

The Triterpenoid CDDO-Methyl Ester Reduces Tumor Burden, Reprograms the Immune Microenvironment, and Protects from Chemotherapy-Induced Toxicity in a Preclinical Mouse Model of Established Lung Cancer


Abstract
1. Introduction
2. Materials and Methods
3. Results
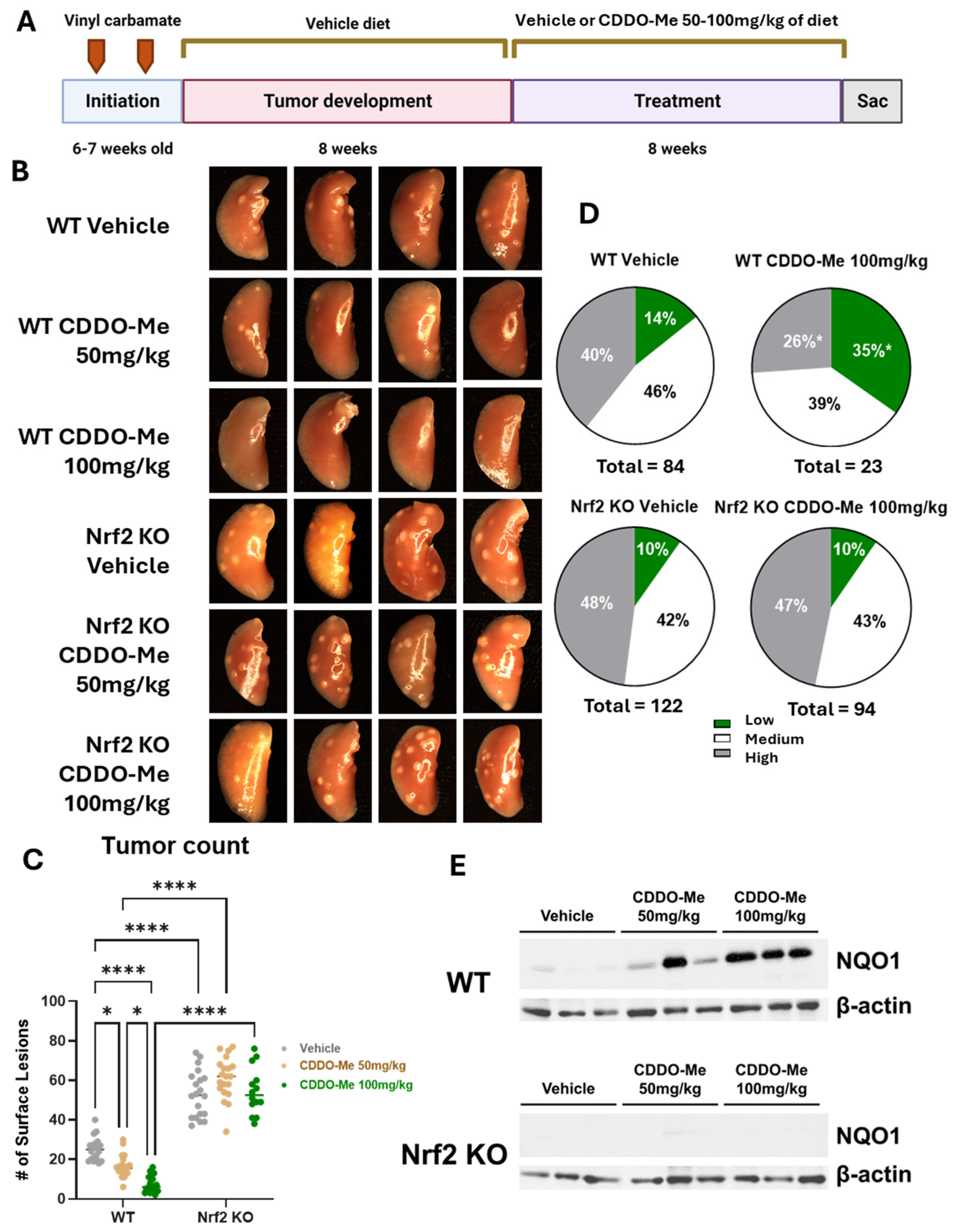
3.1. Study 1: CDDO-Me Reduces Tumor Burden and Histopathological Severity of Established Lung Tumors in a Dose- and Nrf2-Dependent Manner
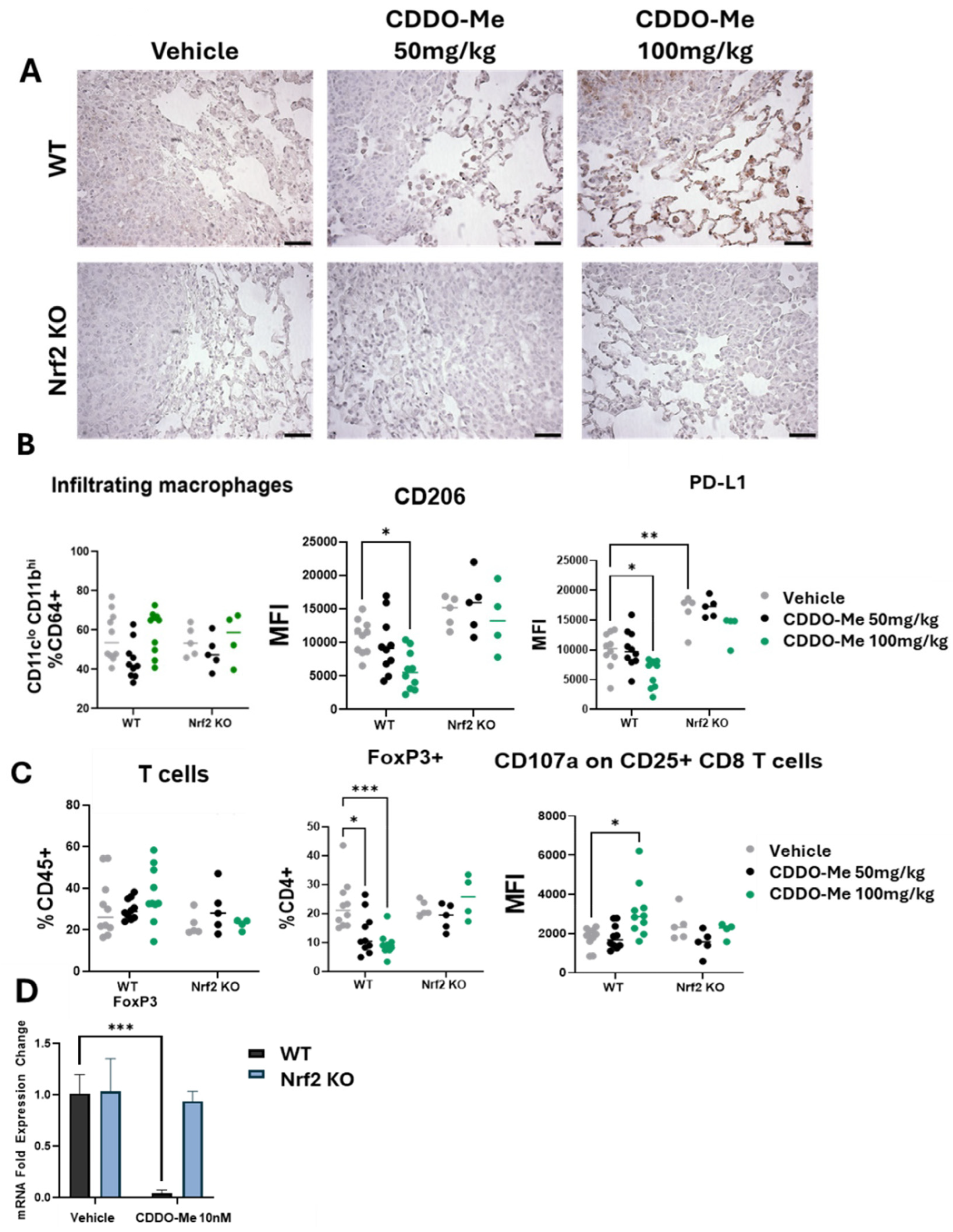
3.2. CDDO-Me Increases NQO1 Expression in the Lungs of WT but Not Nrf2 KO Mice and Modulates Activity but Not Infiltration of Macrophages and T Cells
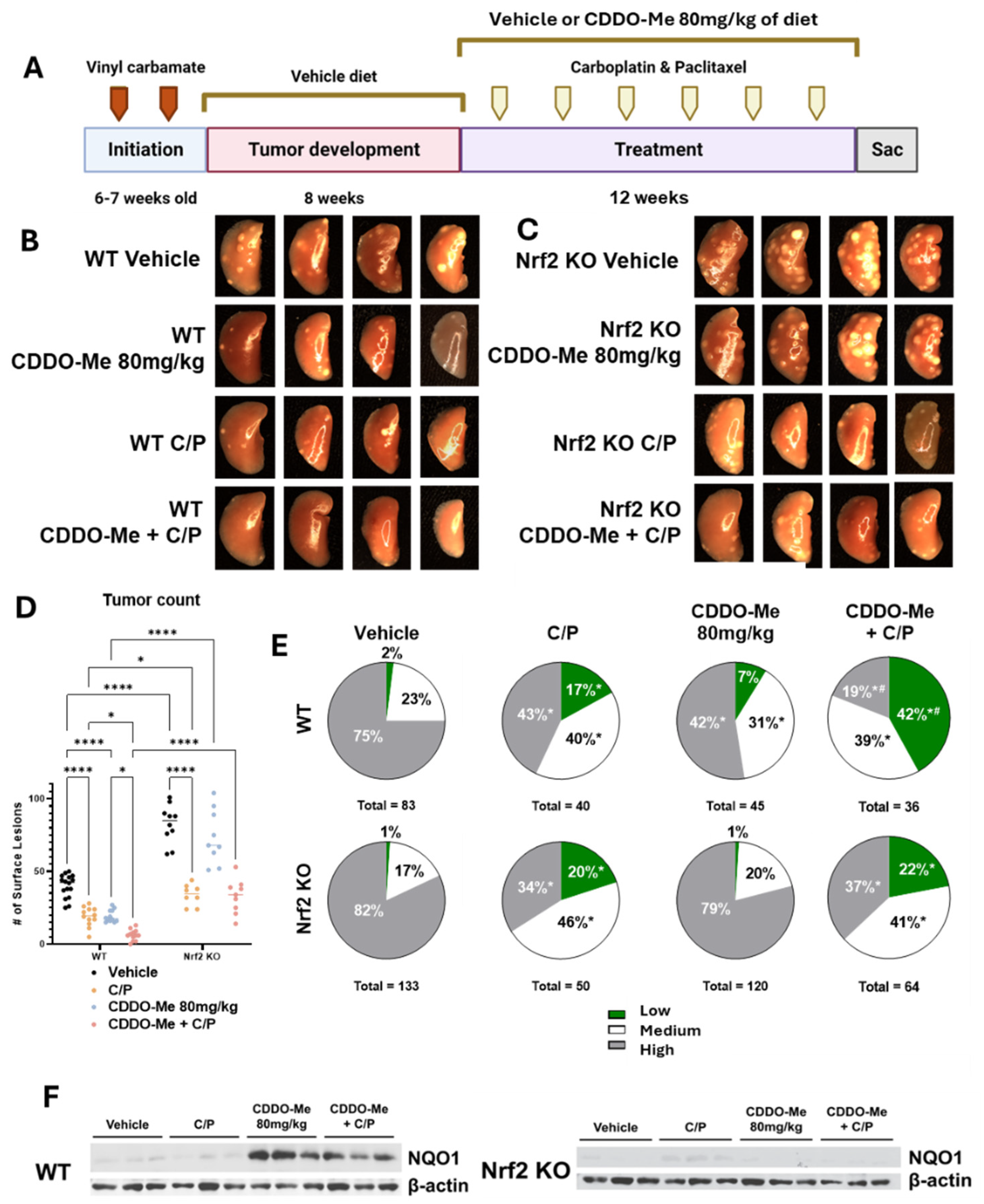
3.3. Study 2: CDDO-Me, Alone and in Combination with Carboplatin and Paclitaxel, Reduces Lung Tumor Burden and the Severity of Tumor Histopathology in a Nrf2-Specific Manner
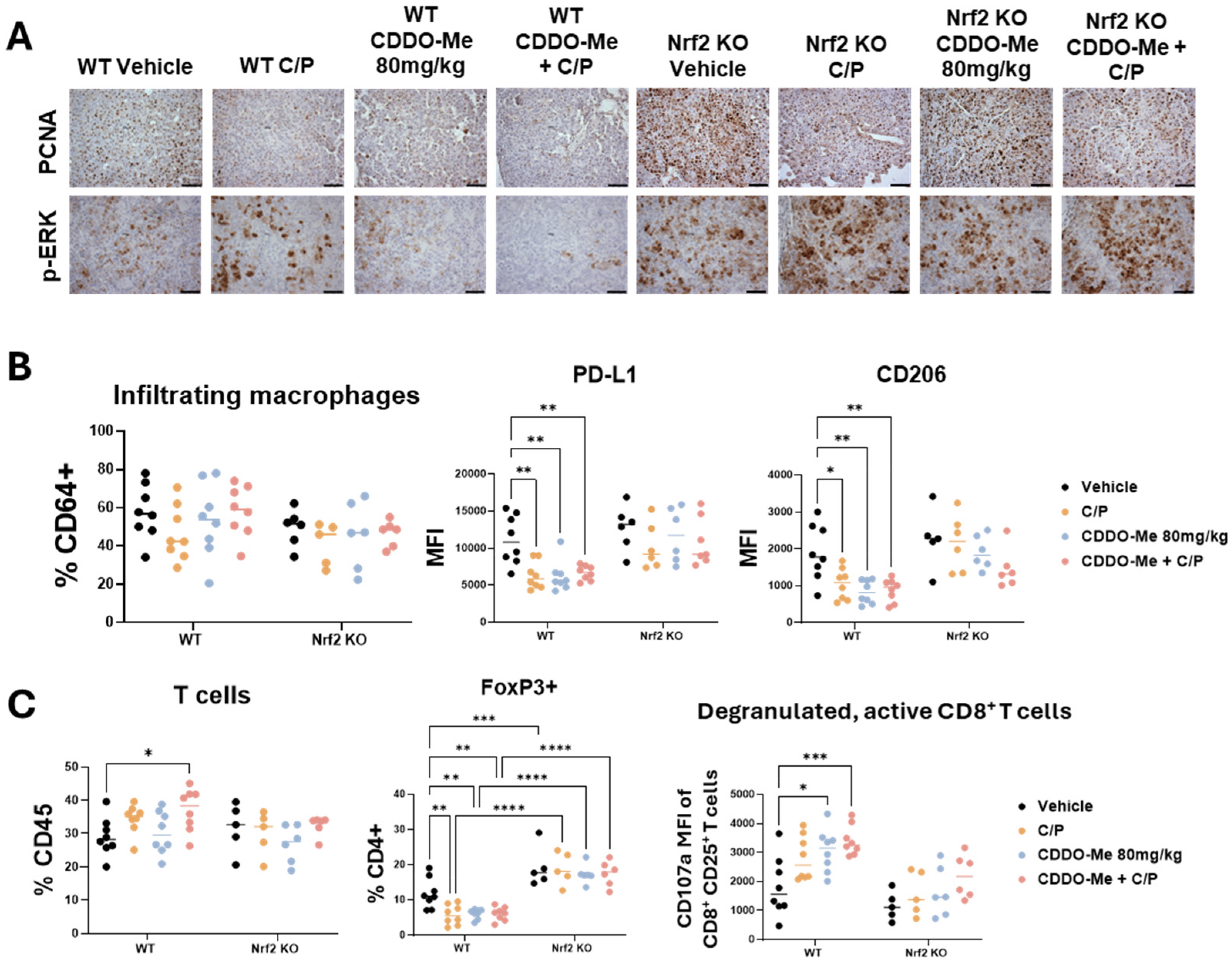
3.4. CDDO-Me and Chemotherapy Alter Immune Cell Phenotype
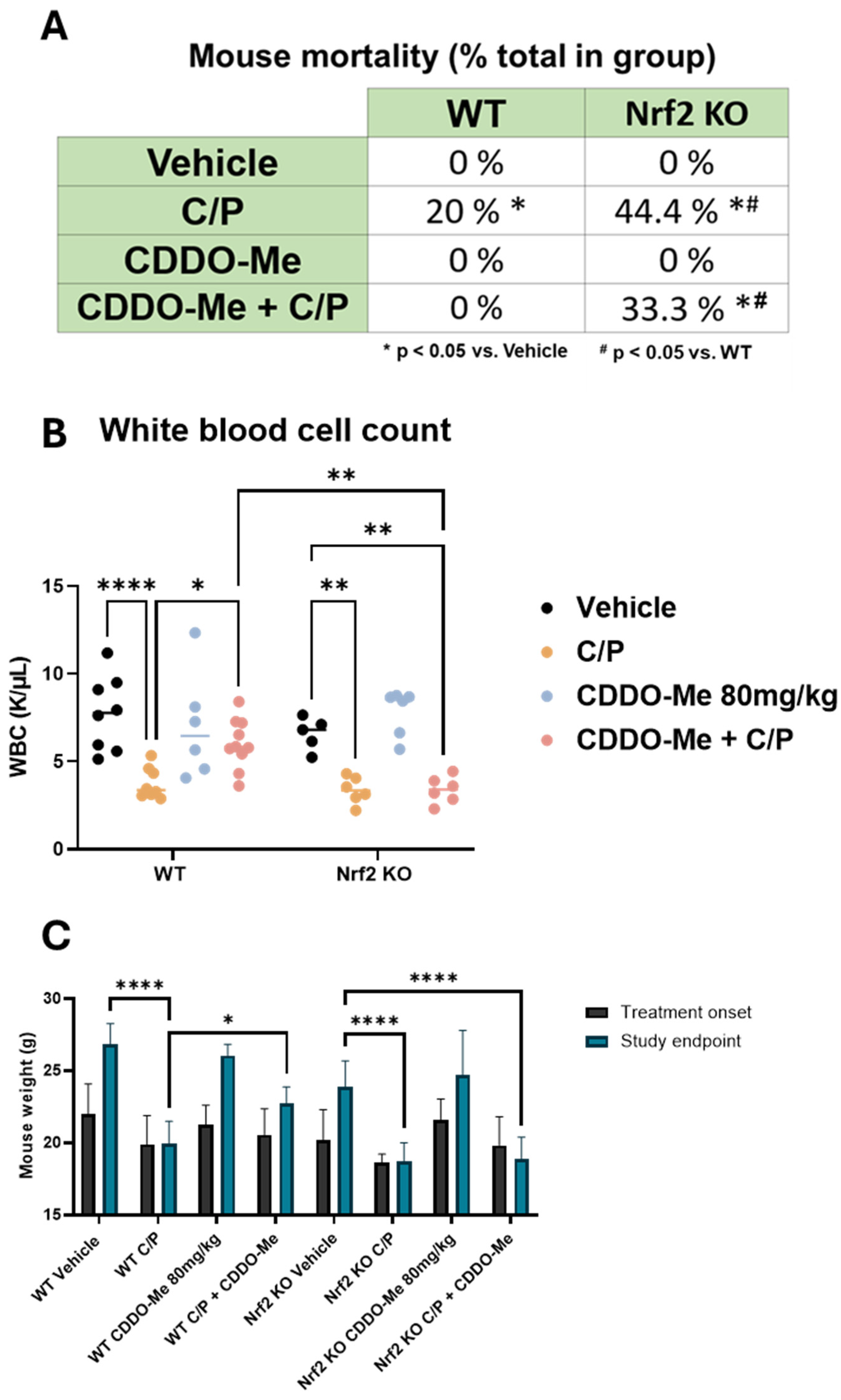
3.5. CDDO-Me Protects from Chemotherapy-Induced Toxicity and Mortality in WT but Not Nrf2 KO Mice
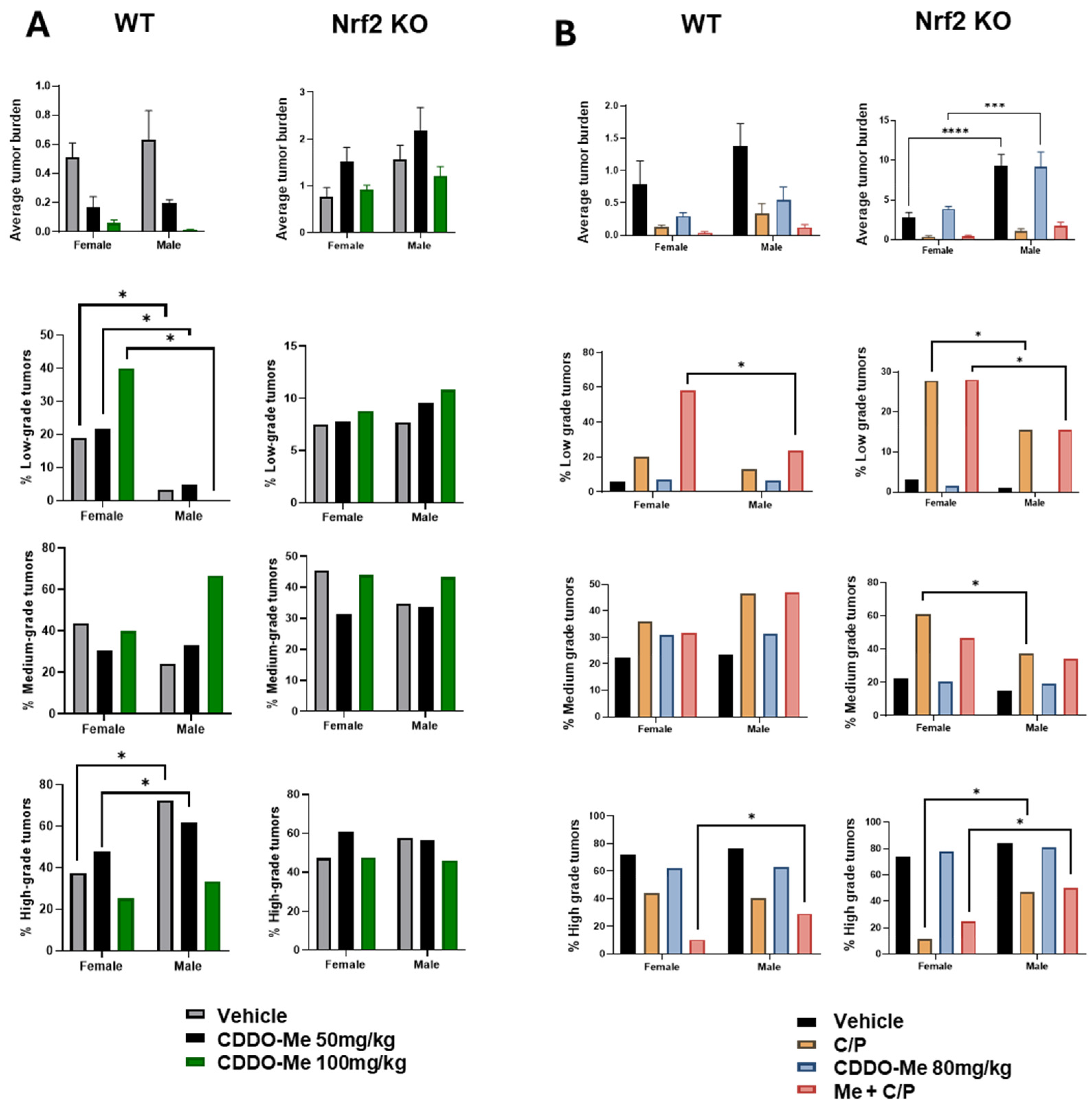
3.6. Increased Tumor Burden and Histopathological Severity in Male vs. Female Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer Statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Nawaz, K.; Webster, R.M. The Non-Small-Cell Lung Cancer Drug Market. Nat. Rev. Drug Discov. 2023, 22, 264–265. [Google Scholar] [CrossRef]
- Casaluce, F.; Gridelli, C. Combined Chemo-Immunotherapy in Advanced Non-Small Cell Lung Cancer: Feasible in the Elderly? Expert. Opin. Emerg. Drugs 2023, 28, 121–127. [Google Scholar] [CrossRef]
- Wang, X.; Qiao, Z.; Aramini, B.; Lin, D.; Li, X.; Fan, J. Potential Biomarkers for Immunotherapy in Non-Small-Cell Lung Cancer. Cancer Metastasis Rev. 2023, 42, 661–675. [Google Scholar] [CrossRef] [PubMed]
- Di Federico, A.; Ricciotti, I.; Favorito, V.; Michelina, S.V.; Scaparone, P.; Metro, G.; De Giglio, A.; Pecci, F.; Lamberti, G.; Ambrogio, C.; et al. Resistance to Kras G12c Inhibition in Non-Small Cell Lung Cancer. Curr. Oncol. Rep. 2023, 25, 1017–1029. [Google Scholar] [CrossRef]
- Altorki, N.K.; Markowitz, G.J.; Gao, D.; Port, J.L.; Saxena, A.; Stiles, B.; McGraw, T.; Mittal, V. The Lung Microenvironment: An Important Regulator of Tumour Growth and Metastasis. Nat. Rev. Cancer 2019, 19, 9–31. [Google Scholar] [CrossRef]
- Shinohara, S.; Takahashi, Y.; Komuro, H.; Matsui, T.; Sugita, Y.; Demachi-Okamura, A.; Muraoka, D.; Takahara, H.; Nakada, T.; Sakakura, N.; et al. New Evaluation of the Tumor Immune Microenvironment of Non-Small Cell Lung Cancer and Its Association with Prognosis. J. ImmunoTherapy Cancer 2022, 10, e003765. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yuan, M.; Wang, C.; Chen, Y.; Zhang, Y.; Zhang, J. T Lymphocyte Cell: A Pivotal Player in Lung Cancer. Front. Immunol. 2023, 14, 1102778. [Google Scholar] [CrossRef] [PubMed]
- Essogmo, F.E.; Zhilenkova, A.V.; Tchawe, Y.S.N.; Owoicho, A.M.; Rusanov, A.S.; Boroda, A.; Pirogova, Y.N.; Sangadzhieva, Z.D.; Sanikovich, V.D.; Bagmet, N.N.; et al. Cytokine Profile in Lung Cancer Patients: Anti-Tumor and Oncogenic Cytokines. Cancers 2023, 15, 5383. [Google Scholar] [CrossRef]
