
IP3R-Mediated Calcium Release Promotes Ferroptotic Death in SH-SY5Y Neuroblastoma Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability
2.3. Cell Morphology
2.4. IP3R1 Downregulation and Mitochondrial Calcium Detection
2.5. Western Blot Analysis
2.6. ROS Detection
2.7. Calcium Change Recordings
2.8. Lipid Peroxidation
2.9. Statistical Analysis
3. Results
3.1. RSL3 Induces Ferroptosis in SH-SY5Y Neuroblastoma Cells
3.1.1. RSL3 Treatment Induces Changes in Cell Morphology and Time- and Concentration-Dependent Cell Death
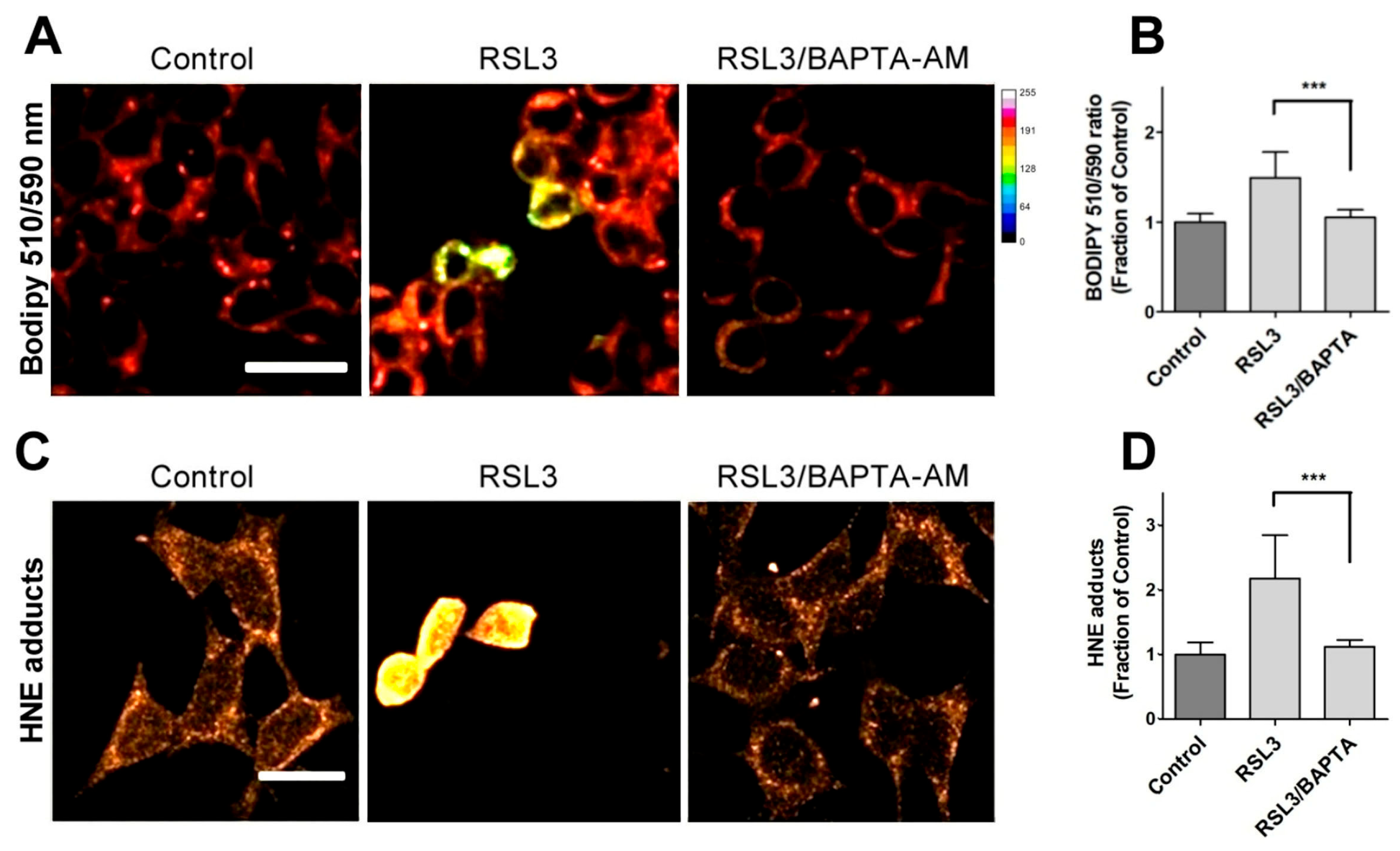
3.1.2. RSL3 Induces ROS Production and Lipid Peroxidation in SH-SY5Y Neuroblastoma Cells
3.1.3. RSL3 Induces Non-Apoptotic Cell Death in SH-SY5Y Neuroblastoma Cells
3.2. Treatment with RSL3 Causes a Delayed Increase in Intracellular Calcium Concentration
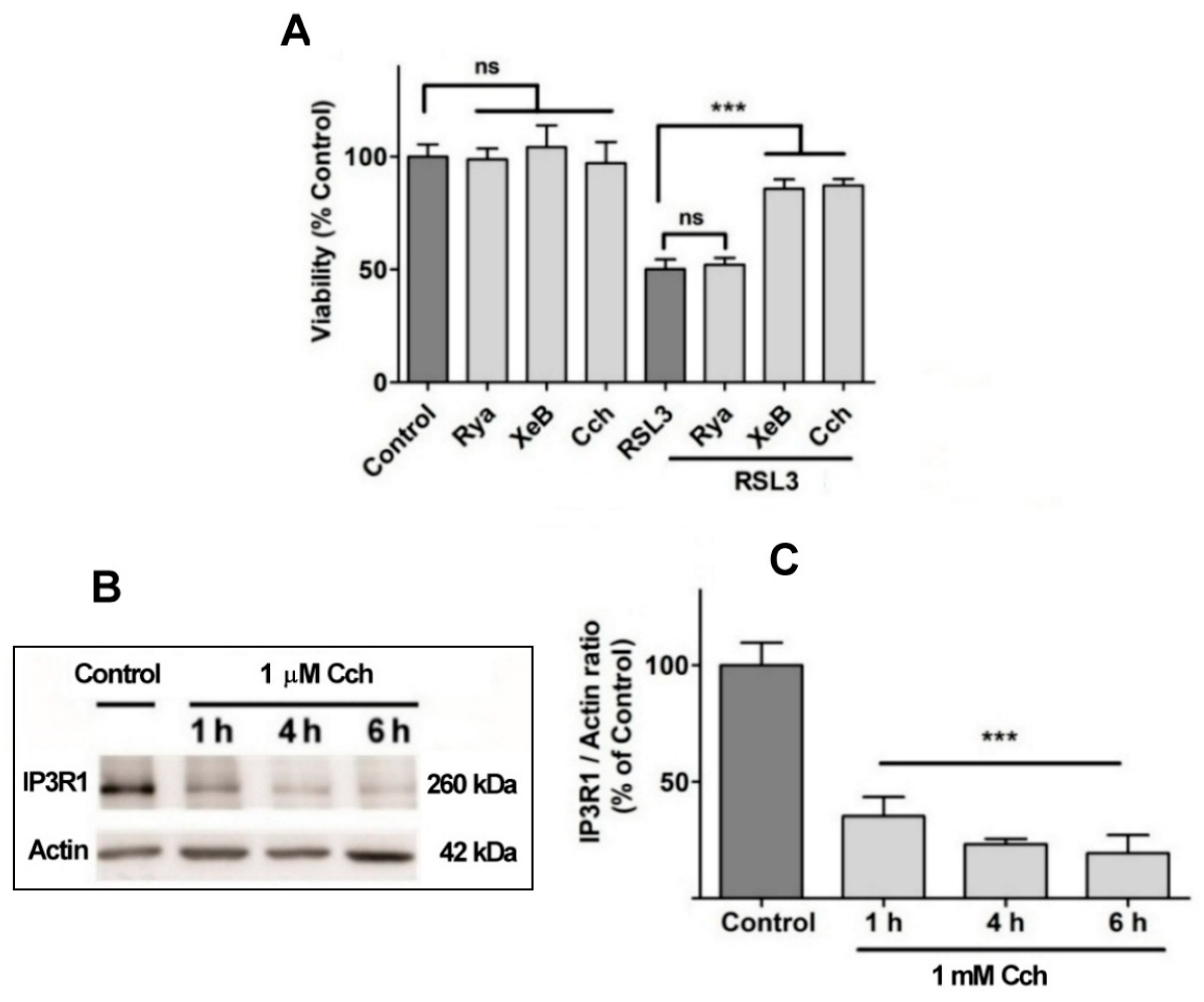
3.3. The IP3R Calcium Channel Mediates RSL3-Induced Increase in Intracellular Calcium
3.4. RSL Treatment Induces an Increase in Mitochondrial Calcium
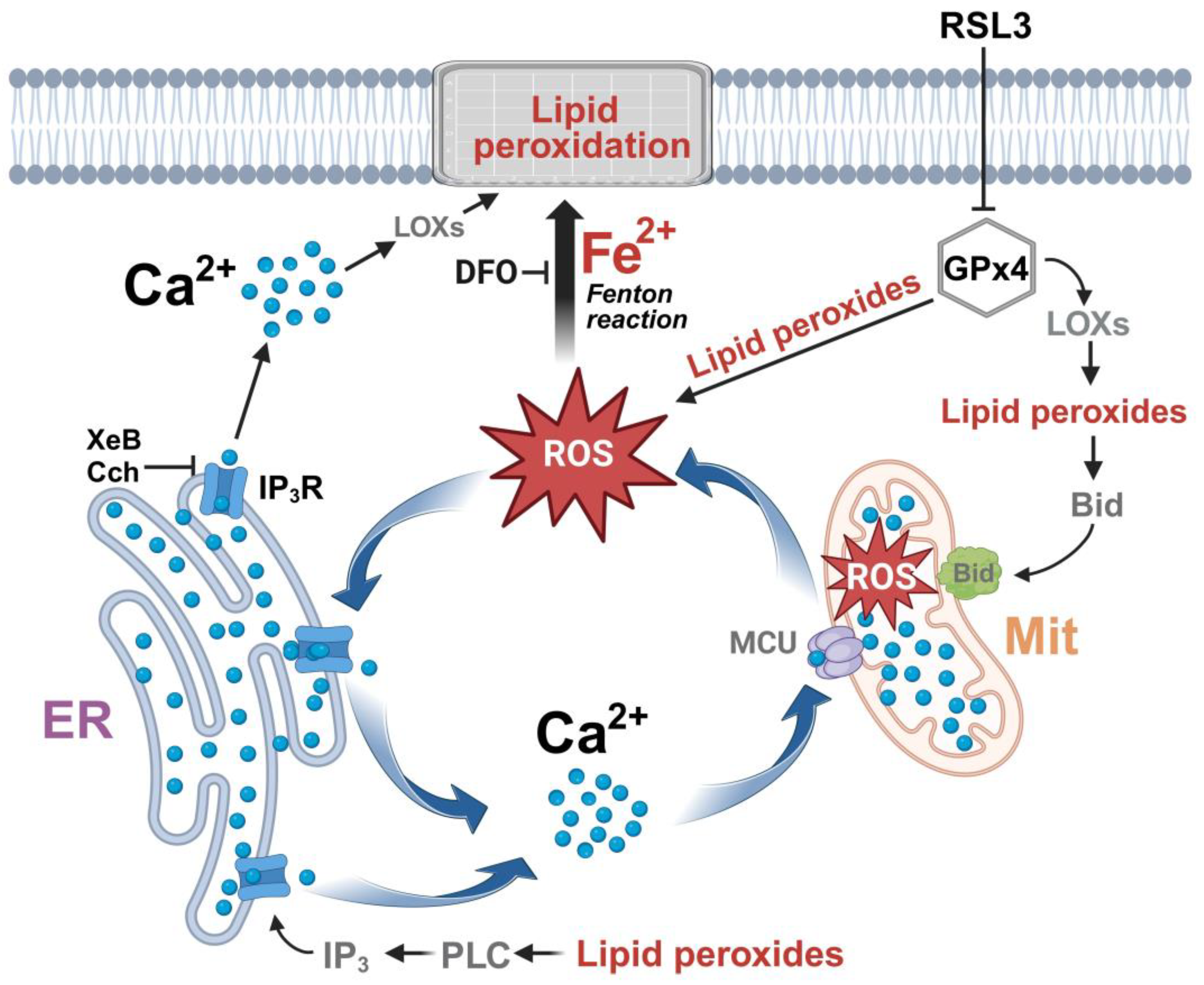
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef]
- Hu, S.; Chu, Y.; Zhou, X.; Wang, X. Recent advances of ferroptosis in tumor: From biological function to clinical application. Biomed. Pharmacother. 2023, 166, 115419. [Google Scholar] [CrossRef]
- Ryan, S.K.; Ugalde, C.L.; Rolland, A.S.; Skidmore, J.; Devos, D.; Hammond, T.R. Therapeutic inhibition of ferroptosis in neurodegenerative disease. Trends Pharmacol. Sci. 2023, 44, 674–688. [Google Scholar] [CrossRef]
- Zhang, X.D.; Liu, Z.Y.; Wang, M.S.; Guo, Y.X.; Wang, X.K.; Luo, K.; Huang, S.; Li, R.F. Mechanisms and regulations of ferroptosis. Front. Immunol. 2023, 14, 1269451. [Google Scholar] [CrossRef]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Murphy, T.H.; Baraban, J.M. Glutamate toxicity in immature cortical neurons precedes development of glutamate receptor currents. Brain Res. Dev. Brain Res. 1990, 57, 146–150. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front. Pharmacol. 2018, 9, 1371. [Google Scholar] [CrossRef]
