
Mechanism of Antiradical Activity of Coumarin-Trihydroxybenzohydrazide Derivatives: A Comprehensive Kinetic DFT Study


 ,
,  ,
,  , ,
, ,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Electron Paramagnetic Resonance (EPR) Spectroscopy Measurement
2.3. Theoretical Calculations
Calculation of the Rate Constants
3. Results
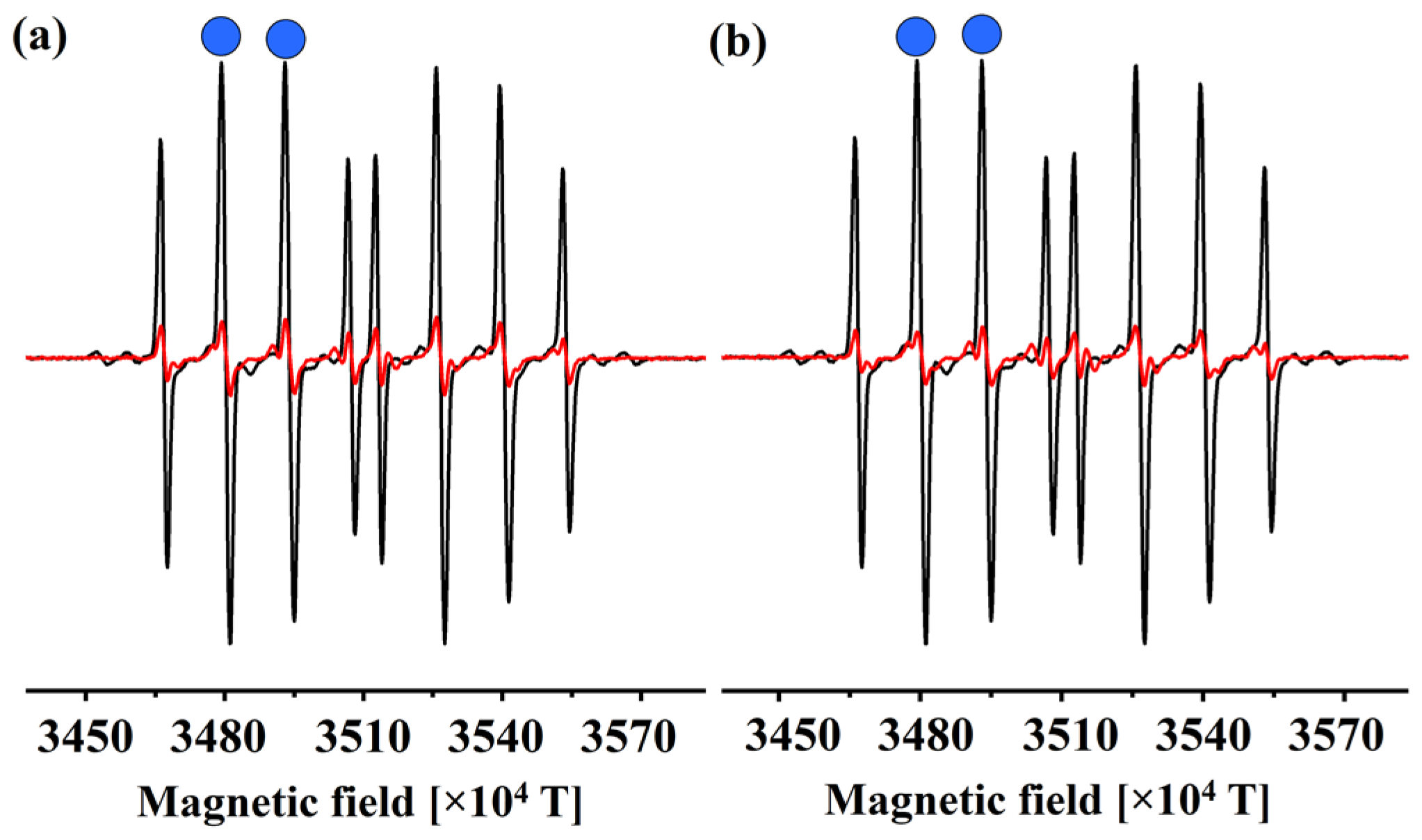
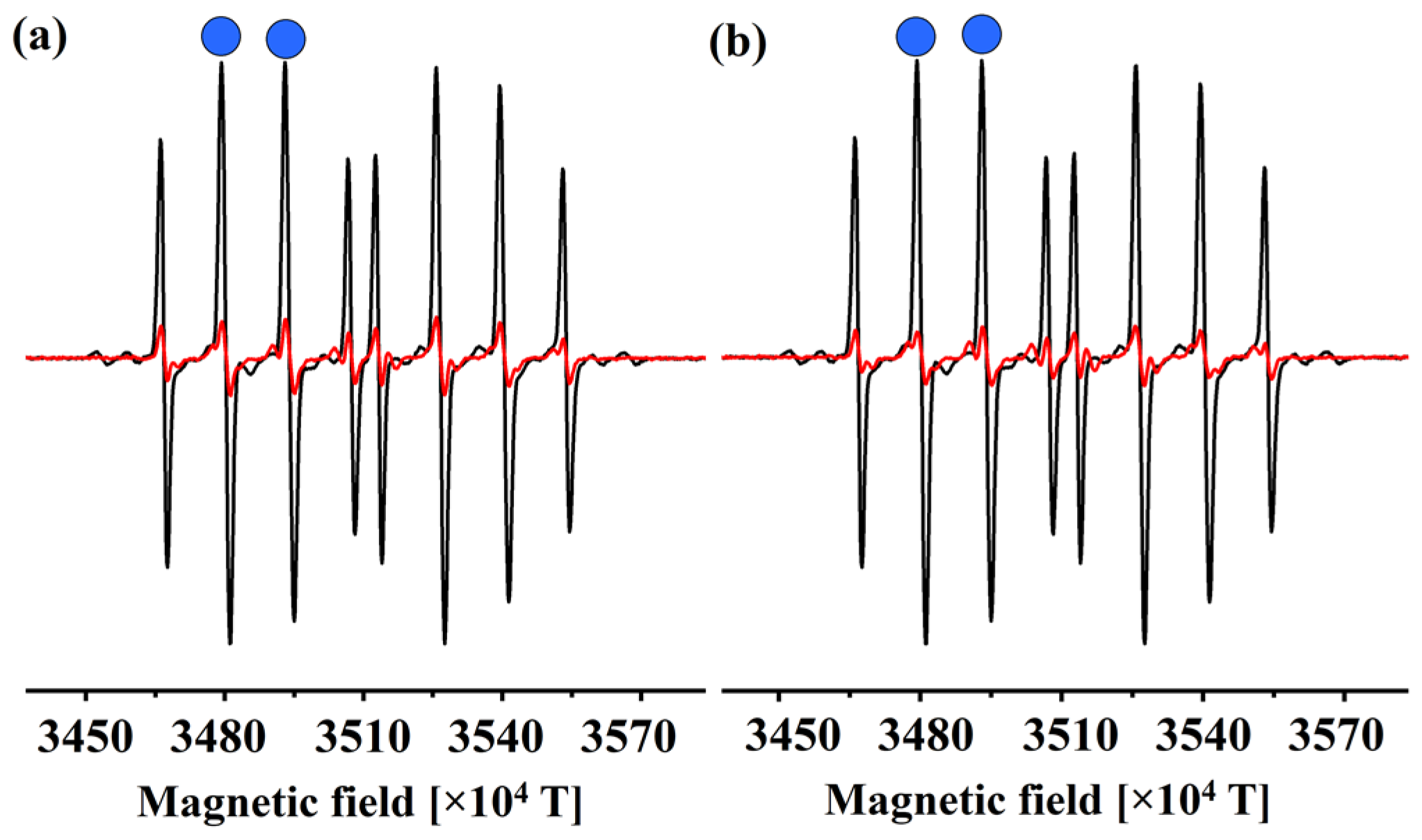
3.1. EPR Determination of the Reactivity of 1 and 2 towards HO•
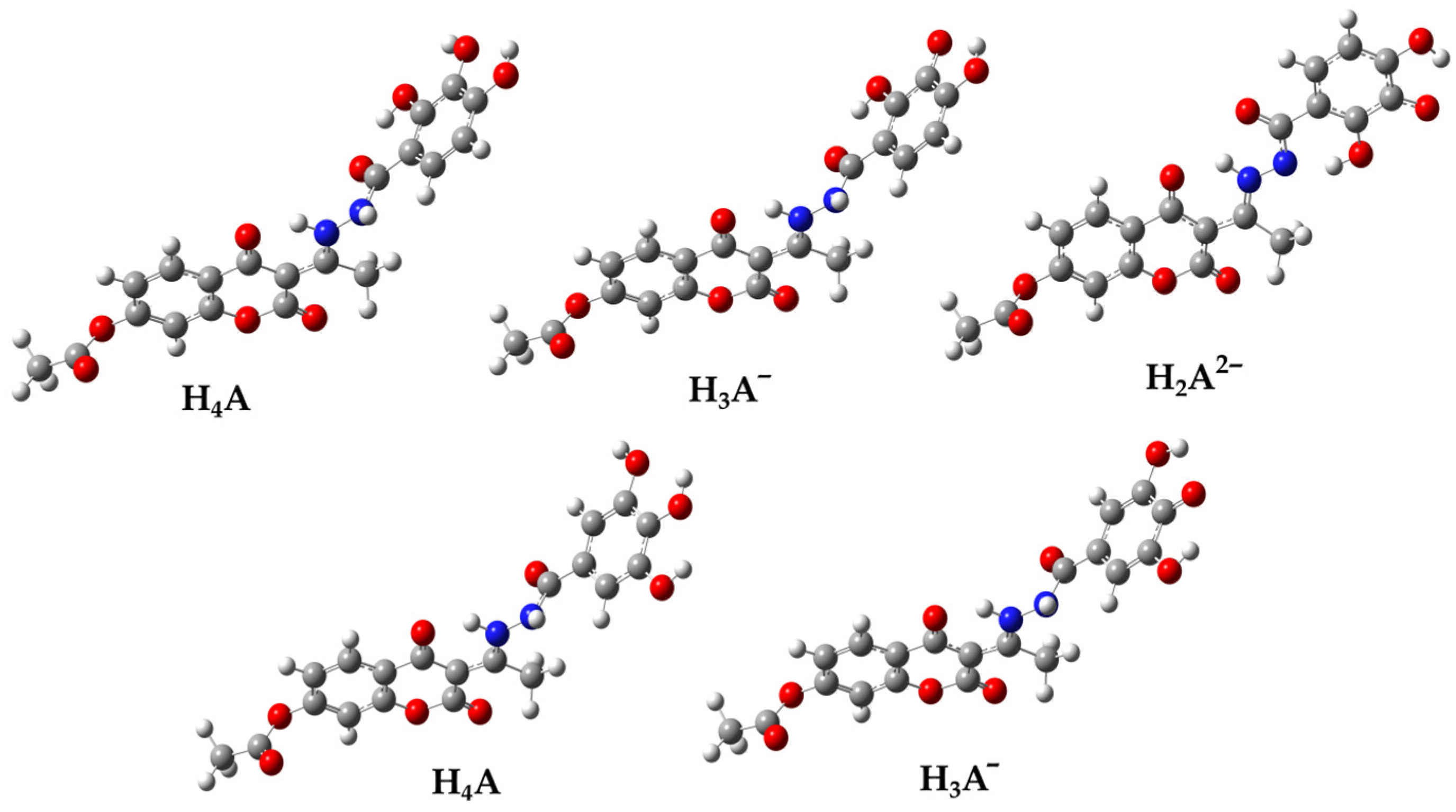
3.2. Thermodynamic Analysis
Thermodynamic Investigation of the Reaction between Anionic Species and HO•
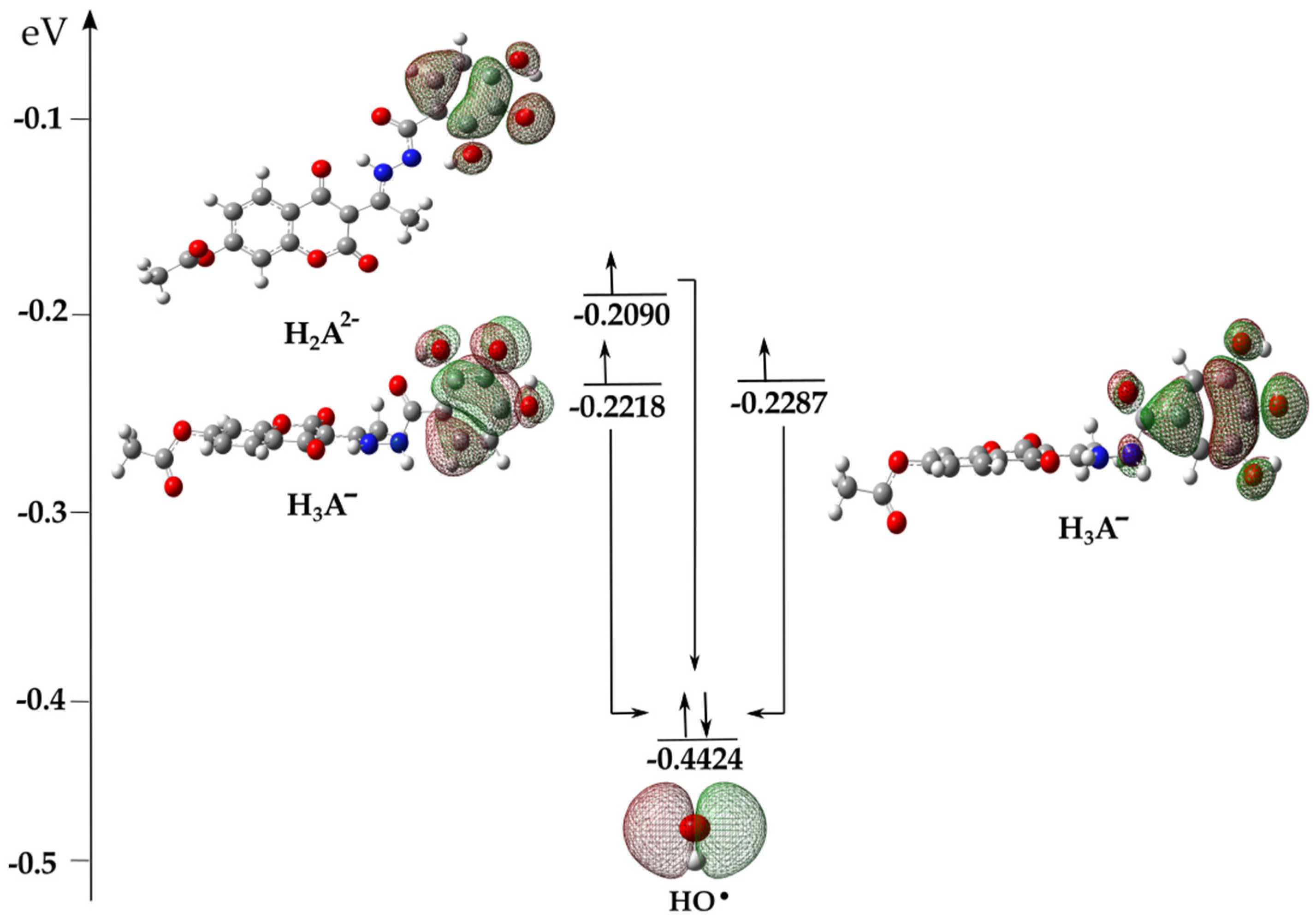
3.3. Kinetic Analysis
Kinetic Investigation of the Reaction between Anionic Species and HO•
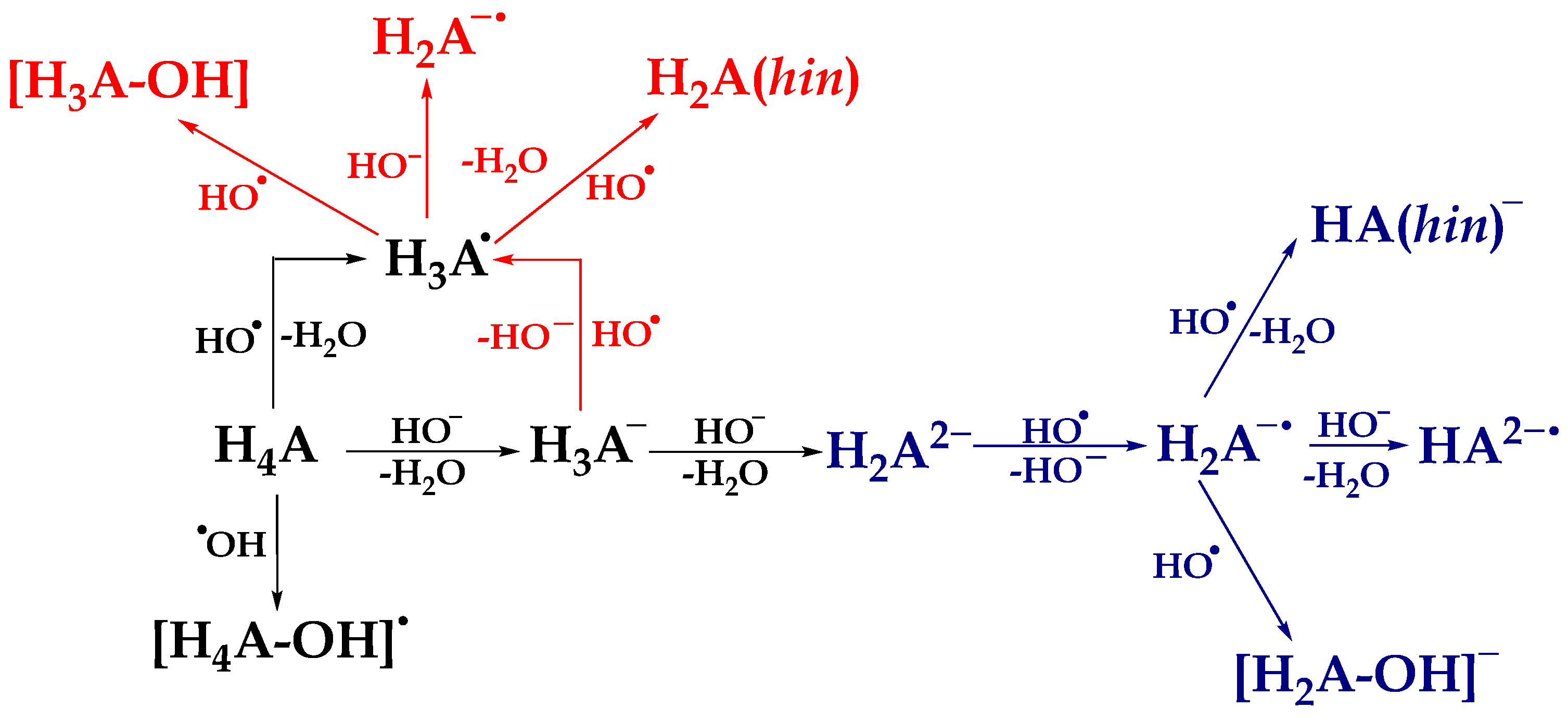
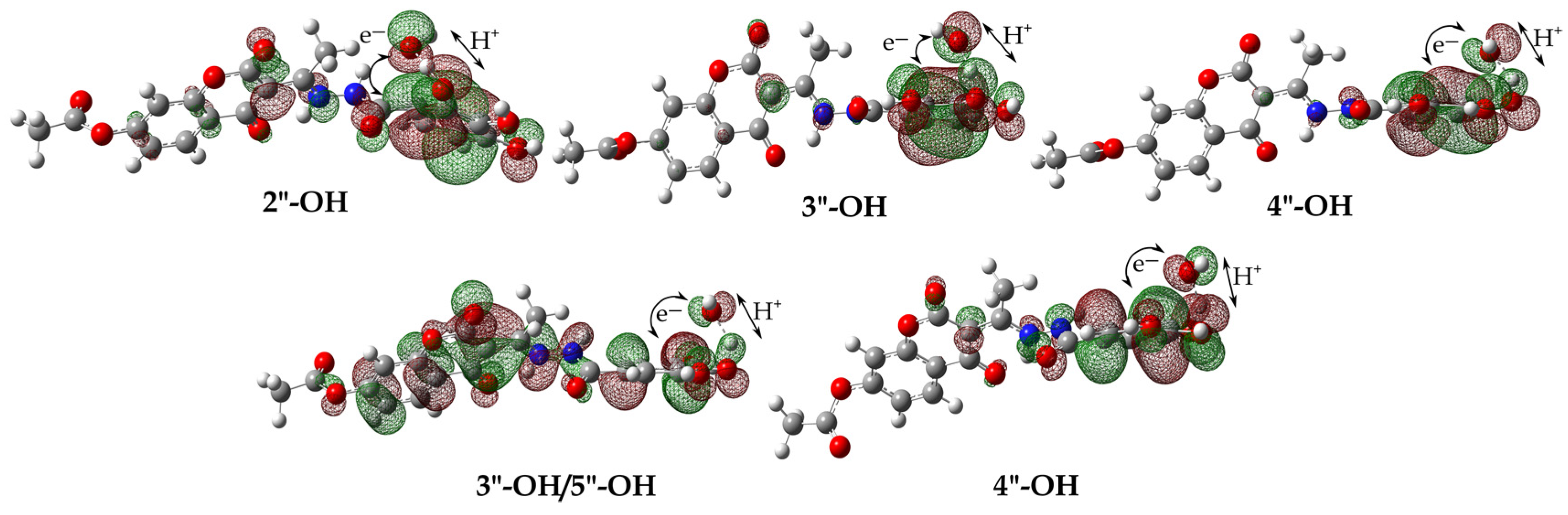
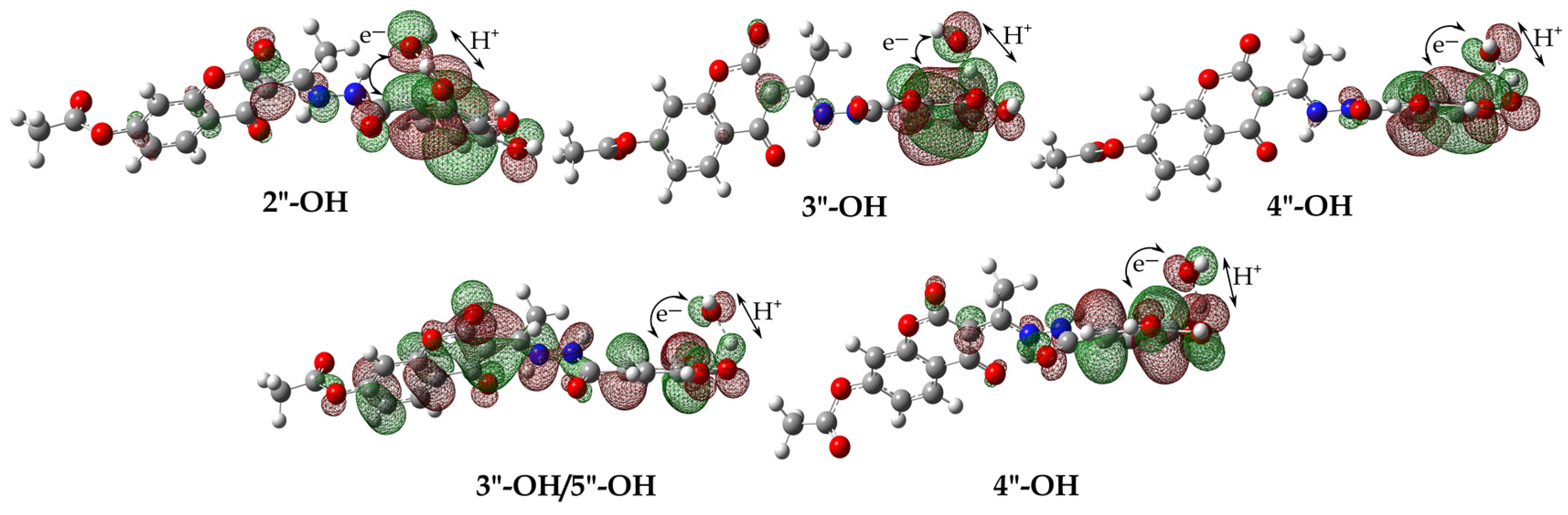
3.4. Defining the Plausible Mechanisms of Antioxidant Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pingret, D.; Fabiano-Tixier, A.S.; Chemat, F. Degradation during application of ultrasound in food processing: A review. Food Control 2013, 31, 593–606. [Google Scholar] [CrossRef]