- Cui, Z.; Ruan, Z.; Li, M.; Ren, R.; Ma, Y.; Zeng, J.; Sun, J.; Ye, W.; Xu, W.; Guo, X.; et al. Intermittent Hypoxia Inhibits Anti-Tumor Immune Response Via Regulating Pd-L1 Expression in Lung Cancer Cells and Tumor-Associated Macrophages. Int. Immunopharmacol. 2023, 122, 110652. [Google Scholar] [CrossRef]
- Yang, Y.; Bazhin, A.V.; Werner, J.; Karakhanova, S. Reactive Oxygen Species in the Immune System. Int. Rev. Immunol. 2013, 32, 249–270. [Google Scholar] [CrossRef] [PubMed]
- Yarosz, E.L.; Chang, C.-H. The Role of Reactive Oxygen Species in Regulating T Cell-Mediated Immunity and Disease. Immune Netw. 2018, 18, e14. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Kuang, Z.; Zhang, D.; Gao, Y.; Ying, M.; Wang, T. Reactive Oxygen Species in Immune Cells: A New Antitumor Target. Biomed. Pharmacother. 2021, 133, 110978. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Takahashi, J.; Yamamoto, M. Molecular Basis of the Keap1-Nrf2 Signaling Pathway. Mol. Cells 2023, 46, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, S.; Patinen, T.; Deen, A.J.; Pitkänen, S.; Härkönen, J.; Kansanen, E.; Küblbeck, J.; Levonen, A.L. The Keap1-Nrf2 Pathway: Targets for Therapy and Role in Cancer. Redox Biol. 2023, 63, 102726. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Ortega, M.; Carrera, A.C.; Garrido, A. Role of Nrf2 in Lung Cancer. Cells 2021, 10, 1879. [Google Scholar] [CrossRef]
- Brüne, B.; Dehne, N.; Grossmann, N.; Jung, M.; Namgaladze, D.; Schmid, T.; von Knethen, A.; Weigert, A. Redox Control of Inflammation in Macrophages. Antioxid. Redox Signal 2013, 19, 595–637. [Google Scholar] [CrossRef]
- Hayashi, M.; Kuga, A.; Suzuki, M.; Panda, H.; Kitamura, H.; Motohashi, H.; Yamamoto, M. Microenvironmental Activation of Nrf2 Restricts the Progression of Nrf2-Activated Malignant Tumors. Cancer Res. 2020, 80, 3331–3344. [Google Scholar] [CrossRef]
- Moerland, J.A.; Leal, A.S.; Lockwood, B.; Demireva, E.Y.; Xie, H.; Krieger-Burke, T.; Liby, K.T. The Triterpenoid Cddo-Methyl Ester Redirects Macrophage Polarization and Reduces Lung Tumor Burden in a Nrf2-Dependent Manner. Antioxidants 2023, 12, 116. [Google Scholar] [CrossRef]
- Liu, L.; Chen, G.; Gong, S.; Huang, R.; Fan, C. Targeting Tumor-Associated Macrophage: An Adjuvant Strategy for Lung Cancer Therapy. Front. Immunol. 2023, 14, 1274547. [Google Scholar] [CrossRef]
- Occhiuto, C.J.; Moerland, J.A.; Leal, A.S.; Gallo, K.A.; Liby, K.T. The Multi-Faceted Consequences of Nrf2 Activation Throughout Carcinogenesis. Mol. Cells 2023, 46, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Robertson, H.; Dinkova-Kostova, A.T.; Hayes, J.D. Nrf2 and the Ambiguous Consequences of Its Activation During Initiation and the Subsequent Stages of Tumourigenesis. Cancers 2020, 12, 3609. [Google Scholar] [CrossRef] [PubMed]
- Liby, K.T.; Sporn, M.B. Synthetic Oleanane Triterpenoids: Multifunctional Drugs with a Broad Range of Applications for Prevention and Treatment of Chronic Disease. Pharmacol. Rev. 2012, 64, 972–1003. [Google Scholar] [CrossRef] [PubMed]
- Liby, K.T. Synthetic Triterpenoids Can Protect against Toxicity without Reducing the Efficacy of Treatment with Carboplatin and Paclitaxel in Experimental Lung Cancer. Dose Response 2014, 12, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Liby, K.; Royce, D.B.; Williams, C.R.; Risingsong, R.; Yore, M.M.; Honda, T.; Gribble, G.W.; Dmitrovsky, E.; Sporn, T.A.; Sporn, M.B. The Synthetic Triterpenoids CDDO-Methyl Ester and CDDO-Ethyl Amide Prevent Lung Cancer Induced by Vinyl Carbamate in A/J Mice. Cancer Res. 2007, 67, 2414–2419. [Google Scholar] [CrossRef]
