Abstract
Animal slaughtering causes the cessation of oxygen delivery and that of nutrients such as cystine, glucose and others to muscle cells. In muscle cells, the changes in oxygen level and pH cause mitochondria, the endoplasmic reticulum, xanthine oxidase and uncoupled NOS to increase the level of O2•−, affecting the generation of H2O2 and the release of iron ions from ferritin. The activation of enzymes that remove and dislocate fatty acids from the membrane affects the sensitivity of muscle cells to peroxidation and ferroptosis. Increasing PUFAs in membrane phospholipids, by feeding animals a diet high in w-3 fatty acids, is a driving factor that increases lipid peroxidation and possible muscle ferroptosis. The activation of lipoxygenases by ROS to Fe3+-lipoxygenase increases hydroperoxide levels in cells. The labile iron pool generated by a “redox cycle” catalyzes phospholipid hydroperoxides to generate lipid electrophiles, proximate executioners of ferroptosis. Ferroptosis in food muscle cells is protected by high concentrations of vitamin E and selenium. In fresh muscle cells, glutathione peroxidase (GSH-PX) and other endogenous antioxidant enzymes are active and prevent lipid peroxidation; however, muscle heating eliminates enzymatic activities, making cells prone to high non-enzymatic lipid peroxidation. In muscle cells, coupled myoglobin and vitamin E act as a hydroperoxidase, preventing the generation of lipid electrophiles. Free iron ion chelators or effectors such as deferoxamine, EDTA, or ceruloplasmin are strong inhibitors of muscle cell lipid peroxidation, proving that muscle ferroptosis is mostly dependent on and catalyzed by the labile iron redox cycle.
1. Introduction
Plants and animals after harvesting or slaughtering for food are organisms in the process of cell death. For years, a great mission for scientists was to explore the mechanism of cell death, since understanding it may open ways for extending life. Ferroptosis is a form of regulatory iron-dependent oxidative stress cell death, affecting membrane integrity by the generation of lipid hydroperoxides and cytotoxic advanced lipid end products (ALEs).
The initiation of lipid oxidation in biological systems at the chemical and biochemical level was divided many years ago into three steps: (1) the activation of oxygen to reactive oxygen species (ROS); (2) the oxidation of fatty acids by ROS and enzymes to hydroperoxides; and (3) the propagation of lipid hydroperoxides by auto-oxidation and iron-redox catalyzers to lipid free radicals, generating cytotoxic compounds [1]. All these steps were observed but not integrated into the unified cell molecular biology process known today, in part, as ferroptosis. Many factors connected to ferroptosis seem to affect lipid peroxidation in animal tissues after slaughtering; however, the most important are the cessation of oxygen delivery (ischemia/anoxia) and blood component transport to organs and cells. Ferroptosis is a process of regulatory cell death [2]. This unique cell death is derived by the obscure generation of lipid peroxidation products claimed to be the proximate executioners of ferroptosis. The specialized cell death program of ferroptosis is triggered at the end by insufficient activity of glutathione peroxidase 4 (GSH-PX4) [3].
2. Ferroptosis Inducers, in General and in Muscle Cells
The ferroptosis process, as known, involves an iron-dependent phospholipid peroxidation to cytotoxic compounds regulated by cellular pathways, including oxygen presence, redox changes, and activation of oxygen to ROS, iron handling, mitochondria activity/dis-activity, endoplasmic reticulum, and lysosomal effectors [4].
The electron transport chain (ETC), localized in the inner mitochondrial membrane, seems to act as the primordial generator of O2•−, H2O2, and HO•. In mitochondria, ETC-I and ETC-III are the main generators of superoxide [5]. Inhibition of ETC-I and ETC-III by rotenone and antimycin A suppresses cysteine deprivation-induced ferroptosis [4]. It seems that ETC-III dominantly plays a primary role in the execution of ferroptosis under conditions of nutrient starvation such as in muscle cells after animal slaughtering [6].
Ischemia and reperfusion are two main phases responsible for muscle injury through an increased generation of ROS such as superoxide anion radicals (O2•−), hydrogen peroxide (H2O2) and hydroxyl radicals (HO•) that trigger oxidative stress-induced cell death. ROS in the muscles are mainly formed from primary sources such as NADPH oxidases (NOXs), redox changes affecting the mitochondria respiratory chain, and secondary sources of activation such as; xanthin dehydrogenase converting to xanthine oxidase and uncoupled nitric oxide synthases (NOSs) [7,8]. ROS could be generated also by oxygen reduction by labile ferrous ions to O2•−and H2O2 [1,9]. In muscle foods, the act of animal slaughtering causes cessation of oxygen to organs and decreases the amount of oxygen for tissues and cells. After death and rigor mortis, cutting the meat into small parts exposes its surface to oxygen and the generation of H2O2 and lipid peroxides [10,11,12] (Figure 1).
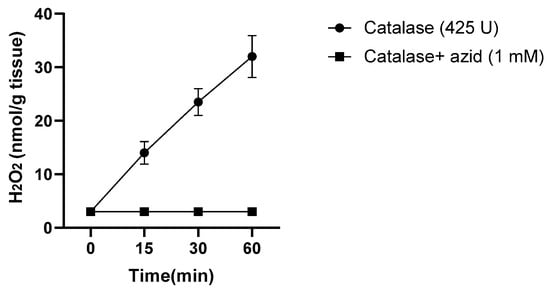
Figure 1.
Generation of H2O2 by turkey dark muscle cells stored at 37 °C, determined by the peroxidase activity of catalase and oxidation of 14C-formate to 14CO2, collected by KOH and detected by a scintillator. Adapted from Harel S. and Kanner J, 1985 [11].
These effects resemble, in part, heart muscle ischemia/reperfusion injury effects [8]. Studies have suggested that heart muscle reperfusion-induced oxidative injury is dependent on critical events such as succinate accumulation in mitochondria during the ischemia stage and generation of superoxide O2•−; cessation of cells due to depletion of amino acids, especially cysteine/glutathione, and accumulation of glutamate. Induction of muscle oxidative injury is also dependent on dislocation of membrane phospholipids such as phosphoetanolamines (PE) and other fatty acids, mostly fatty acyls-arachidonoyl (AA) and adrenoyl (AdA), activation of lipoxygenase 15/12, and generation of lipid hydroperoxides. The hydroperoxides are broken down by the labile iron pool (LIP) to lipid-cytotoxic electrophilic compounds, affecting the integrity of the membranes [13]. Indeed, Ma et al. [14], by using multi-omics and chemogenomic approaches, demonstrated that ischemia triggers a specific redox reaction that induces the increased generation of arachidonic acid 15-lipoxygenase-1 (ALOX15). This enzyme acts during the reperfusion stage with the labile iron pool (LIP) to catalyze phospholipid peroxidation and the breakdown of hydroperoxides into ferroptosis signals.