- Breton, C.V.; Song, A.Y.; Xiao, J.; Kim, S.J.; Mehta, H.H.; Wan, J.; Yen, K.; Sioutas, C.; Lurmann, F.; Xue, S.; et al. Effects of air pollution on mitochondrial function, mitochondrial DNA methylation, and mitochondrial peptide expression. Mitochondrion 2019, 46, 22–29. [Google Scholar] [CrossRef]
- Xu, T.; Ding, W.; Ji, X.; Ao, X.; Liu, Y.; Yu, W.; Wang, J. Molecular mechanisms of ferroptosis and its role in cancer therapy. J. Cell Mol. Med. 2019, 23, 4900–4912. [Google Scholar] [CrossRef]
- Jelinek, A.; Heyder, L.; Daude, M.; Plessner, M.; Krippner, S.; Grosse, R.; Diederich, W.E.; Culmsee, C. Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic. Biol. Med. 2018, 117, 45–57. [Google Scholar] [CrossRef]
- Tan, S.; Schubert, D.; Maher, P. Oxytosis: A novel form of programmed cell death. Curr. Top. Med. Chem. 2001, 1, 497–506. [Google Scholar] [CrossRef]
- Henke, N.; Albrecht, P.; Bouchachia, I.; Ryazantseva, M.; Knoll, K.; Lewerenz, J.; Kaznacheyeva, E.; Maher, P.; Methner, A. The plasma membrane channel ORAI1 mediates detrimental calcium influx caused by endogenous oxidative stress. Cell Death Dis. 2013, 4, e470. [Google Scholar] [CrossRef]
- Fukui, M.; Song, J.H.; Choi, J.; Choi, H.J.; Zhu, B.T. Mechanism of glutamate-induced neurotoxicity in HT22 mouse hippocampal cells. Eur. J. Pharmacol. 2009, 617, 1–11. [Google Scholar] [CrossRef]
- Wolpaw, A.J.; Shimada, K.; Skouta, R.; Welsch, M.E.; Akavia, U.D.; Pe’er, D.; Shaik, F.; Bulinski, J.C.; Stockwell, B.R. Modulatory profiling identifies mechanisms of small molecule-induced cell death. Proc. Natl. Acad. Sci. USA 2011, 108, E771–E780. [Google Scholar] [CrossRef]
- Lewerenz, J.; Ates, G.; Methner, A.; Conrad, M.; Maher, P. Oxytosis/Ferroptosis-(Re-) Emerging Roles for Oxidative Stress-Dependent Non-apoptotic Cell Death in Diseases of the Central Nervous System. Front. Neurosci. 2018, 12, 214. [Google Scholar] [CrossRef]
