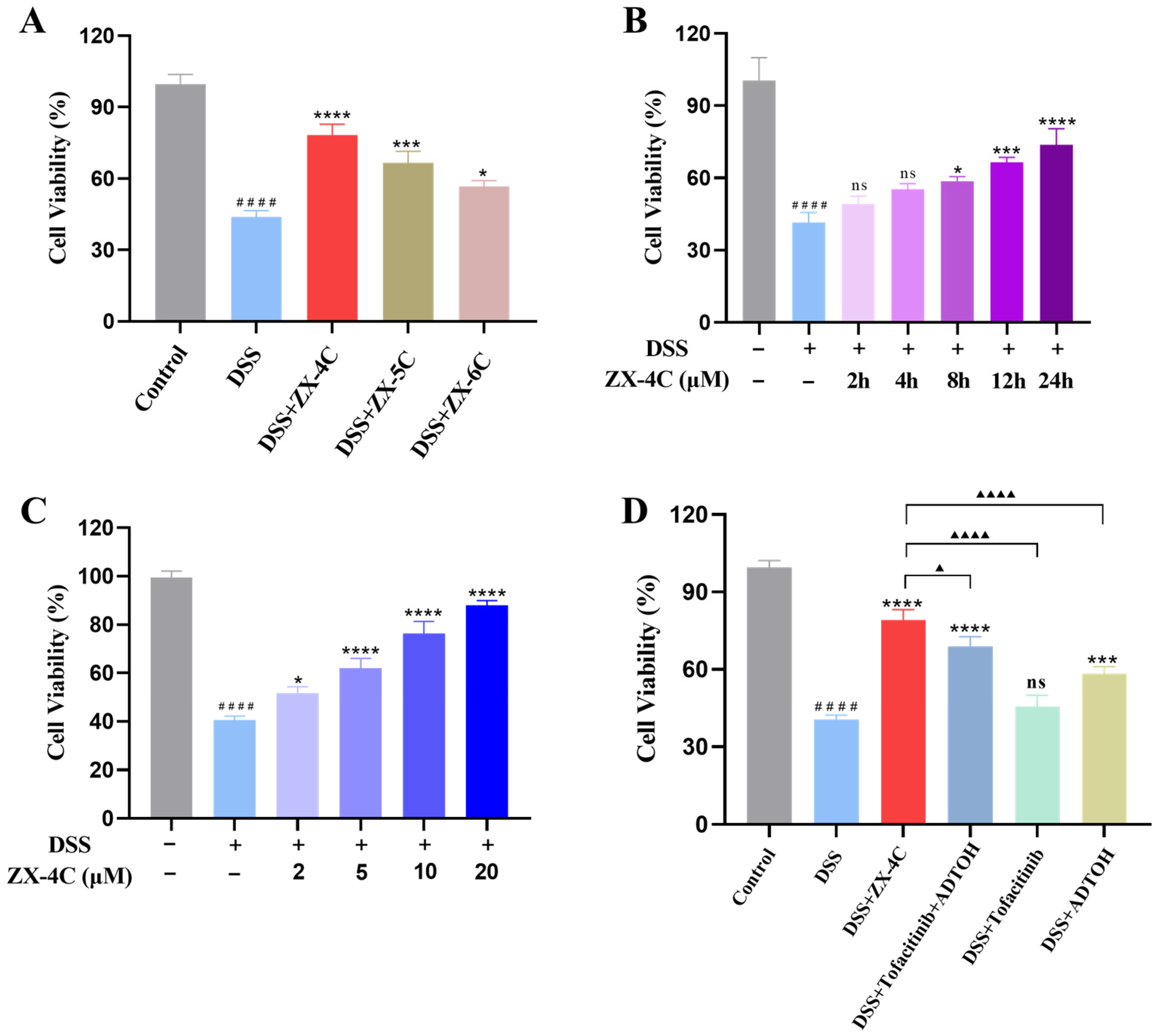
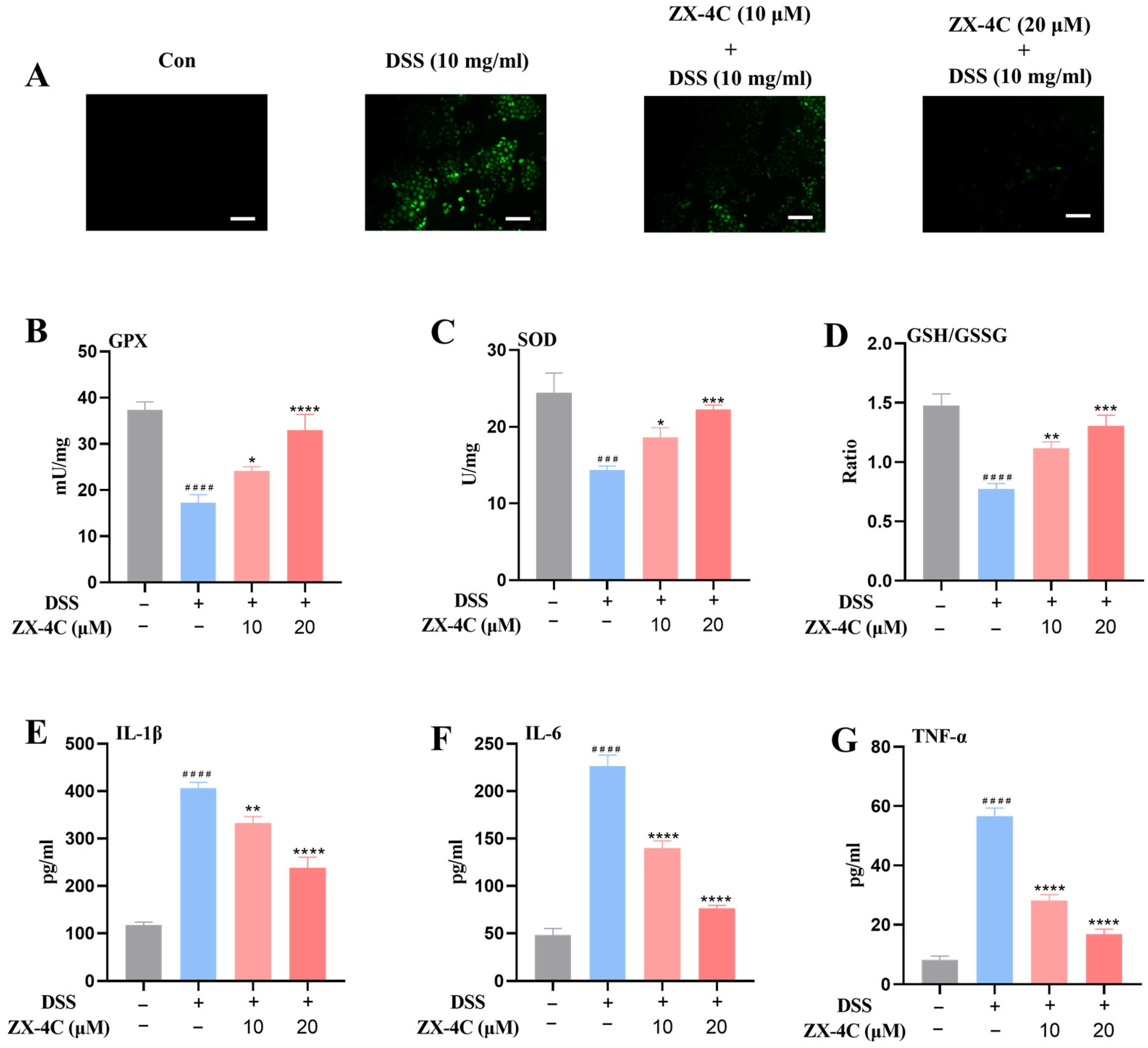
3.4. ZX-4C Exhibited Protective Effects Against DSS-Induced Injury in NCM460 Cells
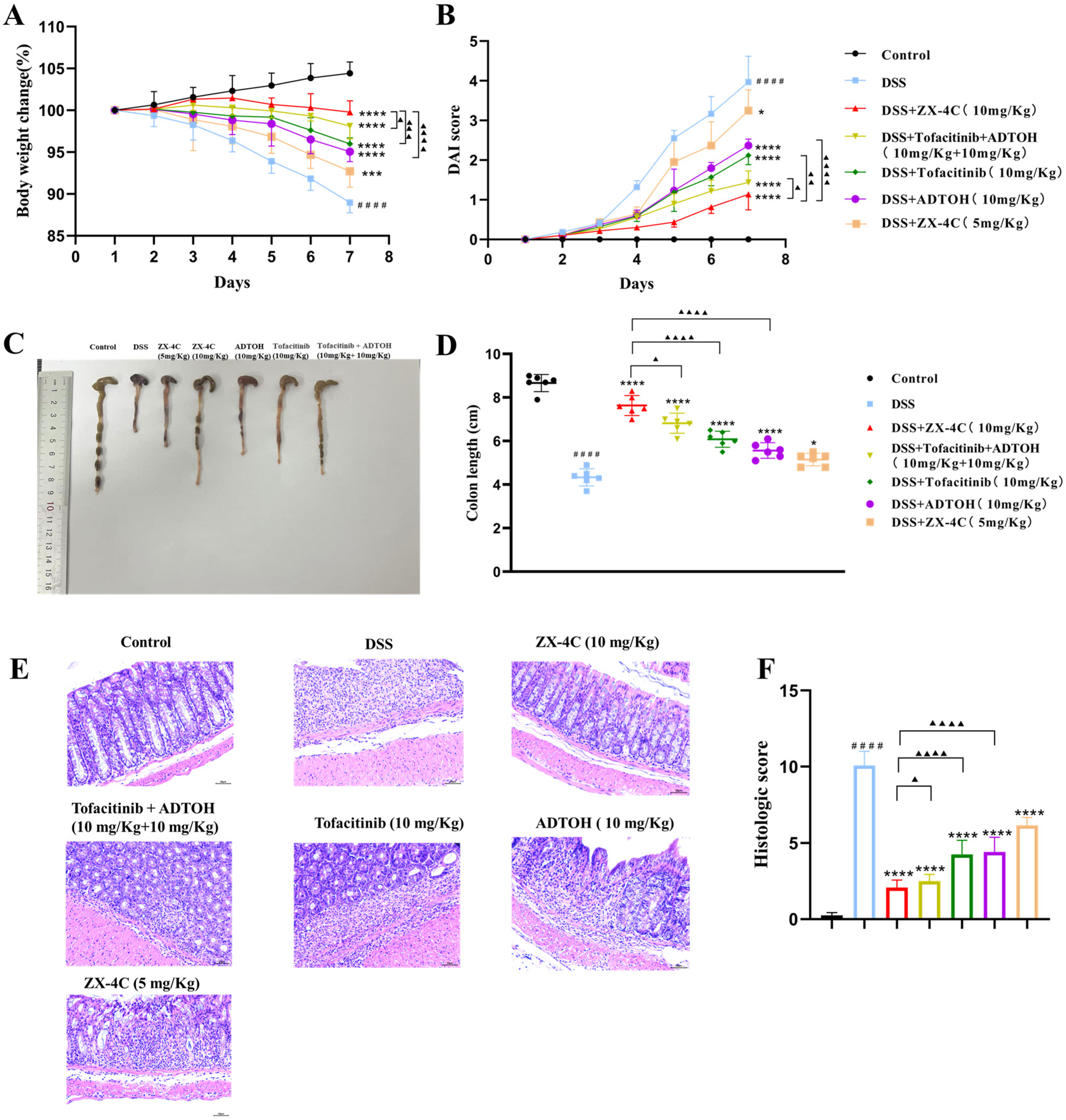
3.5. ZX-4C Alleviated DSS-Induced Colitis in Mice
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Hou, S.; Yang, X.; Tong, Y.; Yang, Y.; Chen, Q.; Wan, B.; Wei, R.; Wang, Y.; Zhang, Y.; Kong, B.; et al. Structure-based discovery of 1H-indole-2-carboxamide derivatives as potent ASK1 inhibitors for potential treatment of ulcerative colitis. Eur. J. Med. Chem. 2021, 211, 113114. [Google Scholar] [CrossRef] [PubMed]
- Junek, R.; Brunova, B.; Kverka, M.; Panajotova, V.; Jandera, A.; Kuchar, M. Antileukotrienic N-arylethyl-2-arylacetamides in the treatment of ulcerative colitis. Eur. J. Med. Chem. 2007, 42, 1084–1094. [Google Scholar] [CrossRef]
- Han, F.; Ning, M.; Wang, K.; Gu, Y.; Qu, H.; Leng, Y.; Shen, J. Design and exploration of gut-restricted bifunctional molecule with TGR5 agonistic and DPP4 inhibitory effects for treating ulcerative colitis. Eur. J. Med. Chem. 2022, 242, 114697. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.H.; Zhang, Q.; Xiao, Y.F.; Fang, Y.C.; Xie, X.; Nan, F.J. Phosphodiesters as GPR84 Antagonists for the Treatment of Ulcerative Colitis. J. Med. Chem. 2022, 65, 3991–4006. [Google Scholar] [CrossRef] [PubMed]
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.F. Ulcerative colitis. Lancet 2017, 389, 1756–1770. [Google Scholar] [CrossRef]
- Kobayashi, T.; Siegmund, B.; Le Berre, C.; Wei, S.C.; Ferrante, M.; Shen, B.; Bernstein, C.N.; Danese, S.; Peyrin-Biroulet, L.; Hibi, T. Ulcerative colitis. Nat. Rev. Dis. Primers 2020, 6, 74. [Google Scholar] [CrossRef]
- Wiggins, J.B.; Rajapakse, R. Balsalazide: A novel 5-aminosalicylate prodrug for the treatment of active ulcerative colitis. Expert. Opin. Drug Metab. Toxicol. 2009, 5, 1279–1284. [Google Scholar] [CrossRef]
