

Crosstalk between Oxidative Stress and Inflammation Caused by Noise and Air Pollution—Implications for Neurodegenerative Diseases




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Air and Noise Pollution—Overview
1.2. Neurodegenerative Disease—Overview
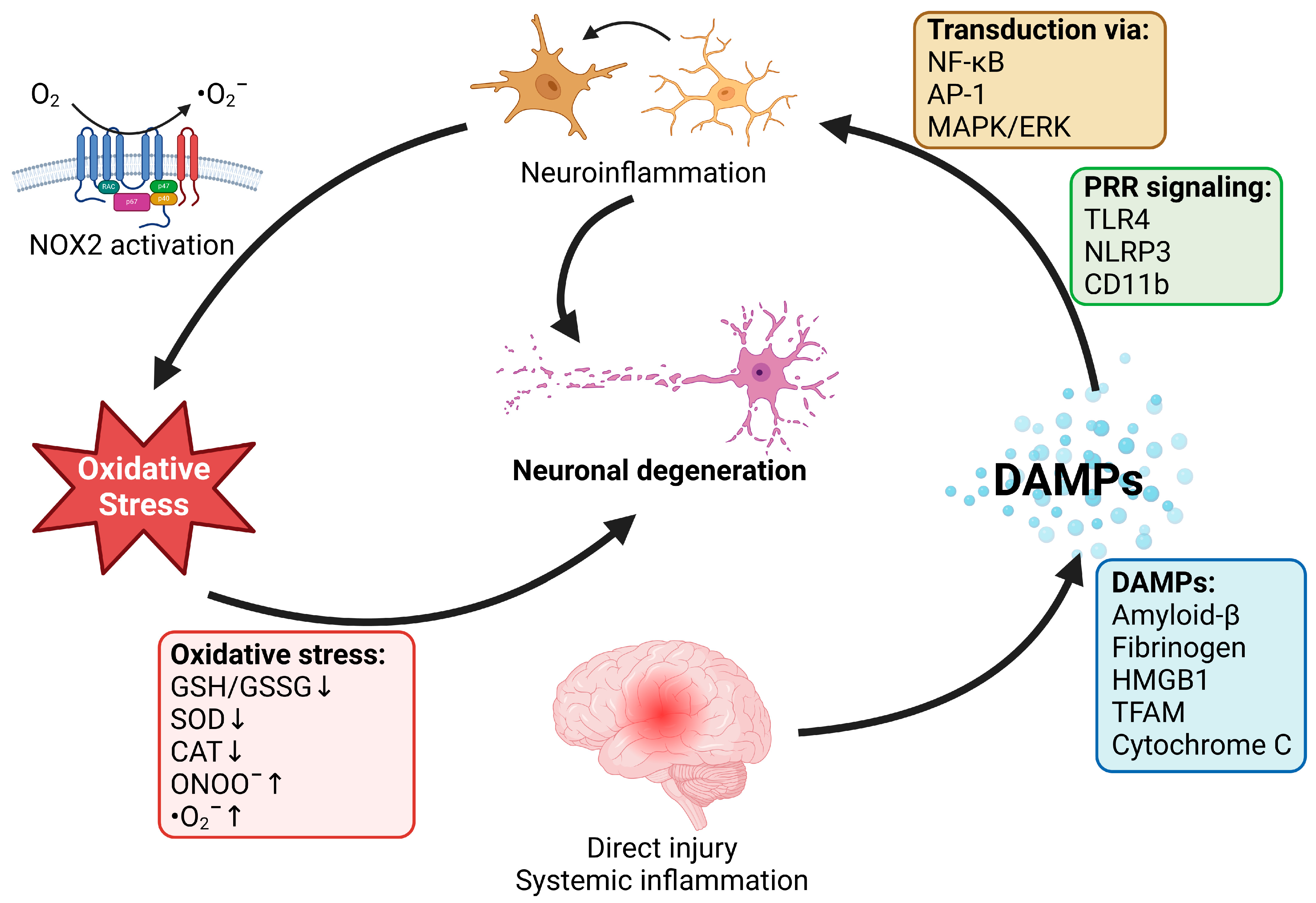
2. Role of Oxidative Stress and Inflammation in Neurodegenerative Diseases
2.1. Neurodegenerative Disease and Oxidative Stress
2.2. Neurodegenerative Diseases and Inflammation
3. Effects of Noise and Air Pollution on Inflammation and Oxidative Stress in Neurodegenerative Disease—Mechanistic Insights from Animal Studies
3.1. Air Pollution
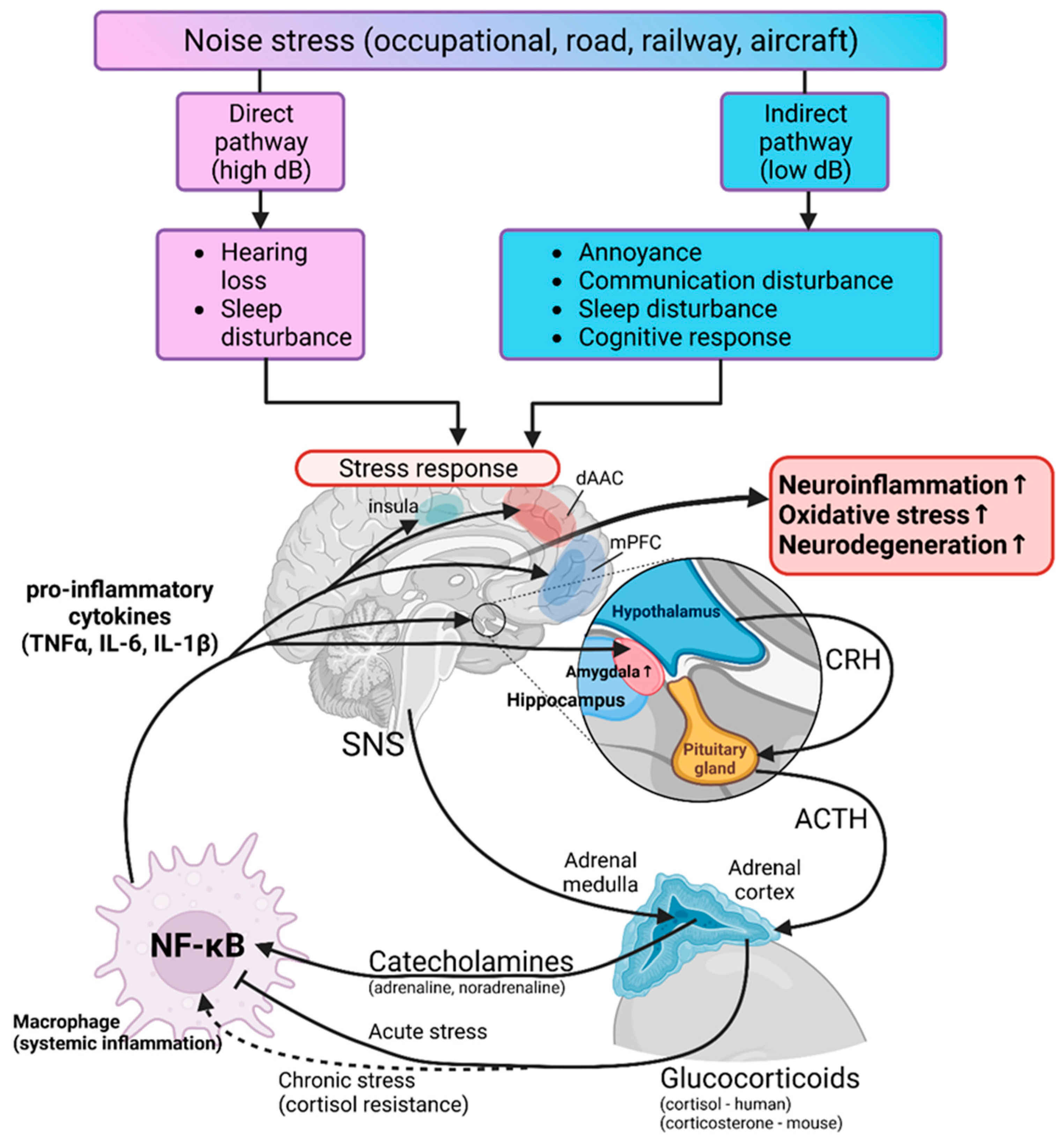
3.2. Transportation Noise
4. Environmental Stressors and Neurodegenerative Disease—Evidence from Epidemiological Studies
4.1. Air Pollution and Neurodegenerative Diseases
4.2. Transportation Noise and Neurodegenerative Diseases
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hahad, O.; Rajagopalan, S.; Lelieveld, J.; Sorensen, M.; Frenis, K.; Daiber, A.; Basner, M.; Nieuwenhuijsen, M.; Brook, R.D.; Munzel, T. Noise and Air Pollution as Risk Factors for Hypertension: Part I-Epidemiology. Hypertension 2023, 80, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Hahad, O.; Rajagopalan, S.; Lelieveld, J.; Sorensen, M.; Kuntic, M.; Daiber, A.; Basner, M.; Nieuwenhuijsen, M.; Brook, R.D.; Munzel, T. Noise and Air Pollution as Risk Factors for Hypertension: Part II-Pathophysiologic Insight. Hypertension 2023, 80, 1384–1392. [Google Scholar] [CrossRef]
- Munzel, T.; Gori, T.; Al-Kindi, S.; Deanfield, J.; Lelieveld, J.; Daiber, A.; Rajagopalan, S. Effects of gaseous and solid constituents of air pollution on endothelial function. Eur. Heart J. 2018, 39, 3543–3550. [Google Scholar] [CrossRef] [PubMed]
- Diseases, G.B.D.; Injuries, C. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- Lelieveld, J.; Klingmuller, K.; Pozzer, A.; Poschl, U.; Fnais, M.; Daiber, A.; Munzel, T. Cardiovascular disease burden from ambient air pollution in Europe reassessed using novel hazard ratio functions. Eur. Heart J. 2019, 40, 1590–1596. [Google Scholar] [CrossRef]
- Vohra, K.; Vodonos, A.; Schwartz, J.; Marais, E.A.; Sulprizio, M.P.; Mickley, L.J. Global mortality from outdoor fine particle pollution generated by fossil fuel combustion: Results from GEOS-Chem. Environ. Res. 2021, 195, 110754. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; Kuntic, M.; Hahad, O.; Delogu, L.G.; Rohrbach, S.; Di Lisa, F.; Schulz, R.; Munzel, T. Effects of air pollution particles (ultrafine and fine particulate matter) on mitochondrial function and oxidative stress—Implications for cardiovascular and neurodegenerative diseases. Arch. Biochem. Biophys. 2020, 696, 108662. [Google Scholar] [CrossRef] [PubMed]
- Pinkerton, K.E.; Green, F.H.; Saiki, C.; Vallyathan, V.; Plopper, C.G.; Gopal, V.; Hung, D.; Bahne, E.B.; Lin, S.S.; Menache, M.G.; et al. Distribution of particulate matter and tissue remodeling in the human lung. Environ. Health Perspect. 2000, 108, 1063–1069. [Google Scholar] [CrossRef]
- Kreyling, W.G.; Semmler-Behnke, M.; Moller, W. Ultrafine particle-lung interactions: Does size matter? J. Aerosol. Med. 2006, 19, 74–83. [Google Scholar] [CrossRef]
- Steven, S.; Oelze, M.; Hausding, M.; Roohani, S.; Kashani, F.; Kroller-Schon, S.; Helmstadter, J.; Jansen, T.; Baum, C.; Iglarz, M.; et al. The Endothelin Receptor Antagonist Macitentan Improves Isosorbide-5-Mononitrate (ISMN) and Isosorbide Dinitrate (ISDN) Induced Endothelial Dysfunction, Oxidative Stress, and Vascular Inflammation. Oxid. Med. Cell Longev. 2018, 2018, 7845629. [Google Scholar] [CrossRef]
- World Health Organization. WHO—Noise. Available online: https://www.who.int/europe/news-room/fact-sheets/item/noise (accessed on 13 February 2024).