- Plehn, S.; Wagle, S.; Rupasinghe, H.P.V. Chaga Mushroom Triterpenoids as Adjuncts to Minimally Invasive Cancer Therapies: A Review. Curr. Res. Toxicol. 2023, 5, 100137. [Google Scholar] [CrossRef] [PubMed]
- Altun, İ.; Sonkaya, A. The Most Common Side Effects Experienced by Patients Were Receiving First Cycle of Chemotherapy. Iran. J. Public Health 2018, 47, 1218–1219. [Google Scholar] [PubMed]
- Jiang, S.; Pan, A.W.; Lin, T.Y.; Zhang, H.; Malfatti, M.; Turteltaub, K.; Henderson, P.T.; Pan, C.X. Paclitaxel Enhances Carboplatin-DNA Adduct Formation and Cytotoxicity. Chem. Res. Toxicol. 2015, 28, 2250–2252. [Google Scholar] [CrossRef] [PubMed]
- Titis, A.P.; Forkert, P.-G. Strain-Related Differences in Bioactivation of Vinyl Carbamate and Formation of DNA Adducts in Lungs of a/J, Cd-1, and C57bl/6 Mice. Toxicol. Sci. 2001, 59, 82–91. [Google Scholar] [CrossRef]
- Yu, Y.R.; O’Koren, E.G.; Hotten, D.F.; Kan, M.J.; Kopin, D.; Nelson, E.R.; Que, L.; Gunn, M.D. A Protocol for the Comprehensive Flow Cytometric Analysis of Immune Cells in Normal and Inflamed Murine Non-Lymphoid Tissues. PLoS ONE 2016, 11, e0150606. [Google Scholar] [CrossRef]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef]
- Jaiswal, A.K. Regulation of genes encoding NAD(P)H:quinone oxidoreductases. Free Rad. Bio Med. 2000, 29, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Debacker, J.M.; Gondry, O.; Lahoutte, T.; Keyaerts, M.; Huvenne, W. The Prognostic Value of CD206 in Solid Malignancies: A Systematic Review and Meta-Analysis. Cancers 2021, 13, 3422. [Google Scholar] [CrossRef]
- Diskin, B.; Adam, S.; Cassini, M.F.; Sanchez, G.; Liria, M.; Aykut, B.; Buttar, C.; Li, E.; Sundberg, B.; Salas, R.D.; et al. Pd-L1 Engagement on T Cells Promotes Self-Tolerance and Suppression of Neighboring Macrophages and Effector T Cells in Cancer. Nat. Immunol. 2020, 21, 442–454. [Google Scholar] [CrossRef]
- Shinchi, Y.; Ishizuka, S.; Komohara, Y.; Matsubara, E.; Mito, R.; Pan, C.; Yoshii, D.; Yonemitsu, K.; Fujiwara, Y.; Ikeda, K.; et al. The expression of PD-1 ligand 1 on macrophages and its clinical impacts and mechanisms in lung adenocarcinoma. Cancer Immunol. Immunother. 2022, 71, 2645–2661. [Google Scholar] [CrossRef]
- Sarkar, T.; Dhar, S.; Sa, G. Tumor-Infiltrating T-Regulatory Cells Adapt to Altered Metabolism to Promote Tumor-Immune Escape. Curr. Res. Immunol. 2021, 2, 132–141. [Google Scholar] [CrossRef]
- Aktas, E.; Kucuksezer, U.C.; Bilgic, S.; Erten, G.; Deniz, G. Relationship between Cd107a Expression and Cytotoxic Activity. Cell. Immunol. 2009, 254, 149–154. [Google Scholar] [CrossRef]
- Zubair, H.M.; Khan, M.A.; Gulzar, F.; Alkholief, M.; Malik, A.; Akhtar, S.; Sharif, A.; Akhtar, M.F.; Abbas, M. Patient Perspectives and Side-Effects Experience on Chemotherapy of Non-Small Cell Lung Cancer: A Qualitative Study. Cancer Manag. Res. 2023, 15, 449–460. [Google Scholar] [CrossRef]
- Sin, C.; Kim, H.; Im, H.S.; Ock, M.; Koh, S.J. Development and Pilot Study of “Smart Cancer Care”: A Platform for Managing Side Effects of Chemotherapy. BMC Health Serv. Res. 2023, 23, 922. [Google Scholar] [CrossRef] [PubMed]
