
Diabetic Retinopathy and Regulation of Mitochondrial Glutathione–Glutathione Peroxidase Axis in Hyperhomocysteinemia


Abstract
1. Introduction
2. Methods
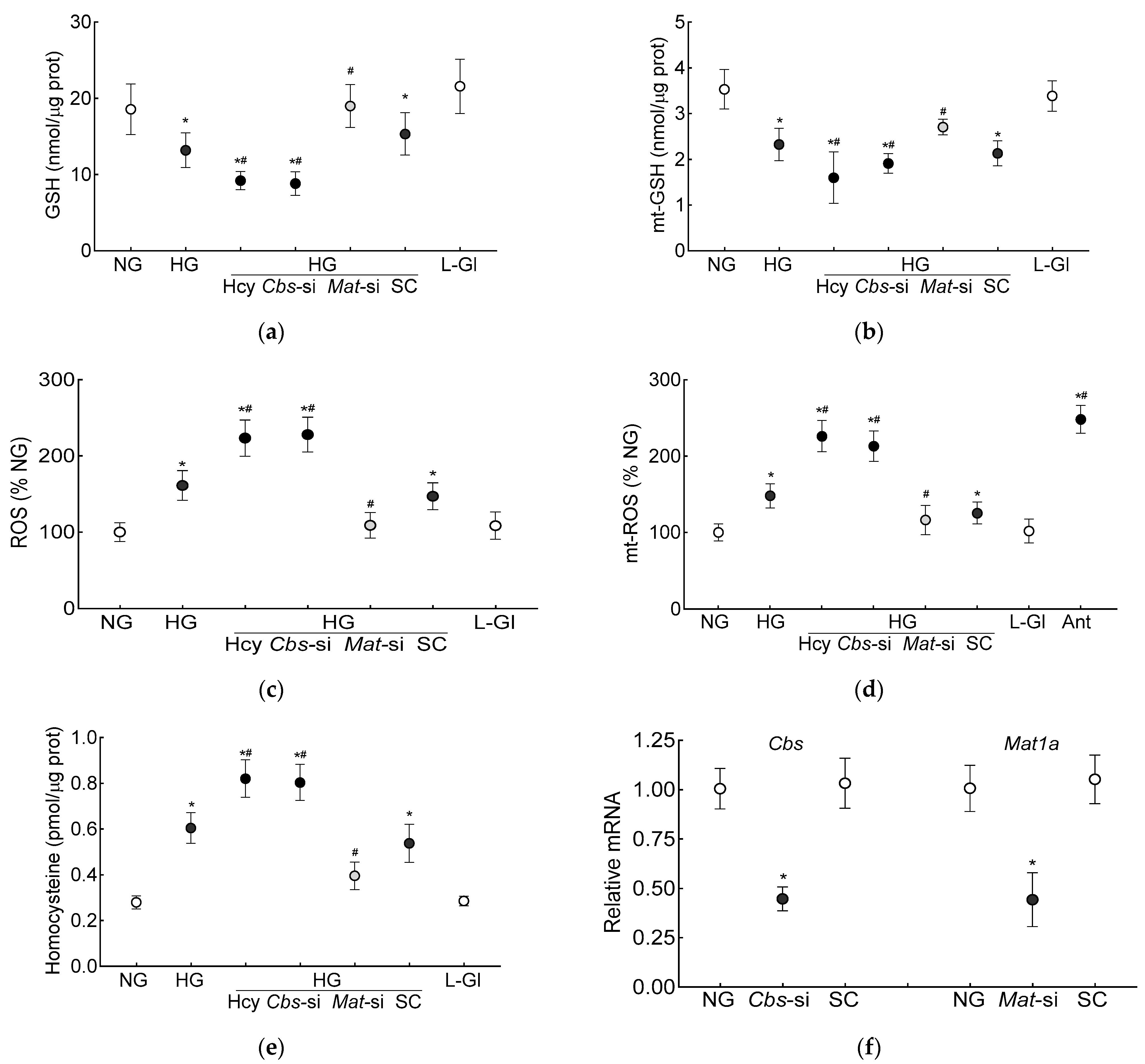
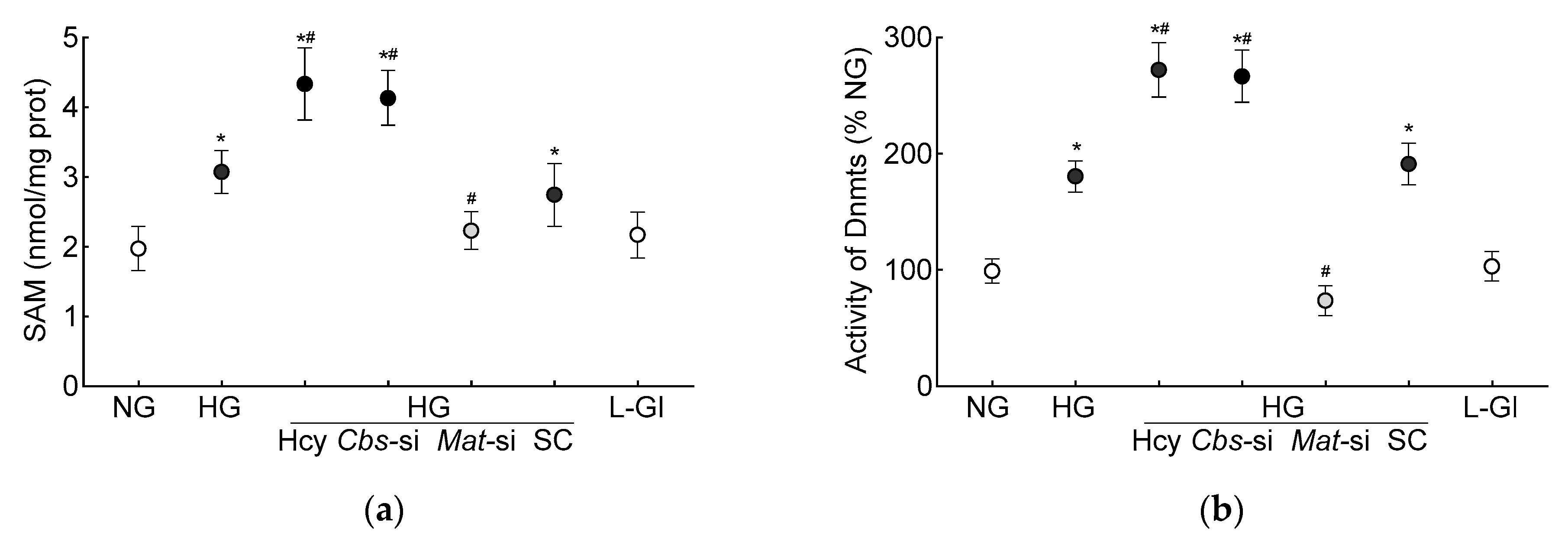
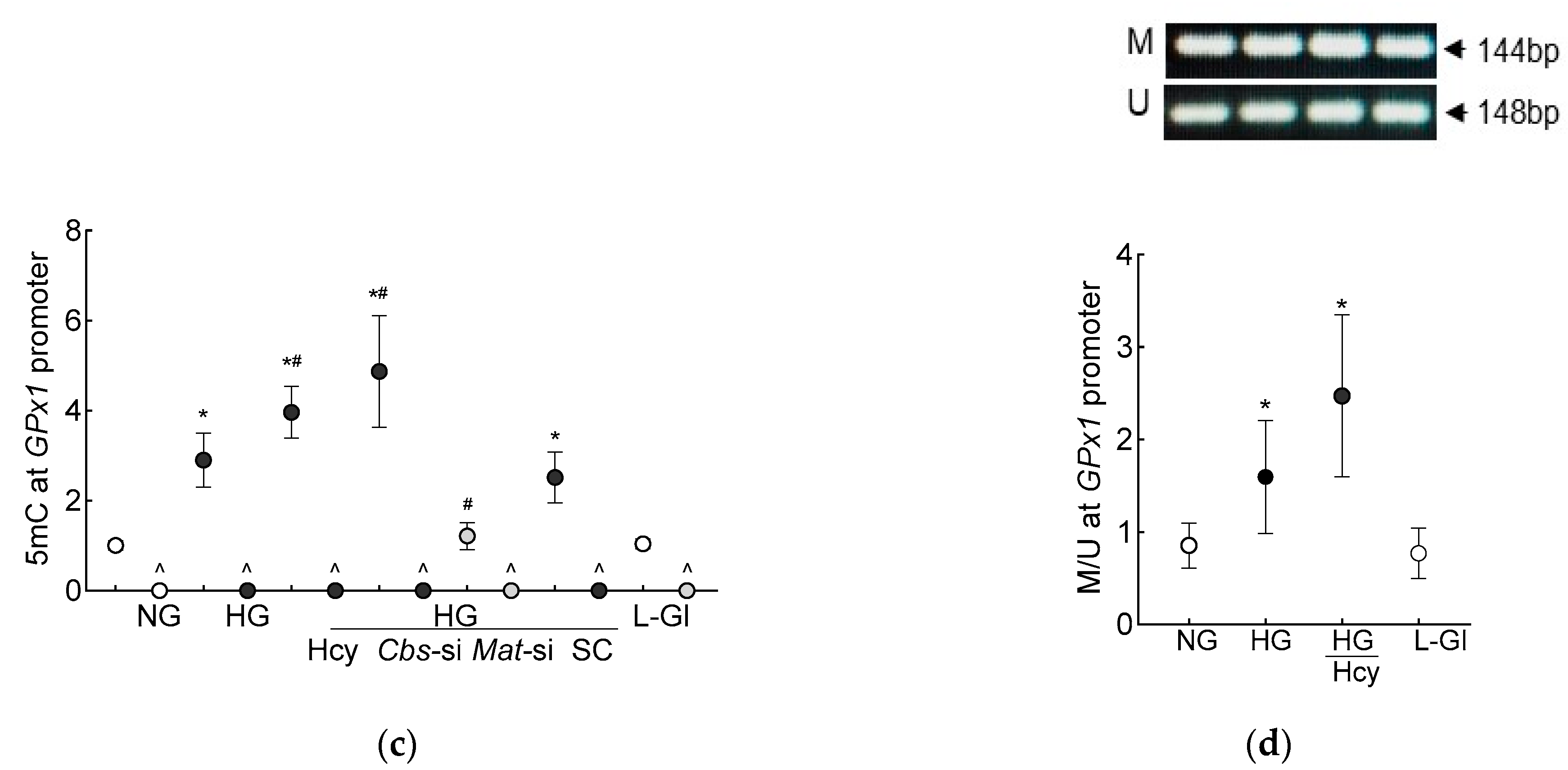
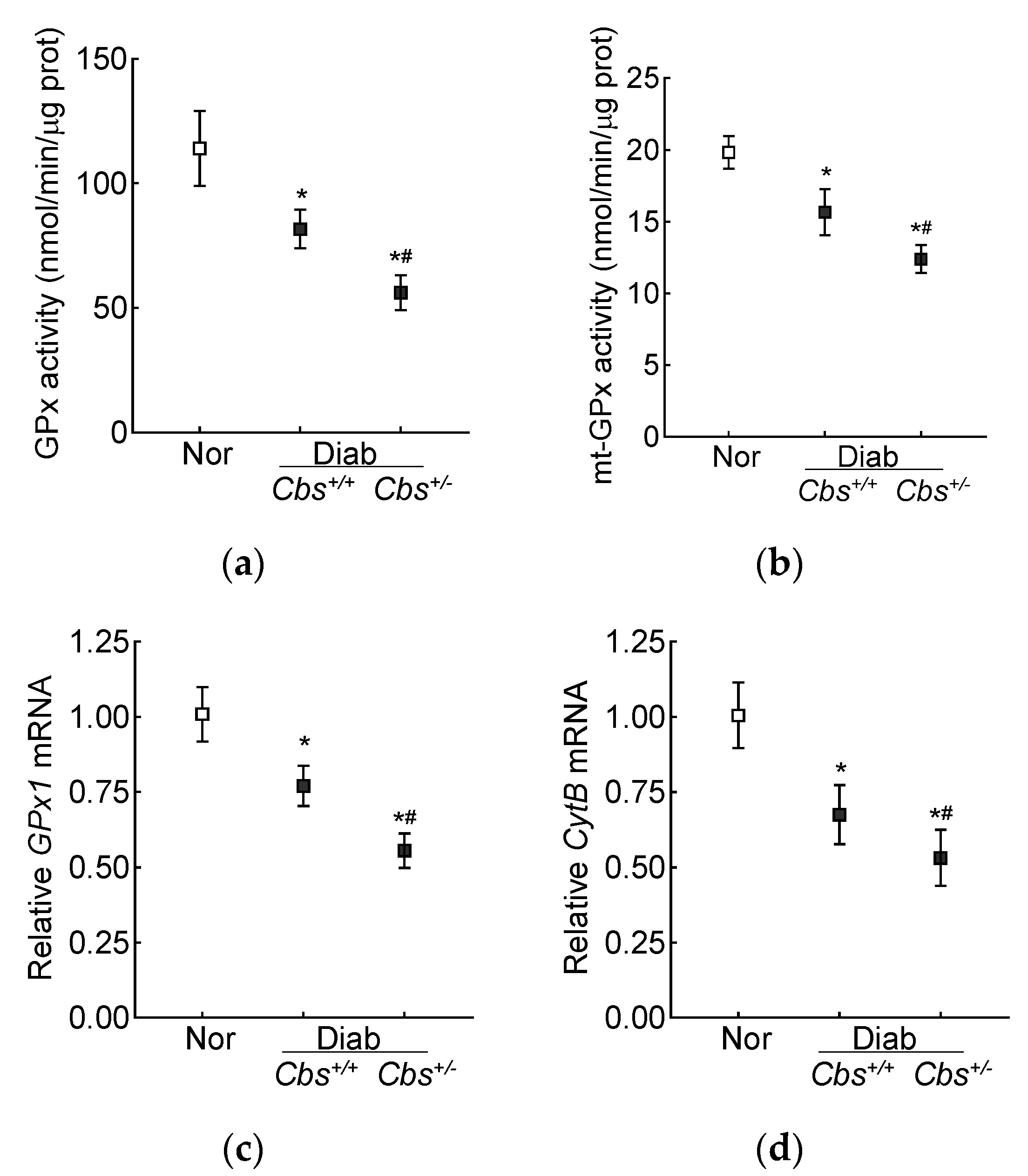
3. Results
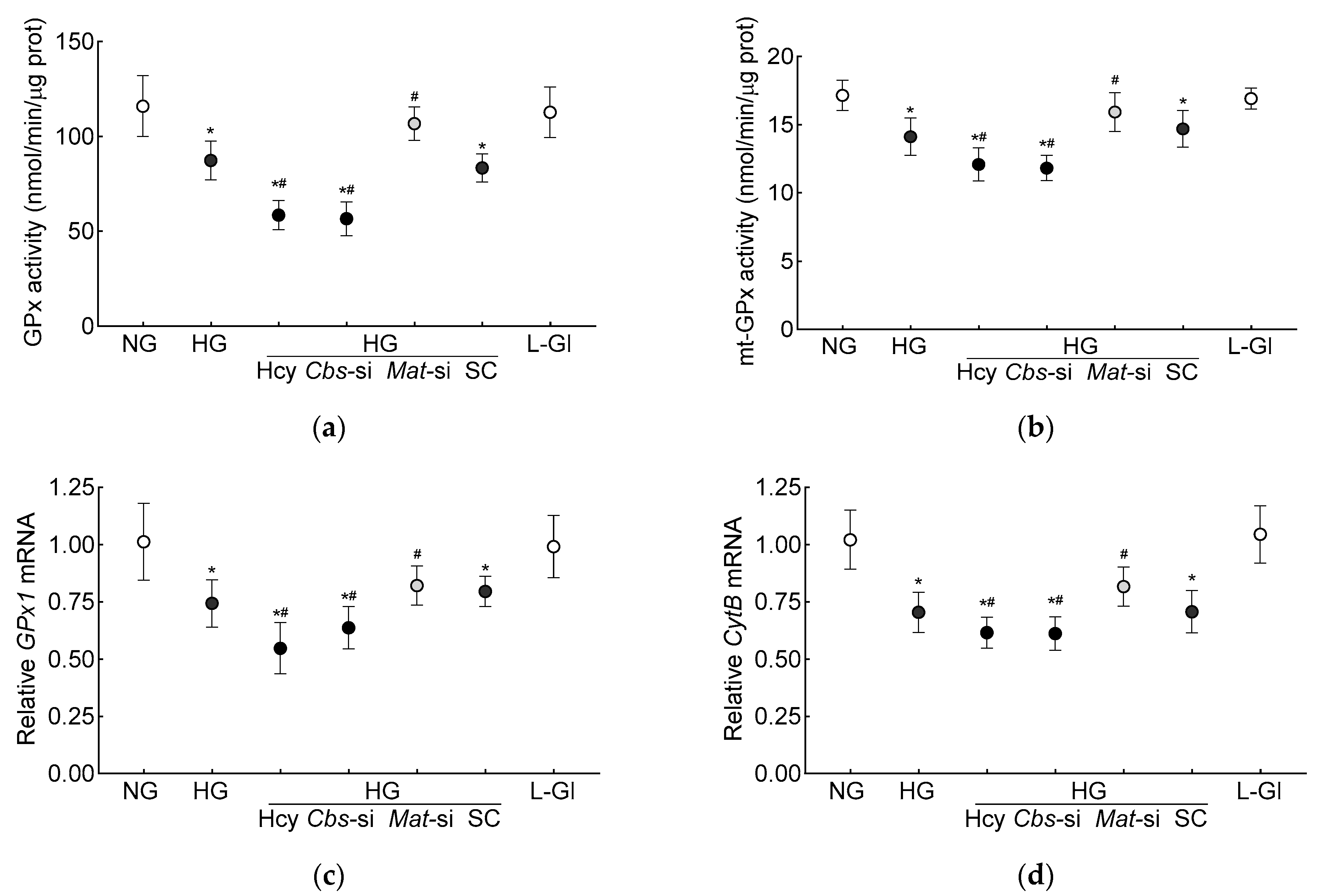
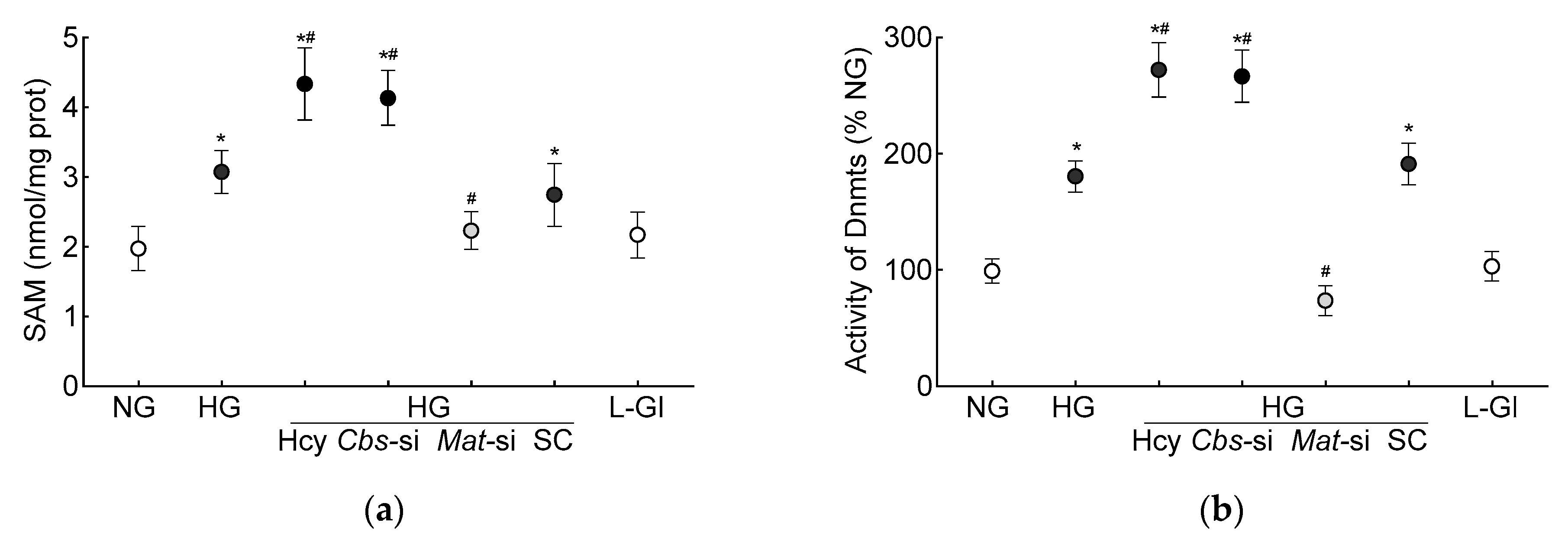
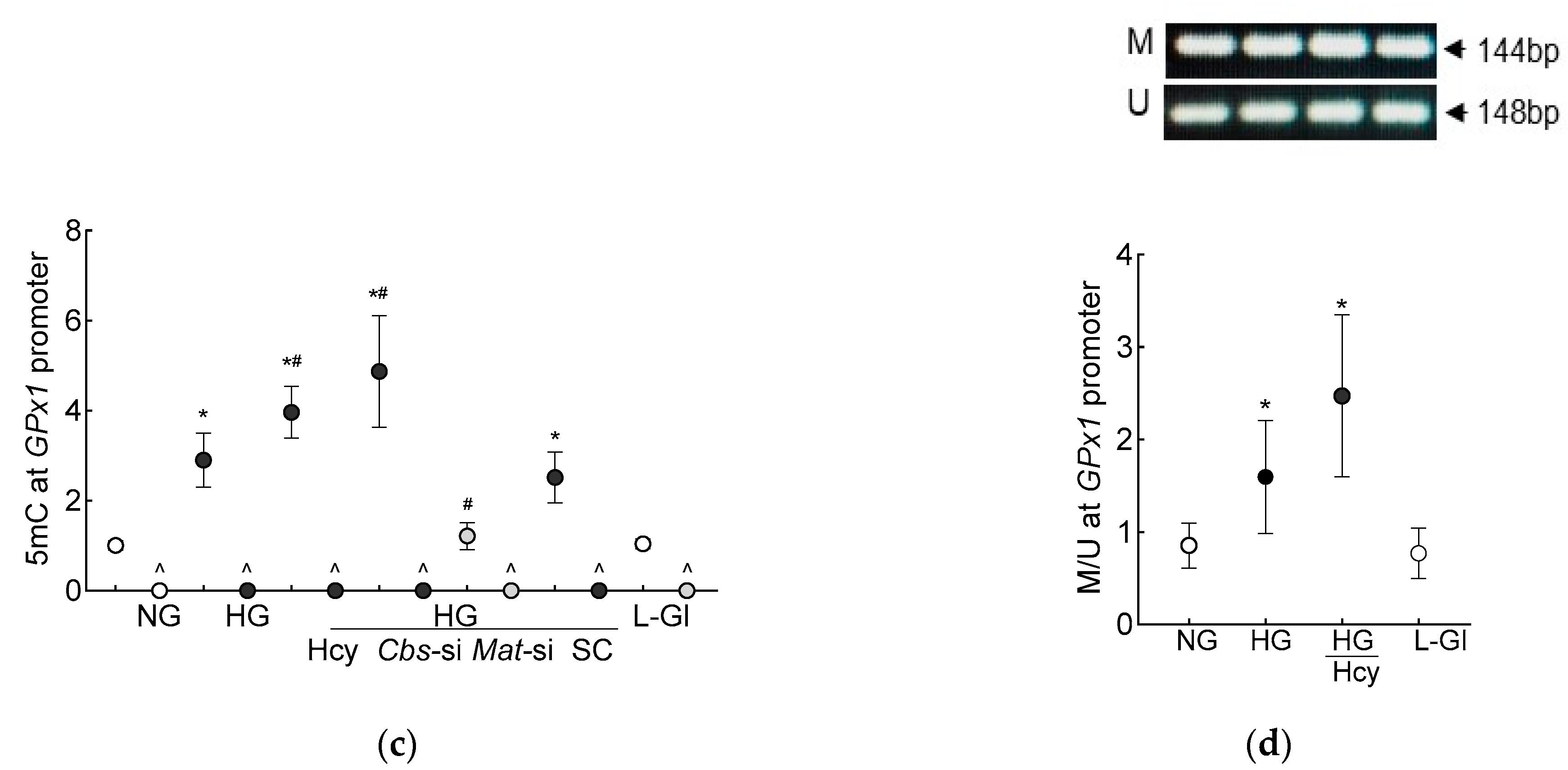
3.1. In Vitro Human Retinal Endothelial Cells
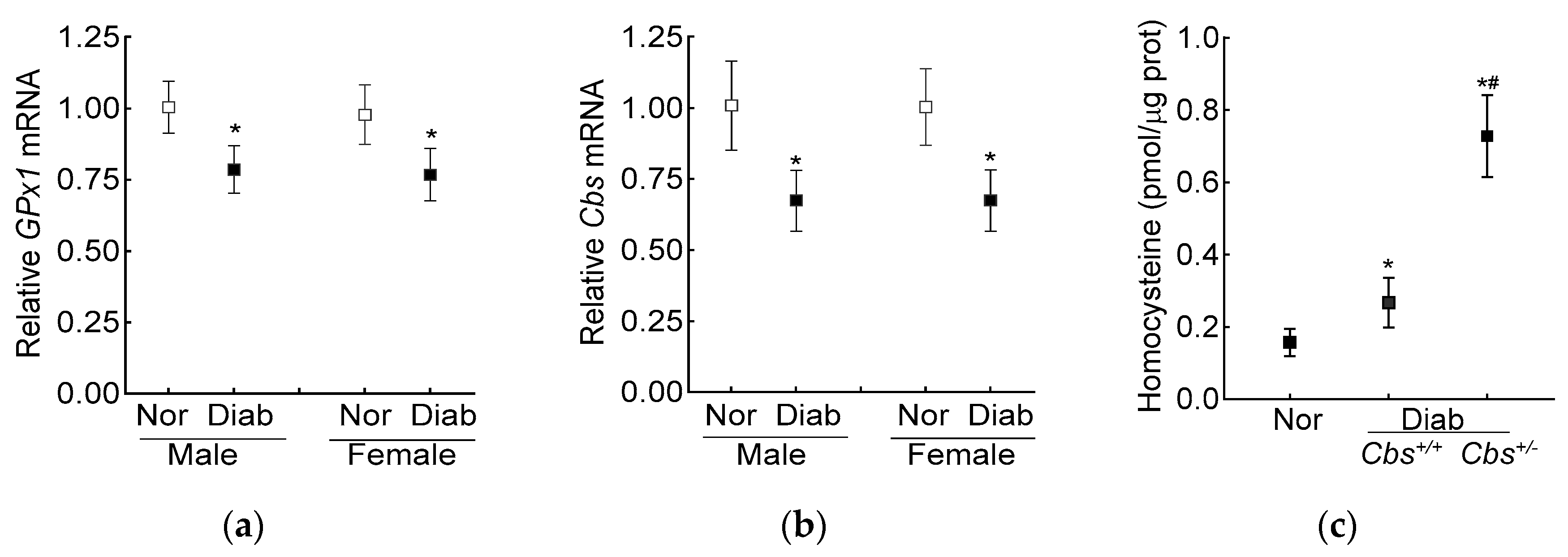
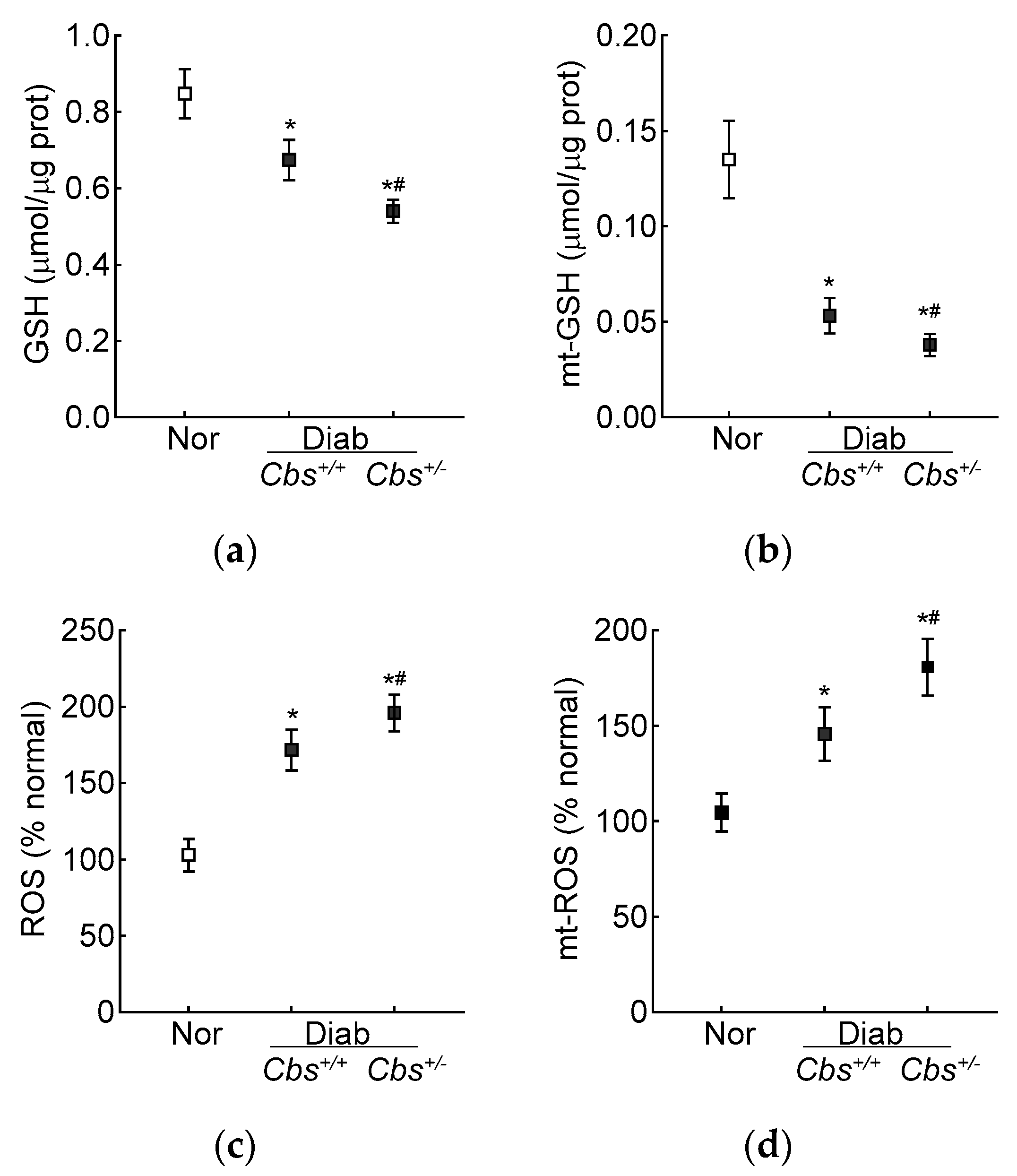
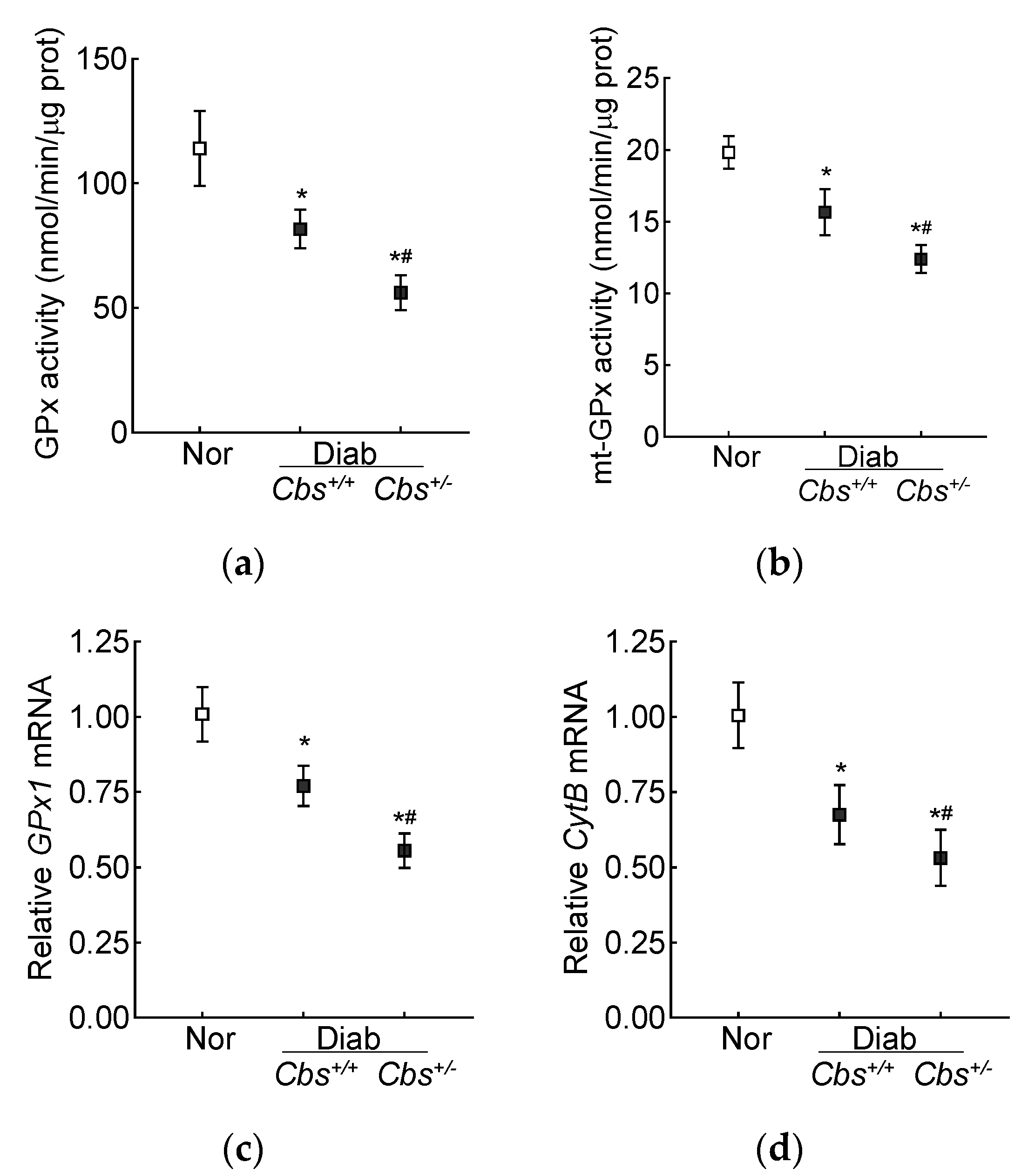
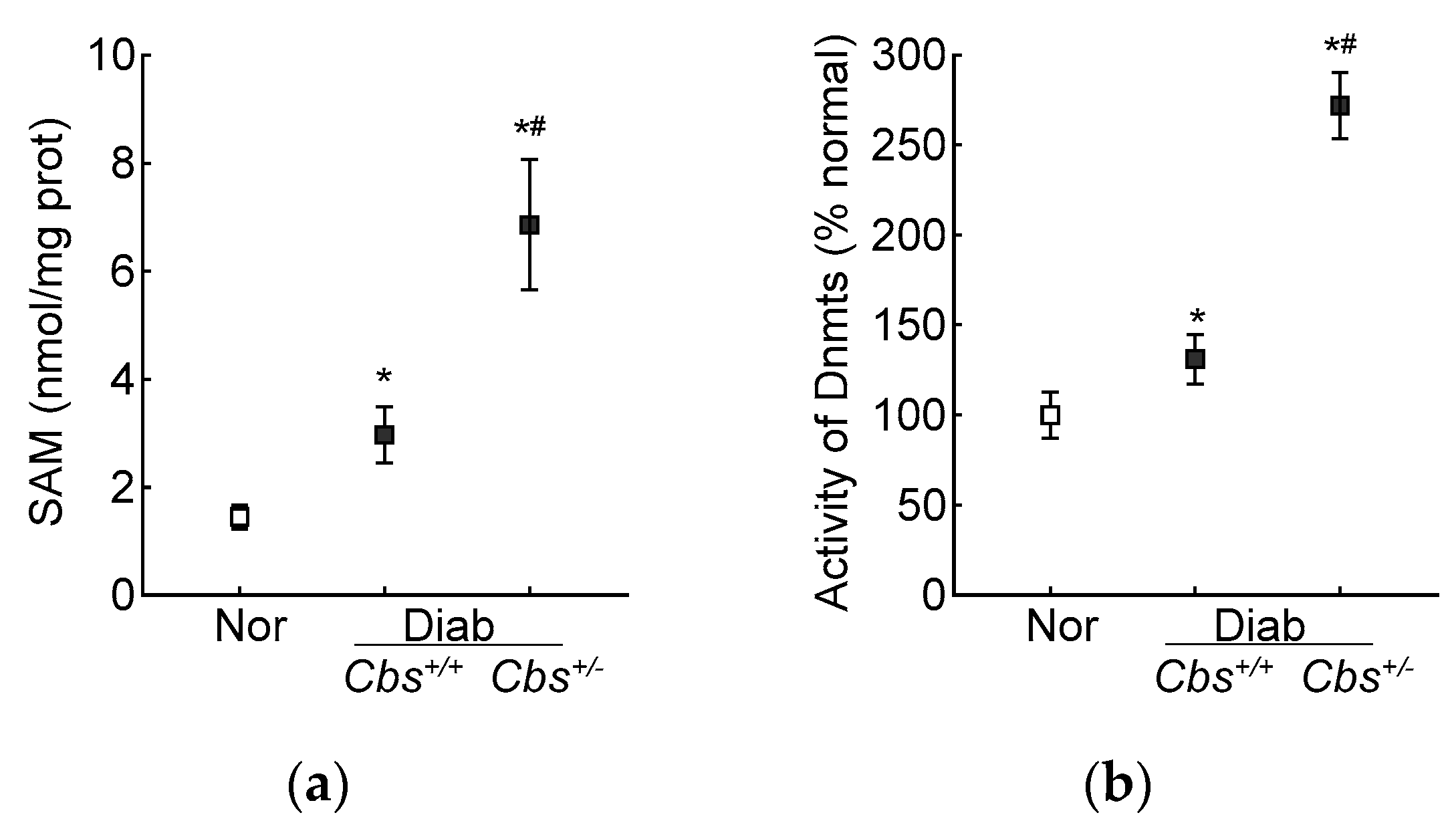
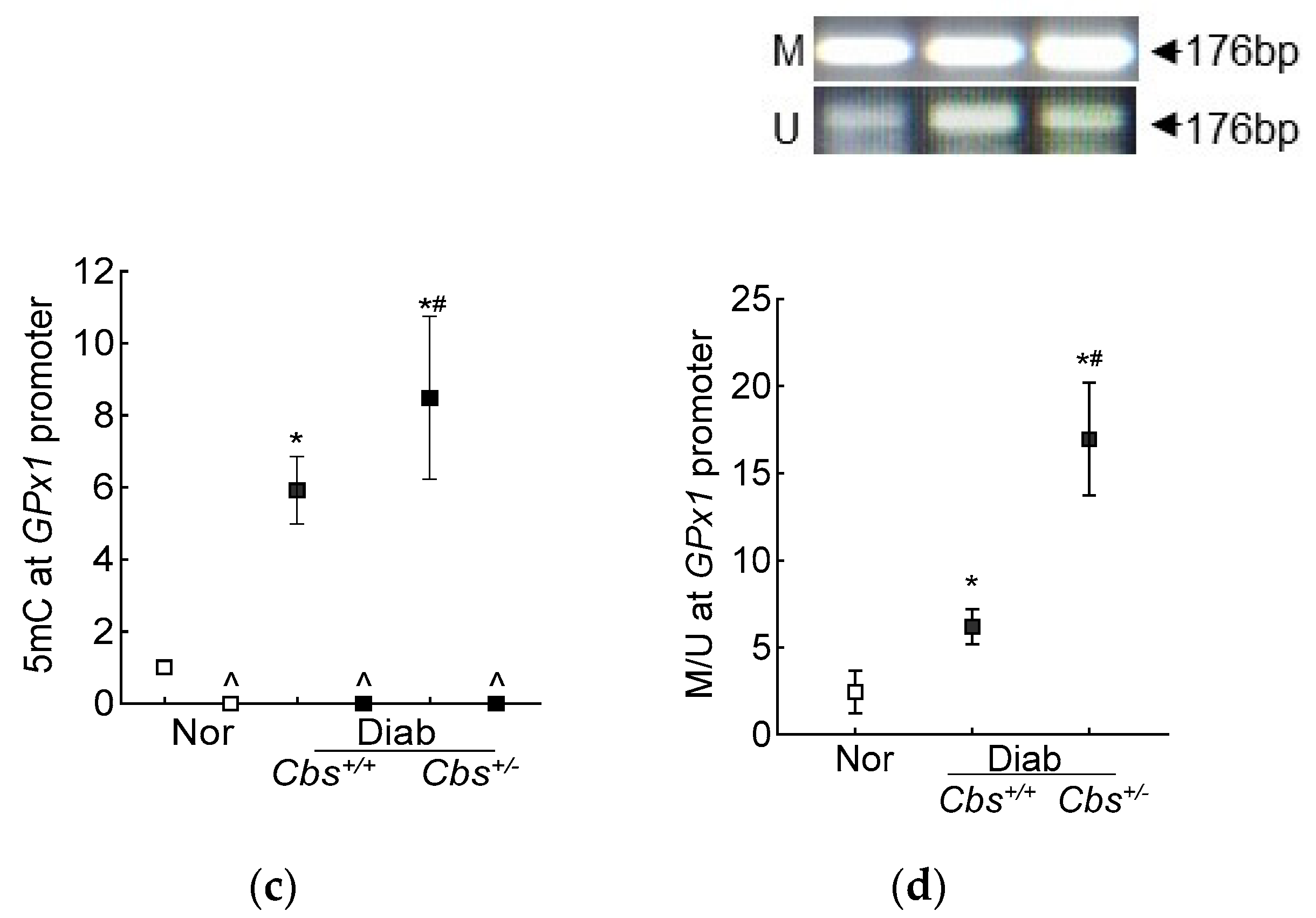
3.2. In Vivo Mouse Model
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Frank, R.N. Diabetic Retinopathy. N. Engl. J. Med. 2004, 350, 48–58. [Google Scholar] [CrossRef]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Mishra, M. Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim. Biophys. Acta 2015, 1852, 2474–2483. [Google Scholar] [CrossRef]
- Kowluru, R.A. Mitochondrial stability in diabetic retinopathy: Lessons learned from epigenetics. Diabetes 2019, 68, 241–247. [Google Scholar] [CrossRef]
- Haydinger, C.D.; Oliver, G.F.; Ashander, L.M.; Smith, J.R. Oxidative Stress and Its Regulation in Diabetic Retinopathy. Antioxidants 2023, 12, 1649. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Kern, T.S.; Engerman, R.L. Abnormalities of retinal metabolism in diabetes or experimental galactosemia. IV. Antioxidant defense system. Free Radic. Biol. Med. 1997, 22, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mohammad, G.; Sahajpal, N. Faulty homocysteine recycling in diabetic retinopathy. Eye Vis. 2020, 7, 4. [Google Scholar] [CrossRef]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef]