- Finley, J.W.; Kong, A.N.; Hintze, K.J.; Jeffery, E.H.; Ji, L.L.; Lei, X.G. Antioxidants in foods: State of the science important to the food industry. J. Agric. Food Chem. 2011, 13, 6837–6846. [Google Scholar] [CrossRef] [PubMed]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010, 8, 118. [Google Scholar] [CrossRef] [PubMed]
- Alkadi, H. A review on free radicals and antioxidants. Infect. Disord. Drug Targets 2020, 1, 16–26. [Google Scholar]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 1, 65–74. [Google Scholar] [CrossRef]
- Chen, A.F.; Chen, D.D.; Daiber, A.; Faraci, F.M.; Li, H.; Rembold, C.M.; Laher, I. Free radical biology of the cardiovascular system. Clin. Sci. 2012, 2, 73–91. [Google Scholar] [CrossRef]
- Patlevič, P.; Vašková, J.; Švorc, P., Jr.; Vaško, L.; Švorc, P. Reactive oxygen species and antioxidant defense in human gastrointestinal diseases. Integr. Med. Res. 2016, 4, 250–258. [Google Scholar] [CrossRef]
- Matés, J.M.; Pérez-Gómez, C.; De Castro, I.N. Antioxidant enzymes and human diseases. Clin. Biochem. 1999, 8, 595–603. [Google Scholar] [CrossRef]