- Dhaneshwar, S.S.; Gairola, N.; Kandpal, M.; Vadnerkar, G.; Bhatt, L.; Rathi, B.; Kadam, S.S. Synthesis, kinetic studies and pharmacological evaluation of mutual azo prodrugs of 5-aminosalicylic acid for colon-specific drug delivery in inflammatory bowel disease. Eur. J. Med. Chem. 2009, 44, 3922–3929. [Google Scholar] [CrossRef]
- Abdalla, M.I.; Herfarth, H. Budesonide for the treatment of ulcerative colitis. Expert. Opin. Pharmacother. 2016, 17, 1549–1559. [Google Scholar] [CrossRef]
- Timmer, A.; Patton, P.H.; Chande, N.; McDonald, J.W.; MacDonald, J.K. Azathioprine and 6-mercaptopurine for maintenance of remission in ulcerative colitis. Cochrane Database Syst. Rev. 2016, 2016, CD000478. [Google Scholar] [CrossRef]
- Bernstein, C.N. Treatment of IBD: Where we are and where we are going. Am. J. Gastroenterol. 2015, 110, 114–126. [Google Scholar] [CrossRef]
- Wang, D.; DuBois, R.N. The role of anti-inflammatory drugs in colorectal cancer. Annu. Rev. Med. 2013, 64, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Ihle, J.N. The Janus protein tyrosine kinases in hematopoietic cytokine signaling. Semin. Immunol. 1995, 7, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Soendergaard, C.; Bergenheim, F.H.; Bjerrum, J.T.; Nielsen, O.H. Targeting JAK-STAT signal transduction in IBD. Pharmacol. Ther. 2018, 192, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Leonard, K.A.; Madge, L.A.; Krawczuk, P.J.; Wang, A.; Kreutter, K.D.; Bacani, G.M.; Chai, W.; Smith, R.C.; Tichenor, M.S.; Harris, M.C.; et al. Discovery of a Gut-Restricted JAK Inhibitor for the Treatment of Inflammatory Bowel Disease. J. Med. Chem. 2020, 63, 2915–2929. [Google Scholar] [CrossRef]
- Izzo, R.; Bevivino, G.; Monteleone, G. Tofacitinib for the treatment of ulcerative colitis. Expert. Opin. Investig. Drugs 2016, 25, 991–997. [Google Scholar] [CrossRef]
