
Role of NLRP3 Inflammasome in Heart Failure Patients Undergoing Cardiac Surgery as a Potential Determinant of Postoperative Atrial Fibrillation and Remodeling: Is SGLT2 Cotransporter Inhibition an Alternative for Cardioprotection?


 ,
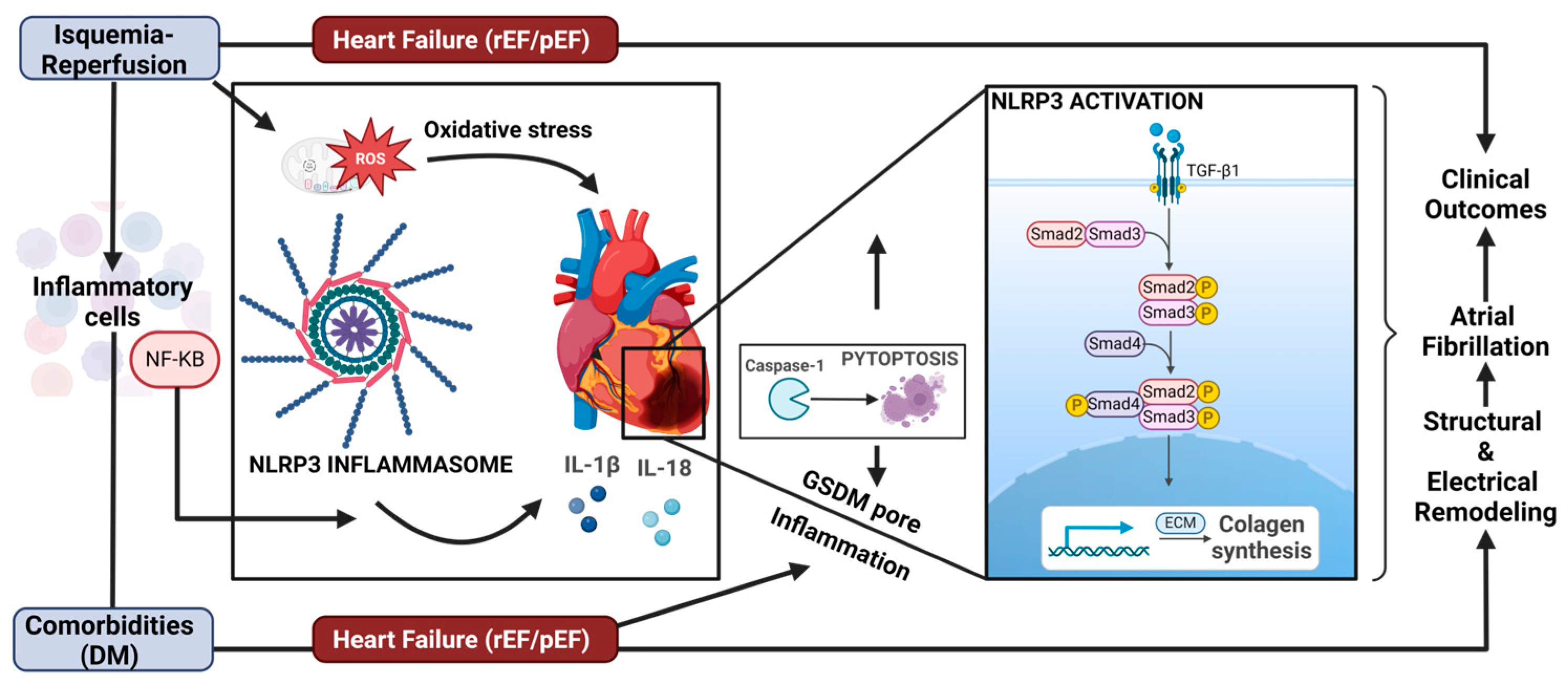
,  , , , , ,
, , , , , 
Abstract
1. Introduction
2. Oxidative Stress and Inflammation as a Mediator of POAF
3. Ischemia–Reperfusion and Cardiac Remodeling
4. NLRP3 Activity as a Mediator of Cardiovascular Damage
5. Role of NLRP3 Inflammasome Activity and Atrial Fibrillation
6. Preventive Therapies for Cardiovascular IR Injury and Remodeling
6.1. SGLT-2 Inhibitors: Pharmacological Properties
6.1.1. Antioxidants and Anti-Inflammatory Effects
Anti-Inflammatory Effects
6.1.2. SGLT2is and Ventricular Remodeling
7. Concluding Remarks
Funding
Conflicts of Interest
References
- Axtell, A.L.; Moonsamy, P.; Melnitchouk, S.; Tolis, G.; Jassar, A.S.; D’Alessandro, D.A.; Villavicencio, M.; Cameron, D.E.; Sundt, T.M. Preoperative predictors of new-onset prolonged atrial fibrillation after surgical aortic valve replacement. J. Thorac. Cardiovasc. Surg. 2020, 159, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Weymann, A.; Popov, A.F.; Sabashnikov, A.; Ali-Hasan-Al-Saegh, S.; Ryazanov, M.; Tse, G.; Mirhosseini, S.J.; Liu, T.; Lotfaliani, M.; Sedaghat, M.; et al. Baseline and postoperative levels of C-reactive protein and interleukins as inflammatory predictors of atrial fibrillation following cardiac surgery: A systematic review and meta-analysis. Kardiol. Pol. 2018, 76, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Tadic, M.; Ivanovic, B.; Zivkovic, N. Predictors of atrial fibrillation following coronary artery bypass surgery. Med. Sci. Monit. 2011, 17, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.G. Atrial fibrillation after cardiac surgery. Ann. Card. Anaesth. 2010, 13, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Dobrev, D.; Aguilar, M.; Heijman, J.; Guichard, J.B.; Nattel, S. Postoperative atrial fibrillation: Mechanisms, manifestations and management. Nat. Rev. Cardiol. 2019, 16, 417–436. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, F.; Wu, Y.; Elliott, M.; Zhou, W.; Deng, Y.; Ren, D.; Zhao, H. Mechanism of IL-6-related spontaneous atrial fibrillation after coronary artery grafting surgery: IL-6 knockout mouse study and human observation. Transl. Res. 2021, 233, 16–31. [Google Scholar] [CrossRef]
- Topkara, V.; Cheema, F.H.; Kesavaramanujam, S.; Mercando, M.L.; Cheema, A.F.; Namerow, P.B.; Argenziano, M.; Naka, Y.; Oz, M.C.; Esrig, B.C. Coronary artery bypass grafting in patients with low ejection fraction. Circulation 2005, 112 (Suppl. S9), I344–I350. [Google Scholar] [CrossRef]
- Dogan, S.M.; Buyukates, M.; Kandemir, O.; Aydin, M.; Gursurer, M.; Acikgoz, S.; Yavuzer, R.; Cam, F.; Dursun, A. Predictors of atrial fibrillation after coronary artery bypass surgery. Coron. Artery Dis. 2007, 18, 327–331. [Google Scholar] [CrossRef]
- Mathis, M.R.; Duggal, N.M.; Janda, A.M.; Fennema, J.L.; Yang, B.O.; Pagani, F.D.; Maile, M.D.; Hofer, R.E.; Jewell, E.S.; Engoren, M.C. Reduced Echocardiographic Inotropy Index after Cardiopulmonary Bypass Is Associated with Complications After Cardiac Surgery: An Institutional Outcomes Study. J. Cardiothorac. Vasc. Anesth. 2021, 35, 2732–2742. [Google Scholar] [CrossRef]
- Russo, C.; Jin, Z.; Sera, F.; Lee, S.E.; Homma, S.; Rundek, T.; Elkind, M.S.; Sacco, R.L.; Di Tullio, M.R. Left Ventricular Systolic Dysfunction by Longitudinal Strain Is an Independent Predictor of Incident Atrial Fibrillation: A Community-Based Cohort Study. Circ. Cardiovasc. Imaging 2015, 8, e003520. [Google Scholar] [CrossRef]
- Thavendiranathan, P.; Negishi, T.; Somerset, E.; Negishi, E.; Penicka, M.; Lemieux, J.; Aakhus, S.; Miyazaki, S.; Shirazi, M.; Galderisi, M.; et al. Strain-Guided Management of Potentially Cardiotoxic Cancer Therapy. J. Am. Coll. Cardiol. 2021, 77, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Kawczynski, M.; Gilbers, M.; Walle, S.; Schalla, S.; Crijns, H.J.; Maessen, J.G.; Schotten, U.; Maesen, B.; Bidar, E. Role of pre-operative transthoracic echocardiography in predicting post-operative atrial fibrillation after cardiac surgery: A systematic review of the literature and meta-analysis. Europace 2021, 23, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Park, J.J.; Park, J.B.; Park, J.H.; Cho, G.Y. Global longitudinal strain to predict mortality in patients with acute heart failure. J. Am. Coll. Cardiol. 2018, 71, 1947–1957. [Google Scholar] [CrossRef] [PubMed]
- Kalam, K.; Otahal, P.; Marwick, T.H. Prognostic implications of global LV dysfunction: A systematic review and meta-analysis of global longitudinal strain and ejection fraction. Heart 2014, 100, 1673–1680. [Google Scholar] [CrossRef] [PubMed]
- Stampehl, M.R.; Mann, D.L.; Nguyen, J.S.; Cota, F.; Colmenares, C.; Dokainish, H. Speckle strain echocardiography predicts outcome in patients with heart failure with both depressed and preserved left ventricular ejection fraction. Echocardiography 2015, 32, 71–78. [Google Scholar] [CrossRef]
- Wang, C.H.; Chan, Y.; Chien-Chia, V.; Lee, H.-F.; Hsiao, F.; Chu, P.-H. Incremental prognostic value of global myocardial work over ejection fraction and global longitudinal strain in patients with heart failure and reduced ejection fraction. Eur. Heart J. Cardiovasc. Imaging 2021, 22, 348–356. [Google Scholar] [CrossRef]
- Buggey, J.; Alenezi, F.; Yoon, H.J.; Phelan, M.; DeVore, A.D.; Khouri, M.G.; Schulte, P.J.; Velazquez, E.J. Left ventricular global longitudinal strain in patients with heart failure with preserved ejection fraction: Outcomes following an acute heart failure hospitalization. ESC Heart Fail. 2017, 4, 432–439. [Google Scholar] [CrossRef]
- Mihm, M.; Yu, F.; Carnes, C.; Reiser, P.J.; McCarthy, P.M.; Van Wagoner, D.R.; Bauer, J.A. Impaired Myofibrillar Energetics and Oxidative Injury During Human Atrial Fibrillation. Circulation 2001, 104, 174–180. [Google Scholar] [CrossRef]
- Carnes, C.; Chung, M.; Nakayama, T.; Nakayama, H.; Baliga, R.S.; Piao, S.; Kanderian, A.; Pavia, S.; Hamlin, R.L.; McCarthy, P.M.; et al. Ascorbate attenuates atrial pacing-induced peroxynitrite formation and electrical remodeling and decreases the incidence of postoperative atrial fibrillation. Circ. Res. 2001, 14, E32–E38. [Google Scholar] [CrossRef]
- Chang, J.; Chen, M.; Liu, W.; Yang, C.; Chen, C.; Chen, Y.L.; Pan, K.; Tsai, T.; Chang, H. Atrial myocardial nox2 containing NADPH oxidase activity contribution to oxidative stress in mitral regurgitation: Potential mechanism for atrial remodeling. Cardiovasc. Pathol. 2011, 20, 99–106. [Google Scholar] [CrossRef]
- Mighiu, A.; Recalde, A.; Ziberna, K.; Carnicer, R.; Tomek, J.; Bub, G.; Brewer, A.C.; Verheule, S.; Shah, A.M.; Simon, J.M.; et al. Inducibility, but not stability, of atrial fibrillation is increased by NOX2 overexpression in mice. Cardiovasc. Res. 2021, 117, 2354–2364. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.; Sadredini, M.; Hasic, A.; Eriksen, M.; Stokke, M.K. Myocardial oxidative stress is increased in early reperfusion, but systemic antioxidative therapy does not prevent ischemia reperfusion arrhythmias in pigs. Front. Cardiovasc. Med. 2023, 26, 1223496. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Hoek, V.T.; Wojcik, K.; Anderson, T.; Li, C.; Shao, Z.; Becker, L.B.; Hamann, K.J. Caspase-dependent cytochrome c release and cell death in chick cardiomyocytes after simulated ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physio. 2004, 286, H2280-6. [Google Scholar] [CrossRef] [PubMed]