- Kostova, I.; Bhatia, S.; Grigorov, P.; Balkansky, S.; Parmar, V.S.; Prasad, A.K.; Saso, L. Coumarins as antioxidants. Curr. Med. Chem. 2011, 25, 3929–3951. [Google Scholar] [CrossRef]
- Al-Majedy, Y.; Al-Amiery, A.; Kadhum, A.A.; BakarMohamad, A. Antioxidant Activity of Coumarins. Syst. Pharm. Rev. 2017, 8, 24–30. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, Z. Coumarin-containing hybrids and their anticancer activities. Eur. J. Med. Chem. 2019, 181, 111587. [Google Scholar] [CrossRef]
- Jameel, E.; Umar, T.; Kumar, J.; Hoda, N. Coumarin: A privileged scaffold for the design and development of antineurodegenerative agents. Chem. Biol. Drug Des. 2016, 1, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, L.C. Coumarin derivatives in inflammatory bowel disease. Molecules 2021, 26, 422. [Google Scholar] [CrossRef] [PubMed]
- Najmanova, I.; Dosedel, M.; Hrdina, R.; Anzenbacher, P.; Filipsky, T.; Riha, M.; Mladenka, P. Cardiovascular effects of coumarins besides their antioxidant activity. Curr. Top. Med. Chem. 2015, 15, 830–849. [Google Scholar] [CrossRef]