- Fernandez-Clotet, A.; Castro-Poceiro, J.; Panes, J. Tofacitinib for the treatment of ulcerative colitis. Expert. Rev. Clin. Immunol. 2018, 14, 881–892. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Ghosh, S.; Panes, J.; Vranic, I.; Su, C.; Rousell, S.; Niezychowski, W.; Study, A.I. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N. Engl. J. Med. 2012, 367, 616–624. [Google Scholar] [CrossRef]
- Deepak, P.; Alayo, Q.A.; Khatiwada, A.; Lin, B.; Fenster, M.; Dimopoulos, C.; Bader, G.; Weisshof, R.; Jacobs, M.; Gutierrez, A.; et al. Safety of Tofacitinib in a Real-World Cohort of Patients With Ulcerative Colitis. Clin. Gastroenterol. Hepatol. 2021, 19, 1592–1601.e3. [Google Scholar] [CrossRef]
- Lightner, A.L.; Vaidya, P.; Holubar, S.; Warusavitarne, J.; Sahnan, K.; Carrano, F.M.; Spinelli, A.; Zaghiyan, K.; Fleshner, P.R. Perioperative safety of tofacitinib in surgical ulcerative colitis patients. Colorectal Dis. 2021, 23, 2085–2090. [Google Scholar] [CrossRef]
- Vakar, V.; Mazumder, R.; Padhi, S.; Tiwari, K.S.; Kinjal, P. Development of Colon Targeting Tablet of a JAK Inhibitor to Combat Chronic Ulcerative Colitis: A Novel Approach for Local Drug Delivery. Indian J. Pharm. Educ. Res. 2021, 55, s414–s427. [Google Scholar] [CrossRef]
- Zhao, G.; Wei, X.; Wu, J.; Eichele, D.D.; Lele, S.M.; Yang, L.; Zhang, F.; Wang, D. A Macromolecular Janus Kinase (JAK) Inhibitor Prodrug Effectively Ameliorates Dextran Sulfate Sodium-Induced Ulcerative Colitis in Mice. Pharm. Res. 2019, 36, 64. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, B.; Mao, Q.; Ping, K.; Zhang, P.; Lin, F.; Liu, D.; Feng, Y.; Sun, M.; Zhang, Y.; et al. Discovery of a Colon-Targeted Azo Prodrug of Tofacitinib through the Establishment of Colon-Specific Delivery Systems Constructed by 5-ASA-PABA-MAC and 5-ASA-PABA-Diamine for the Treatment of Ulcerative Colitis. J. Med. Chem. 2022, 65, 4926–4948. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; Vong, L.; McKnight, W.; Dicay, M.; Martin, G.R. Endogenous and exogenous hydrogen sulfide promotes resolution of colitis in rats. Gastroenterology 2009, 137, 569–578.e1. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; Ferraz, J.G.; Muscara, M.N. Hydrogen sulfide: An endogenous mediator of resolution of inflammation and injury. Antioxid. Redox Signal 2012, 17, 58–67. [Google Scholar] [CrossRef]
- Hirata, I.; Naito, Y.; Takagi, T.; Mizushima, K.; Suzuki, T.; Omatsu, T.; Handa, O.; Ichikawa, H.; Ueda, H.; Yoshikawa, T. Endogenous hydrogen sulfide is an anti-inflammatory molecule in dextran sodium sulfate-induced colitis in mice. Dig. Dis. Sci. 2011, 56, 1379–1386. [Google Scholar] [CrossRef]
- Wallace, J.L. Physiological and pathophysiological roles of hydrogen sulfide in the gastrointestinal tract. Antioxid. Redox Signal 2010, 12, 1125–1133. [Google Scholar] [CrossRef]
- Li, L.; Whiteman, M.; Guan, Y.Y.; Neo, K.L.; Cheng, Y.; Lee, S.W.; Zhao, Y.; Baskar, R.; Tan, C.H.; Moore, P.K. Characterization of a novel, water-soluble hydrogen sulfide-releasing molecule (GYY4137): New insights into the biology of hydrogen sulfide. Circulation 2008, 117, 2351–2360. [Google Scholar] [CrossRef]
- Li, L.; Rossoni, G.; Sparatore, A.; Lee, L.C.; Del Soldato, P.; Moore, P.K. Anti-inflammatory and gastrointestinal effects of a novel diclofenac derivative. Free Radic. Biol. Med. 2007, 42, 706–719. [Google Scholar] [CrossRef]
- Lougiakis, N.; Papapetropoulos, A.; Gikas, E.; Toumpas, S.; Efentakis, P.; Wedmann, R.; Zoga, A.; Zhou, Z.; Iliodromitis, E.K.; Skaltsounis, A.L.; et al. Synthesis and Pharmacological Evaluation of Novel Adenine-Hydrogen Sulfide Slow Release Hybrids Designed as Multitarget Cardioprotective Agents. J. Med. Chem. 2016, 59, 1776–1790. [Google Scholar] [CrossRef]