- Martins, D.; Garcia, L.R.; Queiroz, D.A.; Lazzarin, T.; Rodrigues, C.; da Silva, P.; Polegato, B.F.; de Paiva, S.A.R.; Azevedo, P.S.; Minicucci, M.F.; et al. Oxidative Stress as a Therapeutic Target of Cardiac Remodeling. Antioxidants 2022, 11, 2371. [Google Scholar] [CrossRef] [PubMed]
- Jayaram, R.; Jones, M.; Reilly, S.; Crabtree, M.; Pal, N.; Goodfellow, N.; Nahar, K.; Simon, J.; Carnicer, R.; DeSilva, R.; et al. Atrial nitroso-redox balance and refractoriness following on-pump cardiac surgery: A randomized trial of atorvastatin. Cardiovasc. Res. 2022, 118, 184–195. [Google Scholar] [CrossRef]
- Farias, J.G.; Molina, V.M.; Carrasco, R.; Zepeda, A.B.; Figueroa, E.; Letelier, P.; Castillo, R.L. Antioxidant Therapeutic Strategies for Cardiovascular Conditions Associated with Oxidative Stress. Nutrients 2017, 9, 966. [Google Scholar] [CrossRef]
- Kazzi, M.E.; Rayner, B.; Chami, B.; Dennis, J.M.; Thomas, S. Witting PK Neutrophil-Mediated Cardiac Damage After Acute Myocardial Infarction: Significance of Defining a New Target Cell Type for Developing Cardioprotective Drugs. Antioxid. Redox Signal. 2020, 33, 689–712. [Google Scholar] [CrossRef]
- Castillo, R.L.; Rodrigo, R.; Pérez, F.; Cereceda, M.; Asenjo, R.; Zamorano, J.; Navarrete, R.; Villalabeitia, E.; Sanz, J.; Baeza, C.; et al. Antioxidant therapy reduces oxidative and inflammatory tissue damage in patients subjected to cardiac surgery with extracorporeal circulation. Basic Clin. Pharmacol. Toxicol. 2011, 108, 256–262. [Google Scholar] [CrossRef]
- Sánchez, F.J.; Gonzalez, V.A.; Farrando, M.; Jayat, A.O.; Segovia-Roldan, M.; García-Mendívil, L.; Ordovás, L.; Prado, N.J.; Pueyo, E.; Diez, E.R. Atrial Dyssynchrony Measured by Strain Echocardiography as a Marker of Proarrhythmic Remodeling and Oxidative Stress in Cardiac Surgery Patients. Oxid. Med. Cell Longev. 2020, 30, 8895078. [Google Scholar] [CrossRef]
- Kim, Y.M.; Kattach, H.; Ratnatunga, C.; Pillai, R.; Channon, K.M.; Casadei, B. Association of atrial nicotinamide adenine dinucleotide phosphate oxidase activity with the development of atrial fibrillation after cardiac surgery. J. Am. Coll. Cardiol. 2008, 51, 68–74. [Google Scholar] [CrossRef]
- Rodrigo, R.; Korantzopoulos, P.; Cereceda, M.; Asenjo, R.; Zamorano, J.; Villalabeitia, E.; Baeza, C.; Aguayo, R.; Castillo, R.; Carrasco, R.; et al. A randomized controlled trial to prevent postoperative atrial fibrillation by antioxidant reinforcement. J. Am. Coll. Cardiol. 2013, 62, 1457–1465. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Marchioli, R.; Silletta, M.; Masson, S.; Sellke, F.W.; Libby, P.; Milne, G.L.; Brown, N.J.; Lombardi, F.; Damiano, R.J., Jr.; et al. Oxidative Stress Biomarkers and Incidence of Postoperative Atrial Fibrillation in the Omega-3 Fatty Acids for Prevention of Postoperative Atrial Fibrillation (OPERA) Trial. J. Am. Heart Assoc. 2015, 20, e001886. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Anzai, A.; Katsumata, Y.; Matsuhashi, T.; Ito, K.; Endo, J.; Yamamoto, T.; Takeshima, A.; Shinmura, K.; Shen, W.; et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J. Mol. Cell Cardiol. 2013, 62, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Berezin, A.E.; Berezin, A.A. Adverse Cardiac Remodelling after Acute Myocardial Infarction: Old and New Biomarkers. Dis. Markers 2020, 2020, 1215802. [Google Scholar] [CrossRef] [PubMed]
- van den Berge, J.C.; Vroegindewey, M.M.; Veenis, J.F.; Brugts, J.J.; Caliskan, K.; Manintveld, O.C.; Akkerhuis, K.M.; Boersma, E.; Deckers, J.W.; Constantinescu, A.A. Left ventricular remodelling and prognosis after discharge in new-onset acute heart failure with reduced ejection fraction. ESC Heart Fail. 2021, 8, 2679–2689. [Google Scholar] [CrossRef]
- Lazzerini, P.; Abbate, A.; Boutjdir, M.; Capecchi, P.L. Fir(e)ing the Rhythm Inflammatory Cytokines and Cardiac Arrhythmias. JACC Basic Transl. Sci. 2023, 15, 728–750. [Google Scholar] [CrossRef]
- Lamm, G.; Auer, J.; Weber, T.; Berent, R.; Ng, C.; Eber, B. Postoperative white blood cell count predicts atrial fibrillation after cardiac surgery. J. Cardiothorac. Vasc. Anesth. 2006, 20, 51–56. [Google Scholar] [CrossRef]
- Fontes, M.L.; Amar, D.; Kulak, A.; Koval, K.; Zhang, H.; Shi, W. Increased preoperative white blood cell count predicts postoperative atrial fibrillation after coronary artery bypass surgery. J. Cardiothorac. Vasc. Anesth. 2009, 23, 484–487. [Google Scholar] [CrossRef]
- Friedrichs, K.; Adam, M.; Remane, L. Induction of atrial fibrillation by neutrophils critically depends on CD11b/CD18 integrins. PLoS ONE 2014, 9, e89307. [Google Scholar] [CrossRef]
- Erdolu, B.; Kagan, A.; Engin, M. The Relationship between the HATCH Score, Neutrophil to Lymphocyte Ratio and Postoperative Atrial Fibrillation After Off-Pump Coronary Artery Bypass Graft Surgery. Heart Surg. Forum. 2020, 23, E088–E092. [Google Scholar] [CrossRef]
- Wu, N.; Xu, B.; Xiang, Y.; Wu, L.; Zhang, Y.; Ma, X.; Tong, S.; Shu, M.; Song, Z.; Li, Y.; et al. Association of inflammatory factors with occurrence and recurrence of atrial fibrillation: A meta-analysis. Int. J. Cardiol. 2013, 169, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.A.; Bazzazi, H.; Clark, R.B.; Nygren, A.; Giles, W.R. Actions of emigrated neutrophils on Na(+) and K(+) currents in rat ventricular myocytes. Prog. Biophys. Mol. Biol. 2006, 90, 249–269. [Google Scholar] [CrossRef] [PubMed]
- Holzwirth, E.; Kornej, J.; Erbs, S.; Obradovic, D.; Bollmann, A.; Hindricks, G.; Thiele, H.; Büttner, P. Myeloperoxidase in atrial fibrillation: Association with progression, origin and influence of renin-angiotensin system antagonists. Clin. Res. Cardiol. 2020, 109, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Liu, H.; Liao, J.; Zhao, N.; Tse, G.; Han, B.; Chen, L.; Huang, Z.; Du, Y. Colchicine prevents atrial fibrillation promotion by inhibiting IL-1β-induced IL-6 release and atrial fibrosis in the rat sterile pericarditis model. Biomed. Pharmacother. 2020, 129, 110384. [Google Scholar] [CrossRef] [PubMed]
- Scott, L., Jr.; Fender, L.; Saljic, A.; Li, L.; Chen, X.; Wang, X.; Linz, D.; Lang, J.; Hohl, M.; Twomey, D.; et al. NLRP3 inflammasome is a key driver of obesity-induced atrial arrhythmias. Cardiovasc. Res. 2021, 117, 1746–1759. [Google Scholar] [CrossRef]
- Hemenway, G.; Frishman, W.H. Therapeutic Implications of NLRP3-Mediated Inflammation in Coronary Artery Disease. Cardiol. Rev. 2021, 30, 90–99. [Google Scholar] [CrossRef]
- Shateri, H.; Manafi, B.; Tayebinia, H.; Karimi, J.; Khodadadi, I. Imbalance in thioredoxin system activates NLRP3 inflammasome pathway in epicardial adipose tissue of patients with coronary artery disease. Mol. Biol. Rep. 2021, 48, 1181–1191. [Google Scholar] [CrossRef]