- Borges Bubols, G.; da Rocha Vianna, D.; Medina-Remon, A.; von Poser, G.; Maria Lamuela-Raventos, R.; Lucia Eifler-Lima, V.; Cristina Garcia, S. The antioxidant activity of coumarins and flavonoids. Mini Rev. Med. Chem. 2013, 13, 318–334. [Google Scholar]
- Santos-Sánchez, N.F.; Salas-Coronado, R.; Villanueva-Cañongo, C.; Hernández-Carlos, B. Antioxidant compounds and their antioxidant mechanism. Antioxidants 2019, 10, 1–29. [Google Scholar]
- Avdović, E.H.; Milanović, Ž.; Simijonović, D.; Antonijević, M.; Milutinović, M.; Nikodijević, D.; Filipović, N.; Marković, Z.; Vojinović, R. An Effective, Green Synthesis Procedure for Obtaining Coumarin–Hydroxybenzohydrazide Derivatives and Assessment of Their Antioxidant Activity and Redox Status. Antioxidants 2023, 12, 2070. [Google Scholar] [CrossRef]
- Galano, A.; Alvarez-Idaboy, J.R. A computational methodology for accurate predictions of rate constants in solution: Application to the assessment of primary antioxidant activity. J. Comput. Chem. 2013, 34, 2430–2445. [Google Scholar] [CrossRef]
- Milenković, D.; Đorović, J.; Jeremić, S.; Dimitrić Marković, J.M.; Avdović, E.H.; Marković, Z. Free radical scavenging potency of dihydroxybenzoic acids. J. Chem. 2017, 2017, 5936239. [Google Scholar] [CrossRef]