- Ruszkiewicz, J.; Albrecht, J. Changes in the mitochondrial antioxidant systems in neurodegenerative diseases and acute brain disorders. Neurochem. Int. 2015, 88, 66–72. [Google Scholar] [CrossRef]
- Ekoue, D.N.; He, C.; Diamond, A.M.; Bonini, M.G. Manganese superoxide dismutase and glutathione peroxidase-1 contribute to the rise and fall of mitochondrial reactive oxygen species which drive oncoegenesis. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, H.; Zhou, J.; Shao, Q. Glutathione Peroxidase GPX1 and Its Dichotomous Roles in Cancer. Cancers 2022, 14, 2560. [Google Scholar] [CrossRef]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal 2009, 11, 2685–2700. [Google Scholar] [CrossRef]
- Liang, H.; Ran, Q.; Jang, Y.C.; Holstein, D.; Lechleiter, J.; McDonald-Marsh, T.; Musatov, A.; Song, W.; Van Remmen, H.; Richardson, A. Glutathione peroxidase 4 differentially regulates the release of apoptogenic proteins from mitochondria. Free Radic. Biol. Med. 2009, 47, 312–320. [Google Scholar] [CrossRef]
- He, W.; Chang, L.; Li, X.; Mei, Y. Research progress on the mechanism of ferroptosis and its role in diabetic retinopathy. Front. Endocrinol. 2023, 14, 1155296. [Google Scholar] [CrossRef] [PubMed]
- Frank, R.N. Diabetic retinopathy and systemic factors. Middle East. Afr. J. Ophthalmol. 2015, 22, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, A.; Mohamed, R.; Elsherbiny, N.M.; DeAngelis, M.M.; Bartoli, M.; Al-Shabrawey, M. Homocysteine: A Potential Biomarker for Diabetic Retinopathy. J. Clin. Med. 2019, 8, 121. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Givvimani, S.; Qipshidze, N.; Kundu, S.; Kapoor, S.; Vacek, J.C.; Tyagi, S.C. Hydrogen sulfide mitigates matrix metalloproteinase-9 activity and neurovascular permeability in hyperhomocysteinemic mice. Neurochem. Int. 2010, 56, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, A.; Al-Shabrawey, M.; Roon, P.; Sonne, S.; Covar, J.A.; Matragoon, S.; Ganapathy, P.S.; Atherton, S.S.; El-Remessy, A.; Ganapathy, V.; et al. Alterations of retinal vasculature in cystathionine-Beta-synthase mutant mice, a model of hyperhomocysteinemia. Invest. Ophthalmol. Vis. Sci. 2013, 54, 939–949. [Google Scholar] [CrossRef]
- Kowluru, R.A. Diabetic retinopathy: Mitochondria caught in a muddle of homocysteine. J. Clin. Med. 2020, 9, 3019. [Google Scholar] [CrossRef]
- Malaviya, P.; Kowluru, R.A. Homocysteine and mitochondrial quality control in diabetic retinopathy. Eye Vis. 2024, 11, 5. [Google Scholar] [CrossRef]
- Weber, G.J.; Pushpakumar, S.; Tyagi, S.C.; Sen, U. Homocysteine and hydrogen sulfide in epigenetic, metabolic and microbiota related renovascular hypertension. Pharmacol. Res. 2016, 113, 300–312. [Google Scholar] [CrossRef]
- Perez-Sepulveda, A.; Espana-Perrot, P.P.; Fernandez, X.B.; Ahumada, V.; Bustos, V.; Arraztoa, J.A.; Dobierzewska, A.; Figueroa-Diesel, H.; Rice, G.E.; Illanes, S.E. Levels of key enzymes of methionine-homocysteine metabolism in preeclampsia. Biomed. Res. Int. 2013, 2013, 731962. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, G.; Kowluru, R.A. Homocysteine disrupts balance between MMP-9 and Its tissue inhibitor in diabetic retinopathy: The role of DNA methylation. Int. J. Mol. Sci. 2020, 21, 1771. [Google Scholar] [CrossRef]