- Wen, S.; Cao, C.; Ge, J.; Yang, W.; Wang, Y.; Mou, Y. Research Progress of H(2)S Donors Conjugate Drugs Based on ADTOH. Molecules 2022, 28, 331. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; Caliendo, G.; Santagada, V.; Cirino, G.; Fiorucci, S. Gastrointestinal safety and anti-inflammatory effects of a hydrogen sulfide-releasing diclofenac derivative in the rat. Gastroenterology 2007, 132, 261–271. [Google Scholar] [CrossRef]
- Wallace, J.L.; Caliendo, G.; Santagada, V.; Cirino, G. Markedly reduced toxicity of a hydrogen sulphide-releasing derivative of naproxen (ATB-346). Br. J. Pharmacol. 2010, 159, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- Gugliandolo, E.; Fusco, R.; D’Amico, R.; Militi, A.; Oteri, G.; Wallace, J.L.; Di Paola, R.; Cuzzocrea, S. Anti-inflammatory effect of ATB-352, a H2S -releasing ketoprofen derivative, on lipopolysaccharide-induced periodontitis in rats. Pharmacol. Res. 2018, 132, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Kwapisz, L.; Raffals, L.E.; Bruining, D.H.; Pardi, D.S.; Tremaine, W.J.; Kane, S.V.; Papadakis, K.A.; Coelho-Prabhu, N.; Kisiel, J.B.; Heron, V.; et al. Combination Biologic Therapy in Inflammatory Bowel Disease: Experience From a Tertiary Care Center. Clin. Gastroenterol. Hepatol. 2021, 19, 616–617. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2019, 62, 420–444. [Google Scholar] [CrossRef]
- Blencowe, C.A.; Russell, A.T.; Greco, F.; Hayes, W.; Thornthwaite, D.W. Self-immolative linkers in polymeric delivery systems. Polym. Chem. 2011, 2, 773–790. [Google Scholar] [CrossRef]
- Walther, R.; Rautio, J.; Zelikin, A.N. Prodrugs in medicinal chemistry and enzyme prodrug therapies. Adv. Drug Deliv. Rev. 2017, 118, 65–77. [Google Scholar] [CrossRef]
- Walji, A.M.; Sanchez, R.I.; Clas, S.D.; Nofsinger, R.; de Lera Ruiz, M.; Li, J.; Bennet, A.; John, C.; Bennett, D.J.; Sanders, J.M.; et al. Discovery of MK-8970: An acetal carbonate prodrug of raltegravir with enhanced colonic absorption. ChemMedChem 2015, 10, 245–252. [Google Scholar] [CrossRef]