- Musialik, M.; Kuzmicz, R.; Pawłowski, T.S.; Litwinienko, G. Acidity of hydroxyl groups: An overlooked influence on antiradical properties of flavonoids. J. Org. Chem. 2009, 74, 2699–2709. [Google Scholar] [CrossRef]
- Kumar, Y.A.; Kumar, K.D.; Kim, H.J. A novel electrode for supercapacitors: Efficient PVP-assisted synthesis of Ni3S2 nanostructures grown on Ni foam for energy storage. Dalton Trans. 2020, 49, 4050–4059. [Google Scholar] [CrossRef] [PubMed]
- Yedluri, A.K.; Kulurumotlakatla, D.K.; Sangaraju, S.; Obaidat, I.M.; Kim, H.J. Facile synthesis of novel and highly efficient CoNi2S4-Ni(OH)2 nanosheet arrays as pseudocapacitive-type electrode material for high-performance electrochemical supercapacitors. J. Energy Storage 2020, 31, 101623. [Google Scholar] [CrossRef]
- Jackson, S.K.; Liu, K.J.; Liu, M.; Timmins, G.S. Detection and removal of contaminating hydroxylamines from the spin trap DEPMPO, and re-evaluation of its use to indicate nitrone radical cation formation and SN1 reactions. Free Radic. Biol. Med. 2002, 32, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.W.