Labile iron pool (LIP). Iron transferrin, after entering muscle cells through transferrin, is endocytosed. From endosome, iron is released and mediated by metal-reductase STEAP3 and divalent metal transporter (DMTI) to LIP [15]. Other major sources of LIP are ferritins from cytosol and mitochondria, stored as ferric ions as a complex of 24 subunits. Ferritin-bound iron is released into LIP by nuclear receptor coactivator 4 (NCOA4) dependent auto-phagosome degradation, activated during ischemia [16]. LIP could be released from ferritin through ferritin pores after reduction and solubilization of the ferric to ferrous iron within the ferritin core by reducing agents like ascorbic acid and superoxide anion radical [17] (Figure 2).
In the cytosol, labile iron is coordinated by a pool of small molecules (glutathione) and macro-molecules (iron-chaperones) that transfer the iron ions to different sites of the cell, and is affected by vitamin E [18]. A part of the LIP is an important precursor transferred to mitochondria for myoglobin biosynthesis. Previously, it was shown by us that after slaughtering, muscle cell ferritins are degraded, increasing the LIP in cells [19] Figure 3.

Figure 2.
Loss of iron-ferritin isolated from dark and light turkey muscle after storage at 4 °C. Adapted from Kanner J. and Doll L. 1991) [19].

Figure 3.
Catalytic free iron accumulated during storage of turkey dark muscle at 4 °C. Adapted from Kanner J et al., 1988 [20].
This LIP in muscle foods, in the presence of reducing compounds such as cysteine, glutathione, or ascorbic acid, acts as an “iron redox cycle”, initiating the Fenton reaction and lipid oxidation, generating hydroperoxides, and catalytically breaking down hydroperoxides into cytotoxic reactive aldehydes [9,20,21].
Lipoxygenases (LOXs) are non-heme iron-containing enzymes that play an important role in ferroptosis [22]. Electro-paramagnetic-resonance spectroscopic studies indicate that the native inactive form of the enzyme contains iron in its high-spin ferrous Fe2+state [23]. Lipoxygenase is activated to the ferric state by O2•−, H2O2, or previously formed hydroperoxides (LOOH) [1]. Lipid substrates for the enzymes are free fatty acids and phospholipids containing at least two non-conjugated, 1–4 carbon–carbon double bonds; because of that, oleic acid is not a substrate for lipoxygenases. The enzyme after activation could generate lipid allyl free radicals (L•) [14], which, in the presence of oxygen, generate a peroxyl radical LOO•; this radical is further reduced by the enzyme ferrous state to fatty acid hydroperoxides and back to the ferric active enzyme state. The hydroperoxides are not stable at 37 °C and break down into free radicals of LO•/HO•, inducing lipid auto-oxidation and the generation of many reactive carbonyls, electrophilic cytotoxic compounds, and cell death. Similar and most appropriate compounds are generated by the catalytic decomposition of lipid hydroperoxides by ferrous iron and heme proteins [1,14,24].
Lipids and lipid metabolism affect the sensitivity of ferroptosis. Free fatty acids can be taken into cells by diffusion and by protein transporters. Some lipids such as oleic acid, a mono-unsaturated fatty acid (MUFA), can be synthesized de novo in cells from palmitate by elongation and desaturation enzymes or from diet. The incorporation of MUFA into phospholipids is activated by acyl-CoA synthesized long-chain member 3 (ACSL3) and polyunsaturated fatty acids (PUFA) by ACSL4. Phospholipids can be remodeled by enzymes that remove/add fatty acids from/to membrane phospholipids by phospholipase A2 group and other acyltransferases, affecting the membrane sensitivity to peroxidation and ferroptosis. Increasing PUFA in membrane phospholipids is a driving factor for peroxidation and ferroptosis [22]; however, MUFA-enriched cells exerted no impact on these parameters [14]. A high fat-diet (HFD-PUFA) fed to rats, as an in vivo model of heart-muscle ischemia, had a significant impact on increasing lipid peroxidation parameters such as MDA and diminished GSH content, leading also to a significant increase in mortality [14]. In line with these results, our earlier report on feeding calves with PUFA found, after slaughtering, an increased susceptibility of the muscles to lipid peroxidation. Muscle and extracellular lipid peroxidation further increased with the addition of salt [25] or the inhibition of GSH-PX and other antioxidant enzymes [1,20,26,27].
3. Ferroptosis Prevention, in General and in Food Muscle Cells
Antioxidant Enzymes
Superoxide dismutase, catalase, glutathione peroxidase, ceruloplasmin, and peroxiredoxins are the main enzymes in biological systems, including muscle cells, to fight against ROS [1,28,29,30]. Anther factor in mitochondria that protects against ferroptosis is Coenzyme Q- (CoQ). CoQ can be reduced by dihydroorotate dehydrogenase to CoQH2, which, apart from transferring electrons to ETC III, can act as a local antioxidant, preventing mitochondrial and cell lipid peroxidation and limiting ferroptosis. CoQ is trafficked from mitochondria by a transfer protein (STARD7) to the plasma membrane, enhancing the cell's antioxidant defenses [31]. CoQ or CoQ•. radical can be reduced back to CoQH2 also by the ferroptosis suppressor protein (FSP1) [32]. Ceruloplasmin, an antioxidant enzyme found in high concentrations in animal plasma, reduces ferrous ions to the ferric state, and by this, prevents the progression of lipid peroxidation in minced turkey muscle (Figure 4).
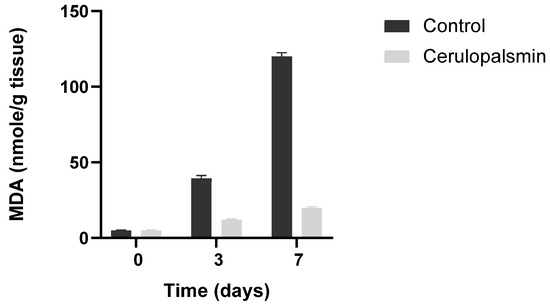
Figure 4.
Ceruloplasmin (150 U/g tissue) inhibits in situ lipid peroxidation of minced turkey muscle tissues. Control, minced muscle 2- Minced muscle in the presence of ceruloplasmin. Adapted from Kanner J et al., 1988 [26].
Tetrahydrobiopterin (BH4), a natural nutrient, is biosynthesized and recycled by animal metabolism and also affects ferroptosis. BH4 acts as a cofactor for several enzymes such as nitric oxide synthase (eNOS) and, by generating •NO, is involved in preventing ferroptosis. In BH4 deficiency, the uncoupled eNOS generates O2•− interacts with •NO to form peroxy-nitrite (HOONO), which decomposes into HO· and •NO2, increasing ferroptosis [33].