- Mattarei, A.; Azzolini, M.; Carraro, M.; Sassi, N.; Zoratti, M.; Paradisi, C.; Biasutto, L. Acetal derivatives as prodrugs of resveratrol. Mol. Pharm. 2013, 10, 2781–2792. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Lee, M.; Xia, J.; Luo, T.; Liu, J.; Rodriguez, M.; Lin, W. Two-Stage SN38 Release from a Core-Shell Nanoparticle Enhances Tumor Deposition and Antitumor Efficacy for Synergistic Combination with Immune Checkpoint Blockade. ACS Nano 2022, 16, 21417–21430. [Google Scholar] [CrossRef] [PubMed]
- Uzzan, M.; Martin, J.C.; Mesin, L.; Livanos, A.E.; Castro-Dopico, T.; Huang, R.; Petralia, F.; Magri, G.; Kumar, S.; Zhao, Q.; et al. Ulcerative colitis is characterized by a plasmablast-skewed humoral response associated with disease activity. Nat. Med. 2022, 28, 766–779. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mou, Y.; Wen, S.; Wang, Y.; Zhao, Y.; Li, Y.-P.; Sha, H.-K.; Gui, L.-J.; Jiang, Z.-Y.; Xu, X.-M. Discovery of Novel Tofacitinib–ADTOH Molecular Hybridization Derivatives for the Treatment of Ulcerative Colitis. Antioxidants 2025, 14, 325. https://doi.org/10.3390/antiox14030325
Mou Y, Wen S, Wang Y, Zhao Y, Li Y-P, Sha H-K, Gui L-J, Jiang Z-Y, Xu X-M. Discovery of Novel Tofacitinib–ADTOH Molecular Hybridization Derivatives for the Treatment of Ulcerative Colitis. Antioxidants. 2025; 14(3):325. https://doi.org/10.3390/antiox14030325
Chicago/Turabian StyleMou, Yi, Shuai Wen, Yan Wang, Yao Zhao, Ying-Ping Li, Hong-Kai Sha, Li-Juan Gui, Zheng-Yu Jiang, and Xiang-Ming Xu. 2025. "Discovery of Novel Tofacitinib–ADTOH Molecular Hybridization Derivatives for the Treatment of Ulcerative Colitis" Antioxidants 14, no. 3: 325. https://doi.org/10.3390/antiox14030325
APA StyleMou, Y., Wen, S., Wang, Y., Zhao, Y., Li, Y.-P., Sha, H.-K., Gui, L.-J., Jiang, Z.-Y., & Xu, X.-M. (2025). Discovery of Novel Tofacitinib–ADTOH Molecular Hybridization Derivatives for the Treatment of Ulcerative Colitis. Antioxidants, 14(3), 325. https://doi.org/10.3390/antiox14030325