
Amphiregulin Exerts Proangiogenic Effects in Developing Murine Lungs

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. In Vivo Experiments
2.1.1. Animals
2.1.2. Hyperoxia Experiments
2.1.3. Lung Tissue Extraction and Real-Time RT-PCR Assays
2.1.4. Flow Cytometry Experiments
2.2. In Vitro Experiments
2.2.1. Cell Culture
2.2.2. Transient Transfection Experiments
2.2.3. Areg Treatment
2.2.4. Hyperoxia (HO) Exposure Experiments
2.2.5. Real-Time RT-PCR Assays
2.2.6. Enzyme-Linked Immunosorbent Assay (ELISA)
2.2.7. Immunoblot Assay
2.2.8. Tubule Formation Assay
2.2.9. Statistical Analyses
3. Results
3.1. Neonatal Murine Lung Expression of Areg mRNA and Areg+ Cells Following HO Exposure
3.2. Fetal Murine Lung Endothelial Cell (EC) Expression of Areg and Its Receptor, Egfr, Following HO Exposure
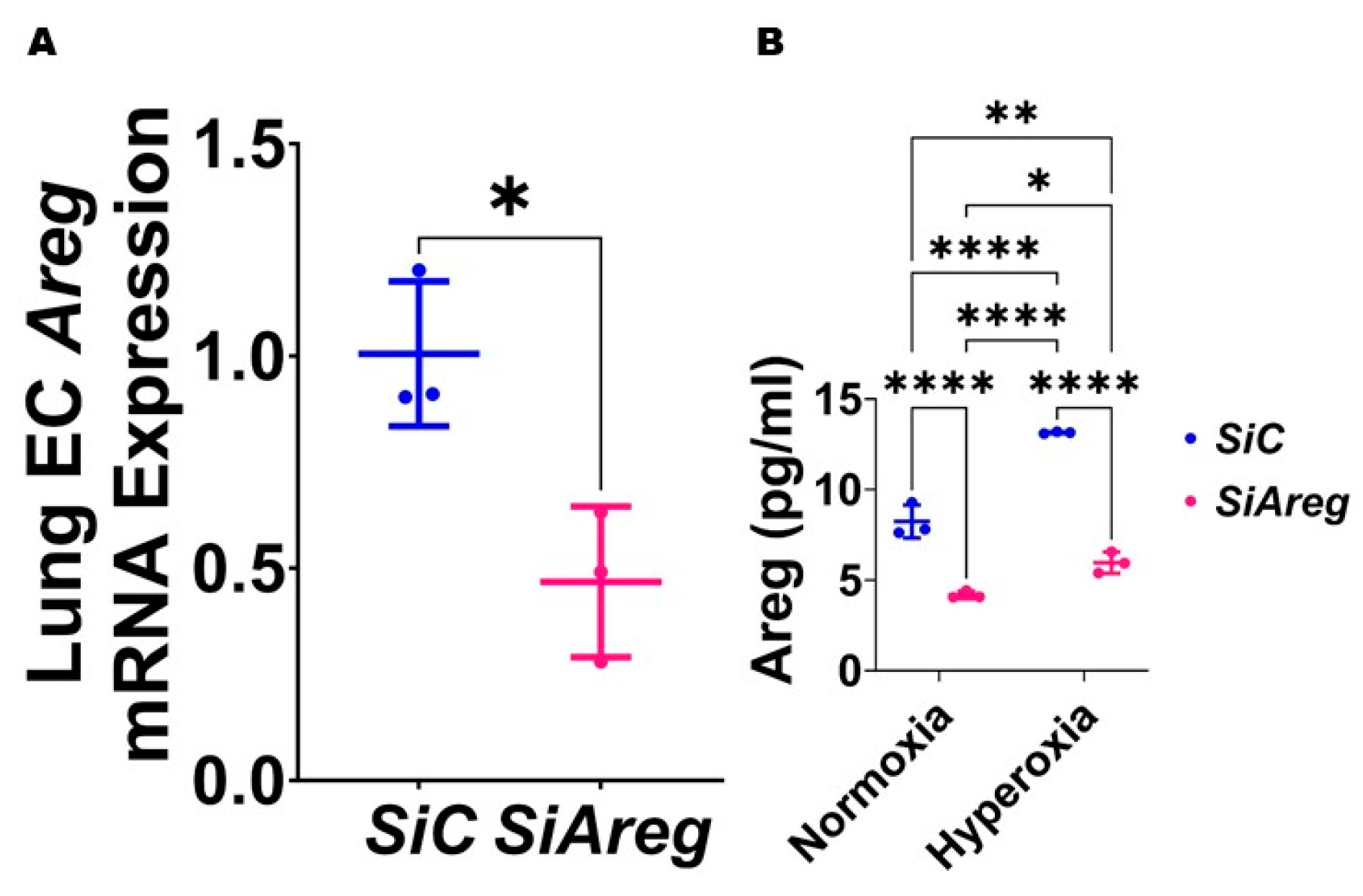
3.3. Areg Knockdown Decreases the Tubule Formation Ability of Murine Lung ECs in HO Conditions


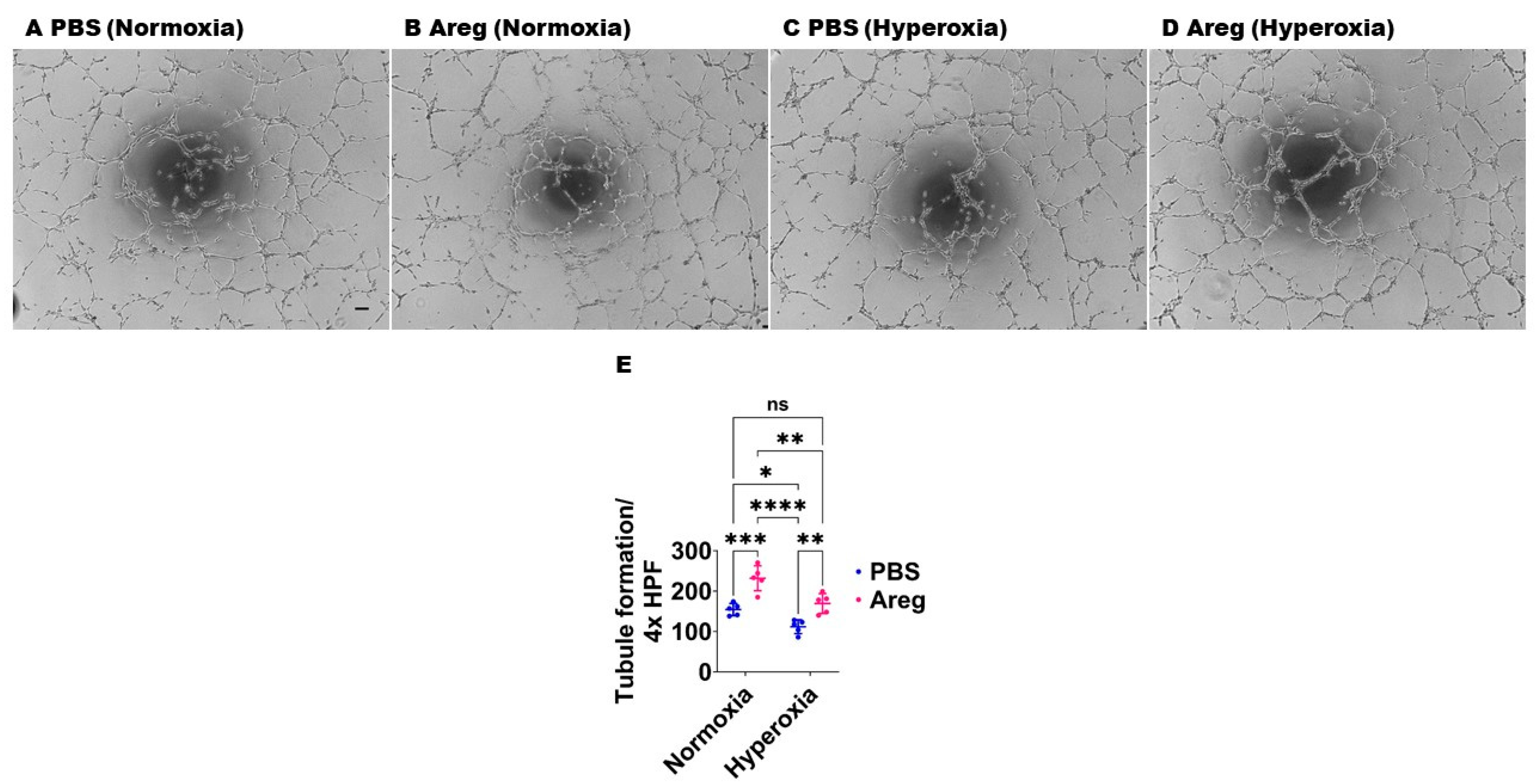
3.4. Recombinant Mouse Areg Protein Increases the Tubule Formation Ability of Murine Lung ECs in HO Conditions


3.5. Recombinant Mouse Areg Protein Increases Total ERK1/2 Activation in Murine Lung ECs in HO Conditions


4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Thébaud, B.; Goss, K.N.; Laughon, M.; Whitsett, J.A.; Abman, S.H.; Steinhorn, R.H.; Aschner, J.L.; Davis, P.G.; McGrath-Morrow, S.A.; Soll, R.F.; et al. Bronchopulmonary dysplasia. Nat. Rev. Dis. Primers 2019, 5, 78. [Google Scholar] [CrossRef] [PubMed]
- Simpson, S.J.; Hall, G.L.; Wilson, A.C. Lung function following very preterm birth in the era of ‘new’ bronchopulmonary dysplasia. Respirology 2015, 20, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Islam, J.Y.; Keller, R.L.; Aschner, J.L.; Hartert, T.V.; Moore, P.E. Understanding the Short- and Long-Term Respiratory Outcomes of Prematurity and Bronchopulmonary Dysplasia. Am. J. Respir. Crit. Care Med. 2015, 192, 134–156. [Google Scholar] [CrossRef] [PubMed]