- Maher, P.; van Leyen, K.; Dey, P.N.; Honrath, B.; Dolga, A.; Methner, A. The role of Ca2+ in cell death caused by oxidative glutamate toxicity and ferroptosis. Cell Calcium 2018, 70, 47–55. [Google Scholar] [CrossRef]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gelé, P.; Pétrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R.; et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef]
- Bogeski, I.; Niemeyer, B.A. Redox regulation of ion channels. Antioxid. Redox Signal. 2014, 21, 859–862. [Google Scholar] [CrossRef]
- Borodkina, A.V.; Shatrova, A.N.; Deryabin, P.I.; Griukova, A.A.; Abushik, P.A.; Antonov, S.M.; Nikolsky, N.N.; Burova, E.B. Calcium alterations signal either to senescence or to autophagy induction in stem cells upon oxidative stress. Aging 2016, 8, 3400–3418. [Google Scholar] [CrossRef]
- Kaplin, A.I.; Ferris, C.D.; Voglmaier, S.M.; Snyder, S.H. Purified reconstituted inositol 1,4,5-trisphosphate receptors. Thiol reagents act directly on receptor protein. J. Biol. Chem. 1994, 269, 28972–28978. [Google Scholar] [CrossRef]
- Marengo, J.J.; Hidalgo, C.; Bull, R. Sulfhydryl oxidation modifies the calcium dependence of ryanodine-sensitive calcium channels of excitable cells. Biophys. J. 1998, 74, 1263–1277. [Google Scholar] [CrossRef]
- Gleitze, S.; Ramírez, O.A.; Vega-Vásquez, I.; Yan, J.; Lobos, P.; Bading, H.; Núñez, M.T.; Paula-Lima, A.; Hidalgo, C. Ryanodine Receptor Mediated Calcium Release Contributes to Ferroptosis Induced in Primary Hippocampal Neurons by GPX4 Inhibition. Antioxidants 2023, 12, 705. [Google Scholar] [CrossRef]
- Suzuki, J.; Kanemaru, K.; Ishii, K.; Ohkura, M.; Okubo, Y.; Iino, M. Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat. Commun. 2014, 5, 4153. [Google Scholar] [CrossRef]
- Arias-Cavieres, A.; Adasme, T.; Sánchez, G.; Muñoz, P.; Hidalgo, C. Aging Impairs Hippocampal-Dependent Recognition Memory and LTP and Prevents the Associated RyR Up-regulation. Front. Aging Neurosci. 2017, 9, 111. [Google Scholar] [CrossRef]