- Jachowski, A.; Marcinkowski, M.; Szydłowski, J.; Grabarczyk, O.; Nogaj, Z.; Marcin, Ł.; Pławski, A.; Jagodziński, P.P.; Słowikowski, B.K. Modern Therapies of Nonsmall Cell Lung Cancer. J. Appl. Genet. 2023, 64, 695–711. [Google Scholar] [CrossRef]
- Skverchinskaya, E.; Levdarovich, N.; Ivanov, A.; Mindukshev, I.; Bukatin, A. Anticancer Drugs Paclitaxel, Carboplatin, Doxorubicin, and Cyclophosphamide Alter the Biophysical Characteristics of Red Blood Cells, in Vitro. Biology 2023, 12, 230. [Google Scholar] [CrossRef]
- Pirker, R.; Pirolli, M.; Quigley, J.; Hulnick, S.; Legg, J.; Collins, H.; Vansteenkiste, J. Hemoglobin Decline in Cancer Patients Receiving Chemotherapy without an Erythropoiesis-Stimulating Agent. Support. Care Cancer 2013, 21, 987–992. [Google Scholar] [CrossRef]
- Groopman, J.E.; Itri, L.M. Chemotherapy-Induced Anemia in Adults: Incidence and Treatment. JNCI J. Natl. Cancer Inst. 1999, 91, 1616–1634. [Google Scholar] [CrossRef]
- Härkönen, J.; Pölönen, P.; Deen, A.J.; Selvarajan, I.; Teppo, H.R.; Dimova, E.Y.; Kietzmann, T.; Ahtiainen, M.; Väyrynen, J.P.; Väyrynen, S.A.; et al. A Pan-Cancer Analysis Shows Immunoevasive Characteristics in Nrf2 Hyperactive Squamous Malignancies. Redox Biol. 2023, 61, 102644. [Google Scholar] [CrossRef]
- Baird, L.; Taguchi, K.; Zhang, A.; Takahashi, Y.; Suzuki, T.; Kensler, T.W.; Yamamoto, M. A Nrf2-Induced Secretory Phenotype Activates Immune Surveillance to Remove Irreparably Damaged Cells. Redox Biol. 2023, 66, 102845. [Google Scholar] [CrossRef]
- Baird, L.; Yamamoto, M. Immunoediting of Keap1-Nrf2 Mutant Tumours Is Required to Circumvent Nrf2-Mediated Immune Surveillance. Redox Biol. 2023, 67, 102904. [Google Scholar] [CrossRef]
- Lin, L.; Wu, Q.; Lu, F.; Lei, J.; Zhou, Y.; Liu, Y.; Zhu, N.; Yu, Y.; Ning, Z.; She, T.; et al. Nrf2 Signaling Pathway: Current Status and Potential Therapeutic Targetable Role in Human Cancers. Front. Oncol. 2023, 13, 1184079. [Google Scholar] [CrossRef]
- Morris, G.; Gevezova, M.; Sarafian, V.; Maes, M. Redox Regulation of the Immune Response. Cell. Mol. Immunol. 2022, 19, 1079–1101. [Google Scholar] [CrossRef]
- Nakamura, K.; Matsunaga, K. Susceptibility of Natural Killer Cells to Reactive Oxygen Species and Their Restoration by the Mimics of Superoxide Dismutase. Cancer Biother. Radiopharm. 1998, 13, 275–290. [Google Scholar] [CrossRef]
- Jiang, S. Mitochondrial Oxidative Phosphorylation Is Linked to T-Cell Exhaustion. Aging 2020, 12, 16665–16666. [Google Scholar] [CrossRef] [PubMed]
- Kesarwani, P.; Murali, A.K.; Al-Khami, A.A.; Mehrotra, S. Redox Regulation of T-Cell Function: From Molecular Mechanisms to Significance in Human Health and Disease. Antioxid. Redox Signal 2013, 18, 1497–1534. [Google Scholar] [CrossRef] [PubMed]
- Subramony, S.H.; Lynch, D.L. A Milestone in the Treatment of Ataxias: Approval of Omaveloxolone for Friedreich Ataxia. Cerebellum 2023, 23, 775–777. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M.; Kanda, H.; Takama, H.; Ichikawa, T.; Hase, H.; Akizawa, T. Randomized Clinical Trial on the Effect of Bardoxolone Methyl on GFR in Diabetic Kidney Disease Patients (TSUBAKI Study). Kidney Int. Rep. 2020, 5, 879–890. [Google Scholar] [CrossRef]