- Twilhaar, E.S.; Wade, R.M.; de Kieviet, J.F.; van Goudoever, J.B.; van Elburg, R.M.; Oosterlaan, J. Cognitive Outcomes of Children Born Extremely or Very Preterm Since the 1990s and Associated Risk Factors: A Meta-analysis and Meta-regression. JAMA Pediatr. 2018, 172, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Haggie, S.; Robinson, P.; Selvadurai, H.; Fitzgerald, D.A. Bronchopulmonary dysplasia: A review of the pulmonary sequelae in the post-surfactant era. J. Paediatr. Child Health 2020, 56, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Hurst, J.R.; Beckmann, J.; Ni, Y.; Bolton, C.E.; McEniery, C.M.; Cockcroft, J.R.; Marlow, N. Respiratory and Cardiovascular Outcomes in Survivors of Extremely Preterm Birth at 19 Years. Am. J. Respir. Crit. Care Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Husain, A.N.; Siddiqui, N.H.; Stocker, J.T. Pathology of arrested acinar development in postsurfactant bronchopulmonary dysplasia. Hum. Pathol. 1998, 29, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Coalson, J.J. Pathology of new bronchopulmonary dysplasia. Semin. Neonatol. SN 2003, 8, 73–81. [Google Scholar] [CrossRef]
- Thébaud, B.; Abman, S.H. Bronchopulmonary dysplasia: Where have all the vessels gone? Roles of angiogenic growth factors in chronic lung disease. Am. J. Respir. Crit. Care Med. 2007, 175, 978–985. [Google Scholar] [CrossRef]
- Abman, S.H. Bronchopulmonary dysplasia: “A vascular hypothesis”. Am. J. Respir. Crit. Care Med. 2001, 164, 1755–1756. [Google Scholar] [CrossRef]
- Shoyab, M.; Plowman, G.D.; McDonald, V.L.; Bradley, J.G.; Todaro, G.J. Structure and function of human amphiregulin: A member of the epidermal growth factor family. Science 1989, 243, 1074–1076. [Google Scholar] [CrossRef] [PubMed]
- Meng, C.; Wang, S.; Wang, X.; Lv, J.; Zeng, W.; Chang, R.; Li, Q.; Wang, X. Amphiregulin inhibits TNF-α-induced alveolar epithelial cell death through EGFR signaling pathway. Biomed. Pharmacother. 2020, 125, 109995. [Google Scholar] [CrossRef] [PubMed]