- Reynolds, I.J.; Hastings, T.G. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J. Neurosci. Off. J. Soc. Neurosci. 1995, 15, 3318–3327. [Google Scholar] [CrossRef]
- Setsukinai, K.; Urano, Y.; Kakinuma, K.; Majima, H.J.; Nagano, T. Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. J. Biol. Chem. 2003, 278, 3170–3175. [Google Scholar] [CrossRef] [PubMed]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [CrossRef]
- Paredes, R.M.; Etzler, J.C.; Watts, L.T.; Zheng, W.; Lechleiter, J.D. Chemical calcium indicators. Methods 2008, 46, 143–151. [Google Scholar] [CrossRef]
- Decuypere, J.P.; Kindt, D.; Luyten, T.; Welkenhuyzen, K.; Missiaen, L.; De Smedt, H.; Bultynck, G.; Parys, J.B. mTOR-Controlled Autophagy Requires Intracellular Ca2+ Signaling. PLoS ONE 2013, 8, e61020. [Google Scholar] [CrossRef]
- Costa, I.; Barbosa, D.J.; Silva, V.; Benfeito, S.; Borges, F.; Remião, F.; Silva, R. Research Models to Study Ferroptosis’s Impact in Neurodegenerative Diseases. Pharmaceutics 2023, 15, 1369. [Google Scholar] [CrossRef]
- Nakatsu, Y.; Kotake, Y.; Komasaka, K.; Hakozaki, H.; Taguchi, R.; Kume, T.; Akaike, A.; Ohta, S. Glutamate excitotoxicity is involved in cell death caused by tributyltin in cultured rat cortical neurons. Toxicol. Sci. 2006, 89, 235–242. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, D.; Li, Y.; Zhang, X. Protective Effects of Verapamil against H2O2-Induced Apoptosis in Human Lens Epithelial Cells. Biomol. Ther. 2014, 22, 553–557. [Google Scholar] [CrossRef]
- Wojcikiewicz, R.J.; Nakade, S.; Mikoshiba, K.; Nahorski, S.R. Inositol 1,4,5-trisphosphate receptor immunoreactivity in SH-SY5Y human neuroblastoma cells is reduced by chronic muscarinic receptor activation. J. Neurochem. 1992, 59, 383–386. [Google Scholar] [CrossRef]
- Marchi, S.; Marinello, M.; Bononi, A.; Bonora, M.; Giorgi, C.; Rimessi, A.; Pinton, P. Selective modulation of subtype III IP3R by Akt regulates ER Ca2+ release and apoptosis. Cell Death Dis. 2012, 3, e304. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Wu, T.; Wang, X.; Cheng, J.; Liang, X.; Li, Y.; Chen, M.; Kong, L.; Tang, M. Nitrogen-doped graphene quantum dots induce ferroptosis through disrupting calcium homeostasis in microglia. Part. Fibre Toxicol. 2022, 19, 22. [Google Scholar] [CrossRef]