- Collaborators, G.B.D.R.F. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1345–1422. [Google Scholar] [CrossRef]
- Wild, C.P. Complementing the genome with an “exposome”: The outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1847–1850. [Google Scholar] [CrossRef]
- Sainani, K. Taking on the exposome—Bringing bioinformatics tools to the environmental side of the health equation. Biomed. Comput. Rev. 2016, 2016, 14–21. [Google Scholar]
- Vrijheid, M. The exposome: A new paradigm to study the impact of environment on health. Thorax 2014, 69, 876–878. [Google Scholar] [CrossRef]
- Hahad, O.; Al-Kindi, S.; Lelieveld, J.; Munzel, T.; Daiber, A. Supporting and implementing the beneficial parts of the exposome: The environment can be the problem, but it can also be the solution. Int. J. Hydrogen Environ. Health 2024, 255, 114290. [Google Scholar] [CrossRef]
- Olden, K.; Wilson, S. Environmental health and genomics: Visions and implications. Nat. Rev. Genet. 2000, 1, 149–153. [Google Scholar] [CrossRef]
- Munzel, T.; Sorensen, M.; Lelieveld, J.; Hahad, O.; Al-Kindi, S.; Nieuwenhuijsen, M.; Giles-Corti, B.; Daiber, A.; Rajagopalan, S. Heart healthy cities: Genetics loads the gun but the environment pulls the trigger. Eur. Heart J. 2021, 42, 2422–2438. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Sorensen, M.; Hahad, O.; Nieuwenhuijsen, M.; Daiber, A. The contribution of the exposome to the burden of cardiovascular disease. Nat. Rev. Cardiol. 2023, 20, 651–669. [Google Scholar] [CrossRef] [PubMed]
- Collaborators, G.B.D.C.o.D. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef]
- Assoc, A.s. 2018 Alzheimer’s Disease Facts and Figures (vol 14, pg 367, 2018). Alzheimers Dement. 2018, 14, 701. [Google Scholar] [CrossRef]
- Hahad, O.; Frenis, K.; Kuntic, M.; Daiber, A.; Munzel, T. Accelerated Aging and Age-Related Diseases (CVD and Neurological) Due to Air Pollution and Traffic Noise Exposure. Int. J. Mol. Sci. 2021, 22, 2419. [Google Scholar] [CrossRef]
- Collaborators, G.B.D.D. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef]
- Prince, M.; Wimo, A.; Guerchet, M.; Ali, G.-C.; Wu, Y.-T.; Prina, M. World Alzheimer Report 2015—The Global Impact of Dementia. Doctoral Dissertation, Alzheimer’s Disease International, London, UK, 2015. [Google Scholar]
- Rasmussen, J.; Langerman, H. Alzheimer’s Disease—Why We Need Early Diagnosis. Degener. Neurol. Neuromuscul. Dis. 2019, 9, 123–130. [Google Scholar] [CrossRef]
- Alzheimer Europe. Dementia in Europe Yearbook 2019: Estimating the Prevalence of Dementia in Europe. Available online: https://www.alzheimer-europe.org/sites/default/files/alzheimer_europe_dementia_in_europe_yearbook_2019.pdf (accessed on 13 February 2024).
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef]
- Peter-Derex, L.; Yammine, P.; Bastuji, H.; Croisile, B. Sleep and Alzheimer’s disease. Sleep Med. Rev. 2015, 19, 29–38. [Google Scholar] [CrossRef]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chetelat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Wolfson, C.; Wolfson, D.B.; Asgharian, M.; M’Lan, C.E.; Ostbye, T.; Rockwood, K.; Hogan, D.B.; Clinical Progression of Dementia Study, G. A reevaluation of the duration of survival after the onset of dementia. N. Engl. J. Med. 2001, 344, 1111–1116. [Google Scholar] [CrossRef]
- Reisberg, B.; Ferris, S.H.; de Leon, M.J.; Crook, T. The Global Deterioration Scale for assessment of primary degenerative dementia. Am. J. Psychiatry 1982, 139, 1136–1139. [Google Scholar] [CrossRef] [PubMed]
- Perl, D.P. Neuropathology of Alzheimer’s disease. Mt. Sinai. J. Med. 2010, 77, 32–42. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Tiiman, A.; Jelic, V.; Jarvet, J.; Jaremo, P.; Bogdanovic, N.; Rigler, R.; Terenius, L.; Graslund, A.; Vukojevic, V. Amyloidogenic Nanoplaques in Blood Serum of Patients with Alzheimer’s Disease Revealed by Time-Resolved Thioflavin T Fluorescence Intensity Fluctuation Analysis. J. Alzheimers. Dis. 2019, 68, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A.; Ekavali. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef]
- Dal Pra, I.; Chiarini, A.; Gui, L.; Chakravarthy, B.; Pacchiana, R.; Gardenal, E.; Whitfield, J.F.; Armato, U. Do astrocytes collaborate with neurons in spreading the “infectious” abeta and Tau drivers of Alzheimer’s disease? Neuroscientist 2015, 21, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef] [PubMed]
- Parkinson’s Foundation. Who Has Parkinson’s? Available online: https://www.parkinson.org/understanding-parkinsons/statistics (accessed on 13 February 2024).
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging Cell 2019, 18, e13031. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013, 9, 13–24. [Google Scholar] [CrossRef]
- Twohig, D.; Nielsen, H.M. alpha-synuclein in the pathophysiology of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 23. [Google Scholar] [CrossRef]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castano-Diez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef]
- National Insitute on Aging. Parkinson’s Disease: Causes, Symptoms, and Treatments. Available online: https://www.nia.nih.gov/health/parkinsons-disease (accessed on 13 February 2024).
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef]
- Parakh, S.; Spencer, D.M.; Halloran, M.A.; Soo, K.Y.; Atkin, J.D. Redox regulation in amyotrophic lateral sclerosis. Oxid. Med. Cell Longev. 2013, 2013, 408681. [Google Scholar] [CrossRef]
- Logroscino, G.; Urso, D.; Tortelli, R. The challenge of amyotrophic lateral sclerosis descriptive epidemiology: To estimate low incidence rates across complex phenotypes in different geographic areas. Curr. Opin. Neurol. 2022, 35, 678–685. [Google Scholar] [CrossRef]
- Hardiman, O.; van den Berg, L.H.; Kiernan, M.C. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 639–649. [Google Scholar] [CrossRef]
- Suzuki, N.; Nishiyama, A.; Warita, H.; Aoki, M. Genetics of amyotrophic lateral sclerosis: Seeking therapeutic targets in the era of gene therapy. J. Hum. Genet. 2023, 68, 131–152. [Google Scholar] [CrossRef]
- Gibson, S.B.; Downie, J.M.; Tsetsou, S.; Feusier, J.E.; Figueroa, K.P.; Bromberg, M.B.; Jorde, L.B.; Pulst, S.M. The evolving genetic risk for sporadic ALS. Neurology 2017, 89, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Carson, M.; Smith, C.D.; Koppenol, W.H. ALS, SOD and peroxynitrite. Nature 1993, 364, 584. [Google Scholar] [CrossRef] [PubMed]
- Goutman, S.A.; Hardiman, O.; Al-Chalabi, A.; Chio, A.; Savelieff, M.G.; Kiernan, M.C.; Feldman, E.L. Emerging insights into the complex genetics and pathophysiology of amyotrophic lateral sclerosis. Lancet Neurol. 2022, 21, 465–479. [Google Scholar] [CrossRef]
- Blokhuis, A.M.; Groen, E.J.; Koppers, M.; van den Berg, L.H.; Pasterkamp, R.J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef]
- Peng, C.; Trojanowski, J.Q.; Lee, V.M. Protein transmission in neurodegenerative disease. Nat. Rev. Neurol. 2020, 16, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, 248. [Google Scholar] [CrossRef]
- Michalska, P.; Leon, R. When It Comes to an End: Oxidative Stress Crosstalk with Protein Aggregation and Neuroinflammation Induce Neurodegeneration. Antioxidants 2020, 9, 740. [Google Scholar] [CrossRef]
- Katsumoto, A.; Takeuchi, H.; Takahashi, K.; Tanaka, F. Microglia in Alzheimer’s Disease: Risk Factors and Inflammation. Front. Neurol. 2018, 9, 978. [Google Scholar] [CrossRef] [PubMed]
- Ischiropoulos, H.; Beckman, J.S. Oxidative stress and nitration in neurodegeneration: Cause, effect, or association? J. Clin. Investig. 2003, 111, 163–169. [Google Scholar] [CrossRef]
- Tonnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Steven, S.; Frenis, K.; Oelze, M.; Kalinovic, S.; Kuntic, M.; Bayo Jimenez, M.T.; Vujacic-Mirski, K.; Helmstadter, J.; Kroller-Schon, S.; Munzel, T.; et al. Vascular Inflammation and Oxidative Stress: Major Triggers for Cardiovascular Disease. Oxid. Med. Cell Longev. 2019, 2019, 7092151. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; Hahad, O.; Andreadou, I.; Steven, S.; Daub, S.; Munzel, T. Redox-related biomarkers in human cardiovascular disease—classical footprints and beyond. Redox Biol. 2021, 42, 101875. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, P.; Kossmann, S.; Munzel, T.; Daiber, A. Redox regulation of cardiovascular inflammation—Immunomodulatory function of mitochondrial and Nox-derived reactive oxygen and nitrogen species. Free Radic. Biol. Med. 2017, 109, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Daiber, A.; Steven, S.; Vujacic-Mirski, K.; Kalinovic, S.; Oelze, M.; Di Lisa, F.; Munzel, T. Regulation of Vascular Function and Inflammation via Cross Talk of Reactive Oxygen and Nitrogen Species from Mitochondria or NADPH Oxidase-Implications for Diabetes Progression. Int. J. Mol. Sci. 2020, 21, 3405. [Google Scholar] [CrossRef] [PubMed]