Antioxidants are largely beneficial for human health [34]. Many natural and synthetic compounds with antioxidant action have been identified and are commercially available. Selenium (Se) is an effective modulator of the antioxidant systems in animals and humans. Selenium is involved in expression and synthesis of many seleno-proteins, including glutathione peroxidase (GSH-Px), thioredoxin (TrxR), and peroxiredoxins (PrxR). More than half of known seleno-proteins are directly or indirectly involved in antioxidant defenses and redox status maintenance. Selenium is the active center element in GPX4 enzyme, extremely important as an anti-ferroptosis enzyme. GSH-Px 1–4, including the membrane GSH-Px, are a family of antioxidant enzymes that decompose H2O2 and LOOH into non-radical compounds such as H2O and hydroxy-fatty acids, through the most effective anti-ferroptosis pathway [35]. GSH-Px acts as a very effective inhibitor of lipid peroxidation in fresh muscle foods [1,36]. However, processing of muscle foods by heating eliminates their activity as antioxidants, allowing the tissues to undergo high lipid peroxidation.
Peroxiredoxin 6 (PRDX6), an important ferroptosis protector, is an enzyme acting as a phospholipid hydroperoxidase, but also as phospholipase A2 and lysophospholipid acyl transferase, an enzyme that can perform lipid detoxification and pathway repair by itself [37]. PRDX6 was found to have the ability to bind phospholipids and uses GSH as its physiological reductant.
Myoglobin (Mb) is known to catalyze the breakdown of lipid hydroperoxides into free radicals, a reaction that can enhance the propagation step and general lipid peroxidation [38,39]. During this reaction, a fraction of the free radicals are auto-reduced by metmyoglobin [40]. Our study demonstrated that metmyoglobin, at a rate concentration of ∼3:1 to hydroperoxides, acted anti-oxidatively and decomposed hydroperoxides, whose concentrations then remained at zero for a long time. This behavior has been recognized in the varying effectiveness of certain hemeproteins that are potent inhibitors at low peroxide levels, very weakly effective at high peroxide levels, and turn into oxidative catalyzers at very high peroxide levels [41]. Catechin, a known polyphenol, supports metmyoglobin antioxidation. The antioxidative activity of the complex metmyoglobin-catechin is stronger at pH 3.0 than at pH 7.0, indicating that this reaction is more dependent on metmyoglobin than on catechin. During this reaction, catechin or other polyphenols such as quercetin not only donate reducing equivalents to prevent lipid peroxidation but also prevent the destruction and polymerization of metmyoglobin. The results of this research highlighted the important and possible reactions of heme proteins and polyphenols as a redox couple, working as hydro-peroxidase or as pseudo-peroxidase. It was found also that d-α-tocopherol acts as a redox couple with metmyoglobin; at low pH, the couple is 150-fold more reactive than catechin [42]. Myoglobin is known to interact well with phospholipids and cell membranes. In tissues where myoglobin presents at high concentration, such as in the heart and muscles, during ferroptosis, it may act as a hydro-peroxidase using vitamin E or other reducing agents as redox cofactors.
Biochemical and tissue-level studies support the idea that Mb is also an important regulator of cellular nitric oxide (•NO) pools, in which Mb is a .NO scavenger under normal cell oxygen levels, but also as a .NO producer in anoxia [43]. Nitric oxide-myoglobin acts as an antioxidant in model systems of muscle lipid peroxidation [44,45], and in most probably cases, can act as an antioxidant in muscle cell ferroptosis.
Vitamin E. Vitamin E (RRR-α-tocopherol) is a potent anti-ferroptotic nutrient and the main chain-breaking lipid-soluble antioxidant in membrane cells, found in all animals but generated by plants. d-α-Tocopherol is a potent peroxyl radical scavenger that interrupts lipid peroxidation in membranes and lipoproteins by free radicals. The oxidized vitamin E is continuously restored by other hydrogen donors such as vitamin C and CoQH2, thereby oxidizing the latter and returning it to a vitamin E reduced state. If additional antioxidants are not present, the vitamin E phenoxy radical can reinitiate lipid peroxidation [46]. Tocopherol-quinone (α-TQ), generated by oxidation of α-tocopherol, can be reduced by reducing agents, NAD(P)H oxidoreductase enzyme, or ferroptosis suppression protein 1 (FSP1) to tocopherol-hydroquinone (α-THQ), a more efficient antioxidant than tocopherol. α-Tocopherol prevents RAS-selective lethal 3 (RSL3) induced ferroptosis cell death; however, the action of vitamin E hydroquinone is ~50-fold stronger. Vitamin E hydroquinone (α-THQ) is an endogenous regulator of ferroptosis, via redox control of 15-lipoxygenase, with very efficient protective activity, ~40-fold higher than that of d-α-tocopherol [23]. The catalytic activity of 15-lipoxygenase requires that the non-heme iron center be in the Fe3+ state; upon addition of α-THQ, a 50% decrease in the Fe3+ to non-active Fe2+ signal was measured. α-Tocotriene-hydroquinol (α-T3HQ), another natural antioxidant, reduced the ferric ion of lipoxygenase even more effectively. The potential of α-T3HQ as an anti-ferroptotic compound was found to be favorable for all ferroptosis measured tests and markers [23].
The effects of vitamin E supplementation [47,48], and high PUFA content in the feed, on calf muscle lipid peroxidation following NaCl addition after slaughtering and storage at 4 °C was studied previously by us [25]. The study found that even though the potential for peroxidation was enhanced by a diet rich in PUFAs or by the addition of NaCl, the high enrichment of muscle tissues with α-tocopherol almost completely inhibited muscle lipid peroxidation and oxymyoglobin oxidation [25], see Figure 5.
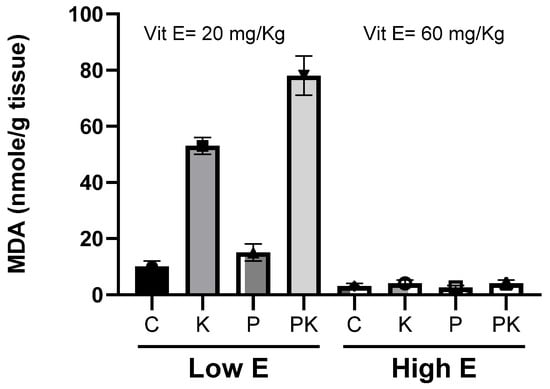
Figure 5.