- Cheff, D.M.; Huang, C.; Scholzen, K.C.; Gencheva, R.; Ronzetti, M.H.; Cheng, Q.; Hall, M.D.; Arnér, E.S.J. The ferroptosis inducing compounds RSL3 and ML162 are not direct inhibitors of GPX4 but of TXNRD1. Redox Biol. 2023, 62, 102703. [Google Scholar] [CrossRef]
- Kemmerling, U.; Muñoz, P.; Müller, M.; Sánchez, G.; Aylwin, M.L.; Klann, E.; Carrasco, M.A.; Hidalgo, C. Calcium release by ryanodine receptors mediates hydrogen peroxide-induced activation of ERK and CREB phosphorylation in N2a cells and hippocampal neurons. Cell Calcium 2007, 41, 491–502. [Google Scholar] [CrossRef]
- Satheesh, N.J.; Büsselberg, D. The role of intracellular calcium for the development and treatment of neuroblastoma. Cancers 2015, 7, 823–848. [Google Scholar] [CrossRef]
- Prins, D.; Michalak, M. Organellar calcium buffers. Cold Spring Harb. Perspect. Biol. 2011, 3, a004069. [Google Scholar] [CrossRef]
- Paschou, M.; Liaropoulou, D.; Kalaitzaki, V.; Efthimiopoulos, S.; Papazafiri, P. Knockdown of Amyloid Precursor Protein Increases Ion Channel Expression and Alters Ca2+ Signaling Pathways. Int. J. Mol. Sci. 2023, 24, 2302. [Google Scholar] [CrossRef]
- Mackrill, J.J.; Challiss, R.A.; O’Connell, D.A.; Lai, F.A.; Nahorski, S.R. Differential expression and regulation of ryanodine receptor and myo-inositol 1,4,5-trisphosphate receptor Ca2+ release channels in mammalian tissues and cell lines. Biochem. J. 1997, 327 Pt 1, 251–258. [Google Scholar] [CrossRef][Green Version]
- Oulès, B.; Del Prete, D.; Greco, B.; Zhang, X.; Lauritzen, I.; Sevalle, J.; Moreno, S.; Paterlini-Bréchot, P.; Trebak, M.; Checler, F.; et al. Ryanodine receptor blockade reduces amyloid-β load and memory impairments in Tg2576 mouse model of Alzheimer disease. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 11820–11834. [Google Scholar] [CrossRef]