- Parent, S.; Vaka, R.; Amant, J.; Kahn, S.; Van Remortel, S.; Bi, C.; Courtman, D.; Stewart, D.J.; Raymond, D.; Davis, D.R. Inactivation of the NLRP3 inflammasome mediates exosome-based prevention of atrial fibrillation. Theranostics 2024, 14, 608–621. [Google Scholar] [CrossRef]
- Parent, S.; Amant, J.; Van Remortel, S.; Kahn, S.; Vaka, R.; Courtman, D.; Stewart, D.J.; Davis, D.R. Atrial Fibrosis and Inflammation in Postoperative Atrial Fibrillation: Comparative Effects of Amiodarone, Colchicine, or Exosomes. JACC Clin. Electrophysiol. 2024, 10, 1037–1049. [Google Scholar] [CrossRef]
- Chau, Y.; Yoo, J.W.; Yuen, H. The impact of post-operative atrial fibrillation on outcomes in coronary artery bypass graft and combined procedures. J. Geriatr. Cardiol. 2021, 18, 319–326. [Google Scholar] [CrossRef]
- Kuppahally, S.; Akoum, N.; Burgon, N.S. Left atrial strain and strain rate in patients with paroxysmal and persistent atrial fibrillation: Relationship to left atrial structural remodeling detected by delayed-enhancement MRI. Circ. Cardiovasc. Imaging 2010, 3, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Gopinathannair, R.; Chen, L.Y.; Chung, M.K.; Cornwell, W.K.; Furie, K.L.; Lakkireddy, D.R.; Marrouche, N.F.; Natale, A.; Olshansky, B.; Joglar, J.A.; et al. Managing Atrial Fibrillation in Patients With Heart Failure and Reduced Ejection Fraction: A Scientific Statement From the American Heart Association. Circ. Arrhythmia Electrophysiol. 2021, 14, HAE0000000000000078. [Google Scholar] [CrossRef] [PubMed]
- Badheka, A.O.; Shah, N.; Grover, P.M.; Patel, N.J.; Chothani, A.; Mehta, K.; Singh, V.; Deshmukh, A.; Savani, G.T.; Rathod, A.; et al. Outcomes in atrial fibrillation patients with and without left ventricular hypertrophy when treated with a lenient rate-control or rhythm-control strategy. Am. J. Cardiol. 2014, 113, 1159–1165. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. HFpEF Is the Substrate for Stroke in Obesity and Diabetes Independent of Atrial Fibrillation. JACC Heart Fail. 2020, 8, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yuan, Y.-H.; Chen, N.-H.; Wang, H.-B. The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson’s disease. Int. Immunopharmacol. 2019, 67, 458–464. [Google Scholar] [CrossRef]
- Yan, Z.; Qi, Z.; Yang, X.; Ji, N.; Wang, Y.; Shi, Q.; Li, M.; Zhang, J.; Zhu, Y. The NLRP3 inflammasome: Multiple activation pathways and its role in primary cells during ventricular remodeling. J. Cell Physiol. 2021, 23, 5547–5563. [Google Scholar] [CrossRef]
- Elliott, E.I.; Sutterwala, F.S. Initiation and perpetuation of NLRP 3 inflammasome activation and assembly. Immunol. Rev. 2015, 265, 35–52. [Google Scholar] [CrossRef]
- Noma, A. ATP-regulated K+ channels in cardiac muscle. Nature 1983, 305, 147–148. [Google Scholar] [CrossRef]
- Rodrigo, G.C. The Na+-dependence of Na+-activated K+-channels (IK(Na)) in guinea pig ventricular myocytes, is different in excised inside/out patches and cell-attached patches. Pflügers Arch. 1993, 422, 530–532. [Google Scholar] [CrossRef]
- Carmeliet, E. Cardiac ionic currents and acute ischemia: From channels to arrhythmias. Physiol. Rev. 1999, 79, 917–1017. [Google Scholar] [CrossRef]
- Mitani, A.; Shattock, M.; Physiology, C. Role of Na-activated K channel, Na-K-Cl cotransport, and NaK pump in [K] e changes during ischemia in rat heart. Am. J. Physiol. Heart Circ. Physiol. 1992, 263, H333–H340. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Wang, Z.; Sun, H.; Ma, L. Role of NLRP3 Inflammasome in Myocardial Ischemia-Reperfusion Injury and Ventricular Remodeling. Med. Sci. Monit. 2022, 28, e934255. [Google Scholar] [CrossRef] [PubMed]
- Amano, F.; Akamatsu, Y. A lipopolysaccharide (LPS)-resistant mutant isolated from a macrophage-like cell line, J774. 1, exhibits an altered activated-macrophage phenotype in response to LPS. Infect. Immun. 1991, 59, 2166–2174. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, H.; Kouadir, M.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Gong, T.; Yang, Y.; Jin, T.; Jiang, W.; Zhou, R. Orchestration of NLRP3 inflammasome activation by ion fluxes. Trends Immunol. 2018, 39, 393–406. [Google Scholar] [CrossRef]
- Murphy, E.; Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Mucke, L. Inflammation in neurodegenerative disease—A double-edged sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef]
- Fioranelli, M.; Roccia, M.G.; Flavin, D.; Cota, L. Regulation of Inflammatory Reaction in Health and Disease. Int. J. Mol. Sci. 2021, 17, 5277. [Google Scholar] [CrossRef]
- Jorquera, G.; Russell, J.; Monsalves-Álvarez, M.; Cruz, G.; Valladares-Ide, D.; Basualto-Alarcón, C.; Barrientos, G.; Estrada, M.; Llanos, P. NLRP3 Inflammasome: Potential Role in Obesity Related Low-Grade Inflammation and Insulin Resistance in Skeletal Muscle. Int. J. Mol. Sci. 2021, 22, 3254. [Google Scholar] [CrossRef]
- Danesh, J.; Whincup, P.; Walker, M.; Lennon, L.; Thomson, A.; Appleby, P.; Gallimore, J.R.; Pepys, M.B. Low grade inflammation and coronary heart disease: Prospective study and updated meta-analyses. BMJ 2000, 321, 199–204. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Cordero, M.D.; Alcocer-Gómez, E.; Ryffel, B. Gain of function mutation and inflammasome driven diseases in human and mouse models. J. Autoimmun. 2018, 91, 13–22. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Lu, A.; Wu, H. Structural mechanisms of inflammasome assembly. FEBS J. 2015, 282, 435–444. [Google Scholar] [CrossRef]
- Masumoto, J.; Taniguchi, S.; Ayukawa, K.; Sarvotham, H.; Kishino, T.; Niikawa, N.; Hidaka, E.; Katsuyama, T.; Higuchi, T.; Sagara, J. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J. Biol. Chem. 1999, 274, 33835–33838. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 8, 477–489. [Google Scholar] [CrossRef]
- Toldo, S.; Mezzaroma, E.; Buckley, L.F.; Potere, N.; Di Nisio, M.; Biondi-Zoccai, G.; Van Tassell, B.; Abbate, A. Targeting the NLRP3 inflammasome in cardiovascular diseases. Pharmacol. Ther. 2022, 236, 108053. [Google Scholar] [CrossRef]
- Bai, B.; Yang, Y.; Wang, Q.; Li, M.; Tian, C.; Liu, Y.; Aung, L.H.; Li, P.; Yu, T.; Chu, X. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis. 2020, 11, 776. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Z.; Luo, M.; Cheng, Z.; Wang, R.; Liu, Q.; Lv, D.; Yan, J.; Shang, F.; Luo, S.; et al. NLRP3 inflammasome contributes to endothelial dysfunction in angiotensin II-induced hypertension in mice. Microvasc. Res. 2022, 143, 104384. [Google Scholar] [CrossRef]
- Sun, H.; Ren, X.; Xiong, X.; Chen, Y.; Zhao, M.; Wang, J.; Zhou, Y.; Han, Y.; Chen, Q.; Li, Y.; et al. NLRP3 inflammasome activation contributes to VSMC phenotypic transformation and proliferation in hypertension. Cell Death Dis. 2017, 8, e3074. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wang, Y.; Mu, N.; Lou, X.; Li, W.; Chen, Y.; Fan, D.; Tan, H. Activation of NLRP3 inflammasomes contributes to hyperhomocysteinemia-aggravated inflammation and atherosclerosis in apoE-deficient mice. Lab. Invest. 2017, 97, 922–934. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Abbate, A. The NLRP3 inflammasome in acute myocardial infarction. Nat. Rev. Cardiol. 2018, 15, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.M.; Ling, Y.H.; Huuskes, B.M.; Ferens, D.M.; Saini, N.; Chan, C.T.; Diep, H.; Kett, M.M.; Samuel, C.S.; Kemp-Harper, B.K.; et al. Pharmacological inhibition of the NLRP3 inflammasome reduces blood pressure, renal damage, and dysfunction in salt-sensitive hypertension. Cardiovasc. Res. 2019, 115, 776–787. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef]