Lipid peroxidation and MDA accumulation during storage at 4 °C of longissimus dorsi muscle of calves fed diets supplemented with different amounts of vitamin E, with or without high PUFA content in feed: control (C); vitamin E (E); PUFA in feed (P); kosher-salted (K); high PUFA and kosher-salted (PK). Adapted from Granit R et al. 2001 [25].
Polyphenols are a natural class of compounds found to protect against ferroptosis. Polyphenols were found to modulate iron metabolism and nuclear factor erythroid 2-related factor 2 (NRF2) signaling to inhibit ferroptosis [49]. Ferroptosis induced in piglet’s intestine by Diquat was inhibited by supplementing the animals with polyphenols extracted from Ilex latifolia, which contains high levels of phenolic acids and tannic acids [50]. Fisetin, another polyphenol, was found to ameliorate fibrotic kidney in mice via inhibiting ACSL4-mediated tubular ferroptosis [51]. Polyphenols were found by many authors to act as inhibitors of muscle food lipid peroxidation and were reviewed [32,52,53].
Vitamins K and K2 are well known for their canonical function as cofactors for γ-glutamyl carboxylase and generation of vitamin K-dependent protein factors for blood coagulation [54]. Vitamins K and K2 are redox-active quinones converted to corresponding hydroquinone (VKH2) by the vitamin K cycle. VKH2 was reported to be a potent radical trapping agent preventing lipid peroxidation [55]. FSPI, which reduces ubiquinone in the presence of NADPH, was found to also reduce vitamin K to VKH2, both acting as ferroptosis suppressor [56]. It is possible that supplementing animals and poultry with high vitamin K before slaughtering can improve protection against muscle lipid peroxidation, but such an experiment has never been performed.
Nitric oxide (•NO) is a free radical gaseous molecule with multiple biochemical and physiological functions [57]. The molecule is generated by enzymes in the nitric oxide (NOS) family, by converting arginine to citrulline using tetrahydrobiopterin (BH4) as a coupled factor liberating •NO [58]. •NO is also produced non-enzymatically by ferrous hemoglobin or myoglobin in the presence of nitrite [44,59,60],or from nitrite in stomach conditions (low pH and ascorbic acid) [61,62], NOS uncoupled with BH4 has a pro-oxidative activity because it generates superoxide radical and •NO to produce peroxynitrous acid (ONOOH). Peroxynitrous acid is very unstable and decomposes into •NO2 and HO• radicals, both known as initiators of lipid peroxidation [63]. However, its pro-oxidative functions do not seem to play a role in ferroptosis [64].
The role of •NO as an antioxidant in food lipid peroxidation and in biological systems was first demonstrated by us [44,60,65]. The antioxidative role of NO/NOS in ferroptosis has been reported [66]. Our studies found that •NO acts as an antioxidant, suppressing the Fenton reaction by scavenging HO• radicals (in a reaction of “iron redox cycled” H2O2), but also (in a hydroperoxide-dependent lipid peroxidation) by scavenging LO• and LOO•, to LONO and LOONO. •NO is known also as an excellent ligand to ferrous-hemeproteins and iron complexes [67]. We found that both complexes act as very active antioxidants by rapid decomposition of H2O2 and LOOH into non-radical compounds such as HNO2 and LONO [59,60]. It was also demonstrated that •NO, which acts as a very good ligand to iron, can inhibit enzymes such as lipoxygenase and cyclooxygenase, both containing iron ions at the active site of the enzyme [68,69]. N-acetylcysteine-NO (NAC-SNO) an •NO donor, was found to replace nitrite in curing muscle foods as an anti-clostridial preservative, pigment developer, and antioxidant, almost without generating nitrosamines [61] (Figure 6).
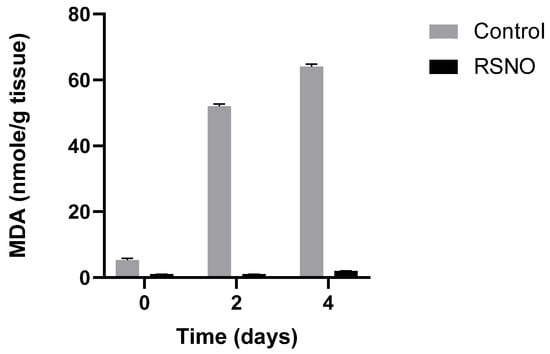
Figure 6.
Lipid peroxidation and MDA accumulation in minced muscle, with and without the presence of NAC-SNO (RSNO), stored at 4 °C. Adapted from Kanner J et al., 2019 [61].
NAC-SNO at relatively low concentration (12 µM), was found to inhibit ferroptosis induced by RLS3 in a culture of hepatic cells. This effect was achieved most probably because of the biochemically broad antioxidant activity of •NO [58,60,70], (see Figure 7).
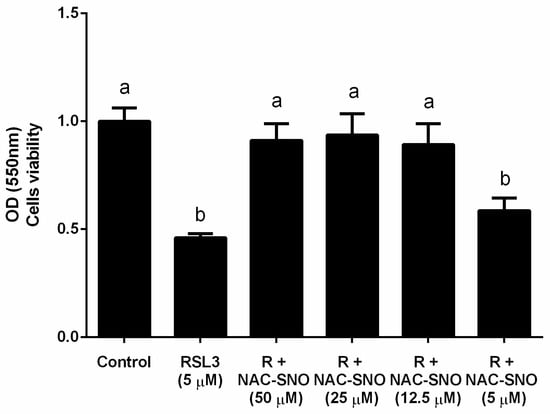
Figure 7.
The effect of NAC−SNO on cell viability in ferroptosis by RSL3. Cell viability was measured by MTT assay of AML12 cells treated with 5 µM RSL3 and different concentrations of NAC-SNO for 6 h. a and b indicate significant statistical differences. Adapted from Shpaizer, A. PhD Thesis 2023 [70].
Synthetic antioxidants that trap lipid radicals and suppress lipid peroxidation in ferroptosis are represented by ferrostatin-1 and liproxstatin-1 [71]. Ferrostatin-1 in the presence of ferrous ion acts similarly to GSH-PX4 by catalytic decomposition of lipid hydroperoxides with ~350-fold more activity than Trolox [72]. Butylated hydroxy toluene (BHT), a synthetic antioxidant known for preventing lipid peroxidation in foods, was found to act also as an inhibitor of ferroptosis in vitro and in vivo [73].