- Robertson, K.D.; Wolffe, A.P. DNA methylation in health and disease. Nat. Rev. Genet. 2000, 1, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, X.; Meng, X.; Kong, Y.; Li, Y.; Yang, C.; Guo, Y.; Wang, X.; Yang, H.; Liu, Z.; et al. Selenium Supplementation Improved Cardiac Functions by Suppressing DNMT2-Mediated GPX1 Promoter DNA Methylation in AGE-Induced Heart Failure. Oxid. Med. Cell Longev. 2022, 2022, 5402997. [Google Scholar] [CrossRef]
- Min, S.Y.; Kim, H.S.; Jung, E.J.; Jung, E.J.; Jee, C.D.; Kim, W.H. Prognostic significance of glutathione peroxidase 1 (GPX1) down-regulation and correlation with aberrant promoter methylation in human gastric cancer. Anticancer Res. 2012, 32, 3169–3175. [Google Scholar] [PubMed]
- Jin, B.; Robertson, K.D. DNA methyltransferases, DNA damage repair, and cancer. Adv. Exp. Med. Biol. 2013, 754, 3–29. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mohammad, G. Mitochondrial Fragmentation in a High Homocysteine Environment in Diabetic Retinopathy. Antioxidants 2022, 11, 365. [Google Scholar] [CrossRef] [PubMed]
- Elsherbiny, N.M.; Sharma, I.; Kira, D.; Alhusban, S.; Samra, Y.A.; Jadeja, R.; Martin, P.; Al-Shabrawey, M.; Tawfik, A. Homocysteine Induces Inflammation in Retina and Brain. Biomolecules 2020, 10, 393. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Abbas, S.N. Diabetes-induced mitochondrial dysfunction in the retina. Investig. Ophthalmol. Vis. Sci. 2003, 44, 5327–5334. [Google Scholar] [CrossRef]
- Stockman, S.L.; Kight, K.E.; Bowers, J.M.; McCarthy, M.M. Neurogenesis in the neonatal rat hippocampus is regulated by sexually dimorphic epigenetic modifiers. Biol. Sex. Differ. 2022, 13, 9. [Google Scholar] [CrossRef]