- Van Acker, K.; Kasri, N.N.; De Smet, P.; Parys, J.B.; De Smedt, H.; Missiaen, L.; Callewaert, G. IP3-mediated Ca2+ signals in human neuroblastoma SH-SY5Y cells with exogenous overexpression of type 3 IP3 receptor. Cell Calcium 2002, 32, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.P.; Bultynck, G.; Aromolaran, A.S.; Zhong, F.; Parys, J.B.; De Smedt, H.; Mignery, G.A.; Roderick, H.L.; Bootman, M.D.; Distelhorst, C.W. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc. Natl. Acad. Sci. USA 2009, 106, 14397–14402. [Google Scholar] [CrossRef] [PubMed]
- Bánsághi, S.; Golenár, T.; Madesh, M.; Csordás, G.; RamachandraRao, S.; Sharma, K.; Yule, D.I.; Joseph, S.K.; Hajnóczky, G. Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species. J. Biol. Chem. 2014, 289, 8170–8181. [Google Scholar] [CrossRef] [PubMed]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e353. [Google Scholar] [CrossRef]
- Nakamura, T.; Ogawa, M.; Kojima, K.; Takayanagi, S.; Ishihara, S.; Hattori, K.; Naguro, I.; Ichijo, H. The mitochondrial Ca2+ uptake regulator, MICU1, is involved in cold stress-induced ferroptosis. EMBO Rep. 2021, 22, e51532. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, W.; Li, Y.; Xiao, Y.; Cheng, J.; Jia, J. The 5-Lipoxygenase Inhibitor Zileuton Confers Neuroprotection against Glutamate Oxidative Damage by Inhibiting Ferroptosis. Biol. Pharm. Bull. 2015, 38, 1234–1239. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Brinckmann, R.; Schnurr, K.; Heydeck, D.; Rosenbach, T.; Kolde, G.; Kühn, H. Membrane translocation of 15-lipoxygenase in hematopoietic cells is calcium-dependent and activates the oxygenase activity of the enzyme. Blood 1998, 91, 64–74. [Google Scholar] [CrossRef]
- Rohlik, D.L.; Patel, E.; Gilbert, N.C.; Offenbacher, A.R.; Garcia, B.L. Investigating membrane-binding properties of lipoxygenases using surface plasmon resonance. Biochem. Biophys. Res. Commun. 2023, 670, 47–54. [Google Scholar] [CrossRef]