Animal slaughtering causes cessation of the flow of nutrients (cystine, glucose), oxygen depletion, and glutamate accumulation in cells. In muscle cells, the change in oxygen level, glutamate levels, and pH induce mitochondria to generate O2•− and H2O2, (see Figure 8). The release of iron from ferritin increases the labile iron pool (LIP) and is mediated by the endosomal metal-transferase STEAP3, divalent metal transporter (DMT1), and nuclear receptor coactivator 4 (NCOA4). Hypoxia inducible factor (HIF) system and perilipin 2 increase the amount of lipid droplets and lipolysis to fatty acid (FA) and metabolism of PUFA by long-chain-fatty-acid-CoA ligase 4, (ACSL4) and lysophospholipid acyltransferase 3, (LPCAT3) to membrane-rich phospholipids (PL), and PUFA, which is oxidized by arachidonic lipoxygenase (ALOX) to phospholipid hydroperoxides (PUFA-OOH). Activation of enzymes that remove or dislocate fatty acids from membrane affects the sensitivity of muscle cells to peroxidation and ferroptosis. Glutamate cysteine ligase (GCL) and glutathione synthetase (GSS) generate glutathione (GSH), which act as a redox couple with glutathione peroxidase (GSH-PX) for degradation of H2O2 and PUFA-OOH to H2O and the non-radical hydroxy fatty acids (LOH), preventing progression of lipid oxidation. Due to the cessation in the supply of cystine after slaughtering, GSH/GSH-PX couple activity decreases the antioxidative defense of muscle cells. The increase in PUFA in membrane phospholipids, by nutritional feeding, is a driving factor that increases lipid peroxidation and muscle ferroptosis. Food muscle cells are protected from ferroptosis by high concentrations of vitamin E, selenium, butylated hydroxy-toluene (BHT), and other radical- trapping antioxidants (RTA), and possibly by ferroptosis suppressor protein 1 (FSP1). Free iron ions chelators such as deferoxamine, EDTA, or ceruloplasmin (CP) act as strong inhibitors of muscle cell lipid peroxidation, proving also that muscle ferroptosis is mostly dependent on and catalyzed by the labile iron redox cycle.
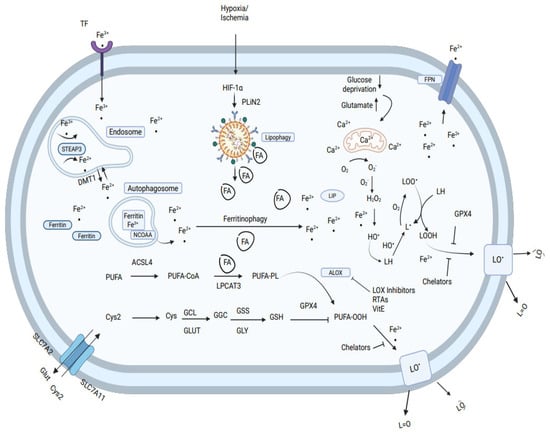
Figure 8.
Molecular mechanism of ferroptosis in cells. Ferroptosis in muscle food is typically triggered by iron-dependent lipid peroxidation.
4. Conclusions
Animal slaughtering causes cessation of the oxygen supply to the organs, decreases the level of oxygen in tissues and cells, and also causes cessation of the flow of nutrients such as glucose, cystine, BH4, and others to cells in muscle foods. After death and rigor mortis, cutting the meat into small parts exposes its surface to high oxygen level (ischemia/reperfusion)-induced generation of H2O2 and lipid peroxides. During muscle ischemia, the level of lactic acid increases, and the cell’s pH decreases, affecting the release of iron ions from ferritin. In muscle cells, the change in oxygen level and pH induces mitochondria, endoplasmic reticulum, xanthine oxidase and uncoupled NOS to increase the levels of O2•− and H2O2 [74]. Both activate lipoxygenases, which act with the labile iron pool to catalyze phospholipid peroxidation. Ferroptosis in muscle food could be ameliorated by supplementing feed with increased levels of vitamin E and selenium, elevating GSH-PX4 and other protein selenium antioxidants, and also adding muscle food antioxidants such as: •NO (NAC-SNO), polyphenols, and iron chelators (polyphosphates, EDTA, and ferrioxamine) [75]. We suggest, for the first time, that post-mortem metabolism, which is altered in the muscle environment after slaughtering, will dictate an imminent process of ferroptosis and that the lipid peroxidation rate within the muscle cells is the key factor for muscle food quality. Inhibition of this imminent ferroptosis rate by nutritional antemortem and post-mortem antioxidant treatments, within the muscle, is therefore essential for retaining food quality and human health. We hope that the review contributes to the possibility of fostering new scientific research connected to antemortem and post-mortem ferroptosis in muscle food.
Author Contributions
Conceptualized the manuscript J.K.; methodology; J.K. and A.S., contributed results from her Ph.D. Thesis. O.T. software; J.K., A.S. and O.T. validation; J.K., A.S. and O.T. formal analysis; J.K., A.S. and O.T. investigation; J.K., A.S. and O.T. writing—original draft preparation, J.K. writing—review and editing; J.K. and O.T. visualization; A.S. supervision, project administration, J.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors thank Irena Peri for editing and Eylon Asido for the technical help with this manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
ACSL4: long-chain-fatty-acid-CoA ligase 4, AIFM2: apoptosis inducing factor mitochondria associated 2, ALOX: polyunsaturated fatty acid lipoxygenase, ALEs: advanced lipid oxidation end products, AMPK: 5 ′-AMP-activated protein kinase, ACO1: aconitase1, ATF4: activating transcription factor 4, α-KG: alpha-ketoglutarate, CoQ: coenzyme Q, CoQH2: reduced CoQ, cPLA2: cytosolic phospholipase A2, CISD1: CDGSH iron-sulfur domain-containing protein 1, CP: ceruloplasmin, CA9: carbonic anhydrase 9, DMT1: divalent metal transporter 1, ETC: electron transport chain, FA-CoA: fatty acyl-CoA, FA-PL: 1-acyl phospholipid, FSP1: ferroptosis suppressor protein 1, Fe 2þ: reduced iron, Fe 3þ: oxidized iron, Fe–S cluster: iron sulfur cluster, FPN: ferroportin, FTH: ferritin heavy chain, FTMT: mitochondrial ferritin, FA-PL: 1-acyl phospholipid, yGCS: glutamate-cysteine ligase, GPX4: glutathione peroxidase 4, GRX: glutaredoxin, GSSG: glutathione disulfide/oxidized glutathione, GSH: glutathione, GS: glutathione synthetase, GLS: glutaminase, Gln: glutamine, Glu: glutamate, GLUD: glutamate dehydrogenase, GOT1: Glutamic-oxaloacetic transaminase 1 cytoplasmic, GPX4: glutathione peroxidase 4, •OH: hydroxyl radical, PLOO • HO-1: heme oxygenase-1, HIF: hypoxia inducible factor, HILPDA: hypoxia-inducible lipid droplet-associated protein, HSPA5: heat shock protein family A member 5, Keap1: Kelch-like ECH- associated protein 1, LPCAT3: lysophospholipid acyltransferase 3, LIP: labile iron pool, Mfn2: mitofusin 2, miR: microRNA, MFRN: mitoferrin, MUFA: monounsaturated fatty acid, Nf2: merlin, NCOA4: nuclear receptor coactivator 4, Nrf2: nuclear factor erythroid 2-related factor 2, PRDX6: peroxiredoxin 6, PDSS: all trans-polyprenyl-diphosphate synthase, PUFA: polyunsaturated fatty acid, PLOH: hydroxy phospholipid, PLOO•: phospholipid hydroperoxyl radical, PLO•: phospholipid alkyl radical, PL•: phospholipid allyl radical, PLOOH: phospholipid hydroperoxide, PLIN2: perilipin 2, ROS: reactive oxygen species, RTA: radical trapping antioxidants, SLC7A11: cystine/glutamate transporter, SLC1A5: neutral amino acid transporter B, STEAP3: six transmembrane epithelial antigen of prostate metalloreductase, SCD1: stearoyl-CoA desaturase 1, TfR: transferrin receptor, TCA: tricarboxylic acid, Tf: transferrin, SLC7A11: cystine/glutamate transporter, YAP: Yes1 associated transcriptional regulator.