- Ahlskog, J.E. Challenging conventional wisdom: The etiologic role of dopamine oxidative stress in Parkinson’s disease. Mov. Disord. 2005, 20, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Bader, V.; Winklhofer, K.F. Mitochondria at the interface between neurodegeneration and neuroinflammation. Semin. Cell Dev. Biol. 2020, 99, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, M.; Pershagen, G.; Thacher, J.D.; Lanki, T.; Wicki, B.; Roosli, M.; Vienneau, D.; Cantuaria, M.L.; Schmidt, J.H.; Aasvang, G.M.; et al. Health position paper and redox perspectives—Disease burden by transportation noise. Redox Biol. 2024, 69, 102995. [Google Scholar] [CrossRef]
- Rink, C.; Khanna, S. Significance of brain tissue oxygenation and the arachidonic acid cascade in stroke. Antioxid. Redox Signal. 2011, 14, 1889–1903. [Google Scholar] [CrossRef] [PubMed]
- Hallermann, S.; de Kock, C.P.; Stuart, G.J.; Kole, M.H. State and location dependence of action potential metabolic cost in cortical pyramidal neurons. Nat. Neurosci. 2012, 15, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Popa-Wagner, A.; Mitran, S.; Sivanesan, S.; Chang, E.; Buga, A.M. ROS and brain diseases: The good, the bad, and the ugly. Oxid. Med. Cell Longev. 2013, 2013, 963520. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; Kroller-Schon, S.; Oelze, M.; Hahad, O.; Li, H.; Schulz, R.; Steven, S.; Munzel, T. Oxidative stress and inflammation contribute to traffic noise-induced vascular and cerebral dysfunction via uncoupling of nitric oxide synthases. Redox Biol. 2020, 34, 101506. [Google Scholar] [CrossRef]
- Simic, G.; Lucassen, P.J.; Krsnik, Z.; Kruslin, B.; Kostovic, I.; Winblad, B.; Bogdanovi. nNOS expression in reactive astrocytes correlates with increased cell death related DNA damage in the hippocampus and entorhinal cortex in Alzheimer’s disease. Exp. Neurol. 2000, 165, 12–26. [Google Scholar] [CrossRef]
- Saha, R.N.; Pahan, K. Signals for the induction of nitric oxide synthase in astrocytes. Neurochem. Int. 2006, 49, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Adibhatla, R.M.; Hatcher, J.F. Lipid oxidation and peroxidation in CNS health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2010, 12, 125–169. [Google Scholar] [CrossRef] [PubMed]
- Martins, R.N.; Harper, C.G.; Stokes, G.B.; Masters, C.L. Increased cerebral glucose-6-phosphate dehydrogenase activity in Alzheimer’s disease may reflect oxidative stress. J. Neurochem. 1986, 46, 1042–1045. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Butterfield, D.A. Role of oxidative stress in the progression of Alzheimer’s disease. J. Alzheimers Dis. 2010, 19, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Ringman, J.M.; Frautschy, S.A.; Teng, E.; Begum, A.N.; Bardens, J.; Beigi, M.; Gylys, K.H.; Badmaev, V.; Heath, D.D.; Apostolova, L.G.; et al. Oral curcumin for Alzheimer’s disease: Tolerability and efficacy in a 24-week randomized, double blind, placebo-controlled study. Alzheimers Res. Ther. 2012, 4, 43. [Google Scholar] [CrossRef]
- Sutherland, G.T.; Chami, B.; Youssef, P.; Witting, P.K. Oxidative stress in Alzheimer’s disease: Primary villain or physiological by-product? Redox. Rep. 2013, 18, 134–141. [Google Scholar] [CrossRef]
- Ansari, M.A.; Scheff, S.W. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 2010, 69, 155–167. [Google Scholar] [CrossRef]
- Markesbery, W.R.; Lovell, M.A. DNA oxidation in Alzheimer’s disease. Antioxid. Redox Signal. 2006, 8, 2039–2045. [Google Scholar] [CrossRef]
- Luth, H.J.; Munch, G.; Arendt, T. Aberrant expression of NOS isoforms in Alzheimer’s disease is structurally related to nitrotyrosine formation. Brain Res. 2002, 953, 135–143. [Google Scholar] [CrossRef]
- Luth, H.J.; Holzer, M.; Gartner, U.; Staufenbiel, M.; Arendt, T. Expression of endothelial and inducible NOS-isoforms is increased in Alzheimer’s disease, in APP23 transgenic mice and after experimental brain lesion in rat: Evidence for an induction by amyloid pathology. Brain Res. 2001, 913, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Massaad, C.A. Neuronal and vascular oxidative stress in Alzheimer’s disease. Curr. Neuropharmacol. 2011, 9, 662–673. [Google Scholar] [CrossRef]
- Casado, A.; Encarnacion Lopez-Fernandez, M.; Concepcion Casado, M.; de La Torre, R. Lipid peroxidation and antioxidant enzyme activities in vascular and Alzheimer dementias. Neurochem. Res. 2008, 33, 450–458. [Google Scholar] [CrossRef]
- Marcus, D.L.; Thomas, C.; Rodriguez, C.; Simberkoff, K.; Tsai, J.S.; Strafaci, J.A.; Freedman, M.L. Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer’s disease. Exp. Neurol. 1998, 150, 40–44. [Google Scholar] [CrossRef]
- Sultana, R.; Piroddi, M.; Galli, F.; Butterfield, D.A. Protein levels and activity of some antioxidant enzymes in hippocampus of subjects with amnestic mild cognitive impairment. Neurochem. Res. 2008, 33, 2540–2546. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.E.; de Vasconcelos, A.S.; da Costa Vilhena, T.; da Silva, T.L.; da Silva Barbosa, A.; Gomes, A.R.; Dolabela, M.F.; Percario, S. Oxidative Stress in Alzheimer’s Disease: Should We Keep Trying Antioxidant Therapies? Cell Mol. Neurobiol. 2015, 35, 595–614. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Carter, C.J.; Wells, F.R.; Javoy-Agid, F.; Agid, Y.; Lees, A.; Jenner, P.; Marsden, C.D. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J. Neurochem. 1989, 52, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Mythri, R.B.; Venkateshappa, C.; Harish, G.; Mahadevan, A.; Muthane, U.B.; Yasha, T.C.; Srinivas Bharath, M.M.; Shankar, S.K. Evaluation of markers of oxidative stress, antioxidant function and astrocytic proliferation in the striatum and frontal cortex of Parkinson’s disease brains. Neurochem. Res. 2011, 36, 1452–1463. [Google Scholar] [CrossRef] [PubMed]
- Castellani, R.J.; Perry, G.; Siedlak, S.L.; Nunomura, A.; Shimohama, S.; Zhang, J.; Montine, T.; Sayre, L.M.; Smith, M.A. Hydroxynonenal adducts indicate a role for lipid peroxidation in neocortical and brainstem Lewy bodies in humans. Neurosci. Lett. 2002, 319, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Yoritaka, A.; Hattori, N.; Uchida, K.; Tanaka, M.; Stadtman, E.R.; Mizuno, Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc. Natl. Acad. Sci. USA 1996, 93, 2696–2701. [Google Scholar] [CrossRef]
- Shamoto-Nagai, M.; Maruyama, W.; Hashizume, Y.; Yoshida, M.; Osawa, T.; Riederer, P.; Naoi, M. In parkinsonian substantia nigra, alpha-synuclein is modified by acrolein, a lipid-peroxidation product, and accumulates in the dopamine neurons with inhibition of proteasome activity. J. Neural. Transm. 2007, 114, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.; Gronnemose, A.L.; Pedersen, J.N.; Nowak, J.S.; Christiansen, G.; Nielsen, J.; Mulder, F.A.A.; Otzen, D.E.; Jorgensen, T.J.D. Lipid Peroxidation Products HNE and ONE Promote and Stabilize Alpha-Synuclein Oligomers by Chemical Modifications. Biochemistry 2021, 60, 3644–3658. [Google Scholar] [CrossRef]
- Schildknecht, S.; Gerding, H.R.; Karreman, C.; Drescher, M.; Lashuel, H.A.; Outeiro, T.F.; Di Monte, D.A.; Leist, M. Oxidative and nitrative alpha-synuclein modifications and proteostatic stress: Implications for disease mechanisms and interventions in synucleinopathies. J. Neurochem. 2013, 125, 491–511. [Google Scholar] [CrossRef]
- Przedborski, S.; Chen, Q.; Vila, M.; Giasson, B.I.; Djaldatti, R.; Vukosavic, S.; Souza, J.M.; Jackson-Lewis, V.; Lee, V.M.; Ischiropoulos, H. Oxidative post-translational modifications of alpha-synuclein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson’s disease. J. Neurochem. 2001, 76, 637–640. [Google Scholar] [CrossRef]
- Zhang, J.; Perry, G.; Smith, M.A.; Robertson, D.; Olson, S.J.; Graham, D.G.; Montine, T.J. Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am. J. Pathol. 1999, 154, 1423–1429. [Google Scholar] [CrossRef]
- Alam, Z.I.; Daniel, S.E.; Lees, A.J.; Marsden, D.C.; Jenner, P.; Halliwell, B. A generalised increase in protein carbonyls in the brain in Parkinson’s but not incidental Lewy body disease. J. Neurochem. 1997, 69, 1326–1329. [Google Scholar] [CrossRef] [PubMed]
- Ogata, M.; Kaneya, D.; Shin-Ya, K.; Li, L.; Abe, Y.; Katoh, H.; Seki, S.; Seki, Y.; Gonda, R.; Urano, S.; et al. Trapping effect of eugenol on hydroxyl radicals induced by L-DOPA in vitro. Chem. Pharm. Bull 2005, 53, 1167–1170. [Google Scholar] [CrossRef] [PubMed]