- Ogata-Suetsugu, S.; Yanagihara, T.; Hamada, N.; Ikeda-Harada, C.; Yokoyama, T.; Suzuki, K.; Kawaguchi, T.; Maeyama, T.; Kuwano, K.; Nakanishi, Y. Amphiregulin suppresses epithelial cell apoptosis in lipopolysaccharide-induced lung injury in mice. Biochem. Biophys. Res. Commun. 2017, 484, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Green, J.A.; Moltedo, B.; Arvey, A.; Hemmers, S.; Yuan, S.; Treuting, P.M.; Rudensky, A.Y. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell 2015, 162, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Schuger, L.; Johnson, G.R.; Gilbride, K.; Plowman, G.D.; Mandel, R. Amphiregulin in lung branching morphogenesis: Interaction with heparan sulfate proteoglycan modulates cell proliferation. Development 1996, 122, 1759–1767. [Google Scholar] [CrossRef]
- Ali, N.; Zirak, B.; Rodriguez, R.S.; Pauli, M.L.; Truong, H.A.; Lai, K.; Ahn, R.; Corbin, K.; Lowe, M.M.; Scharschmidt, T.C.; et al. Regulatory T Cells in Skin Facilitate Epithelial Stem Cell Differentiation. Cell 2017, 169, 1119–1129.e1111. [Google Scholar] [CrossRef]
- Ito, M.; Komai, K.; Mise-Omata, S.; Iizuka-Koga, M.; Noguchi, Y.; Kondo, T.; Sakai, R.; Matsuo, K.; Nakayama, T.; Yoshie, O.; et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 2019, 565, 246–250. [Google Scholar] [CrossRef]
- Xia, N.; Lu, Y.; Gu, M.; Li, N.; Liu, M.; Jiao, J.; Zhu, Z.; Li, J.; Li, D.; Tang, T.; et al. A Unique Population of Regulatory T Cells in Heart Potentiates Cardiac Protection From Myocardial Infarction. Circulation 2020, 142, 1956–1973. [Google Scholar] [CrossRef]
- Harb, H.; Benamar, M.; Lai, P.S.; Contini, P.; Griffith, J.W.; Crestani, E.; Schmitz-Abe, K.; Chen, Q.; Fong, J.; Marri, L.; et al. Notch4 signaling limits regulatory T-cell-mediated tissue repair and promotes severe lung inflammation in viral infections. Immunity 2021, 54, 1186–1199.e1187. [Google Scholar] [CrossRef]