- Walther, M.; Wiesner, R.; Kuhn, H. Investigations into calcium-dependent membrane association of 15-lipoxygenase-1. Mechanistic roles of surface-exposed hydrophobic amino acids and calcium. J. Biol. Chem. 2004, 279, 3717–3725. [Google Scholar] [CrossRef]
- Pedrera, L.; Espiritu, R.A.; Ros, U.; Weber, J.; Schmitt, A.; Stroh, J.; Hailfinger, S.; von Karstedt, S.; García-Sáez, A.J. Ferroptotic pores induce Ca2+ fluxes and ESCRT-III activation to modulate cell death kinetics. Cell Death Differ. 2021, 28, 1644–1657. [Google Scholar] [CrossRef] [PubMed]
- Takemura, H.; Hughes, A.R.; Thastrup, O.; Putney, J.W., Jr. Activation of calcium entry by the tumor promoter thapsigargin in parotid acinar cells. Evidence that an intracellular calcium pool and not an inositol phosphate regulates calcium fluxes at the plasma membrane. J. Biol. Chem. 1989, 264, 12266–12271. [Google Scholar] [CrossRef]
- Domijan, A.M.; Kovac, S.; Abramov, A.Y. Lipid peroxidation is essential for phospholipase C activity and the inositol-trisphosphate-related Ca2+ signal. J. Cell Sci. 2014, 127, 21–26. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos, J.; Gleitze, S.; Hidalgo, C.; Núñez, M.T. IP3R-Mediated Calcium Release Promotes Ferroptotic Death in SH-SY5Y Neuroblastoma Cells. Antioxidants 2024, 13, 196. https://doi.org/10.3390/antiox13020196
Campos J, Gleitze S, Hidalgo C, Núñez MT. IP3R-Mediated Calcium Release Promotes Ferroptotic Death in SH-SY5Y Neuroblastoma Cells. Antioxidants. 2024; 13(2):196. https://doi.org/10.3390/antiox13020196
Chicago/Turabian StyleCampos, Joaquín, Silvia Gleitze, Cecilia Hidalgo, and Marco T. Núñez. 2024. "IP3R-Mediated Calcium Release Promotes Ferroptotic Death in SH-SY5Y Neuroblastoma Cells" Antioxidants 13, no. 2: 196. https://doi.org/10.3390/antiox13020196
APA StyleCampos, J., Gleitze, S., Hidalgo, C., & Núñez, M. T. (2024). IP3R-Mediated Calcium Release Promotes Ferroptotic Death in SH-SY5Y Neuroblastoma Cells. Antioxidants, 13(2), 196. https://doi.org/10.3390/antiox13020196