- Group, P.D.M.C.; Gray, R.; Ives, N.; Rick, C.; Patel, S.; Gray, A.; Jenkinson, C.; McIntosh, E.; Wheatley, K.; Williams, A.; et al. Long-term effectiveness of dopamine agonists and monoamine oxidase B inhibitors compared with levodopa as initial treatment for Parkinson’s disease (PD MED): A large, open-label, pragmatic randomised trial. Lancet 2014, 384, 1196–1205. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Yi, L.X.; Wang, D.Q.; Lim, T.M.; Tan, E.K. Role of dopamine in the pathophysiology of Parkinson’s disease. Transl. Neurodegener. 2023, 12, 44. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Lim, T.M. Roles of glutathione (GSH) in dopamine (DA) oxidation studied by improved tandem HPLC plus ESI-MS. Neurochem. Res. 2009, 34, 316–326. [Google Scholar] [CrossRef]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994, 36, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Marttila, R.J.; Lorentz, H.; Rinne, U.K. Oxygen toxicity protecting enzymes in Parkinson’s disease. Increase of superoxide dismutase-like activity in the substantia nigra and basal nucleus. J. Neurol. Sci. 1988, 86, 321–331. [Google Scholar] [CrossRef]
- Grunewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Haas, R.H.; Nasirian, F.; Nakano, K.; Ward, D.; Pay, M.; Hill, R.; Shults, C.W. Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson’s disease. Ann. Neurol. 1995, 37, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.R.; Caudle, W.M.; Guillot, T.S.; Watson, J.L.; Nakamaru-Ogiso, E.; Seo, B.B.; Sherer, T.B.; Greenamyre, J.T.; Yagi, T.; Matsuno-Yagi, A.; et al. Obligatory role for complex I inhibition in the dopaminergic neurotoxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Toxicol. Sci. 2007, 95, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Shults, C.W.; Oakes, D.; Kieburtz, K.; Beal, M.F.; Haas, R.; Plumb, S.; Juncos, J.L.; Nutt, J.; Shoulson, I.; Carter, J.; et al. Effects of coenzyme Q10 in early Parkinson disease: Evidence of slowing of the functional decline. Arch. Neurol. 2002, 59, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.G.; Sun, M.X.; Zhang, W.L.; Wang, W.W.; Jin, Y.M.; Xie, C.L. The efficacy and safety of coenzyme Q10 in Parkinson’s disease: A meta-analysis of randomized controlled trials. Neurol. Sci. 2017, 38, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H., Jr. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 2004, 43, 19–30. [Google Scholar] [CrossRef]
- Forsberg, K.; Jonsson, P.A.; Andersen, P.M.; Bergemalm, D.; Graffmo, K.S.; Hultdin, M.; Jacobsson, J.; Rosquist, R.; Marklund, S.L.; Brannstrom, T. Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS ONE 2010, 5, e11552. [Google Scholar] [CrossRef]
- Tian, Y.P.; Che, F.Y.; Su, Q.P.; Lu, Y.C.; You, C.P.; Huang, L.M.; Wang, S.G.; Wang, L.; Yu, J.X. Effects of mutant TDP-43 on the Nrf2/ARE pathway and protein expression of MafK and JDP2 in NSC-34 cells. Genet. Mol. Res. 2017, 16, gmr16029638. [Google Scholar] [CrossRef]
- Duan, W.; Li, X.; Shi, J.; Guo, Y.; Li, Z.; Li, C. Mutant TAR DNA-binding protein-43 induces oxidative injury in motor neuron-like cell. Neuroscience 2010, 169, 1621–1629. [Google Scholar] [CrossRef]
- Andrade, N.S.; Ramic, M.; Esanov, R.; Liu, W.; Rybin, M.J.; Gaidosh, G.; Abdallah, A.; Del’Olio, S.; Huff, T.C.; Chee, N.T.; et al. Dipeptide repeat proteins inhibit homology-directed DNA double strand break repair in C9ORF72 ALS/FTD. Mol. Neurodegener. 2020, 15, 13. [Google Scholar] [CrossRef]
- Deng, Q.; Holler, C.J.; Taylor, G.; Hudson, K.F.; Watkins, W.; Gearing, M.; Ito, D.; Murray, M.E.; Dickson, D.W.; Seyfried, N.T.; et al. FUS is phosphorylated by DNA-PK and accumulates in the cytoplasm after DNA damage. J. Neurosci. 2014, 34, 7802–7813. [Google Scholar] [CrossRef]
- Tohgi, H.; Abe, T.; Yamazaki, K.; Murata, T.; Ishizaki, E.; Isobe, C. Remarkable increase in cerebrospinal fluid 3-nitrotyrosine in patients with sporadic amyotrophic lateral sclerosis. Ann. Neurol. 1999, 46, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.G.; Henry, Y.K.; Mattson, M.P.; Appel, S.H. Presence of 4-hydroxynonenal in cerebrospinal fluid of patients with sporadic amyotrophic lateral sclerosis. Ann. Neurol. 1998, 44, 696–699. [Google Scholar] [CrossRef]
- Bogdanov, M.; Brown, R.H.; Matson, W.; Smart, R.; Hayden, D.; O’Donnell, H.; Flint Beal, M.; Cudkowicz, M. Increased oxidative damage to DNA in ALS patients. Free Radic. Biol. Med. 2000, 29, 652–658. [Google Scholar] [CrossRef]
- Ihara, Y.; Nobukuni, K.; Takata, H.; Hayabara, T. Oxidative stress and metal content in blood and cerebrospinal fluid of amyotrophic lateral sclerosis patients with and without a Cu, Zn-superoxide dismutase mutation. Neurol. Res. 2005, 27, 105–108. [Google Scholar] [CrossRef]
- Ikawa, M.; Okazawa, H.; Tsujikawa, T.; Matsunaga, A.; Yamamura, O.; Mori, T.; Hamano, T.; Kiyono, Y.; Nakamoto, Y.; Yoneda, M. Increased oxidative stress is related to disease severity in the ALS motor cortex: A PET study. Neurology 2015, 84, 2033–2039. [Google Scholar] [CrossRef]
- D’Amico, E.; Factor-Litvak, P.; Santella, R.M.; Mitsumoto, H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2013, 65, 509–527. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Oliveira, T.; Montezinho, L.; Mendes, C.; Firuzi, O.; Saso, L.; Oliveira, P.J.; Silva, F.S.G. Oxidative Stress in Amyotrophic Lateral Sclerosis: Pathophysiology and Opportunities for Pharmacological Intervention. Oxid. Med. Cell Longev. 2020, 2020, 5021694. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Itoyama, Y.; Sobue, G.; Tsuji, S.; Aoki, M.; Doyu, M.; Hamada, C.; Kondo, K.; Yoneoka, T.; Akimoto, M.; et al. Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Front. Degener 2014, 15, 610–617. [Google Scholar] [CrossRef]
- Liu, S.; Liu, Y.; Hao, W.; Wolf, L.; Kiliaan, A.J.; Penke, B.; Rube, C.E.; Walter, J.; Heneka, M.T.; Hartmann, T.; et al. TLR2 is a primary receptor for Alzheimer’s amyloid beta peptide to trigger neuroinflammatory activation. J. Immunol. 2012, 188, 1098–1107. [Google Scholar] [CrossRef]
- Amor, S.; Puentes, F.; Baker, D.; van der Valk, P. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Bsibsi, M.; Ravid, R.; Gveric, D.; van Noort, J.M. Broad expression of Toll-like receptors in the human central nervous system. J. Neuropathol. Exp. Neurol. 2002, 61, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- van Noort, J.M.; Bsibsi, M. Toll-like receptors in the CNS: Implications for neurodegeneration and repair. Prog. Brain Res. 2009, 175, 139–148. [Google Scholar] [CrossRef]
- Hsieh, H.L.; Yang, C.M. Role of redox signaling in neuroinflammation and neurodegenerative diseases. Biomed. Res. Int. 2013, 2013, 484613. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflammation 2014, 11, 98. [Google Scholar] [CrossRef]
- Daiber, A.; Di Lisa, F.; Oelze, M.; Kroller-Schon, S.; Steven, S.; Schulz, E.; Munzel, T. Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br. J. Pharmacol. 2017, 174, 1670–1689. [Google Scholar] [CrossRef]
- Hensley, K.; Maidt, M.L.; Yu, Z.; Sang, H.; Markesbery, W.R.; Floyd, R.A. Electrochemical analysis of protein nitrotyrosine and dityrosine in the Alzheimer brain indicates region-specific accumulation. J. Neurosci. 1998, 18, 8126–8132. [Google Scholar] [CrossRef]
- Castegna, A.; Thongboonkerd, V.; Klein, J.B.; Lynn, B.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of nitrated proteins in Alzheimer’s disease brain. J. Neurochem. 2003, 85, 1394–1401. [Google Scholar] [CrossRef]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lu, M.; Du, R.H.; Qiao, C.; Jiang, C.Y.; Zhang, K.Z.; Ding, J.H.; Hu, G. MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol. Neurodegener 2016, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef]
- Lassmann, H. Review: The architecture of inflammatory demyelinating lesions: Implications for studies on pathogenesis. Neuropathol. Appl. Neurobiol. 2011, 37, 698–710. [Google Scholar] [CrossRef]
- Babbe, H.; Roers, A.; Waisman, A.; Lassmann, H.; Goebels, N.; Hohlfeld, R.; Friese, M.; Schroder, R.; Deckert, M.; Schmidt, S.; et al. Clonal expansions of CD8+ T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J. Exp. Med. 2000, 192, 393–404. [Google Scholar] [CrossRef]
- Marik, C.; Felts, P.A.; Bauer, J.; Lassmann, H.; Smith, K.J. Lesion genesis in a subset of patients with multiple sclerosis: A role for innate immunity? Brain 2007, 130, 2800–2815. [Google Scholar] [CrossRef]
- Lassmann, H.; van Horssen, J. The molecular basis of neurodegeneration in multiple sclerosis. FEBS Lett. 2011, 585, 3715–3723. [Google Scholar] [CrossRef] [PubMed]
- Beland, L.C.; Markovinovic, A.; Jakovac, H.; De Marchi, F.; Bilic, E.; Mazzini, L.; Kriz, J.; Munitic, I. Immunity in amyotrophic lateral sclerosis: Blurred lines between excessive inflammation and inefficient immune responses. Brain Commun. 2020, 2, fcaa124. [Google Scholar] [CrossRef]
- Staats, K.A.; Borchelt, D.R.; Tansey, M.G.; Wymer, J. Blood-based biomarkers of inflammation in amyotrophic lateral sclerosis. Mol. Neurodegener 2022, 17, 11. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.S.; Patel, P.; Dutta, K.; Julien, J.P. Inflammation Induces TDP-43 Mislocalization and Aggregation. PLoS ONE 2015, 10, e0140248. [Google Scholar] [CrossRef]