- Kalamkar, S.; Acharya, J.; Kolappurath Madathil, A.; Gajjar, V.; Divate, U.; Karandikar-Iyer, S.; Goel, P.; Ghaskadbi, S. Randomized Clinical Trial of How Long-Term Glutathione Supplementation Offers Protection from Oxidative Damage and Improves HbA1c in Elderly Type 2 Diabetic Patients. Antioxidants 2022, 11, 1026. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.R.; Saldana-Caboverde, A.; Garcia, S.; Diaz, F. The Organization of Mitochondrial Supercomplexes is Modulated by Oxidative Stress In Vivo in Mouse Models of Mitochondrial Encephalopathy. Int. J. Mol. Sci. 2018, 19, 1582. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, C.A.; Legart, C.; Møllerhøj, M.B.; Madsen, M.R.; Hansen, H.H.; Kønig, M.J.; Helgstrand, F.; Hjørne, F.P.; Toxværd, A.; Langhoff, J.L.; et al. A liver secretome gene signature-based approach for determining circulating biomarkers of NAFLD severity. PLoS ONE 2022, 17, e0275901. [Google Scholar] [CrossRef] [PubMed]
- Li, L.C.; Dahiya, R. MethPrimer: Designing primers for methylation PCRs. Bioinformatics 2002, 18, 1427–1431. [Google Scholar] [CrossRef] [PubMed]
- Tewari, S.; Zhong, Q.; Santos, J.M.; Kowluru, R.A. Mitochondria DNA replication and DNA methylation in the metabolic memory associated with continued progression of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4881–4888. [Google Scholar] [CrossRef] [PubMed]
- Kurdyukov, S.; Bullock, M. DNA Methylation Analysis: Choosing the Right Method. Biology 2016, 5, 3. [Google Scholar] [CrossRef]
- Perry, N.; Wasko, K.; Cheng, J.; Tabbaa, D.; Marco, E.; Giannoukos, G.; Albright, C.F.; Borges, C.M. Methylation-Sensitive Restriction Enzyme Quantitative Polymerase Chain Reaction Enables Rapid, Accurate, and Precise Detection of Methylation Status of the Regulatory T Cell (Treg)-Specific Demethylation Region in Primary Human Tregs. J. Immunol. 