References
- Kanner, J.; German, J.B.; Kinsella, J.E. Initiation of Lipid Peroxidation in Biological Systems. Crit. Rev. Food Sci. Nutr. 1987, 25, 317–364. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in Ferroptosis and Its Pharmacological Implication. Free Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and Function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial Formation of Reactive Oxygen Species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Homma, T.; Kobayashi, S.; Sato, H.; Fujii, J. Superoxide Produced by Mitochondrial Complex III Plays a Pivotal Role in the Execution of Ferroptosis Induced by Cysteine Starvation. Arch. Biochem. Biophys. 2021, 700, 108775. [Google Scholar] [CrossRef]
- Chazelas, P.; Steichen, C.; Favreau, F.; Trouillas, P.; Hannaert, P.; Thuillier, R.; Giraud, S.; Hauet, T.; Guillard, J. Oxidative Stress Evaluation in Ischemia Reperfusion Models: Characteristics, Limits and Perspectives. Int. J. Mol. Sci. 2021, 22, 2366. [Google Scholar] [CrossRef]
- Jin, B.; Zhang, Z.; Zhang, Y.; Yang, M.; Wang, C.; Xu, J.; Zhu, Y.; Mi, Y.; Jiang, J.; Sun, Z. Ferroptosis and Myocardial Ischemia-Reperfusion: Mechanistic Insights and New Therapeutic Perspectives. Front. Pharmacol. 2024, 15, 1482986. [Google Scholar] [CrossRef] [PubMed]
- Kanner, J. Mechanism of Nonenzymic Lipid-Peroxidation in Muscle Foods. ACS Symp. Ser. 1992, 500, 55–73. [Google Scholar] [CrossRef]
- Harel, S.; Kanner, J. Hydrogen-Peroxide Generation in Ground Muscle Tissues. J. Agric. Food Chem. 1985, 33, 1186–1188. [Google Scholar] [CrossRef]
- Kanner, J.; Harel, S. Initiation of Membranal Lipid-Peroxidation by Activated Metmyoglobin and Methemoglobin. Arch. Biochem. Biophys. 1985, 237, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Kohen, R.; Kakunda, A.; Rubinstein, A. The Role of Cationized Catalase and Cationized Glucose Oxidase in Mucosal Oxidative Damage Induced in the Rat Jejunum. J. Biol. Chem. 1992, 267, 21349–21354. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic Accumulation of Succinate Controls Reperfusion Injury through Mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Ma, X.H.; Liu, J.H.Z.; Liu, C.Y.; Sun, W.Y.; Duan, W.J.; Wang, G.; Kurihara, H.; He, R.R.; Li, Y.F.; Chen, Y.; et al. ALOX15-Launched PUFA-Phospholipids Peroxidation Increases the Susceptibility of Ferroptosis in Ischemia-Induced Myocardial Damage. Signal Transduct. Target. Ther. 2022, 7, 288. [Google Scholar] [CrossRef]
- Rochette, L.; Dogon, G.; Rigal, E.; Zeller, M.; Cottin, Y.; Vergely, C. Lipid Peroxidation and Iron Metabolism: Two Corner Stones in the Homeostasis Control of Ferroptosis. Int. J. Mol. Sci. 2023, 24, 449. [Google Scholar] [CrossRef]
- Fuhrmann, D.C.; Brüne, B. A Graphical Journey through Iron Metabolism, MicroRNAs, and Hypoxia in Ferroptosis. Redox Biol. 2022, 54, 102365. [Google Scholar] [CrossRef]
- Badu-Boateng, C.; Naftalin, R.J. Ascorbate and Ferritin Interactions: Consequences for Iron Release in Vitro and in Vivo and Implications for Inflammation. Free Radic. Biol. Med. 2019, 133, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, W.; Chow, C.K. Dietary Vitamin E Reduces Labile Iron in Rat Tissues. J. Biochem. Mol. Toxicol. 2005, 19, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Kanner, J.; Doll, L. Ferritin in Turkey Muscle-Tissue—A Source of Catalytic Iron Ions for Lipid-Peroxidation. J. Agric. Food Chem. 1991, 39, 247–249. [Google Scholar] [CrossRef]
- Kanner, J.; Salan, M.A.; Harel, S.; Shegalovich, J. Lipid-Peroxidation of Muscle Food—The Role of the Cytosolic Fraction. J. Agric. Food Chem. 1991, 39, 242–246. [Google Scholar] [CrossRef]
- Bechaux, J.; de La Pomélie, D.; Théron, L.; Santé-Lhoutellier, V.; Gatellier, P. Iron-Catalysed Chemistry in the Gastrointestinal Tract: Mechanisms, Kinetics and Consequences. A Review. Food Chem. 2018, 268, 27–39. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized Arachidonic and Adrenic PEs Navigate Cells to Ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Hinman, A.; Holst, C.R.; Latham, J.C.; Bruegger, J.J.; Ulas, G.; McCusker, K.P.; Amagata, A.; Davis, D.; Hoff, K.G.; Kahn-Kirby, A.H.; et al. Vitamin E Hydroquinone Is an Endogenous Regulator of Ferroptosis via Redox Control of 15-Lipoxygenase. PLoS ONE 2018, 13, e0201369. [Google Scholar] [CrossRef]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Granit, R.; Angel, S.; Akiri, B.; Holzer, Z.; Aharoni, Y.; Orlov, A.; Kanner, J. Effects of Vitamin E Supplementation on Lipid Peroxidation and Color Retention of Salted Calf Muscle from a Diet Rich in Polyunsaturated Fatty Acids. J. Agric. Food Chem. 2001, 49, 5951–5956. [Google Scholar] [CrossRef]
- Kanner, J.; Sofer, F.; Harel, S.; Doll, L. Antioxidant Activity of Ceruloplasmin in Muscle Membrane and Insitu Lipid-Peroxidation. J. Agric. Food Chem. 1988, 36, 415–417. [Google Scholar] [CrossRef]