- Swarup, V.; Phaneuf, D.; Dupre, N.; Petri, S.; Strong, M.; Kriz, J.; Julien, J.P. Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor kappaB-mediated pathogenic pathways. J. Exp. Med. 2011, 208, 2429–2447. [Google Scholar] [CrossRef] [PubMed]
- Kia, A.; McAvoy, K.; Krishnamurthy, K.; Trotti, D.; Pasinelli, P. Astrocytes expressing ALS-linked mutant FUS induce motor neuron death through release of tumor necrosis factor-alpha. Glia 2018, 66, 1016–1033. [Google Scholar] [CrossRef]
- Brettschneider, J.; Toledo, J.B.; Van Deerlin, V.M.; Elman, L.; McCluskey, L.; Lee, V.M.; Trojanowski, J.Q. Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e39216. [Google Scholar] [CrossRef]
- Lino, M.M.; Schneider, C.; Caroni, P. Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J. Neurosci. 2002, 22, 4825–4832. [Google Scholar] [CrossRef]
- Clement, A.M.; Nguyen, M.D.; Roberts, E.A.; Garcia, M.L.; Boillee, S.; Rule, M.; McMahon, A.P.; Doucette, W.; Siwek, D.; Ferrante, R.J.; et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 2003, 302, 113–117. [Google Scholar] [CrossRef]
- Spiller, K.J.; Restrepo, C.R.; Khan, T.; Dominique, M.A.; Fang, T.C.; Canter, R.G.; Roberts, C.J.; Miller, K.R.; Ransohoff, R.M.; Trojanowski, J.Q.; et al. Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat. Neurosci. 2018, 21, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Wang, K.; Deng, L.; Chen, Y.; Nice, E.C.; Huang, C. Redox regulation of inflammation: Old elements, a new story. Med. Res. Rev. 2015, 35, 306–340. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, P.; Knorr, M.; Kossmann, S.; Stratmann, J.; Hausding, M.; Schuhmacher, S.; Karbach, S.H.; Schwenk, M.; Yogev, N.; Schulz, E.; et al. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation 2011, 124, 1370–1381. [Google Scholar] [CrossRef] [PubMed]
- Kossmann, S.; Schwenk, M.; Hausding, M.; Karbach, S.H.; Schmidgen, M.I.; Brandt, M.; Knorr, M.; Hu, H.; Kroller-Schon, S.; Schonfelder, T.; et al. Angiotensin II-induced vascular dysfunction depends on interferon-gamma-driven immune cell recruitment and mutual activation of monocytes and NK-cells. Arterioscler Thromb. Vasc. Biol. 2013, 33, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.; Weyand, C.; Harrison, D.G. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 2007, 204, 2449–2460. [Google Scholar] [CrossRef] [PubMed]
- Qiao, M.; Zhao, Q.; Lee, C.F.; Tannock, L.R.; Smart, E.J.; LeBaron, R.G.; Phelix, C.F.; Rangel, Y.; Asmis, R. Thiol oxidative stress induced by metabolic disorders amplifies macrophage chemotactic responses and accelerates atherogenesis and kidney injury in LDL receptor-deficient mice. Arterioscler Thromb Vasc. Biol. 2009, 29, 1779–1786. [Google Scholar] [CrossRef]
- Kim, H.S.; Ullevig, S.L.; Zamora, D.; Lee, C.F.; Asmis, R. Redox regulation of MAPK phosphatase 1 controls monocyte migration and macrophage recruitment. Proc. Natl. Acad. Sci. USA 2012, 109, E2803–E2812. [Google Scholar] [CrossRef]
- Kim, H.S.; Ullevig, S.L.; Nguyen, H.N.; Vanegas, D.; Asmis, R. Redox regulation of 14-3-3zeta controls monocyte migration. Arterioscler Thromb Vasc. Biol. 2014, 34, 1514–1521. [Google Scholar] [CrossRef]
- Ullevig, S.; Kim, H.S.; Asmis, R. S-glutathionylation in monocyte and macrophage (dys)function. Int. J. Mol. Sci. 2013, 14, 15212–15232. [Google Scholar] [CrossRef]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in neurodegenerative diseases: Mechanism and potential therapeutic targets. Signal Transduct. Target Ther. 2023, 8, 359. [Google Scholar] [CrossRef]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Gerrits, E.; Brouwer, N.; Kooistra, S.M.; Woodbury, M.E.; Vermeiren, Y.; Lambourne, M.; Mulder, J.; Kummer, M.; Moller, T.; Biber, K.; et al. Distinct amyloid-beta and tau-associated microglia profiles in Alzheimer’s disease. Acta Neuropathol. 2021, 141, 681–696. [Google Scholar] [CrossRef]
- Smith, A.M.; Davey, K.; Tsartsalis, S.; Khozoie, C.; Fancy, N.; Tang, S.S.; Liaptsi, E.; Weinert, M.; McGarry, A.; Muirhead, R.C.J.; et al. Diverse human astrocyte and microglial transcriptional responses to Alzheimer’s pathology. Acta Neuropathol. 2022, 143, 75–91. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Wang, K.; Hu, G.; Wang, X.; Miao, Z.; Azevedo, J.A.; Suh, E.; Van Deerlin, V.M.; Choi, D.; Roeder, K.; et al. APOE and TREM2 regulate amyloid-responsive microglia in Alzheimer’s disease. Acta Neuropathol. 2020, 140, 477–493. [Google Scholar] [CrossRef] [PubMed]
- Smajic, S.; Prada-Medina, C.A.; Landoulsi, Z.; Ghelfi, J.; Delcambre, S.; Dietrich, C.; Jarazo, J.; Henck, J.; Balachandran, S.; Pachchek, S.; et al. Single-cell sequencing of human midbrain reveals glial activation and a Parkinson-specific neuronal state. Brain 2022, 145, 964–978. [Google Scholar] [CrossRef] [PubMed]
- Kylkilahti, T.M.; Berends, E.; Ramos, M.; Shanbhag, N.C.; Toger, J.; Markenroth Bloch, K.; Lundgaard, I. Achieving brain clearance and preventing neurodegenerative diseases-A glymphatic perspective. J. Cereb Blood Flow. Metab. 2021, 41, 2137–2149. [Google Scholar] [CrossRef] [PubMed]
- Brandebura, A.N.; Paumier, A.; Onur, T.S.; Allen, N.J. Astrocyte contribution to dysfunction, risk and progression in neurodegenerative disorders. Nat. Rev. Neurosci. 2023, 24, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Al-Dalahmah, O.; Sosunov, A.A.; Shaik, A.; Ofori, K.; Liu, Y.; Vonsattel, J.P.; Adorjan, I.; Menon, V.; Goldman, J.E. Single-nucleus RNA-seq identifies Huntington disease astrocyte states. Acta Neuropathol. Commun. 2020, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Habib, N.; McCabe, C.; Medina, S.; Varshavsky, M.; Kitsberg, D.; Dvir-Szternfeld, R.; Green, G.; Dionne, D.; Nguyen, L.; Marshall, J.L.; et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 2020, 23, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.F.; Cao, H.; Fu, A.K.Y.; Ip, N.Y. Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 25800–25809. [Google Scholar] [CrossRef] [PubMed]
- Leng, K.; Li, E.; Eser, R.; Piergies, A.; Sit, R.; Tan, M.; Neff, N.; Li, S.H.; Rodriguez, R.D.; Suemoto, C.K.; et al. Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat. Neurosci. 2021, 24, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.; Araujo, J.A.; Li, H.; Sioutas, C.; Kleinman, M. Particulate matter induced enhancement of inflammatory markers in the brains of apolipoprotein E knockout mice. J. Nanosci. Nanotechnol. 2009, 9, 5099–5104. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.; Oldham, M.; Becaria, A.; Bondy, S.C.; Meacher, D.; Sioutas, C.; Misra, C.; Mendez, L.B.; Kleinman, M. Particulate matter in polluted air may increase biomarkers of inflammation in mouse brain. Neurotoxicology 2005, 26, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Hogan, M.K.; Kovalycsik, T.; Sun, Q.; Rajagopalan, S.; Nelson, R.J. Combined effects of exposure to dim light at night and fine particulate matter on C3H/HeNHsd mice. Behav. Brain Res. 2015, 294, 81–88. [Google Scholar] [CrossRef]
- Cheng, H.; Saffari, A.; Sioutas, C.; Forman, H.J.; Morgan, T.E.; Finch, C.E. Nanoscale Particulate Matter from Urban Traffic Rapidly Induces Oxidative Stress and Inflammation in Olfactory Epithelium with Concomitant Effects on Brain. Environ. Health Perspect. 2016, 124, 1537–1546. [Google Scholar] [CrossRef]
- Tyler, C.R.; Zychowski, K.E.; Sanchez, B.N.; Rivero, V.; Lucas, S.; Herbert, G.; Liu, J.; Irshad, H.; McDonald, J.D.; Bleske, B.E.; et al. Surface area-dependence of gas-particle interactions influences pulmonary and neuroinflammatory outcomes. Part Fibre Toxicol. 2016, 13, 64. [Google Scholar] [CrossRef]
- Herr, D.; Jew, K.; Wong, C.; Kennell, A.; Gelein, R.; Chalupa, D.; Raab, A.; Oberdorster, G.; Olschowka, J.; O’Banion, M.K.; et al. Effects of concentrated ambient ultrafine particulate matter on hallmarks of Alzheimer’s disease in the 3xTgAD mouse model. Neurotoxicology 2021, 84, 172–183. [Google Scholar] [CrossRef]
- Calderon-Garciduenas, L.; Herrera-Soto, A.; Jury, N.; Maher, B.A.; Gonzalez-Maciel, A.; Reynoso-Robles, R.; Ruiz-Rudolph, P.; van Zundert, B.; Varela-Nallar, L. Reduced repressive epigenetic marks, increased DNA damage and Alzheimer’s disease hallmarks in the brain of humans and mice exposed to particulate urban air pollution. Environ. Res. 2020, 183, 109226. [Google Scholar] [CrossRef]
- Bhatt, D.P.; Puig, K.L.; Gorr, M.W.; Wold, L.E.; Combs, C.K. A pilot study to assess effects of long-term inhalation of airborne particulate matter on early Alzheimer-like changes in the mouse brain. PLoS ONE 2015, 10, e0127102. [Google Scholar] [CrossRef]
- Cacciottolo, M.; Wang, X.; Driscoll, I.; Woodward, N.; Saffari, A.; Reyes, J.; Serre, M.L.; Vizuete, W.; Sioutas, C.; Morgan, T.E.; et al. Particulate air pollutants, APOE alleles and their contributions to cognitive impairment in older women and to amyloidogenesis in experimental models. Transl. Psychiatry 2017, 7, e1022. [Google Scholar] [CrossRef]