2021, 206, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Pacana, T.; Cazanave, S.; Verdianelli, A.; Patel, V.; Min, H.K.; Mirshahi, F.; Quinlivan, E.; Sanyal, A.J. Dysregulated Hepatic Methionine Metabolism Drives Homocysteine Elevation in Diet-Induced Nonalcoholic Fatty Liver Disease. PLoS ONE 2015, 10, e0136822. [Google Scholar] [CrossRef]
- Mishra, M.; Duraisamy, A.J.; Kowluru, R.A. Sirt1—A guardian of the development of diabetic retinopathy. Diabetes 2018, 67, 745–754. [Google Scholar] [CrossRef]
- Chew, E.Y.; Davis, M.D.; Danis, R.P.; Lovato, J.F.; Perdue, L.H.; Greven, C.; Genuth, S.; Goff, D.C.; Leiter, L.A.; Ismail-Beigi, F.; et al. The effects of medical management on the progression of diabetic retinopathy in persons with type 2 diabetes: The Action to Control Cardiovascular Risk in Diabetes (ACCORD) Eye Study. Ophthalmology 2014, 121, 2443–2451. [Google Scholar] [CrossRef]
- Kowluru, R.A. Diabetic retinopathy and NADPH oxidase-2: A sweet slippery road. Antioxidants 2021, 10, 783. [Google Scholar] [CrossRef]
- Lei, X.; Zeng, G.; Zhang, Y.; Li, Q.; Zhang, J.; Bai, Z.; Yang, K. Association between homocysteine level and the risk of diabetic retinopathy: A systematic review and meta-analysis. Diabetol. Metab. Syndr. 2018, 10, 61. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, P.; Tatarkova, Z.; Sivonova, M.K.; Racay, P.; Lehotsky, J. Homocysteine and Mitochondria in Cardiovascular and Cerebrovascular Systems. Int. J. Mol. Sci. 2020, 21, 7698. [Google Scholar] [CrossRef]
- Toledano, M.B.; Huang, M.E. The Unfinished Puzzle of Glutathione Physiological Functions, an Old Molecule That Still Retains Many Enigmas. Antioxid. Redox Signal 2017, 27, 1127–1129. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Aw, T.Y. Glutathione and modulation of cell apoptosis. Biochim. Biophys. Acta 2012, 1823, 1767–1777. [Google Scholar] [CrossRef] [PubMed]