- Koeppen, M.; Lee, J.W.; Seo, S.W.; Brodsky, K.S.; Kreth, S.; Yang, I.V.; Buttrick, P.M.; Eckle, T.; Eltzschig, H.K. Hypoxia-inducible factor 2-alpha-dependent induction of amphiregulin dampens myocardial ischemia-reperfusion injury. Nat. Commun. 2018, 9, 816. [Google Scholar] [CrossRef]
- Liu, J.; Pan, L.; Hong, W.; Chen, S.; Bai, P.; Luo, W.; Sun, X.; He, F.; Jia, X.; Cai, J.; et al. GPR174 knockdown enhances blood flow recovery in hindlimb ischemia mice model by upregulating AREG expression. Nat. Commun. 2022, 13, 7519. [Google Scholar] [CrossRef]
- Florentin, J.; Zhao, J.; Tai, Y.Y.; Sun, W.; Ohayon, L.L.; O’Neil, S.P.; Arunkumar, A.; Zhang, X.; Zhu, J.; Al Aaraj, Y.; et al. Loss of Amphiregulin drives inflammation and endothelial apoptosis in pulmonary hypertension. Life Sci. Alliance 2022, 5. [Google Scholar] [CrossRef]
- Yuan, W.; Xu, W.; Li, Y.; Jiang, W.; Li, Y.; Huang, Q.; Chen, B.; Wu, S.; Wang, Y.; Song, W.; et al. TAZ sensitizes EGFR wild-type non-small-cell lung cancer to gefitinib by promoting amphiregulin transcription. Cell Death Dis. 2019, 10, 283. [Google Scholar] [CrossRef]
- Shan, S.; Li, Y.; Wang, J.; Lv, Z.; Yi, D.; Huang, Q.; Corrigan, C.J.; Wang, W.; Quangeng, Z.; Ying, S. Nasal administration of interleukin-33 induces airways angiogenesis and expression of multiple angiogenic factors in a murine asthma surrogate. Immunology 2016, 148, 83–91. [Google Scholar] [CrossRef]
- Yao, X.; Wang, W.; Li, Y.; Huang, P.; Zhang, Q.; Wang, J.; Wang, W.; Lv, Z.; An, Y.; Qin, J.; et al. IL-25 induces airways angiogenesis and expression of multiple angiogenic factors in a murine asthma model. Respir. Res. 2015, 16, 39. [Google Scholar] [CrossRef]
- Aslam, M.; Baveja, R.; Liang, O.D.; Fernandez-Gonzalez, A.; Lee, C.; Mitsialis, S.A.; Kourembanas, S. Bone marrow stromal cells attenuate lung injury in a murine model of neonatal chronic lung disease. Am. J. Respir. Crit. Care Med. 2009, 180, 1122–1130. [Google Scholar] [CrossRef]