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Tošović, J.; Marković, S. Antioxidative activity of chlorogenic acid relative to trolox in aqueous solution–DFT study. Food Chem. 2019, 278, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Milanović, Ž.; Dimić, D.; Antonijević, M.; Žižić, M.; Milenković, D.; Avdović, E.; Marković, Z. Influence of acid-base equilibria on the rate of the chemical reaction in the Advanced Oxidation Processes: Coumarin derivatives and hydroxyl radical. Chem. Eng. J. 2023, 453, 139648. [Google Scholar] [CrossRef]
- Alvareda, E.; Denis, P.A.; Iribarne, F.; Paulino, M. Bond dissociation energies and enthalpies of formation of flavonoids: A G4 and M06-2X investigation. Comput. Theor. Chem. 2016, 1091, 18–23. [Google Scholar] [CrossRef]
- Pérez-González, A.; Alvarez-Idaboy, J.R.; Galano, A. Dual antioxidant/pro-oxidant behavior of the tryptophan metabolite 3-hydroxyanthranilic acid: A theoretical investigation of reaction mechanisms and kinetics. New J. Chem. 2017, 10, 3829–3845. [Google Scholar] [CrossRef]
- Advanced Chemistry Development Inc. (ACD/Labs) ACD/PERCEPTA Version 2015 Frankfurt Am Main. 2016. Available online: www.acdlabs.com/pka (accessed on 10 January 2023).