- Usui, F.; Shirasuna, K.; Kimura, H.; Tatsumi, K.; Kawashima, A.; Karasawa, T.; Hida, S.; Sagara, J.; Taniguchi, S.; Takahashi, M. Critical role of caspase-1 in vascular inflammation and development of atherosclerosis in Western diet-fed apolipoprotein E-deficient mice. Biochem. Biophys. Res. Commun. 2012, 425, 162–168. [Google Scholar] [CrossRef]
- Menu, P.; Pellegrin, M.; Aubert, J.F.; Bouzourene, K.; Tardivel, A.; Mazzolai, L.; Tschopp, J. Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis. 2011, 2, e137. [Google Scholar] [CrossRef]
- Zheng, Y.; Xu, L.; Dong, N.; Li, F. NLRP3 inflammasome: The rising star in cardiovascular diseases. Front. Cardiovasc. Med. 2022, 9, 927061. [Google Scholar] [CrossRef]
- Varghese, G.P.; Folkersen, L.; Strawbridge, R.J.; Halvorsen, B.; Yndestad, A.; Ranheim, T.; Krohg-Sørensen, K.; Skjelland, M.; Espevik, T.; Aukrust, P.; et al. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. J. Am. Heart Assoc. 2016, 5, e00303. [Google Scholar] [CrossRef]
- Toldo, S.; Mezzaroma, E.; Mauro, A.G.; Salloum, F.; Van Tassell, B.W.; Abbate, A. The inflammasome in myocardial injury and cardiac remodeling. Antioxid. Redox Signal. 2015, 22, 1146–1161. [Google Scholar] [CrossRef]
- Toldo, S.; Mezzaroma, E.; McGeough, M.D.; Peña, C.A.; Marchetti, C.; Sonnino, C.; Van Tassell, B.W.; Salloum, F.N.; Voelkel, N.F.; Hoffman, H.M.; et al. Independent roles of the priming and the triggering of the NLRP3 inflammasome in the heart. Cardiovasc Res. 2015, 105, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Higashikuni, Y.; Liu, W.; Numata, G.; Tanaka, K.; Fukuda, D.; Tanaka, Y.; Hirata, Y.; Imamura, T.; Takimoto, E.; Komuro, I.; et al. NLRP3 Inflammasome Activation Through Heart-Brain Interaction Initiates Cardiac Inflammation and Hypertrophy During Pressure Overload. Circulation 2023, 147, 338–355. [Google Scholar] [CrossRef] [PubMed]
- Van Tassell, B.W.; Trankle, C.R.; Canada, J.M.; Carbone, S.; Buckley, L.; Kadariya, D.; Del Buono, M.G.; Billingsley, H.; Wohlford, G.; Viscusi, M.; et al. IL-1 Blockade in Patients with Heart Failure with Preserved Ejection Fraction. Circ. Heart Fail. 2018, 11, e005036. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef]
- Gao, R.; Li, X.; Xiang, H.; Yang, H.; Lv, C.; Sun, X.; Chen, H.; Gao, Y.; Yang, J.; Luo, W.; et al. The covalent NLRP3-inflammasome inhibitor Oridonin relieves myocardial infarction induced myocardial fibrosis and cardiac remodeling in mice. Int. Immunopharmacol. 2021, 90, 107133. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, L.; Wang, S.; Cheng, H.; Xu, L.; Pei, G.; Wang, Y.; Fu, C.; Jiang, Y.; He, C.; et al. Signaling pathways and targeted therapy for myocardial infarction. Signal Transduct. Target. Ther. 2022, 7, 78. [Google Scholar] [CrossRef]
- Tardif, J.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. CANTOS Trial Group. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Del Buono, M.; Crea, F.; Versaci, F.; Biondi-Zoccai, G. NLRP3 Inflammasome: A New Promising Therapeutic Target to Treat Heart Failure. J. Cardiovasc. Pharmacol. 2021, 77, 159–161. [Google Scholar] [CrossRef]
- Harada, M.; Van Wagoner, D.R.; Nattel, S. Role of inflammation in atrial fibrillation pathophysiology and management. Circ. J. 2015, 79, 495–502. [Google Scholar] [CrossRef]
- Gungor, B.; Ekmekci, A.; Arman, A.; Ozcan, K.S.; Ucer, E.; Alper, A.T.; Calik, N.; Yilmaz, H.; Tezel, T.; Coker, A.; et al. Assessment of interleukin-1 gene cluster polymorphisms in lone atrial fibrillation: New insight into the role of inflammation in atrial fibrillation. Pacing Clin. Electrophysiol. 2013, 36, 1220–1227. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.-A.; Kolpakov, M.A.; Guo, X.; Du, B.; Nguyen, Y.; Wang, T.; Powel, P.; Dell’Italia, L.; Sabri, A. A Intracardiac administration of neutrophil protease cathepsin G activates noncanonical inflammasome pathway and promotes inflammation and pathological remodeling in non-injured heart. J. Mol. Cell Cardiol. 2019, 134, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Veleva, T.; Scott, L.; Cao, S.; Li, L.; Chen, G.; Jeyabal, P.; Pan, X.; Alsina, K.M.; Abu-Taha, I.; et al. Enhanced cardiomyocyte nlrp3 inflammasome signaling promotes atrial fibrillation. Circulation 2018, 138, 2227–2242. [Google Scholar] [CrossRef] [PubMed]
- Heijman, J.; Muna, P.; Veleva, T.; Molina, C.E.; Molina, C.E.; Sutanto, H.; Tekook, M.; Wang, Q.; Abu-Taha, I.H.; Gorka, M.; et al. Atrial Myocyte NLRP3/CaMKII Nexus Forms a Substrate for Postoperative Atrial Fibrillation. Circ. Res. 2020, 127, 1036–1055. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Huo, Y.; Tai, B.; Lin, C.; Yang, H.; Tsai, C. Ziprasidone triggers inflammasome signaling via PI3K-Akt-mTOR pathway to promote atrial fibrillation. Biomed. Pharmacother. 2024, 175, 116649. [Google Scholar] [CrossRef]
- Yang, H.; Zhu, J.; Fu, H.; Shuai, W. Dapansutrile Ameliorates Atrial Inflammation and Vulnerability to Atrial Fibrillation in HFpEF Rats. Heart Lung Circ. 2024, 33, 65–77. [Google Scholar] [CrossRef]
- Lin, A.E.; Bapat, A.C.; Xiao, L.; Niroula, A.; Ye, J.; Wong, W.J. Clonal Hematopoiesis of Indeterminate Potential With Loss of Tet2 Enhances Risk for Atrial Fibrillation Through Nlrp3 Inflammasome Activation. Circulation 2024, 30, 1419–1434. [Google Scholar] [CrossRef]
- Yang, T.C.; Chang, P.Y.; Lu, S.C. L5-LDL from ST-elevation myocardial infarction patients induces IL-1beta production via LOX-1 and NLRP3 inflammasome activation in macrophages. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H265–H274. [Google Scholar] [CrossRef]
- Sokolova, M.; Sjaastad, I.; Louwe, M.C.; Alfsnes, K.; Aronsen, J.M.; Zhang, L.; Haugstad, S.B.; Bendiksen, B.A.; Øgaard, J.; Bliksøen, M.; et al. NLRP3 inflammasome promotes myocardial remodeling during diet induced obesity. Front. Immunol. 2019, 10, 1621. [Google Scholar] [CrossRef]
- Suetomi, T.; Willeford, A.; Brand, C.S.; Cho, Y.; Ross, R.S.; Miyamoto, S.; Brown, J.H. Inflammation and NLRP3 inflammasome activation initiated in response to pressure overload by Ca2+/calmodulin dependent protein kinase II delta signaling in cardiomyocytes are essential for adverse cardiac remodeling. Circulation 2018, 138, 2530–2544. [Google Scholar] [CrossRef]