- Nagaraj, S.; Youn, J.I.; Weber, H.; Iclozan, C.; Lu, L.; Cotter, M.J.; Meyer, C.; Becerra, C.R.; Fishman, M.; Antonia, S.; et al. Anti-Inflammatory Triterpenoid Blocks Immune Suppressive Function of MDSCs and Improves Immune Response in Cancer. Clin. Cancer Res. 2010, 16, 1812–1823. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.S.; Bhandari, R.; Torres, G.M.; Martyanov, V.; ElTanbouly, M.A.; Archambault, K.; Whitfield, M.L.; Liby, K.T.; Pioli, P.A. Cddo-Me Alters the Tumor Microenvironment in Estrogen Receptor Negative Breast Cancer. Sci. Rep. 2020, 10, 6560. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Y.; Zhe, H.; Zhao, R. Preclinical Evidences toward the Use of Triterpenoid CDDO-Me for Solid Cancer Prevention and Treatment. Mol. Cancer 2014, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A.; Ahmed, S.; Brankov, N.; Perloff, M. Triterpenoids as Potential Agents for the Chemoprevention and Therapy of Breast Cancer. Front. Biosci. 2011, 16, 980–996. [Google Scholar] [CrossRef]
- Politi, K.; Cruz, C.S.D.; Homer, R. Thoracic Neoplasia: Carcinoma. In Pathobiology of Human Disease; McManus, L.M., Mitchell, R.N., Eds.; Elsevier, Inc.: San Diego, CA, USA, 2014; pp. 2677–2689. [Google Scholar]
- Dahl, G.A.; Miller, J.A.; Miller, E.C. Vinyl Carbamate as a Promutagen and a More Carcinogenic Analog of Ethyl Carbamate. Cancer Res. 1978, 38, 3793–3804. [Google Scholar] [PubMed]
- Hamarsheh, S.; Groß, O.; Brummer, T.; Zeiser, R. Immune Modulatory Effects of Oncogenic Kras in Cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef] [PubMed]
- Okano, J.; Rustgi, A.K. Paclitaxel Induces Prolonged Activation of the Ras/Mek/Erk Pathway Independently of Activating the Programmed Cell Death Machinery. J. Biol. Chem. 2001, 276, 19555–19564. [Google Scholar] [CrossRef]
- To, C.; Ringelberg, C.S.; Royce, D.B.; Williams, C.R.; Risingsong, R.; Sporn, M.B.; Liby, K.T. Dimethyl Fumarate and the Oleanane Triterpenoids, CDDO-Imidazolide and CDDO-Methyl Ester, Both Activate the Nrf2 Pathway but Have Opposite Effects in the A/J Model of Lung Carcinogenesis. Carcinogenesis 2015, 36, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Pouremamali, F.; Pouremamali, A.; Dadashpour, M.; Soozangar, N.; Jeddi, F. An Update of Nrf2 Activators and Inhibitors in Cancer Prevention/Promotion. Cell Commun. Signal. 2022, 20, 100. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Copple, I.M. Advances and Challenges in Therapeutic Targeting of Nrf2. Trends Pharmacol. Sci. 2023, 44, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Sedighzadeh, S.S.; Khoshbin, A.P.; Razi, S.; Keshavarz-Fathi, M.; Rezaei, N. A Narrative Review of Tumor-Associated Macrophages in Lung Cancer: Regulation of Macrophage Polarization and Therapeutic Implications. Transl. Lung Cancer Res. 2021, 10, 1889–1916. [Google Scholar] [CrossRef] [PubMed]
- Roux, C.; Jafari, S.M.; Shinde, R.; Duncan, G.; Cescon, D.W.; Silvester, J.; Chu, M.F.; Hodgson, K.; Berger, T.; Wakeham, A.; et al. Reactive Oxygen Species Modulate Macrophage Immunosuppressive Phenotype through the up-Regulation of Pd-L1. Proc. Natl. Acad. Sci. USA 2019, 116, 4326–4335. [Google Scholar] [CrossRef] [PubMed]