- Dragoev, S.G. Lipid Peroxidation in Muscle Foods: Impact on Quality, Safety and Human Health. Foods 2024, 13, 797. [Google Scholar] [CrossRef] [PubMed]
- Kanner, J.; Harel, S.; Hazan, B. Muscle Membranal Lipid-Peroxidation by an Iron Redox Cycle System—Initiation by Oxy Radicals and Site-Specific Mechanism. J. Agric. Food Chem. 1986, 34, 506–510. [Google Scholar] [CrossRef]
- Rhee, S.G.; Woo, H.A. Multiple Functions of 2-Cys Peroxiredoxins, I and II, and Their Regulations via Post-Translational Modifications. Free Radic. Biol. Med. 2020, 152, 107–115. [Google Scholar] [CrossRef]
- Halliwell, B. Understanding Mechanisms of Antioxidant Action in Health and Disease. Nat. Rev. Mol. Cell Biol. 2024, 25, 13–33. [Google Scholar] [CrossRef]
- Fujii, J.; Yamada, K.I. Defense Systems to Avoid Ferroptosis Caused by Lipid Peroxidation-Mediated Membrane Damage. Free Radic. Res. 2023, 57, 353–372. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 Is a Glutathione-Independent Ferroptosis Suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Zheng, D.; Liu, J.; Piao, H.; Zhu, Z.; Wei, R.; Liu, K. ROS-Triggered Endothelial Cell Death Mechanisms: Focus on Pyroptosis, Parthanatos, and Ferroptosis. Front. Immunol. 2022, 13, 1039241. [Google Scholar] [CrossRef]
- Kanner, J. Food Polyphenols as Preventive Medicine. Antioxidants 2023, 12, 2103. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M. Lipid Peroxidation and Ferroptosis: The Role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.M.; Decker, E.A. Endogenous Skeletal Muscle Antioxidants. Crit. Rev. Food Sci. Nutr. 1994, 34, 403–426. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lange, M.; Dixon, S.J.; Olzmann, J.A. Lipid Quality Control and Ferroptosis: From Concept to Mechanism. Annu. Rev. Biochem. 2024, 93, 499–528. [Google Scholar] [CrossRef]
- Lapidot, T.; Granit, R.; Kanner, J. Lipid Hydroperoxidase Activity of Myoglobin and Phenolic Antioxidants in Simulated Gastric Fluid. J. Agric. Food Chem. 2005, 53, 3391–3396. [Google Scholar] [CrossRef]
- Reeder, B.J.; Wilson, M.T. The Effects of pH on the Mechanism of Hydrogen Peroxide and Lipid Hydroperoxide Consumption by Myoglobin: A Role for the Protonated Ferryl Species. Free Rad. Biol. Med. 2001, 30, 1311–1318. [Google Scholar] [CrossRef]
- Kröger-Ohlsen, M.V.; Andersen, M.L.; Skibsted, L.H. Acid-Catalysed Autoreduction of Ferrylmyoglobin in Aqueous Solution Studied by Freeze Quenching and ESR Spectroscopy. Free Radic. Res. 1999, 30, 305–314. [Google Scholar] [CrossRef]
- Flögel, U.; Gödecke, A.; Klotz, L.; Schrader, J. Role of Myoglobin in the Antioxidant Defense of the Heart. FASEB J. 2004, 18, 1156–1158. [Google Scholar] [CrossRef] [PubMed]
- Rabkin, B.; Tirosh, O.; Kanner, J. Reactivity of Vitamin e as an Antioxidant in Red Meat and the Stomach Medium. J. Agric. Food Chem. 2022, 70, 12172–12179. [Google Scholar] [CrossRef]
- Adepu, K.K.; Anishkin, A.; Adams, S.H.; Chintapalli, S. V A Versatile Delivery Vehicle for Cellular Oxygen and Fuels, or Metabolic Sensor?—A Review and Perspective on the Functions of Myoglobin. Physiol. Rev. 2024, 104, 1611–1642. [Google Scholar] [CrossRef] [PubMed]
- Kanner, J.; Bengera, I.; Berman, S. Nitric-Oxide Myoglobin as an Inhibitor of Lipid Oxidation. Lipids 1980, 15, 944–948. [Google Scholar] [CrossRef]
- Baron, C.P.; Møller, J.K.S.; Skibsted, L.H.; Andersen, H.J. Nitrosylmyoglobin as Antioxidant-Kinetics and Proposed Mechanism for Reduction of Hydroperoxides. Free Radic. Res. 2007, 41, 892–902. [Google Scholar] [CrossRef]
- Thomas, S.R.; Stocker, R. Forum: Role of Oxidation in Atherosclerosis Molecular Action of Vitamin E in Lipoprotein Oxidation: Implications for Atherosclerosis. Free Radic. Biol. Med. 2000, 28, 1795–1805. [Google Scholar] [CrossRef]
- Arnold, R.N.; Scheller, K.K.; Arp, S.C.; Williamsz, S.N.; Buege, D.R.; Schaefer3, D.M. Effect of Long-or Short-Term Feeding of a-Tocopheryl Acetate to Holstein and Crossbred Beef Steers on Performance, Carcass Characteristics, and Beef Color Stability. Animal. Sci. 1992, 70, 3055–3065. [Google Scholar] [CrossRef]
- Buckley, D.J.; Morrissey, P.A.; Gray, J.I. Influence of Dietary Vitamin E on the Oxidative Stability and Quality of Pig Meat. J. Anim. Sci. 1994, 73, 3122–3130. [Google Scholar] [CrossRef]
- Zheng, K.; Dong, Y.; Yang, R.; Liang, Y.; Wu, H.; He, Z. Regulation of Ferroptosis by Bioactive Phytochemicals: Implications for Medical Nutritional Therapy. Pharmacol. Res. 2021, 168, 105580. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wei, Y.; Hua, H.; Jing, X.; Zhu, H.; Xiao, K.; Zhao, J.; Liu, Y. Polyphenols Sourced from Ilex latifolia Thunb. Relieve Intestinal Injury via Modulating Ferroptosis in Weanling Piglets under Oxidative Stress. Antioxidants 2022, 11, 966. [Google Scholar] [CrossRef]
- Wang, B.; Yang, L.N.; Yang, L.T.; Liang, Y.; Guo, F.; Fu, P.; Ma, L. Fisetin Ameliorates Fibrotic Kidney Disease in Mice via Inhibiting ACSL4-Mediated Tubular Ferroptosis. Acta Pharmacol. Sin. 2024, 45, 150–165. [Google Scholar] [CrossRef]