- Cai, S.-M.; Yang, R.-Q.; Li, Y.; Ning, Z.; Zhang, L.; Zhou, G.; Luo, W.; Li, D.; Chen, Y.; Pan, M.; et al. Angiotensin-(1–7) improves liver fibrosis by regulating the NLRP3 inflammasome via redox balance modulation. Antioxid. Redox Signal. 2016, 24, 795–812. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Pan, T.; Zheng, B.; Chen, Y.; Li, W. Autophagy attenuates angiotensin II induced pulmonary fibrosis by inhibiting redox imbalance-mediated NOD-like receptor family pyrin domain containing 3 inflammasome activation. Antioxid. Redox Signal. 2019, 30, 520–541. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.R.; Kumar, K.M.; Behera, B.P.; Patra, S.; Sekhar, C.; Panigrahi, D.P.; Praharaj, P.P.; Singh, A.; Patil, S.; Dhiman, R.; et al. Mitochondrial dysfunction as a driver of NLRP3 inflammasome activation and its modulation through mitophagy for potential therapeutics. Int. J. Biochem. Cell Biol. 2021, 136, 106013. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.Y.; Li, M.; Feng, X.; Wang, J.; Cao, L.; Shen, X.; Chen, J.; Sun, M.; Sheng, R.; Han, F.; et al. Combined NADPH and the NOX inhibitor apocynin provides greater anti-inflammatory and neuroprotective effects in a mouse model of stroke. Free Radic. Biol. Med. 2017, 104, 333–345. [Google Scholar] [CrossRef]
- Zhang, H.; Kim, H.; Park, B.W.; Noh, M.; Kim, Y.; Park, J.; Park, J.; Kim, J.; Sim, W.; Ban, K.; et al. CU06-1004 enhances vascular integrity and improves cardiac remodeling by suppressing edema and inflammation in myocardial ischemia-reperfusion injury. Exp. Mol. Med. 2022, 54, 23–34. [Google Scholar] [CrossRef]
- Bujak, M.; Kweon, H.J.; Chatila, K.; Li, N.; Taffet, G.; Frangogiannis, N.G. Aging-related defects are associated with adverse cardiac remodeling in a mouse model of reperfused myocardial infarction. J. Am. Coll. Cardiol. 2008, 51, 1384–1392. [Google Scholar] [CrossRef]
- Tarone, G.; Balligand, J.L.; Bauersachs, J.; Clerk, A.; De Windt, L.; Heymans, S.; Hilfiker-Kleiner, D.; Hirsch, E.; Iaccarino, G.; Knöll, R.; et al. Targeting myocardial remodelling to develop novel therapies for heart failure: A position paper from the Working Group on Myocardial Function of the European Society of Cardiology. Eur. J. Heart Fail. 2014, 16, 494–508. [Google Scholar] [CrossRef]
- de Boer, R.A.; van der Velde, A.R.; Mueller, C.; van Veldhuisen, D.J.; Anker, S.D.; Peacock, W.F.; Adams, K.F.; Maisel, A. Galectin-3: A modifiable risk factor in heart failure. Cardiovasc. Drugs Ther. 2014, 28, 237–246. [Google Scholar] [CrossRef]
- Lu, Q.; Li, X.; Liu, J.; Sun, X.; Rousselle, T.; Ren, D.; Tong, N.; Li, J. AMPK is associated with the beneficial effects of antidiabetic agents on cardiovascular diseases. Biosci. Rep. 2019, 39, BSR20181995. [Google Scholar] [CrossRef]
- Čater, M.; Bombek, L.K. Protective Role of Mitochondrial Uncoupling Proteins against Age-Related Oxidative Stress in Type 2 Diabetes Mellitus. Antioxidants 2022, 11, 1473. [Google Scholar] [CrossRef]
- Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Bou-Teen, D.; Kaludercic, N.; Weissman, D.; Turan, B.; Maack, C.; Di Lisa, F.; Ruiz-Meana, M. Mitochondrial ROS and mitochondria-targeted antioxidants in the aged heart. Free Radic. Biol. Med. 2021, 167, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.; Shao, P.; Wallace, C.; Chua, S.; Sung, P.; Ko, S.; Chai, H.; Chung, S.; Chen, K.; Lu, K.; et al. Combined Therapy with SS31 and Mitochondria Mitigates Myocardial Ischemia-Reperfusion Injury in Rats. Int. J. Mol. Sci. 2018, 19, 2782. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhang, F.; Yan, P.; Zhang, S.; Lou, Y.; Geng, Z.; Li, Z.; Zhang, Y.; Xu, Y.; Lu, Y.; et al. LARP7 Protects Against Heart Failure by Enhancing Mitochondrial Biogenesis. Circulation 2021, 143, 2007–2022. [Google Scholar] [CrossRef]
- Talasaz, A.H.; Salehiomran, A.; Heidary, Z.; Gholami, K.; Aryannejad, H.; Jalali, A.; Daei, M. The effects of vitamin D supplementation on postoperative atrial fibrillation after coronary artery bypass grafting in patients with vitamin D deficiency. J. Card. Surg. 2022, 37, 2219–2224. [Google Scholar] [CrossRef]
- Rodrigo, R.; Cereceda, M.; Castillo, R.; Asenjo, R.; Zamorano, J.; Araya, J.; Castillo-Koch, R.; Espinoza, J.; Larraín, E. Prevention of atrial fibrillation following cardiac surgery: Basis for a novel therapeutic strategy based on non-hypoxic myocardial preconditioning. Pharmacol. Ther. 2008, 118, 104–127. [Google Scholar] [CrossRef]
- Menezes-Rodrigues, F.S.; Errante, P.R.; Araújo, E.A.; Fernandes, M.P.P.; Silva, M.M.D.; Pires-Oliveira, M.; Scorza, C.A.; Scorza, F.A.; Taha, M.O.; Caricati-Neto, A. Cardioprotection stimulated by resveratrol and grape products prevents lethal cardiac arrhythmias in an animal model of ischemia and reperfusion. Acta Cir. Bras. 2021, 36, e360306. [Google Scholar] [CrossRef]
- Hedayati, N.; Yaghoobi, A.; Salami, M.; Gholinezhad, Y.; Aghadavood, F.; Eshraghi, R. Impact of polyphenols on heart failure and cardiac hypertrophy: Clinical effects and molecular mechanisms. Front. Cardiovasc. Med. 2023, 10, 1174816. [Google Scholar] [CrossRef]
- Martins-Marques, T.; Rodriguez-Sinovas, A.; Girao, H. Cellular crosstalk in cardioprotection: Where and when do reactive oxygen species play a role? Free Radic. Biol. Med. 2021, 169, 397–409. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. EMPA-REG OUTCOME Investigators. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Filippatos, G.; Anker, S.D.; Butler, J.; Farmakis, D.; Ferreira, J.P.; Gollop, N.D.; Brueckmann, M.; Iwata, T.; Pocock, S.; Zannad, F.; et al. Effects of empagliflozin on cardiovascular and renal outcomes in heart failure with reduced ejection fraction according to age: A secondary analysis of EMPEROR-Reduced. Eur. J. Heart Fail. 2022, 24, 2297–2304. [Google Scholar] [CrossRef] [PubMed]
- Saisho, Y. SGLT2 Inhibitors: The Star in the Treatment of Type 2 Diabetes? Diseases 2020, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Madonna, R.; Moscato, S.; Cufaro, M.C.; Pieragostino, D.; Mattii, L.; Boccio, P.; Ghelardoni, S.; Zucchi, R.; Caterina, R. Empagliflozin inhibits excessive autophagy through the AMPK/GSK3β signalling pathway in diabetic cardiomyopathy. Cardiovasc. Res. 2023, 119, 1175–1189. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Shiou, Y.; Jhuo, S.; Chang, C.; Liu, P.; Jhuang, W.-J. The sodium-glucose co-transporter 2 inhibitor empagliflozin attenuates cardiac fibrosis and improves ventricular hemodynamics in hypertensive heart failure rats. Cardiovasc. Diabetol. 2019, 18, 45. [Google Scholar] [CrossRef]
- Li, X.; Lu, Q.; Qiu, Y.; Carmo, J.M.; Wang, Z.; da Silva, A.A. Direct Cardiac Actions of the Sodium Glucose Co-Transporter 2 Inhibitor Empagliflozin Improve Myocardial Oxidative Phosphorylation and Attenuate Pressure-Overload Heart Failure. J. Am. Heart Assoc. 2021, 10, e018298. [Google Scholar] [CrossRef]
- Hsieh, P.; Chu, M.; Ching, H.; Huang, Y.; Chou, W.; Tsai, K.; Chan, S. Dapagliflozin Mitigates Doxorubicin-Caused Myocardium Damage by Regulating AKT-Mediated Oxidative Stress, Cardiac Remodeling, and Inflammation. Int. J. Mol. Sci. 2022, 23, 10146. [Google Scholar] [CrossRef]
- Castelvecchio, S.; Frigelli, M.; Sturla, F.; Milani, V.; Pappalardo, O.A.; Citarella, M.; Menicanti, L.; Votta, E. Elucidating the mechanisms underlying left ventricular function recovery in patients with ischemic heart failure undergoing surgical remodeling: A 3-dimensional ultrasound analysis. J. Thorac. Cardiovasc. Surg. 2023, 165, 1418–1429.e4. [Google Scholar] [CrossRef]
- Ma, S.; Chen, L.; Yan, J.; Shen, M.; Zhang, R.; Li, M. Dapagliflozin attenuates residual cardiac remodeling after surgical ventricular reconstruction in mice with an enlarged heart after myocardial infarction. Biomed. Pharmacother. 2022, 156, 113765. [Google Scholar] [CrossRef]