- Canton, M.; Sánchez-Rodríguez, R.; Spera, I.; Venegas, F.C.; Favia, M.; Viola, A.; Castegna, A. Reactive Oxygen Species in Macrophages: Sources and Targets. Front. Immunol. 2021, 12, 734229. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Geng, J.; Gao, J.; Zhao, H.; Li, J.; Shi, Y.; Yang, B.; Xiao, C.; Linghu, Y.; Sun, X.; et al. Macrophage Achieves Self-Protection against Oxidative Stress-Induced Ageing through the Mst-Nrf2 Axis. Nat. Commun. 2019, 10, 755. [Google Scholar] [CrossRef]
- Gnanaprakasam, J.N.R.; Kushwaha, B.; Liu, L.; Chen, X.; Kang, S.; Wang, T.; Cassel, T.A.; Adams, C.M.; Higashi, R.M.; Scott, D.A.; et al. Asparagine Restriction Enhances Cd8+ T Cell Metabolic Fitness and Antitumoral Functionality through an Nrf2-Dependent Stress Response. Nat. Metab. 2023, 5, 1423–1439. [Google Scholar] [CrossRef]
- Morzadec, C.; Macoch, M.; Sparfel, L.; Kerdine-Römer, S.; Fardel, O.; Vernhet, L. Nrf2 Expression and Activity in Human T Lymphocytes: Stimulation by T Cell Receptor Activation and Priming by Inorganic Arsenic and Tert-Butylhydroquinone. Free Radic. Biol. Med. 2014, 71, 133–145. [Google Scholar] [CrossRef]
- Turley, A.E.; Zagorski, J.; Rockwell, C.E. The Role of Nrf2 in Primary Human Cd4 T Cell Activation and Differentiation. FASEB J. 2017, 31, lb621. [Google Scholar] [CrossRef]
- Pant, A.; Dasgupta, D.; Tripathi, A.; Pyaram, K. Beyond Antioxidation: Keap1-Nrf2 in the Development and Effector Functions of Adaptive Immune Cells. Immunohorizons 2023, 7, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Nk Cells in the Tumor Microenvironment and Thioredoxin Activity. J. Clin. Investig. 2020, 130, 5115–5117. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Tian, Y.; Bai, Z.; Liu, J.; Zhang, Y.; Qi, J.; Jin, M.; Zhu, J.; Li, X. Increased Oxidative Stress Contributes to Impaired Peripheral Cd56(Dim)Cd57(+) Nk Cells from Patients with Systemic Lupus Erythematosus. Arthritis Res. Ther. 2022, 24, 48. [Google Scholar] [CrossRef] [PubMed]
- Stefanie, R.; Takahiro, N.; Isabelle, M.; Jonas, M.; Andreas, L.; Elias, S.J.A.; Rolf, K.; Linnea, W.S. Targeting of Nrf2 Improves Antitumoral Responses by Human Nk Cells, Til and Car T Cells During Oxidative Stress. J. Immunother. Cancer 2022, 10, e004458. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef]
- Yoh, K.; Itoh, K.; Enomoto, A.; Hirayama, A.; Yamaguchi, N.; Kobayashi, M.; Morito, N.; Koyama, A.; Yamamoto, M.; Takahashi, S. Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney Int. 2001, 60, 1343–1353. [Google Scholar] [CrossRef]
- Klemm, P.; Rajendiran, A.; Fragoulis, A.; Wruck, C.; Schippers, A.; Wagner, N.; Bopp, T.; Tenbrock, K.; Ohl, K. Nrf2 expression driven by Foxp3 specific deletion of Keap1 results in loss of immune tolerance in mice. Eur. J. Immunol. 2020, 50, 515–524. [Google Scholar] [CrossRef]
- May, L.; Shows, K.; Nana-Sinkam, P.; Li, H.; Landry, J.W. Sex Differences in Lung Cancer. Cancers 2023, 15, 3111. [Google Scholar] [CrossRef]
- Poleri, C. Sex-Based Differences in Lung Cancer: Does It Matter? J. Thorac. Oncol. 2022, 17, 599–601. [Google Scholar] [CrossRef]
- Stabellini, N.; Bruno, D.S.; Dmukauskas, M.; Barda, A.J.; Cao, L.; Shanahan, J.; Waite, K.; Montero, A.J.; Barnholtz-Sloan, J.S. Sex Differences in Lung Cancer Treatment and Outcomes at a Large Hybrid Academic-Community Practice. JTO Clin. Res. Rep. 2022, 3, 100307. [Google Scholar] [CrossRef]