- Griffith, O.W.; Meister, A. Origin and turnover of mitochondrial glutathione. Proc. Natl. Acad. Sci. USA 1985, 82, 4668–4672. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Checa, J.C.; García-Ruiz, C.; Colell, A.; Morales, A.; Marí, M.; Miranda, M.; Ardite, E. Oxidative stress: Role of mitochondria and protection by glutathione. Biofactors 1998, 8, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Loscalzo, J. The role of glutathione peroxidase-1 in health and disease. Free Radic. Biol. Med. 2022, 188, 146–161. [Google Scholar] [CrossRef]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Homocysteine and glutathione peroxidase-1. Antioxid. Redox Signal 2007, 9, 1923–1940. [Google Scholar] [CrossRef] [PubMed]
- Mandaviya, P.R.; Stolk, L.; Heil, S.G. Homocysteine and DNA methylation: A review of animal and human literature. Mol. Genet. Metab. 2014, 113, 243–252. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5′-3′) | Accession No. | Amplicon (bp) |
|---|---|---|---|
| Human | |||
| GPx1 | Fwd-CAACCAGTTTGGGCATCAGG Rev-AGAGAGTAGCCAGACTCTCCG | NM_201397.3 | 134 |
| Cbs | Fwd-GACCAAGTTCCTGAGCGACA Rev-CGGAGGATCTCGATGGTGTG | NM_000071.3 | 171 |
| Mat1a | Fwd-GCGTGAGTGGAGAAGTGTGA Rev-CCAGTCAGTCCAAGACAGCC | NM_000429.3 | 142 |
| CytB | Fwd-TCACCAGACGCCTCAACCGC Rev-GCCTCGCCCGATGTGTAGGA | ENSG00000198727 | 138 |
| B-actin | Fwd-AGCCTCGCCTTTGCCGATCCG Rev-TCTCTTGCTCTGGGCCTCGTCG | NM_001101.5 | 237 |
| GPx1 Promoter (MeDIP) | Fwd-GGGGGCCGGATGAGGCGGGA Rev-AACTGGCCGGCGGCGGGTCAC | NM_201397 | 71 |
| GPx1 Promoter (MS-PCR) | Methyl Fwd-GTTTTTTTCGGTTTAGGAGGAGTAC Rev-CGTAACACAAACAATTTTCCGA | NM_201397 | 144 |