- Kostin, S.; Krizanic, F.; Kelesidis, T.; Pagonas, N. The role of NETosis in heart failure. Heart Fail. Rev. 2024, 29, 1097–1106. [Google Scholar] [CrossRef]
- Ehrenkranz, J.R.; Lewis, N.G.; Kahn, C.R.; Roth, J. Phlorizin: A review. Diabetes Metab. Res. Rev. 2005, 21, 31–38. [Google Scholar] [CrossRef]
- Xie, Y.; Wei, Y.; Li, D.; Pu, J.; Ding, H.; Zhang, X. Mechanisms of SGLT2 Inhibitors in Heart Failure and Their Clinical Value. J. Cardiovasc. Pharmacol. 2023, 81, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Madaan, T.; Akhtar, M.; Najmi, A.K. Sodium glucose CoTransporter 2 (SGLT2) inhibitors: Current status and future perspective. Eur. J. Pharm. Sci. 2016, 93, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Rathore, A.; Parwani, D.; Mallick, C.; Asati, V.; Agarwal, S.; Rajoriya, V.; Das, R.; Kashaw, S.K. An exhaustive perspective on structural insights of SGLT2 inhibitors: A novel class of antidiabetic agent. Eur. J. Med. Chem. 2020, 204, 112523. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, D.; Esposito, K. Class effect for SGLT-2 inhibitors: A tale of 9 drugs. Cardiovasc. Diabetol. 2019, 18, 94. [Google Scholar] [CrossRef] [PubMed]
- Devineni, D.; Morrow, L.; Hompesch, M.; Skee, D.; Vandebosch, A.; Murphy, J.; Ways, K.; Schwartz, S. Canagliflozin improves glycaemic control over 28 days in subjects with type 2 diabetes not optimally controlled on insulin. Diabetes Obes. Metab. 2012, 14, 539–545. [Google Scholar] [CrossRef]
- Devineni, D.; Curtin, C.R.; Polidori, D.; Gutierrez, M.J.; Murphy, J.; Rusch, S.; Rothenberg, P.L. Pharmacokinetics and pharmacodynamics of canagliflozin, a sodium glucose co-transporter 2 inhibitor, in subjects with type 2 diabetes mellitus. J. Clin. Pharmacol. 2013, 53, 601–610. [Google Scholar] [CrossRef]
- Yang, L.; Li, H.; Li, H.; Bui, A.; Chang, M.; Liu, X.; Kasichayanula, S.; Griffen, S.C.; Lacreta, F.P.; Boulton, D.W. Pharmacokinetic and pharmacodynamic properties of single- and multiple-dose of dapagliflozin, a selective inhibitor of SGLT2, in healthy Chinese subjects. Clin. Ther. 2013, 35, 1211–1222.e2. [Google Scholar] [CrossRef]
- Tang, W.; Reele, S.; Hamer-Maansson, J.E.; Parikh, S.; de Bruin, T.W. Dapagliflozin twice daily or once daily: Effect on pharmacokinetics and urinary glucose excretion in healthy subjects. Diabetes Obes. Metab. 2015, 17, 423–425. [Google Scholar] [CrossRef]
- Ayoub, B.M.; Mowaka, S.; Elzanfaly, E.S.; Ashoush, N.; Elmazar, M.M.; Mousa, S.A. Pharmacokinetic Evaluation of Empagliflozin in Healthy Egyptian Volunteers Using LC-MS/MS and Comparison with Other Ethnic Populations. Sci. Rep. 2017, 7, 2583. [Google Scholar] [CrossRef]
- Sarashina, A.; Koiwai, K.; Seman, L.J.; Yamamura, N.; Taniguchi, A.; Negishi, T.; Sesoko, S.; Woerle, H.J.; Dugi, K.A. Safety, tolerability, pharmacokinetics and pharmacodynamics of single doses of empagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor, in healthy Japanese subjects. Drug Metab. Pharmacokinet. 2013, 28, 213–219. [Google Scholar] [CrossRef]
- Sahasrabudhe, V.; Fediuk, D.J.; Matschke, K.; Shi, H.; Liang, Y.; Hickman, A.; Bass, A.; Terra, S.G.; Zhou, S.; Krishna, R.; et al. Effect of Food on the Pharmacokinetics of Ertugliflozin and Its Fixed-Dose Combinations Ertugliflozin/Sitagliptin and Ertugliflozin/Metformin. Clin. Pharmacol. Drug Dev. 2019, 8, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Mu, Y.; Shi, H.; Liang, Y.; Liu, Z.; Matschke, K.; Hickman, A.; Krishna, R.; Sahasrabudhe, V. Pharmacokinetic Properties of Single and Multiple Doses of Ertugliflozin, a Selective Inhibitor of SGLT2, in Healthy Chinese Subjects. Clin. Pharmacol. Drug Dev. 2020, 9, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Kadokura, T.; Akiyama, N.; Kashiwagi, A.; Utsuno, A.; Kazuta, K.; Yoshida, S.; Nagase, I.; Smulders, R.; Kageyama, S. Pharmacokinetic and pharmacodynamic study of ipragliflozin in Japanese patients with type 2 diabetes mellitus: A randomized, double-blind, placebo-controlled study. Diabetes Res. Clin. Pract. 2014, 106, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Kaku, K.; Isaka, H.; Toyoshima, J.; Sakatani, T. Clinical pharmacology study of ipragliflozin in Japanese patients with type 1 diabetes mellitus: A phase 2, randomized, placebo-controlled trial. Diabetes Obes. Metab. 2019, 21, 1445–1454. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Takano, Y.; Schwab, D.; Portron, A.; Kasahara-Ito, N.; Saito, T.; Iida, S. Effect of Renal Impairment on the Pharmacokinetics and Pharmacodynamics of Tofogliflozin (A SELECTIVE SGLT2 Inhibitor) in Patients with Type 2 Diabetes Mellitus. Drug Res. 2019, 69, 314–322. [Google Scholar] [CrossRef]
- Rosenwasser, R.F.; Rosenwasser, J.N.; Sutton, D.; Choksi, R.; Epstein, B. Tofogliflozin: A highly selective SGLT2 inhibitor for the treatment of type 2 diabetes. Drugs Today 2014, 50, 739–745. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Atkin, S.L.; Butler, A.E.; Sahebkar, A. Sodium-glucose cotransporter inhibitors and oxidative stress: An update. J. Cell Physiol. 2019, 234, 3231–3237. [Google Scholar] [CrossRef]
- Mylonas, N.; Nikolaou, P.E.; Karakasis, P.; Stachteas, P.; Fragakis, N.; Andreadou, I. Endothelial Protection by Sodium-Glucose Cotransporter 2 Inhibitors: A Literature Review of In Vitro and In Vivo Studies. Int. J. Mol. Sci. 2024, 25, 7274. [Google Scholar] [CrossRef]
- Sa-Nguanmoo, P.; Tanajak, P.; Kerdphoo, S.; Jaiwongkam, T.; Pratchayasa-kul, W.; Chattipakorn, N.; Chattipakorn, S.C. SGLT2-inhibitor and DPP-4 inhibitor improve brain function via attenuatingmitochondrial dysfunction, insulin resistance, inflammation, andapoptosis in HFD-induced obese rats. Toxicol. Appl. Pharmacol. 2017, 333, 43–50. [Google Scholar] [CrossRef]
- Pignatelli, P.; Baratta, F.; Buzzetti, R.; D’Amico, A.; Castellani, V.; Bartimoccia, S. The Sodium-Glucose Co-Transporter-2 (SGLT2) Inhibitors Reduce Platelet Activation and Thrombus Formation by Lowering NOX2-Related Oxidative Stress: A Pilot Study. Antioxidants 2022, 11, 1878. [Google Scholar] [CrossRef]
- Li, C.; Zhang, J.; Xue, M.; Li, X.; Han, F.; Liu, X.; Xu, L.; Lu, Y.; Cheng, Y.; Li, T.; et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc. Diabetol. 2019, 18, 15. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Akoumianakis, I.; Badi, I.; Akawi, N.; Kotanidis, C.P.; Polkinghorne, M.; Stadiotti, I.; Sommariva, E.; Antonopoulos, A.S.; Carena, M.C.; et al. Effects of canagliflozin on human myocardial redox signalling: Clinical implications. Eur. Heart J. 2021, 42, 4947–4960. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lu, J.; Wang, Y.; Sun, P.; Gao, T.; Xu, N. Canagliflozin Attenuates Lipotoxicity in Cardiomyocytes by Inhibiting Inflammation and Ferroptosis through Activating AMPK Pathway. Int. J. Mol. Sci. 2023, 24, 858. [Google Scholar] [CrossRef] [PubMed]
- Liu, I.F.; Lin, T.; Wang, S.; Yen, C.; Li, C.; Kuo, H.; Hsieh, C.; Chang, C.; Chang, C.; Chen, Y.; et al. Long-term administration of Western diet induced metabolic syndrome in mice and causes cardiac microvascular dysfunction, cardiomyocyte mitochondrial damage, and cardiac remodeling involving caveolae and caveolin-1 expression. Biol. Direct. 2023, 189, 9. [Google Scholar] [CrossRef] [PubMed]