- Allegra, A.; Caserta, S.; Genovese, S.; Pioggia, G.; Gangemi, S. Gender Differences in Oxidative Stress in Relation to Cancer Susceptibility and Survival. Antioxidants 2023, 12, 1255. [Google Scholar] [CrossRef]
- Ali, I.; Högberg, J.; Hsieh, J.-H.; Auerbach, S.; Korhonen, A.; Stenius, U.; Silins, I. Gender Differences in Cancer Susceptibility: Role of Oxidative Stress. Carcinogenesis 2016, 37, 985–992. [Google Scholar] [CrossRef]
- Martínez de Toda, I.; González-Sánchez, M.; Cerro, E.D.-D.; Valera, G.; Carracedo, J.; Guerra-Pérez, N. Sex Differences in Markers of Oxidation and Inflammation. Implications for Ageing. Mech. Ageing Dev. 2023, 211, 111797. [Google Scholar] [CrossRef] [PubMed]
- Zabłocka-Słowińska, K.; Płaczkowska, S.; Skórska, K.; Prescha, A.; Pawełczyk, K.; Porębska, I.; Kosacka, M.; Grajeta, H. Oxidative Stress in Lung Cancer Patients Is Associated with Altered Serum Markers of Lipid Metabolism. PLoS ONE 2019, 14, e0215246. [Google Scholar] [CrossRef] [PubMed]
- Rakshith, H.T.; Lohita, S.; Rebello, A.P.; Goudanavar, P.S.; Naveen, N.R. Sex Differences in Drug Effects and/or Toxicity in Oncology. Curr. Res. Pharmacol. Drug Discov. 2023, 4, 100152. [Google Scholar] [CrossRef] [PubMed]
- Rampen, F.H.J. Malignant Melanoma: Sex Differences in Response to Chemotherapy? Eur. J. Cancer Clin. Oncol. 1982, 18, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Ramos, C.M.; Quackenbush, J.; DeMeo, D.L. Genome-Wide Sex and Gender Differences in Cancer. Front. Oncol. 2020, 10, 597788. [Google Scholar] [CrossRef]
- Kamble, S.M.; Goyal, S.N.; Patil, C.R. Multifunctional Pentacyclic Triterpenoids as Adjuvants in Cancer Chemotherapy: A Review. RSC Adv. 2014, 4, 33370–33382. [Google Scholar] [CrossRef]
- Parikh, K.; Banna, G.; Liu, S.V.; Friedlaender, A.; Desai, A.; Subbiah, V.; Addeo, A. Drugging KRAS: Current perspectives and state-of-art review. J. Hematol. Oncol. 2022, 15, 152. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | WT Control | WT CDDO-Me 50 mg/kg | Nrf2 KO Control | Nrf2 KO CDDO-Me 50 mg/kg |
|---|---|---|---|---|
| Total surface tumors | 352 | 231 | 812 | 856 |
| Average # of tumors per mouse (% WT Control) | 25.14 ± 1.3 (100%) | 16.5 ± 1.3 (65.6%) * | 62.46 ± 2.6 (248.4%) #### | 61.14 ± 3.2 (243.2%) #### |
| # of mice per group | 14 | 14 | 13 | 14 |
| Average # tumors/slide (% WT Control) | 2.18 ± 0.2 (100%) | 1.57 ± 0.3 (72.1%) | 5.04 ± 0.2 (231.3%) #### | 5.54 ± 0.2 (254.1%) #### |
| Average Tumor Size (mm3) (% WT Control) | 0.26 ± 0.08 (100%) | 0.12 ± 0.05 (45.2%) | 0.35 ± 0.1 (133.1%) | 0.33 ± 0.1 (127.6%) # |
| Average Tumor Burden (mm3) (% WT Control) | 0.57 ± 0.3 (100%) | 0.19 ± 0.3 (32.6%) | 1.75 ± 0.5 (307.7%) # | 1.85 ± 0.6 (324.11%) # |
| Low Grade (% total) | 11.5 | 13.6 | 7.6 | 9.0 |
| Medium Grade (% total) | 34.4 | 31.8 | 38.9 | 32.9 |
| High Grade (% total) | 54.1 | 54.6 | 53.5 | 58.1 |
| B | WT Control | WT CDDO-Me 100 mg/kg | Nrf2 KO Control | Nrf2 KO CDDO-Me 100 mg/kg |
| Total surface tumors | 403 | 125 | 799 | 650 |
| Average # of tumors per mouse (% WT Control) | 22.4 ± 1.3 (100%) | 6.9 ± 1.0 (31%) **** | 53.3 ± 2.6 (237.9%) #### | 54.17 ± 2.4 (241.9%) #### |
| # of mice per group | 18 | 18 | 15 | 12 |
| Average # tumors/slide (% WT Control) | 2.3 ± 0.3 (100%) | 0.64 ± 0.2 (27.4%) *** | 4.07 ± 0.5 (174.3%) ### | 3.92 ± 0.4 (167.9%) #### |