- Patten, K.T.; Valenzuela, A.E.; Wallis, C.; Berg, E.L.; Silverman, J.L.; Bein, K.J.; Wexler, A.S.; Lein, P.J. The Effects of Chronic Exposure to Ambient Traffic-Related Air Pollution on Alzheimer’s Disease Phenotypes in Wildtype and Genetically Predisposed Male and Female Rats. Environ. Health Perspect. 2021, 129, 57005. [Google Scholar] [CrossRef]
- Sahu, B.; Mackos, A.R.; Floden, A.M.; Wold, L.E.; Combs, C.K. Particulate Matter Exposure Exacerbates Amyloid-beta Plaque Deposition and Gliosis in APP/PS1 Mice. J. Alzheimer’s Dis. JAD 2021, 80, 761–774. [Google Scholar] [CrossRef]
- Lee, S.H.; Chen, Y.H.; Chien, C.C.; Yan, Y.H.; Chen, H.C.; Chuang, H.C.; Hsieh, H.I.; Cho, K.H.; Kuo, L.W.; Chou, C.C.; et al. Three month inhalation exposure to low-level PM2.5 induced brain toxicity in an Alzheimer’s disease mouse model. PLoS ONE 2021, 16, e0254587. [Google Scholar] [CrossRef] [PubMed]
- Ku, T.; Li, B.; Gao, R.; Zhang, Y.; Yan, W.; Ji, X.; Li, G.; Sang, N. NF-kappaB-regulated microRNA-574-5p underlies synaptic and cognitive impairment in response to atmospheric PM(2.5) aspiration. Part Fibre Toxicol. 2017, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Fonken, L.K.; Xu, X.; Weil, Z.M.; Chen, G.; Sun, Q.; Rajagopalan, S.; Nelson, R.J. Air pollution impairs cognition, provokes depressive-like behaviors and alters hippocampal cytokine expression and morphology. Mol. Psychiatry 2011, 16, 987–995, 973. [Google Scholar] [CrossRef]
- Rivas-Arancibia, S.; Zimbron, L.F.; Rodriguez-Martinez, E.; Maldonado, P.D.; Borgonio Perez, G.; Sepulveda-Parada, M. Oxidative stress-dependent changes in immune responses and cell death in the substantia nigra after ozone exposure in rat. Front. Aging Neurosci. 2015, 7, 65. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Lopez, D.; Bautista-Martinez, J.A.; Reyes-Hernandez, C.I.; Aguilar-Martinez, M.; Rivas-Arancibia, S. Oxidative stress, progressive damage in the substantia nigra and plasma dopamine oxidation, in rats chronically exposed to ozone. Toxicol. Lett. 2010, 197, 193–200. [Google Scholar] [CrossRef]
- Mumaw, C.L.; Levesque, S.; McGraw, C.; Robertson, S.; Lucas, S.; Stafflinger, J.E.; Campen, M.J.; Hall, P.; Norenberg, J.P.; Anderson, T.; et al. Microglial priming through the lung-brain axis: The role of air pollution-induced circulating factors. Faseb. J. 2016, 30, 1880–1891. [Google Scholar] [CrossRef]
- Hernandez-Zimbron, L.F.; Rivas-Arancibia, S. Oxidative stress caused by ozone exposure induces beta-amyloid 1-42 overproduction and mitochondrial accumulation by activating the amyloidogenic pathway. Neuroscience 2015, 304, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Calderon-Garciduenas, L.; Azzarelli, B.; Acuna, H.; Garcia, R.; Gambling, T.M.; Osnaya, N.; Monroy, S.; MR, D.E.L.T.; Carson, J.L.; Villarreal-Calderon, A.; et al. Air pollution and brain damage. Toxicol. Pathol. 2002, 30, 373–389. [Google Scholar] [CrossRef]
- Calderon-Garciduenas, L.; Maronpot, R.R.; Torres-Jardon, R.; Henriquez-Roldan, C.; Schoonhoven, R.; Acuna-Ayala, H.; Villarreal-Calderon, A.; Nakamura, J.; Fernando, R.; Reed, W.; et al. DNA damage in nasal and brain tissues of canines exposed to air pollutants is associated with evidence of chronic brain inflammation and neurodegeneration. Toxicol. Pathol. 2003, 31, 524–538. [Google Scholar] [CrossRef] [PubMed]
- Calderon-Garciduenas, L.; Mora-Tiscareno, A.; Gomez-Garza, G.; Carrasco-Portugal Mdel, C.; Perez-Guille, B.; Flores-Murrieta, F.J.; Perez-Guille, G.; Osnaya, N.; Juarez-Olguin, H.; Monroy, M.E.; et al. Effects of a cyclooxygenase-2 preferential inhibitor in young healthy dogs exposed to air pollution: A pilot study. Toxicol. Pathol. 2009, 37, 644–660. [Google Scholar] [CrossRef]
- Levesque, S.; Taetzsch, T.; Lull, M.E.; Kodavanti, U.; Stadler, K.; Wagner, A.; Johnson, J.A.; Duke, L.; Kodavanti, P.; Surace, M.J.; et al. Diesel exhaust activates and primes microglia: Air pollution, neuroinflammation, and regulation of dopaminergic neurotoxicity. Environ. Health Perspect. 2011, 119, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Gerlofs-Nijland, M.E.; van Berlo, D.; Cassee, F.R.; Schins, R.P.; Wang, K.; Campbell, A. Effect of prolonged exposure to diesel engine exhaust on proinflammatory markers in different regions of the rat brain. Part Fibre Toxicol. 2010, 7, 12. [Google Scholar] [CrossRef]
- Oppenheim, H.A.; Lucero, J.; Guyot, A.C.; Herbert, L.M.; McDonald, J.D.; Mabondzo, A.; Lund, A.K. Exposure to vehicle emissions results in altered blood brain barrier permeability and expression of matrix metalloproteinases and tight junction proteins in mice. Part Fibre Toxicol. 2013, 10, 62. [Google Scholar] [CrossRef]
- Mumaw, C.L.; Surace, M.; Levesque, S.; Kodavanti, U.P.; Kodavanti, P.R.S.; Royland, J.E.; Block, M.L. Atypical microglial response to biodiesel exhaust in healthy and hypertensive rats. Neurotoxicology 2017, 59, 155–163. [Google Scholar] [CrossRef]
- Kilian, J.G.; Mejias-Ortega, M.; Hsu, H.W.; Herman, D.A.; Vidal, J.; Arechavala, R.J.; Renusch, S.; Dalal, H.; Hasen, I.; Ting, A.; et al. Exposure to quasi-ultrafine particulate matter accelerates memory impairment and Alzheimer’s disease-like neuropathology in the AppNL-G-F knock-in mouse model. Toxicol. Sci. 2023, 193, 175–191. [Google Scholar] [CrossRef]
- Allen, J.L.; Liu, X.; Weston, D.; Conrad, K.; Oberdorster, G.; Cory-Slechta, D.A. Consequences of developmental exposure to concentrated ambient ultrafine particle air pollution combined with the adult paraquat and maneb model of the Parkinson’s disease phenotype in male mice. Neurotoxicology 2014, 41, 80–88. [Google Scholar] [CrossRef]
- Yuan, X.; Yang, Y.; Liu, C.; Tian, Y.; Xia, D.; Liu, Z.; Pan, L.; Xiong, M.; Xiong, J.; Meng, L.; et al. Fine Particulate Matter Triggers alpha-Synuclein Fibrillization and Parkinson-like Neurodegeneration. Mov. Disord. 2022, 37, 1817–1830. [Google Scholar] [CrossRef]
- Jayaraj, R.L.; Rodriguez, E.A.; Wang, Y.; Block, M.L. Outdoor Ambient Air Pollution and Neurodegenerative Diseases: The Neuroinflammation Hypothesis. Curr. Environ. Health Rep. 2017, 4, 166–179. [Google Scholar] [CrossRef]
- Calderon-Garciduenas, L.; Solt, A.C.; Henriquez-Roldan, C.; Torres-Jardon, R.; Nuse, B.; Herritt, L.; Villarreal-Calderon, R.; Osnaya, N.; Stone, I.; Garcia, R.; et al. Long-term air pollution exposure is associated with neuroinflammation, an altered innate immune response, disruption of the blood-brain barrier, ultrafine particulate deposition, and accumulation of amyloid beta-42 and alpha-synuclein in children and young adults. Toxicol. Pathol. 2008, 36, 289–310. [Google Scholar] [CrossRef]
- Muhlfeld, C.; Rothen-Rutishauser, B.; Blank, F.; Vanhecke, D.; Ochs, M.; Gehr, P. Interactions of nanoparticles with pulmonary structures and cellular responses. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L817–L829. [Google Scholar] [CrossRef] [PubMed]
- Oberdorster, G.; Sharp, Z.; Atudorei, V.; Elder, A.; Gelein, R.; Kreyling, W.; Cox, C. Translocation of inhaled ultrafine particles to the brain. Inhal. Toxicol. 2004, 16, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Kuntic, M.; Kuntic, I.; Krishnankutty, R.; Gericke, A.; Oelze, M.; Junglas, T.; Bayo Jimenez, M.T.; Stamm, P.; Nandudu, M.; Hahad, O.; et al. Co-exposure to urban particulate matter and aircraft noise adversely impacts the cerebro-pulmonary-cardiovascular axis in mice. Redox Biol. 2023, 59, 102580. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Calderon-Garciduenas, L. Air pollution: Mechanisms of neuroinflammation and CNS disease. Trends Neurosci. 2009, 32, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Calderon-Garciduenas, L.; Cross, J.V.; Franco-Lira, M.; Aragon-Flores, M.; Kavanaugh, M.; Torres-Jardon, R.; Chao, C.K.; Thompson, C.; Chang, J.; Zhu, H.; et al. Brain immune interactions and air pollution: Macrophage inhibitory factor (MIF), prion cellular protein (PrP(C)), Interleukin-6 (IL-6), interleukin 1 receptor antagonist (IL-1Ra), and interleukin-2 (IL-2) in cerebrospinal fluid and MIF in serum differentiate urban children exposed to severe vs. low air pollution. Front. Neurosci. 2013, 7, 183. [Google Scholar] [CrossRef] [PubMed]
- Tamagawa, E.; van Eeden, S.F. Impaired lung function and risk for stroke: Role of the systemic inflammation response? Chest 2006, 130, 1631–1633. [Google Scholar] [CrossRef] [PubMed]
- Kodavanti, U.P. Stretching the stress boundary: Linking air pollution health effects to a neurohormonal stress response. Biochim. Biophys. Acta 2016, 1860, 2880–2890. [Google Scholar] [CrossRef] [PubMed]
- Babisch, W. The Noise/Stress Concept, Risk Assessment and Research Needs. Noise Health 2002, 4, 1–11. [Google Scholar]
- Campos-Rodriguez, R.; Godinez-Victoria, M.; Abarca-Rojano, E.; Pacheco-Yepez, J.; Reyna-Garfias, H.; Barbosa-Cabrera, R.E.; Drago-Serrano, M.E. Stress modulates intestinal secretory immunoglobulin A. Front. Integr. Neurosci. 2013, 7, 86. [Google Scholar] [CrossRef]