- Lou, X.; Zhang, Y.; Guo, J.; Gao, L.; Ding, Y.; Zhuo, X.; Lei, Q.; Bian, J.; Lei, R.; Gong, W.; et al. What is the impact of ferroptosis on diabetic cardiomyopathy: A systematic review. Heart Fail. Rev. 2024, 29, 1–11. [Google Scholar] [CrossRef]
- Hartupee, J.; Mann, D.L. Neurohormonal activation in heart failure with reduced ejection fraction. Nat. Rev. Cardiol. 2017, 14, 30–38. [Google Scholar] [CrossRef]
- Stumpf, C.; Seybold, K.; Petzi, S.; Wasmeier, G.; Raaz, D.; Yilmaz, A.; Anger, T.; Daniel, W.G.; Garlichs, C.D. Interleukin-10 improves left ventricular function in rats with heart failure subsequent to myocardial infarction. Eur. J. Heart Fail. 2008, 10, 733–739. [Google Scholar] [CrossRef]
- Packer, M. Leptin-Aldosterone-Neprilysin Axis: Identification of Its Distinctive Role in the Pathogenesis of the Three Phenotypes of Heart Failure in People with Obesity. Circulation 2018, 137, 1614–1631. [Google Scholar] [CrossRef]
- De Angelis, E.; Pecoraro, M.; Rusciano, M.R.; Ciccarelli, M.; Popolo, A. Cross-Talk between Neurohormonal Pathways and the Immune System in Heart Failure: A Review of the Literature. Int. J. Mol. Sci. 2019, 20, 1698. [Google Scholar] [CrossRef]
- Sugi, Y.; Yasukawa, H.; Kai, H.; Fukui, D.; Futamata, N.; Mawatari, K. Reduction and activation of circulating dendritic cells in patients with decompensated heart failure. Int. J. Cardiol. 2011, 147, 258–264. [Google Scholar] [CrossRef]
- Martini, E.; Kunderfranco, P.; Peano, C.; Carullo, P.; Cremonesi, M.; Schorn, T.; Carriero, R.; Termanini, A.; Colombo, F.S.; Jachetti, E.; et al. Single-Cell Sequencing of Mouse Heart Immune Infiltrate in Pressure Overload-Driven Heart Failure Reveals Extent of Immune Activation. Circulation 2019, 140, 2089–2107. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.M.; Chang, N.C.; Lin, S.Z. Dapagliflozin, a selective SGLT2 Inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via STAT3 signaling in infarcted rat hearts. Free Radic. Biol. Med. 2017, 104, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, M.; Moustafa, Y.M.; Mehanna, E.T.; Elrayess, R.A.; El-Sayed, N.M.; Hazem, R.M. Empagliflozin protects against isoprenaline-induced fibrosis in rat heart through modulation of TGF-β/SMAD pathway. Life Sci. 2024, 337, 122354. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, Y.; Wang, Z.; Tan, M.; Lin, J.; Qian, X.; Li, H.; Jiang, T. Dapagliflozin alleviates myocardial ischemia/reperfusion injury by reducing ferroptosis via MAPK signaling inhibition. Front. Pharmacol. 2023, 20, 1078205. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Nakamura, T.; Inouye, K.; Li, J.; Tang, Y.; Lundbäck, P.; Valdes-Ferrer, S.I.; Olofsson, P.S.; Kalb, T.; Roth, J.; et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 2012, 488, 670–674. [Google Scholar] [CrossRef]
- Coll, R.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Mueller, J.L.; Vitari, A.C.; Misaghi, S.; Fedorova, A.; Deshayes, K.; Lee, W.P.; Hoffman, H.M.; Dixit, V.M. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J. Cell Biol. 2009, 187, 61–70. [Google Scholar] [CrossRef]
- He, H.; Jiang, H.; Chen, Y.; Ye, J.; Wang, A.; Wang, C. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat. Commun. 2018, 9, 2550. [Google Scholar] [CrossRef]
- Marchetti, C.; Swartzwelter, B.; Gamboni, F.; Neff, C.P.; Richter, K.; Azam, T.; Carta, S.; Tengesdal, I.; Nemkov, T.; D’Alessandro, A.; et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, E1530–E1539. [Google Scholar] [CrossRef]
- Jiang, H.; He, H.; Chen, Y.; Huang, W.; Cheng, J.; Ye, J. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 2017, 214, 3219–3238. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, Y.; Wang, X.; Yang, Y.; Tao, Y.J. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol. Med. 2018, 10, e8689. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Peng, Q. NLRP3 Inhibitor Tranilast Attenuates Gestational Diabetes Mellitus in a Genetic Mouse Model. Drugs R D 2022, 22, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Missmahl, H.P.; Riemann, J. Simple determination of microangiopathy of the capillaries of the rectal mucosa in diabetics. Wien. Klin. Wochenschr. 1968, 46, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, X.; Wang, Q. Effects and mechanisms of SGLT2 inhibitors on the NLRP3 inflammasome, with a focus on atherosclerosis. Front. Endocrinol. 2022, 13, 992937. [Google Scholar] [CrossRef]
- Andreadou, I.; Daiber, A.; Baxter, G.F.; Brizzi, M.F.; Di Lisa, F.; Kaludercic, N.; Lazou, A.; Varga, Z.V.; Zuurbier, C.J.; Schulz, R. Influence of cardiometabolic comorbidities on myocardial function, infarction, and cardioprotection: Role of cardiac redox signaling. Free Radic. Biol. Med. 2021, 166, 33–52. [Google Scholar] [CrossRef]
- Brown, A.J.; Gandy, S.; McCrimmon, R.; Houston, J.G.; Struthers, A.D.; Lang, C.C. A randomized controlled trial of dapagliflozin on left ventricular hypertrophy in people with type two diabetes: The DAPA-LVH trial. Eur. Heart J. 2020, 41, 3421–3432. [Google Scholar] [CrossRef]
- Guo, W.; Zhao, L.; Huang, W.; Chen, J.; Zhong, T.; Yan, S. Sodium-glucose cotransporter 2 inhibitors, inflammation, and heart failure: A two-sample Mendelian randomization study. J. Cardiovasc. Diabetol. 2024, 23, 118. [Google Scholar] [CrossRef]
- Stachteas, P.; Nasoufidou, A.; Karagiannidis, E.; Patoulias, D.; Karakasis, P.; Alexiou, S.; Samaras, A.; Zormpas, G.; Stavropoulos, G.; Tsalikakis, D.; et al. The Role of Sodium Glucose Co-Transporter 2 Inhibitors in Atrial Fibrillation: A Comprehensive Review. J. Clin. Med. 2024, 13, 5408. [Google Scholar] [CrossRef]
- Zarei, B.; Fazli, B.; Tayyebi, M.; Teshnizi, M.A.; Moeinipour, A.; Javedanfar, O.; Bayaz, R.J.D.; Rahmati, M.; Ghavami, V.; Amini, S.; et al. Evaluation of the effect of empagliflozin on prevention of atrial fibrillation after coronary artery bypass grafting: A double-blind, randomized, placebo-controlled trial. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2024; online ahead of print. [Google Scholar] [CrossRef]
- Aghakouchakzadeh, M.; Hosseini, K.; Haghjoo, M.; Mirzabeigi, P.; Tajdini, M.; Talasaz, A.H.; Jalali, A.; Askarinejad, A.; Kohansal, E.; Hedayat, B.; et al. Empagliflozin to prevent post-operative atrial fibrillation in patients undergoing coronary artery bypass graft surgery: Rationale and design of the EMPOAF trial. Pacing Clin. Electrophysiol. 2024, 47, 1087–1095. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Butler, A.E.; Atkin, S.L.; Katsiki, N.; Sahebkar, A. Sodium-glucose cotransporter 2 inhibitors and inflammation in chronic kidney disease: Possible molecular pathways. J. Cell Physiol. 2018, 234, 223–230. [Google Scholar] [CrossRef]
- Mozaffarian, D.; Marchioli, R.; Macchia, A.; Silletta, M.G.; Ferrazzi, P.; Gardner, T.J.; Latini, R.; Libby, P.; Lombardi, F.; O’Gara, P.T.; et al. Fish oil and postoperative atrial fibrillation: The Omega-3 Fatty Acids for Prevention of Post-operative Atrial Fibrillation (OPERA) randomized trial. JAMA 2012, 308, 2001–2011. [Google Scholar] [CrossRef] [PubMed]