- Serra, V.; Salvatori, G.; Pastorelli, G. Dietary Polyphenol Supplementation in Food Producing Animals: Effects on the Quality of Derived Products. Animals 2021, 11, 401. [Google Scholar] [CrossRef]
- Mihaylova, D.; Dimitrova-Dimova, M.; Popova, A. Dietary Phenolic Compounds—Wellbeing and Perspective Applications. Int. J. Mol. Sci. 2024, 25, 4769. [Google Scholar] [CrossRef]
- Shearer, M.J.; Okano, T. Annual Review of Nutrition Key Pathways and Regulators of Vitamin K Function and Intermediary Metabolism. Annu. Rev. Nutr. 2018, 38, 127–151. [Google Scholar] [CrossRef]
- Vervoort, L.M.T.; Ronden~, J.E.; Thijssen, H.H.W. The Potent Antioxidant Activity of the Vitamin K Cycle in Microsomal Lipid. Peroxidation 1997, 54, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Mishima, E.; Ito, J.; Wu, Z.; Nakamura, T.; Wahida, A.; Doll, S.; Tonnus, W.; Nepachalovich, P.; Eggenhofer, E.; Aldrovandi, M.; et al. A Non-Canonical Vitamin K Cycle Is a Potent Ferroptosis Suppressor. Nature 2022, 608, 778–783. [Google Scholar] [CrossRef]
- Poeggeler, B.; Singh, S.K.; Sambamurti, K.; Pappolla, M.A. Nitric Oxide as a Determinant of Human Longevity and Health Span. Int. J. Mol. Sci. 2023, 24, 14533. [Google Scholar] [CrossRef] [PubMed]
- Gantner, B.N.; LaFond, K.M.; Bonini, M.G. Nitric Oxide in Cellular Adaptation and Disease. Redox Biol. 2020, 34, 101550. [Google Scholar] [CrossRef]
- Kanner, J.; Harel, S.; Shagalovich, J.; Berman, S. Antioxidative Effect of Nitrite in Cured Meat-Products—Nitric-Oxide Iron Complexes of Low-Molecular Weight. J. Agric. Food Chem. 1984, 32, 512–515. [Google Scholar] [CrossRef]
- Kanner, J.; Harel, S.; Rina, G. Nitric Oxide as an Antioxidant. Arch. Biochem. Biophys. 1991, 289, 130–136. [Google Scholar] [CrossRef]
- Kanner, J.; Shpaizer, A.; Nelgas, L.; Tirosh, O. S-Nitroso-N-Acetylcysteine (NAC-SNO) as an Antioxidant in Cured Meat and Stomach Medium. J. Agric. Food Chem. 2019, 67, 10930–10936. [Google Scholar] [CrossRef]
- Volk, J.; Gorelik, S.; Granit, R.; Kohen, R.; Kanner, J. The Dual Function of Nitrite under Stomach Conditions Is Modulated by Reducing Compounds. Free Radic. Biol. Med. 2009, 47, 496–502. [Google Scholar] [CrossRef]
- Radi, R. Oxygen Radicals, Nitric Oxide, and Peroxynitrite: Redox Pathways in Molecular Medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef]
- Soula, M.; Weber, R.A.; Zilka, O.; Alwaseem, H.; La, K.; Yen, F.; Molina, H.; Garcia-Bermudez, J.; Pratt, D.A.; Birsoy, K. Metabolic Determinants of Cancer Cell Sensitivity to Canonical Ferroptosis Inducers. Nat. Chem. Biol. 2020, 16, 1351–1360. [Google Scholar] [CrossRef]
- Skibsted, L.H. Nitric Oxide and Quality and Safety of Muscle Based Foods. Nitric Oxide 2011, 24, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Homma, T.; Kobayashi, S.; Conrad, M.; Konno, H.; Yokoyama, C.; Fujii, J. Nitric Oxide Protects against Ferroptosis by Aborting the Lipid Peroxidation Chain Reaction. Nitric Oxide 2021, 115, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Møller, J.K.S.; Skibsted, L.H. Nitric Oxide and Myoglobins. Chem. Rev. 2002, 102, 1167–1178. [Google Scholar] [CrossRef]
- Kanner, J.; Harel, S.; Granit, R. Nitric-Oxide, an Inhibitor of Lipid Oxidation by Lipoxygenase, Cyclooxygenase and Hemoglobin. Lipids 1992, 27, 46–49. [Google Scholar] [CrossRef]
- Wood, I.; Trostchansky, A.; Rubbo, H. Structural Considerations on Lipoxygenase Function, Inhibition and Crosstalk with Nitric Oxide Pathways. Biochimie 2020, 178, 170–180. [Google Scholar] [CrossRef]
- Abu-Halaka, D.; Shpaizer, A.; Zeigerman, H.; Kanner, J.; Tirosh, O. DMF-Activated Nrf2 Ameliorates Palmitic Acid Toxicity While Potentiates Ferroptosis Mediated Cell Death: Protective Role of the NO-Donor S-Nitroso-N-Acetylcysteine. Antioxidants 2023, 12, 512. [Google Scholar] [CrossRef] [PubMed]
- Zilka, O.; Shah, R.; Li, B.; Friedmann Angeli, J.P.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vučković, A.M.; Bosello Travain, V.; Zaccarin, M.; Zennaro, L.; et al. Insight into the Mechanism of Ferroptosis Inhibition by Ferrostatin-1. Redox Biol. 2020, 28, 101328. [Google Scholar] [CrossRef] [PubMed]
- Faraji, P.; Borchert, A.; Ahmadian, S.; Kuhn, H. Butylated Hydroxytoluene (BHT) Protects SH-SY5Y Neuroblastoma Cells from Ferroptotic Cell Death: Insights from In Vitro and In Vivo Studies. Antioxidants 2024, 13, 242. [Google Scholar] [CrossRef]
- Yu, H.; Chen, X.; Guo, X.; Chen, D.; Jiang, L.; Qi, Y.; Shao, J.; Tao, L.; Hang, J.; Lu, G.; et al. The Clinical Value of Serum Xanthine Oxidase Levels in Patients with Acute Ischemic Stroke. Redox Biol. 2023, 60, 102623. [Google Scholar] [CrossRef]
- Kanner, J. Oxidative Processes in Meat and Meat-Products—Quality Implications. Meat Sci. 1994, 36, 169–189. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).