- Munzel, T.; Sorensen, M.; Daiber, A. Transportation noise pollution and cardiovascular disease. Nat. Rev. Cardiol. 2021, 18, 619–636. [Google Scholar] [CrossRef]
- Daiber, A.; Kroller-Schon, S.; Frenis, K.; Oelze, M.; Kalinovic, S.; Vujacic-Mirski, K.; Kuntic, M.; Bayo Jimenez, M.T.; Helmstadter, J.; Steven, S.; et al. Environmental noise induces the release of stress hormones and inflammatory signaling molecules leading to oxidative stress and vascular dysfunction-Signatures of the internal exposome. Biofactors 2019, 45, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Manukyan, A.L. Noise as a cause of neurodegenerative disorders: Molecular and cellular mechanisms. Neurol. Sci. 2022, 43, 2983–2993. [Google Scholar] [CrossRef] [PubMed]
- Jafari, Z.; Kolb, B.E.; Mohajerani, M.H. Noise exposure accelerates the risk of cognitive impairment and Alzheimer’s disease: Adulthood, gestational, and prenatal mechanistic evidence from animal studies. Neurosci. Biobehav. Rev. 2020, 117, 110–128. [Google Scholar] [CrossRef] [PubMed]
- Kroller-Schon, S.; Daiber, A.; Steven, S.; Oelze, M.; Frenis, K.; Kalinovic, S.; Heimann, A.; Schmidt, F.P.; Pinto, A.; Kvandova, M.; et al. Crucial role for Nox2 and sleep deprivation in aircraft noise-induced vascular and cerebral oxidative stress, inflammation, and gene regulation. Eur. Heart J. 2018, 39, 3528–3539. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Daiber, A.; Steven, S.; Tran, L.P.; Ullmann, E.; Kossmann, S.; Schmidt, F.P.; Oelze, M.; Xia, N.; Li, H.; et al. Effects of noise on vascular function, oxidative stress, and inflammation: Mechanistic insight from studies in mice. Eur. Heart J. 2017, 38, 2838–2849. [Google Scholar] [CrossRef] [PubMed]
- Gai, Z.; Li, K.; Sun, H.; She, X.; Cui, B.; Wang, R. Effects of chronic noise on mRNA and protein expression of CRF family molecules and its relationship with p-tau in the rat prefrontal cortex. J. Neurol. Sci. 2016, 368, 307–313. [Google Scholar] [CrossRef]
- Cui, B.; Zhu, L.; She, X.; Wu, M.; Ma, Q.; Wang, T.; Zhang, N.; Xu, C.; Chen, X.; An, G.; et al. Chronic noise exposure causes persistence of tau hyperphosphorylation and formation of NFT tau in the rat hippocampus and prefrontal cortex. Exp. Neurol. 2012, 238, 122–129. [Google Scholar] [CrossRef]
- Zheng, P.; She, X.; Wang, C.; Zhu, Y.; Fu, B.; Ma, K.; Yang, H.; Gao, X.; Li, X.; Wu, F.; et al. Around-the-Clock Noise Induces AD-like Neuropathology by Disrupting Autophagy Flux Homeostasis. Cells 2022, 11, 2742. [Google Scholar] [CrossRef]
- Li, Q.; Liu, Y.; Sun, M. Autophagy and Alzheimer’s Disease. Cell Mol. Neurobiol. 2017, 37, 377–388. [Google Scholar] [CrossRef]
- Su, D.; Li, W.; Chi, H.; Yang, H.; She, X.; Wang, K.; Gao, X.; Ma, K.; Zhang, M.; Cui, B. Transcriptome analysis of the hippocampus in environmental noise-exposed SAMP8 mice reveals regulatory pathways associated with Alzheimer’s disease neuropathology. Environ. Health Prev. Med. 2020, 25, 3. [Google Scholar] [CrossRef]
- Cui, B.; Su, D.; Li, W.; She, X.; Zhang, M.; Wang, R.; Zhai, Q. Effects of chronic noise exposure on the microbiome-gut-brain axis in senescence-accelerated prone mice: Implications for Alzheimer’s disease. J. Neuroinflammation 2018, 15, 190. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Li, W.; She, X.; Chen, X.; Zhai, Q.; Cui, B.; Wang, R. Chronic noise exposure exacerbates AD-like neuropathology in SAMP8 mice in relation to Wnt signaling in the PFC and hippocampus. Sci. Rep. 2018, 8, 14622. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Cao, W.; Zhang, M.; Su, D.; Yang, H.; Li, Z.; Li, C.; She, X.; Wang, K.; Gao, X.; et al. Environmental noise stress disturbs commensal microbiota homeostasis and induces oxi-inflammmation and AD-like neuropathology through epithelial barrier disruption in the EOAD mouse model. J. Neuroinflammation 2021, 18, 9. [Google Scholar] [CrossRef] [PubMed]
- Jafari, Z.; Okuma, M.; Karem, H.; Mehla, J.; Kolb, B.E.; Mohajerani, M.H. Prenatal noise stress aggravates cognitive decline and the onset and progression of beta amyloid pathology in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2019, 77, 66–86. [Google Scholar] [CrossRef]
- Saljo, A.; Bao, F.; Shi, J.; Hamberger, A.; Hansson, H.A.; Haglid, K.G. Expression of c-Fos and c-Myc and deposition of beta-APP in neurons in the adult rat brain as a result of exposure to short-lasting impulse noise. J. Neurotrauma 2002, 19, 379–385. [Google Scholar] [CrossRef]
- Paciello, F.; Rinaudo, M.; Longo, V.; Cocco, S.; Conforto, G.; Pisani, A.; Podda, M.V.; Fetoni, A.R.; Paludetti, G.; Grassi, C. Auditory sensory deprivation induced by noise exposure exacerbates cognitive decline in a mouse model of Alzheimer’s disease. eLife 2021, 10, e70908. [Google Scholar] [CrossRef]
- Paciello, F.; Pisani, A.; Rinaudo, M.; Cocco, S.; Paludetti, G.; Fetoni, A.R.; Grassi, C. Noise-induced auditory damage affects hippocampus causing memory deficits in a model of early age-related hearing loss. Neurobiol. Dis. 2023, 178, 106024. [Google Scholar] [CrossRef]
- Qian, M.; Wang, Q.; Wang, Z.; Ma, Q.; Wang, X.; Han, K.; Wu, H.; Huang, Z. Dose-Dependent Pattern of Cochlear Synaptic Degeneration in C57BL/6J Mice Induced by Repeated Noise Exposure. Neural. Plast. 2021, 2021, 9919977. [Google Scholar] [CrossRef] [PubMed]
- Hahad, O.; Bayo Jimenez, M.T.; Kuntic, M.; Frenis, K.; Steven, S.; Daiber, A.; Munzel, T. Cerebral consequences of environmental noise exposure. Environ. Int. 2022, 165, 107306. [Google Scholar] [CrossRef] [PubMed]
- Hahad, O.; Lelieveld, J.; Birklein, F.; Lieb, K.; Daiber, A.; Munzel, T. Ambient Air Pollution Increases the Risk of Cerebrovascular and Neuropsychiatric Disorders through Induction of Inflammation and Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 4306. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Lu, Y.; Cheng, H.; Wang, C.; Chan, P. The impact of long-term exposure to ambient air pollution and second-hand smoke on the onset of Parkinson disease: A review and meta-analysis. Public Health 2020, 179, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Kasdagli, M.I.; Katsouyanni, K.; Dimakopoulou, K.; Samoli, E. Air pollution and Parkinson’s disease: A systematic review and meta-analysis up to 2018. Int. J. Hydrogen Environ. Health 2019, 222, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.Y.; Fang, Y.; Li, F.L.; Dong, B.; Hua, X.G.; Jiang, W.; Zhang, H.; Lyu, Y.; Zhang, X.J. Association between ambient air pollution and Parkinson’s disease: Systematic review and meta-analysis. Environ. Res. 2019, 168, 448–459. [Google Scholar] [CrossRef]
- Tsai, T.L.; Lin, Y.T.; Hwang, B.F.; Nakayama, S.F.; Tsai, C.H.; Sun, X.L.; Ma, C.; Jung, C.R. Fine particulate matter is a potential determinant of Alzheimer’s disease: A systemic review and meta-analysis. Environ. Res. 2019, 177, 108638. [Google Scholar] [CrossRef]
- Fu, P.; Guo, X.; Cheung, F.M.H.; Yung, K.K.L. The association between PM2.5 exposure and neurological disorders: A systematic review and meta-analysis. Sci. Total Environ. 2019, 655, 1240–1248. [Google Scholar] [CrossRef]
- Grande, G.; Ljungman, P.L.S.; Eneroth, K.; Bellander, T.; Rizzuto, D. Association Between Cardiovascular Disease and Long-term Exposure to Air Pollution With the Risk of Dementia. JAMA Neurol. 2020, 77, 801–809. [Google Scholar] [CrossRef]
- Ilango, S.D.; Chen, H.; Hystad, P.; van Donkelaar, A.; Kwong, J.C.; Tu, K.; Martin, R.V.; Benmarhnia, T. The role of cardiovascular disease in the relationship between air pollution and incident dementia: A population-based cohort study. Int. J. Epidemiol. 2020, 49, 36–44. [Google Scholar] [CrossRef]
- Cerza, F.; Renzi, M.; Gariazzo, C.; Davoli, M.; Michelozzi, P.; Forastiere, F.; Cesaroni, G. Long-term exposure to air pollution and hospitalization for dementia in the Rome longitudinal study. Environ. Health A Glob. Access Sci. Source 2019, 18, 72. [Google Scholar] [CrossRef]
- Li, C.Y.; Li, C.H.; Martini, S.; Hou, W.H. Association between air pollution and risk of vascular dementia: A multipollutant analysis in Taiwan. Environ. Int. 2019, 133, 105233. [Google Scholar] [CrossRef]
- Oudin, A.; Forsberg, B.; Adolfsson, A.N.; Lind, N.; Modig, L.; Nordin, M.; Nordin, S.; Adolfsson, R.; Nilsson, L.G. Traffic-Related Air Pollution and Dementia Incidence in Northern Sweden: A Longitudinal Study. Environ. Health Perspect. 2016, 124, 306–312. [Google Scholar] [CrossRef]