- Corradi, D.; Saffitz, J.; Novelli, D.; Asimaki, A.; Simon, C.; Oldoni, E.; Masson, S.; Meessen, J.M.; Monaco, R.; Manuguerra, R.; et al. Prospective Evaluation of Clinico-Pathological Predictors of Postoperative Atrial Fibrillation: An Ancillary Study From the OPERA Trial. Circ. Arrhythm. Electrophysiol. 2020, 13, e008382. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; De Zeeuw, D.; Fulcher, G.; Erondu, N.; Wayne Shaw, D.S.L.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Uthman, L.; Baartscheer, A.; Schumacher, C.A.; Fiolet, J.W.; Kuschma, M.C.; Hollmann, M.W.; Coronel, R.; Weber, N.C.; Zuurbier, C.J. Direct Cardiac Actions of Sodium Glucose Cotransporter 2 Inhibitors Target Pathogenic Mechanisms Underlying Heart Failure in Diabetic Patients. Front. Physiol. 2018, 9, 1575. [Google Scholar] [CrossRef] [PubMed]
- Chilton, R.J. Effects of sodium-glucose cotransporter-2 inhibitors on the cardiovascular and renal complications of type 2 diabetes Diabetes. Obes. Metab. 2020, 22, 16–29. [Google Scholar] [CrossRef]
- Palmiero, G.; Cesaro, A.; Vetrano, E.; Pafundi, P.C.; Galiero, R.; Caturano, A.; Moscarella, E.; Gragnano, F.; Salvatore, T.; Rinaldi, L.; et al. Impact of SGLT2 Inhibitors on Heart Failure: From Pathophysiology to Clinical Effects. Int. J. Mol. Sci. 2021, 22, 5863. [Google Scholar] [CrossRef]
- Kang, S.; Verma, S.; Hassanabad, A.F.; Teng, G.; Belke, D.D.; Dundas, J.A.; Guzzardi, D.G.; Svystonyuk, D.A.; Pattar, S.S.; Park, D.S.; et al. Direct Effects of Empagliflozin on Extracellular Matrix Remodelling in Human Cardiac Myofibroblasts: Novel Translational Clues to Explain EMPA-REG OUTCOME Results. Can. J. Cardiol. 2020, 36, 543–553. [Google Scholar] [CrossRef]
- Byrne, N.J.; Matsumura, N.; Maayah, Z.H.; Ferdaoussi, M.; Takahara, S.; Darwesh, A.M.; Levasseur, J.L.; Jahng, J.W.; Vos, D.; Parajuli, N.; et al. Empagliflozin Blunts Worsening Cardiac Dysfunction Associated with Reduced NLRP3 (Nucleotide-Binding Domain-Like Receptor Protein 3) Inflammasome Activation in Heart Failure. Circ. Heart Fail. 2020, 13, e006277. [Google Scholar] [CrossRef]
- Inzucchi, S.E.; Zinman, B.; Fitchett, D.; Wanner, C.; Ferrannini, E.; Schumacher, M.; Schmoor, C.; Ohneberg, K.; Johansen, O.E.; George, J.T.; et al. How Does Empagliflozin Reduce Cardiovascular Mortality? Insights From a Mediation Analysis of the EMPA-REG OUTCOME Trial. Diabetes Care. 2018, 41, 356–363. [Google Scholar] [CrossRef]
- Ceriello, A.; Ofstad, A.; Zwiener, I.; Kaspers, S.; George, J.; Nicolucci, A. Empagliflozin reduced long-term HbA1c variability and cardiovascular death: Insights from the EMPA-REG OUTCOME trial. Cardiovasc. Diabetol. 2020, 19, 176. [Google Scholar] [CrossRef]
- Anker, S.; Butler, J.; Filippatos, G.S.; Jamal, W.; Salsali, A.; Schnee, J.; Kimura, K.; Zeller, C.; George, J.; Brueckmann, M.; et al. Evaluation of the effects of sodium-glucose co-transporter 2 inhibition with empagliflozin on morbidity and mortality in patients with chronic heart failure and a preserved ejection fraction: Rationale for and design of the EMPEROR-Preserved Trial. Eur. J. Heart Fail. 2019, 21, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Hundertmark, M.; Agbaje, O.F.; Coleman, R. Design and rationale of the EMPA-VISION trial: Investigating the metabolic effects of empagliflozin in patients with heart failure. ESC Heart Fail. 2021, 8, 2580–2590. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Lee, S.G.; Kim, S.H.; Kim, J.H.; Choi, E.; Cho, W.; Rim, J.H.; Hwang, I.; Lee, C.J.; Lee, M.; et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat. Commun. 2020, 11, 2127. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Drug | Dose | Tmax | Half-Life (t1/2) | References |
|---|---|---|---|---|
| Canagliflozin | 25–400 mg | 1.0–4.0 h | 10.22–16.30 h | [145,146] |
| Dapagliflozin | 25–50 mg | 0.5–1.5 h | 8.1–16.4 h | [147,148] |
| Empagliflozin | 1–100 mg | 1.0–2.5 h | 6.1–16.5 h | [149,150] |
| Ertugliflozin | 5–25 mg | 1.0–4.0 h | 9.91–16.87 h | [151,152] |
| Ipragliflozin | 25–100 mg | 0.5–0.9 h | 10.5–11.3 | [153,154] |
| Tofogliflozin | 10–640 mg | 0.5–2.0 h | 3.81–6.21 h | [155,156] |
| Inhibitor | Mechanism of Inhibition | Effect on SGLT2 | Reference |
|---|---|---|---|
| MCC950 | Blocks NLRP3 activation by inhibiting ATPase activity, preventing inflammasome assembly | No effect on SGLT2 identified | [176] |
| Glyburide | Inhibits NLRP3 activation through K+ efflux blockade | No effect on SGLT2 identified | [177] |
| Oridonin | Covalently binds to NLRP3, disrupting inflammasome assembly | No effect on SGLT2 identified | [178] |
| OLT1177 (Dapansutrile) | Inhibits ASC oligomerization, a key step in inflammasome activation | No effect on SGLT2 identified | [179] |
| CY-09 | Binds to NACHT domain, blocking NLRP3 ATPase activity | Inhibits NLRP3 oligomerization by binding NACHT domain | [180] |
| Tranilast | Inhibits NLRP3 oligomerization by binding NACHT domain | No effect on SGLT2 identified | [181,182] |
| beta-hydroxybutyrate (β-OHB) | Inhibits oxidative stress and it can protect the function of mitochondria and exert an anti-inflammatory effect, as it is an endogenous NLRP3 inflammasome inhibitor. | An increase in β-OHB levels by the administration of SGLT-2 inhibitors | [183] |
| SGLT2 inhibitors (e.g., Empagliflozin) | Reduce NLRP3 activation indirectly by suppressing NF-κB signaling and promoting autophagy | Yes, as primary function | [184] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castillo, R.L.; Farías, J.; Sandoval, C.; González-Candia, A.; Figueroa, E.; Quezada, M.; Cruz, G.; Llanos, P.; Jorquera, G.; Kostin, S.; et al. Role of NLRP3 Inflammasome in Heart Failure Patients Undergoing Cardiac Surgery as a Potential Determinant of Postoperative Atrial Fibrillation and Remodeling: Is SGLT2 Cotransporter Inhibition an Alternative for Cardioprotection? Antioxidants 2024, 13, 1388. https://doi.org/10.3390/antiox13111388
Castillo RL, Farías J, Sandoval C, González-Candia A, Figueroa E, Quezada M, Cruz G, Llanos P, Jorquera G, Kostin S, et al. Role of NLRP3 Inflammasome in Heart Failure Patients Undergoing Cardiac Surgery as a Potential Determinant of Postoperative Atrial Fibrillation and Remodeling: Is SGLT2 Cotransporter Inhibition an Alternative for Cardioprotection? Antioxidants. 2024; 13(11):1388. https://doi.org/10.3390/antiox13111388
Chicago/Turabian StyleCastillo, Rodrigo L., Jorge Farías, Cristian Sandoval, Alejandro González-Candia, Esteban Figueroa, Mauricio Quezada, Gonzalo Cruz, Paola Llanos, Gonzalo Jorquera, Sawa Kostin, and et al. 2024. "Role of NLRP3 Inflammasome in Heart Failure Patients Undergoing Cardiac Surgery as a Potential Determinant of Postoperative Atrial Fibrillation and Remodeling: Is SGLT2 Cotransporter Inhibition an Alternative for Cardioprotection?" Antioxidants 13, no. 11: 1388. https://doi.org/10.3390/antiox13111388
APA StyleCastillo, R. L., Farías, J., Sandoval, C., González-Candia, A., Figueroa, E., Quezada, M., Cruz, G., Llanos, P., Jorquera, G., Kostin, S., & Carrasco, R. (2024). Role of NLRP3 Inflammasome in Heart Failure Patients Undergoing Cardiac Surgery as a Potential Determinant of Postoperative Atrial Fibrillation and Remodeling: Is SGLT2 Cotransporter Inhibition an Alternative for Cardioprotection? Antioxidants, 13(11), 1388. https://doi.org/10.3390/antiox13111388