| Average Tumor Size (mm3) (% WT Control) | 0.17 ± 0.09 (100%) | 0.07 ± 0.02 (39.7%) * | 0.29 ± 0.02 (170.7%) | 0.26 ± 0.1 (151.47%) ### |
| Average Tumor Burden (mm3) (% WT Control) | 0.4 ± 0.09 (100%) | 0.04 ± 0.02 (10.9%) *** | 1.19 ± 0.2 (297.5%) #### | 1.01 ± 0.1 (151.5%) #### |
| Low Grade (% total) | 14.3 | 34.8 * | 9.7 | 9.6 # |
| Medium Grade (% total) | 46.4 | 39.1 | 42.3 | 43.6 |
| High Grade (% total) | 39.3 | 26.1 * | 48.0 | 46.8 # |
| WT Control | WT C/P | WT CDDO-Me 80 mg/kg | WT CDDO-Me + C/P | Nrf2 KO Control | Nrf2 KO C/P | Nrf2 KO CDDO-Me 80 mg/kg | Nrf2 KO CDDO-Me + C/P | |
|---|---|---|---|---|---|---|---|---|
| Total surface tumors | 639 | 227 | 231 | 75 | 826 | 271 | 664 | 296 |
| Average per mouse (% WT Control) | 39.9 ± 2.0 (100%) | 18.9 ± 1.9 (47.4) **** | 21 ± 1.2 (52.6%) **** | 5.8 ± 1.1 (14.5%) **** ! | 91.8 ± 4.2 (229.8%) #### | 38.7 ± 2.6 (96.9%) # $$$$ | 73.8 ± 6.2 (184.7%) | 37 ± 3.9 (92.6%) #### $$$$ |
| # of mice per group | 16 | 12 | 10 | 13 | 9 | 7 | 9 | 8 |
| Average # of tumors per slide (% WT Control) | 2.59 ± 0.28 (100%) | 1.67 ± 0.17 (64.3%) * | 2.25 ± 0.32 (86.8%) | 1.38 ± 0.24 (53.38%) * | 7.39 ± 0.78 (284.9%) #### | 3.57 ± 0.64 (137.7%) ### $$$$ | 6.67 ± 0.51 (257%) #### | 4.00 ± 0.54 (154.2%) #### $$$$ |
| Average Tumor Size (mm3/tumor) (% WT Control) | 0.42 ± 0.16 (100%) | 0.12 ± 0.04 (29.1%) ** | 0.17 ± 0.05 (39.7%) * | 0.05 ± 0.02 (12.4%) **** | 0.97 ± 0.45 (232%) ## | 0.19 ± 0.09 (44.9%) * $$$$ | 0.85 ± 0.33 (203.8%) ### | 0.23 ± 0.12 (55.8%) # $$$$ |
| Average Tumor Burden (mm3) (% WT Control) | 1.09 ± 0.25 (100%) | 0.20 ± 0.06 (18.7%) **** | 0.37 ± 0.07 (34.4%) *** | 0.07 ± 0.02 (6.6%) **** ! | 7.17 ± 1.2 (660.9%) #### | 0.67 ± 0.18 (61.8%) # $$$$ | 5.68 ± 0.86 (523.8%) $ #### | 0.93 ± 0.24 (86%) #### |
| Low Grade (% total) | 2.4 | 17.5 * | 6.7 | 41.7 * ! | 1.5 | 20 $ | 0.8# | 21.9# $ |
| Medium Grade (% total) | 22.9 | 40 * | 31.1 * | 38.9 * | 16.5 | 46 $ | 20.0 | 40.6 $ |
| High Grade (% total) | 74.7 | 42.5 * | 62.2 * | 19.4 * ! | 82 | 34 $ | 79.2 | 37.5 # $ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moerland, J.A.; Liby, K.T. The Triterpenoid CDDO-Methyl Ester Reduces Tumor Burden, Reprograms the Immune Microenvironment, and Protects from Chemotherapy-Induced Toxicity in a Preclinical Mouse Model of Established Lung Cancer. Antioxidants 2024, 13, 621. https://doi.org/10.3390/antiox13060621
Moerland JA, Liby KT. The Triterpenoid CDDO-Methyl Ester Reduces Tumor Burden, Reprograms the Immune Microenvironment, and Protects from Chemotherapy-Induced Toxicity in a Preclinical Mouse Model of Established Lung Cancer. Antioxidants. 2024; 13(6):621. https://doi.org/10.3390/antiox13060621
Chicago/Turabian StyleMoerland, Jessica A., and Karen T. Liby. 2024. "The Triterpenoid CDDO-Methyl Ester Reduces Tumor Burden, Reprograms the Immune Microenvironment, and Protects from Chemotherapy-Induced Toxicity in a Preclinical Mouse Model of Established Lung Cancer" Antioxidants 13, no. 6: 621. https://doi.org/10.3390/antiox13060621
APA StyleMoerland, J. A., & Liby, K. T. (2024). The Triterpenoid CDDO-Methyl Ester Reduces Tumor Burden, Reprograms the Immune Microenvironment, and Protects from Chemotherapy-Induced Toxicity in a Preclinical Mouse Model of Established Lung Cancer. Antioxidants, 13(6), 621. https://doi.org/10.3390/antiox13060621