- Oudin, A.; Andersson, J.; Sundstrom, A.; Nordin Adolfsson, A.; Oudin Astrom, D.; Adolfsson, R.; Forsberg, B.; Nordin, M. Traffic-Related Air Pollution as a Risk Factor for Dementia: No Clear Modifying Effects of APOEvarepsilon4 in the Betula Cohort. J. Alzheimer’s Dis. JAD 2019, 71, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kwong, J.C.; Copes, R.; Hystad, P.; van Donkelaar, A.; Tu, K.; Brook, J.R.; Goldberg, M.S.; Martin, R.V.; Murray, B.J.; et al. Exposure to ambient air pollution and the incidence of dementia: A population-based cohort study. Environ. Int. 2017, 108, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kwong, J.C.; Copes, R.; Tu, K.; Villeneuve, P.J.; van Donkelaar, A.; Hystad, P.; Martin, R.V.; Murray, B.J.; Jessiman, B.; et al. Living near major roads and the incidence of dementia, Parkinson’s disease, and multiple sclerosis: A population-based cohort study. Lancet 2017, 389, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Andersson, J.; Oudin, A.; Sundstrom, A.; Forsberg, B.; Adolfsson, R.; Nordin, M. Road traffic noise, air pollution, and risk of dementia—results from the Betula project. Environ. Res. 2018, 166, 334–339. [Google Scholar] [CrossRef]
- Carey, I.M.; Anderson, H.R.; Atkinson, R.W.; Beevers, S.D.; Cook, D.G.; Strachan, D.P.; Dajnak, D.; Gulliver, J.; Kelly, F.J. Are noise and air pollution related to the incidence of dementia? A cohort study in London, England. BMJ Open 2018, 8, e022404. [Google Scholar] [CrossRef] [PubMed]
- Malek, A.M.; Arena, V.C.; Song, R.; Whitsel, E.A.; Rager, J.R.; Stewart, J.; Yanosky, J.D.; Liao, D.; Talbott, E.O. Long-term air pollution and risk of amyotrophic lateral sclerosis mortality in the Women’s Health Initiative cohort. Environ. Res. 2023, 216, 114510. [Google Scholar] [CrossRef]
- Malek, A.M.; Barchowsky, A.; Bowser, R.; Heiman-Patterson, T.; Lacomis, D.; Rana, S.; Ada, Y.; Talbott, E.O. Exposure to hazardous air pollutants and the risk of amyotrophic lateral sclerosis. Environ. Pollut. 2015, 197, 181–186. [Google Scholar] [CrossRef]
- Seelen, M.; Toro Campos, R.A.; Veldink, J.H.; Visser, A.E.; Hoek, G.; Brunekreef, B.; van der Kooi, A.J.; de Visser, M.; Raaphorst, J.; van den Berg, L.H.; et al. Long-Term Air Pollution Exposure and Amyotrophic Lateral Sclerosis in Netherlands: A Population-based Case-control Study. Environ. Health Perspect. 2017, 125, 097023. [Google Scholar] [CrossRef]
- Parks, R.M.; Nunez, Y.; Balalian, A.A.; Gibson, E.A.; Hansen, J.; Raaschou-Nielsen, O.; Ketzel, M.; Khan, J.; Brandt, J.; Vermeulen, R.; et al. Long-term Traffic-related Air Pollutant Exposure and Amyotrophic Lateral Sclerosis Diagnosis in Denmark: A Bayesian Hierarchical Analysis. Epidemiology 2022, 33, 757–766. [Google Scholar] [CrossRef]
- Myung, W.; Lee, H.; Kim, H. Short-term air pollution exposure and emergency department visits for amyotrophic lateral sclerosis: A time-stratified case-crossover analysis. Environ. Int. 2019, 123, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Antonioni, A.; Govoni, V.; Brancaleoni, L.; Dona, A.; Granieri, E.; Bergamini, M.; Gerdol, R.; Pugliatti, M. Amyotrophic Lateral Sclerosis and Air Pollutants in the Province of Ferrara, Northern Italy: An Ecological Study. Int. J. Environ. Res. Public Health 2023, 20, 5591. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Peters, S.; van Boxmeer, L.; Downward, G.S.; Hoek, G.; Kioumourtzoglou, M.A.; Weisskopf, M.G.; Hansen, J.; van den Berg, L.H.; Vermeulen, R.C.H. Long-Term Exposure to Ultrafine Particles and Particulate Matter Constituents and the Risk of Amyotrophic Lateral Sclerosis. Environ. Health Perspect. 2021, 129, 97702. [Google Scholar] [CrossRef]
- Heydarpour, P.; Amini, H.; Khoshkish, S.; Seidkhani, H.; Sahraian, M.A.; Yunesian, M. Potential impact of air pollution on multiple sclerosis in Tehran, Iran. Neuroepidemiology 2014, 43, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Angelici, L.; Piola, M.; Cavalleri, T.; Randi, G.; Cortini, F.; Bergamaschi, R.; Baccarelli, A.A.; Bertazzi, P.A.; Pesatori, A.C.; Bollati, V. Effects of particulate matter exposure on multiple sclerosis hospital admission in Lombardy region, Italy. Environ. Res. 2016, 145, 68–73. [Google Scholar] [CrossRef]
- Vojinovic, S.; Savic, D.; Lukic, S.; Savic, L.; Vojinovic, J. Disease relapses in multiple sclerosis can be influenced by air pollution and climate seasonal conditions. Vojnosanit Pregl. 2015, 72, 44–49. [Google Scholar] [CrossRef]
- Weuve, J.; D’Souza, J.; Beck, T.; Evans, D.A.; Kaufman, J.D.; Rajan, K.B.; de Leon, C.F.M.; Adar, S.D. Long-term community noise exposure in relation to dementia, cognition, and cognitive decline in older adults. Alzheimers Dement. 2021, 17, 525–533. [Google Scholar] [CrossRef]
- Tzivian, L.; Dlugaj, M.; Winkler, A.; Weinmayr, G.; Hennig, F.; Fuks, K.B.; Vossoughi, M.; Schikowski, T.; Weimar, C.; Erbel, R.; et al. Long-Term Air Pollution and Traffic Noise Exposures and Mild Cognitive Impairment in Older Adults: A Cross-Sectional Analysis of the Heinz Nixdorf Recall Study. Environ. Health Perspect. 2016, 124, 1361–1368. [Google Scholar] [CrossRef]
- Tzivian, L.; Dlugaj, M.; Winkler, A.; Hennig, F.; Fuks, K.; Sugiri, D.; Schikowski, T.; Jakobs, H.; Erbel, R.; Jockel, K.H.; et al. Long-term air pollution and traffic noise exposures and cognitive function:A cross-sectional analysis of the Heinz Nixdorf Recall study. J. Toxicol. Environ. Health A 2016, 79, 1057–1069. [Google Scholar] [CrossRef]
- Tzivian, L.; Jokisch, M.; Winkler, A.; Weimar, C.; Hennig, F.; Sugiri, D.; Soppa, V.J.; Dragano, N.; Erbel, R.; Jockel, K.H.; et al. Associations of long-term exposure to air pollution and road traffic noise with cognitive function-An analysis of effect measure modification. Environ. Int. 2017, 103, 30–38. [Google Scholar] [CrossRef]
- Fuks, K.B.; Wigmann, C.; Altug, H.; Schikowski, T. Road Traffic Noise at the Residence, Annoyance, and Cognitive Function in Elderly Women. Int. J. Environ. Res. Public Health 2019, 16, 1790. [Google Scholar] [CrossRef]
- Mac Domhnaill, C.; Douglas, O.; Lyons, S.; Murphy, E.; Nolan, A. Road traffic noise and cognitive function in older adults: A cross-sectional investigation of The Irish Longitudinal Study on Ageing. BMC Public Health 2021, 21, 1814. [Google Scholar] [CrossRef]
- Yu, Y.; Mayeda, E.R.; Paul, K.C.; Lee, E.; Jerrett, M.; Su, J.; Wu, J.; Shih, I.F.; Haan, M.; Ritz, B. Traffic-related Noise Exposure and Late-life Dementia and Cognitive Impairment in Mexican-Americans. Epidemiology 2020, 31, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Haan, M.; Paul, K.C.; Mayeda, E.R.; Jerrett, M.; Wu, J.; Lee, E.; Su, J.; Shih, I.F.; Inoue, K.; et al. Metabolic dysfunction modifies the influence of traffic-related air pollution and noise exposure on late-life dementia and cognitive impairment: A cohort study of older Mexican-Americans. Environ. Epidemiol. 2020, 4, e122. [Google Scholar] [CrossRef]
- Linares, C.; Culqui, D.; Carmona, R.; Ortiz, C.; Diaz, J. Short-term association between environmental factors and hospital admissions due to dementia in Madrid. Environ. Res. 2017, 152, 214–220. [Google Scholar] [CrossRef]
- Yuchi, W.; Sbihi, H.; Davies, H.; Tamburic, L.; Brauer, M. Road proximity, air pollution, noise, green space and neurologic disease incidence: A population-based cohort study. Environ. Health A Glob. Access Sci. Source 2020, 19, 8. [Google Scholar] [CrossRef] [PubMed]
- Cantuaria, M.L.; Waldorff, F.B.; Wermuth, L.; Pedersen, E.R.; Poulsen, A.H.; Thacher, J.D.; Raaschou-Nielsen, O.; Ketzel, M.; Khan, J.; Valencia, V.H.; et al. Residential exposure to transportation noise in Denmark and incidence of dementia: National cohort study. BMJ 2021, 374, n1954. [Google Scholar] [CrossRef]
- Cole-Hunter, T.; So, R.; Amini, H.; Backalarz, C.; Brandt, J.; Brauner, E.V.; Hertel, O.; Jensen, S.S.; Jorgensen, J.T.; Ketzel, M.; et al. Long-term exposure to road traffic noise and all-cause and cause-specific mortality: A Danish Nurse Cohort study. Sci. Total Environ. 2022, 820, 153057. [Google Scholar] [CrossRef] [PubMed]
- Parra, K.L.; Alexander, G.E.; Raichlen, D.A.; Klimentidis, Y.C.; Furlong, M.A. Exposure to air pollution and risk of incident dementia in the UK Biobank. Environ. Res. 2022, 209, 112895. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuntić, M.; Hahad, O.; Münzel, T.; Daiber, A. Crosstalk between Oxidative Stress and Inflammation Caused by Noise and Air Pollution—Implications for Neurodegenerative Diseases. Antioxidants 2024, 13, 266. https://doi.org/10.3390/antiox13030266
Kuntić M, Hahad O, Münzel T, Daiber A. Crosstalk between Oxidative Stress and Inflammation Caused by Noise and Air Pollution—Implications for Neurodegenerative Diseases. Antioxidants. 2024; 13(3):266. https://doi.org/10.3390/antiox13030266
Chicago/Turabian StyleKuntić, Marin, Omar Hahad, Thomas Münzel, and Andreas Daiber. 2024. "Crosstalk between Oxidative Stress and Inflammation Caused by Noise and Air Pollution—Implications for Neurodegenerative Diseases" Antioxidants 13, no. 3: 266. https://doi.org/10.3390/antiox13030266
APA StyleKuntić, M., Hahad, O., Münzel, T., & Daiber, A. (2024). Crosstalk between Oxidative Stress and Inflammation Caused by Noise and Air Pollution—Implications for Neurodegenerative Diseases. Antioxidants, 13(3), 266. https://doi.org/10.3390/antiox13030266