- Fernández-Ramos, A.; Miller, J.A.; Klippenstein, S.J.; Truhlar, D.G. Modeling the kinetics of bimolecular reactions. Chem. Rev. 2006, 11, 4518–4584. [Google Scholar] [CrossRef]
- Eyring, H. The activated complex in chemical reactions. J. Chem. Phys. 1935, 2, 107–115. [Google Scholar] [CrossRef]
- Duncan, W.T.; Bell, R.L.; Truong, T.N.J. TheRate: Program for ab initio direct dynamics calculations of thermal and vibrational-state-selected rate constants. Comput. Chem. 1998, 19, 1039–1052. [Google Scholar] [CrossRef]
- Eckart, C. The penetration of a potential barrier by electrons. Phys. Rev. 1930, 11, 1303. [Google Scholar] [CrossRef]
- Marcus, R.A. Electron transfer reactions in chemistry. Theory and experiment. Rev. Mod. Phys. 1993, 65, 599–610. [Google Scholar] [CrossRef]
- Amić, A.; Dimitrić Marković, J.M.; Marković, Z.; Milenković, D.; Milanović, Ž.; Antonijević, M.; Mastiľák Cagardová, D.; Rodríguez-Guerra Pedregal, J. Theoretical Study of Radical Inactivation, LOX Inhibition, and Iron Chelation: The Role of Ferulic Acid in Skin Protection against UVA Induced Oxidative Stress. Antioxidants 2021, 10, 1303. [Google Scholar] [CrossRef]
- Milanović, Ž.; Tošović, J.; Marković, S.; Marković, Z. Comparison of the scavenging capacities of phloroglucinol and 2,4,6-trihydroxypyridine towards HO radical: A computational study. RSC Adv. 2020, 10, 43262–43272. [Google Scholar] [CrossRef]
- Marino, T.; Galano, A.; Russo, N. Radical scavenging ability of gallic acid toward OH and OOH radicals. Reaction mechanism and rate constants from the density functional theory. J. Phys. Chem. B 2014, 35, 10380–10389. [Google Scholar] [CrossRef]
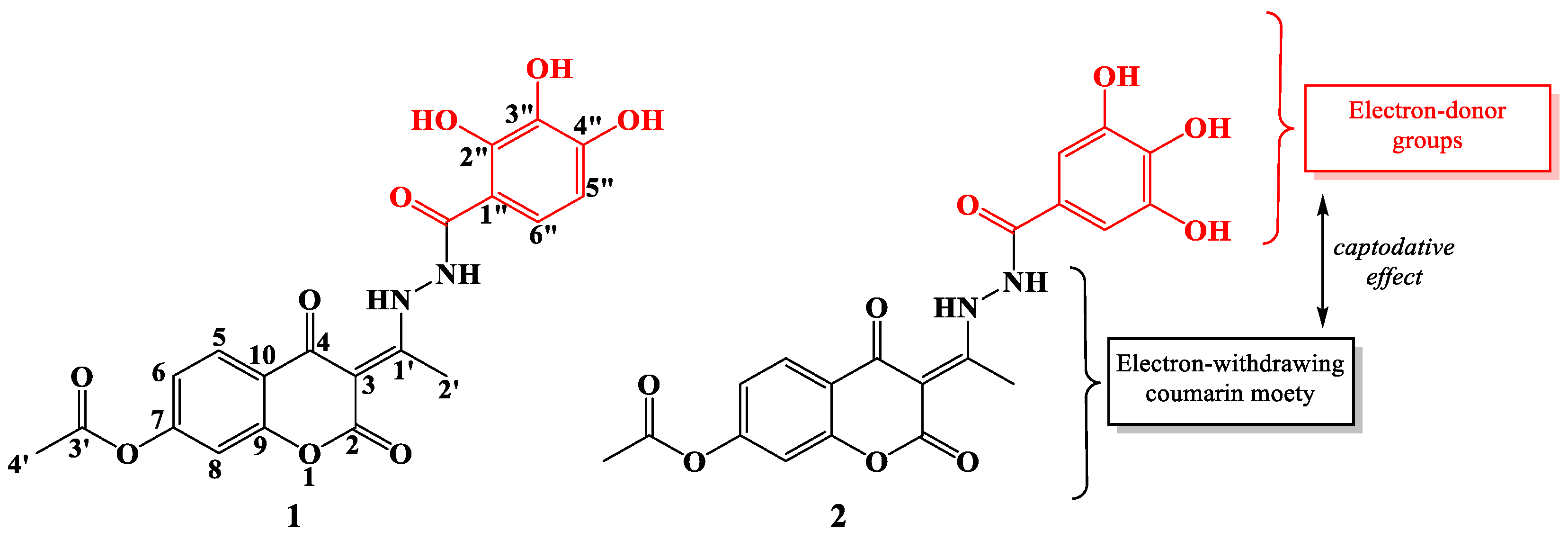



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Acid-Base Species | Position | HO• | ||||
|---|---|---|---|---|---|---|---|
| HAT/PCET | SET-PT | SPLET | |||||
| ΔrGHAT/PCET | ΔrGSET | ΔrGPT | ΔrGSPL | ΔrGET | |||
| 1 | H4A | 2″-OH | −152 | 127 | −280 | −156 | 4 |
| 3″-OH | −157 | −284 | −116 | −41 | |||
| 4″-OH | −134 | −262 | −128 | −6 | |||
| 2-NH | −142 | −269 | −168 | 26 | |||
| H3A− | 2″-OH | −202 | −41 | −187 | / | / | |
| 4″-OH | −194 | −156 | |||||
| 2-NH | −182 | −183 | |||||
| H2A2− | 4″-OH | −214 | −87 | −94 | / | / | |
| 2-NH/2″-OH | −158 | −140 | |||||
| 2 | H4A | 3″-OH/5″-OH | −145 | 133 | −278 | −125 | −20 |
| 4″-OH | −165 | −299 | −142 | −23 | |||
| N-H | −136 | −270 | −146 | 9 | |||
| H3A− | 3″-OH | −184 | −23 | −161 | / | / | |
| 5″-OH | |||||||
| 2-NH | −98 | −165 | |||||
| Position | RAF/RCF | ||||||
| 1 | 2 | ||||||
| H4A | H3A− | H2A2− | H4A | H3A− | |||
| C-3 | −11 | −46 | −75 | −12 | −69 | ||
| C-5 | −40 | −124 | 5 | −44 | −143 | ||
| C-6 | −20 | 5 | 34 | −28 | −6 | ||
| C-7 | −43 | 102 | −20 | −42 | 75 | ||
| C-8 | −27 | 9 | 40 | −35 | −14 | ||
| C-9 | −37 | 120 | 1 | −36 | 91 | ||
| C-10 | 1 | 46 | 76 | 3 | 21 | ||
| C-1′ | −59 | −63 | −107 | −60 | 5 | ||
| C-1″ | 1 | −175 | −124 | −8 | −172 | ||
| C-2″ | −50 | −187 | −161 | −42 | −108 | ||
| C-3″ | −38 | −194 | −120 | −54 | −167 | ||
| C-4″ | −57 | −169 | −163 | −49 | −70 | ||