| Unmethyl Fwd-TTTTTTTTGGTTTAGGAGTATG Rev-TCACATAACACAAACAATTTTCCA | 148 | ||
| Mouse | |||
| GPx1 | Fwd-GACACCAGGTATATGGGGCG Rev-AGAAAGTAAGCGGTGTCCCG | NM_008160.6 | 149 |
| Cbs | Fwd-CGTGCTCTCCATCCTAGCAC Rev-GTTGGCTCTTGAGTCCCCTC | NM_144855.4 | 137 |
| Mat1a | Fwd-GCAGAAGTCATCTCCTCGGG Rev-ACAGGTCCATTCATTGTGCCA | NM_133653.3 | 146 |
| CytB | Fwd-ACCCGCCCCATCCAACATCTCAT Rev-TTGAGGCTCCGTTTGCGTGT | AB819918.1 | 203 |
| 18S rRNA | Fwd-GCCCTGTAATTGGAATGAGTCCACTT Rev-CTCCCCAAGATCCAACTACGAGCTTT | NR_003278.3 | 148 |
| GPx1 Promoter (MeDIP) | Fwd-AACATCTCCAGTATGTGTG Rev-GACATTCTCAATGAGCAG | NM_008160 | 141 |
| GPx1 Promoter (MS-PCR) | Methyl Fwd-GTAGGGTTTTGTGAGCGTTAGTAC Rev-AATAAACAACACCTTACCCCG | NM_008160 | 176 |
| Unmethyl Fwd-TAGGGTTTTGTGAGTGTTAGTATGG Rev-CAATAAACAACACCTTACCCCAC | 176 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malaviya, P.; Kowluru, R.A. Diabetic Retinopathy and Regulation of Mitochondrial Glutathione–Glutathione Peroxidase Axis in Hyperhomocysteinemia. Antioxidants 2024, 13, 254. https://doi.org/10.3390/antiox13030254
Malaviya P, Kowluru RA. Diabetic Retinopathy and Regulation of Mitochondrial Glutathione–Glutathione Peroxidase Axis in Hyperhomocysteinemia. Antioxidants. 2024; 13(3):254. https://doi.org/10.3390/antiox13030254
Chicago/Turabian StyleMalaviya, Pooja, and Renu A. Kowluru. 2024. "Diabetic Retinopathy and Regulation of Mitochondrial Glutathione–Glutathione Peroxidase Axis in Hyperhomocysteinemia" Antioxidants 13, no. 3: 254. https://doi.org/10.3390/antiox13030254
APA StyleMalaviya, P., & Kowluru, R. A. (2024). Diabetic Retinopathy and Regulation of Mitochondrial Glutathione–Glutathione Peroxidase Axis in Hyperhomocysteinemia. Antioxidants, 13(3), 254. https://doi.org/10.3390/antiox13030254