| C-5″ | −42 | −84 | −83 | −26 | −170 | ||
| C-6″ | −54 | −195 | −183 | −63 | −107 | ||
| Compounds | Acid-Base Species | Position | HAT/PCET | SPLET | |||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | H4A | 2″-OH | 58 | 1.94 × 104 | ~0 | 1.91 × 109 | 1 | 8.02 × 109 | |
| 3″-OH | 41 | 2.22 × 107 | 10 | 7.57 × 109 | |||||
| 4″-OH | 55 | 9.22 × 104 | 2 | 8.01 × 109 | |||||
| 2-NH | ~0 | 1.91 × 109 | 26 | 1.47 × 108 | |||||
| 2 | H4A | 3″-OH/ 5″-OH | 59 | 3.15 × 104 | ~0 | 1.91 × 109 | 0 | 8.02 × 109 | |
| 4″-OH | 54 | 3.05 × 105 | 0 | 8.02 × 109 | |||||
| 2-NH | ~0 | 1.91 × 109 | 14 | 5.84 × 109 | |||||
| Compounds | Acid-Base Species | Position | HAT/PCET | SET-PT | |||||
| 1 | H3A− | 2″-OH | ~0 | 1.91 × 109 | 10 | 7.57 × 109 | ~0 | 1.91 × 109 | |
| 4″-OH | |||||||||
| 2-NH | |||||||||
| H2A2− | 4″-OH | ~0 | 1.91 × 109 | 35 | 4.89 × 106 | ~0 | 1.91 × 109 | ||
| 2-NH | |||||||||
| 2 | H3A− | 3″-OH | ~0 | 1.91 × 109 | 0 | 8.02 × 109 | ~0 | 1.91 × 109 | |
| 5″-OH | |||||||||
| 2-NH | |||||||||
| Position | RAF (H4A) | ||||||||
| 1 | 2 | ||||||||
| C-3 | 38 | 4.26 × 107 | 48 | 7.98 × 105 | |||||
| C-5 | 51 | 2.95 × 105 | 56 | 4.30 × 104 | |||||
| C-6 | 38 | 4.20 × 107 | 45 | 2.74 × 106 | |||||
| C-7 | 60 | 3.32 × 103 | 63 | 2.34 × 103 | |||||
| C-8 | 39 | 3.12 × 107 | 41 | 1.13 × 107 | |||||
| C-9 | 50 | 3.54 × 105 | 53 | 1.11 × 105 | |||||
| C-10 | 43 | 5.28 × 106 | 47 | 1.23 × 106 | |||||
| C-1′ | 42 | 9.00 × 106 | 45 | 2.24 × 106 | |||||
| C-1″ | 37 | 6.37 × 107 | 35 | 1.20 × 108 | |||||
| C-2″ | 39 | 3.21 × 107 | 29 | 5.69 × 108 | |||||
| C-3″ | 22 | 1.67 × 108 | 47 | 1.09 × 106 | |||||
| C-4″ | 38 | 3.89 × 107 | 29 | 7.41 × 107 | |||||
| C-5″ | 38 | 3.97 × 107 | 39 | 2.45 × 107 | |||||
| C-6″ | 28 | 7.29 × 107 | 25 | 1.39 × 107 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milanović, Ž.; Dimić, D.; Avdović, E.H.; Simijonović, D.M.; Nakarada, Đ.; Jakovljević, V.; Vojinović, R.; Marković, Z.S. Mechanism of Antiradical Activity of Coumarin-Trihydroxybenzohydrazide Derivatives: A Comprehensive Kinetic DFT Study. Antioxidants 2024, 13, 143. https://doi.org/10.3390/antiox13020143
Milanović Ž, Dimić D, Avdović EH, Simijonović DM, Nakarada Đ, Jakovljević V, Vojinović R, Marković ZS. Mechanism of Antiradical Activity of Coumarin-Trihydroxybenzohydrazide Derivatives: A Comprehensive Kinetic DFT Study. Antioxidants. 2024; 13(2):143. https://doi.org/10.3390/antiox13020143
Chicago/Turabian StyleMilanović, Žiko, Dušan Dimić, Edina H. Avdović, Dušica M. Simijonović, Đura Nakarada, Vladimir Jakovljević, Radiša Vojinović, and Zoran S. Marković. 2024. "Mechanism of Antiradical Activity of Coumarin-Trihydroxybenzohydrazide Derivatives: A Comprehensive Kinetic DFT Study" Antioxidants 13, no. 2: 143. https://doi.org/10.3390/antiox13020143
APA StyleMilanović, Ž., Dimić, D., Avdović, E. H., Simijonović, D. M., Nakarada, Đ., Jakovljević, V., Vojinović, R., & Marković, Z. S. (2024). Mechanism of Antiradical Activity of Coumarin-Trihydroxybenzohydrazide Derivatives: A Comprehensive Kinetic DFT Study. Antioxidants, 13(2), 143. https://doi.org/10.3390/antiox13020143