- Lee, K.J.; Berkelhamer, S.K.; Kim, G.A.; Taylor, J.M.; O’Shea, K.M.; Steinhorn, R.H.; Farrow, K.N. Disrupted Pulmonary Artery cGMP Signaling in Mice with Hyperoxia-Induced Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2013, 50, 369–378. [Google Scholar] [CrossRef]
- Menon, R.T.; Shrestha, A.K.; Reynolds, C.L.; Barrios, R.; Shivanna, B. Long-term pulmonary and cardiovascular morbidities of neonatal hyperoxia exposure in mice. Int. J. Biochem. Cell Biol. 2018, 94, 119–124. [Google Scholar] [CrossRef]
- Reynolds, C.L.; Zhang, S.; Shrestha, A.K.; Barrios, R.; Shivanna, B. Phenotypic assessment of pulmonary hypertension using high-resolution echocardiography is feasible in neonatal mice with experimental bronchopulmonary dysplasia and pulmonary hypertension: A step toward preventing chronic obstructive pulmonary disease. Int. J. Chron. Obs. Pulmon Dis. 2016, 11, 1597–1605. [Google Scholar] [CrossRef]
- Elsaie, A.; Menon, R.T.; Shrestha, A.K.; Gowda, S.H.; Varghese, N.P.; Barrios, R.J.; Blanco, C.L.; Konduri, G.G.; Shivanna, B. Endothelial Adenosine Monophosphate-Activated Protein Kinase-Alpha1 Deficiency Potentiates Hyperoxia-Induced Experimental Bronchopulmonary Dysplasia and Pulmonary Hypertension. Antioxidants 2021, 10, 1913. [Google Scholar] [CrossRef]
- Menon, R.T.; Thapa, S.; Shrestha, A.K.; Barrios, R.; Shivanna, B. Extracellular Signal-Regulated Kinase 1 Alone Is Dispensable for Hyperoxia-Mediated Alveolar and Pulmonary Vascular Simplification in Neonatal Mice. Antioxidants 2022, 11, 1130. [Google Scholar] [CrossRef] [PubMed]
- Shivanna, B.; Zhang, W.; Jiang, W.; Welty, S.E.; Couroucli, X.I.; Wang, L.; Moorthy, B. Functional deficiency of aryl hydrocarbon receptor augments oxygen toxicity-induced alveolar simplification in newborn mice. Toxicol. Appl. Pharmacol. 2013, 267, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Shivanna, B.; Chu, C.; Welty, S.E.; Jiang, W.; Wang, L.; Couroucli, X.I.; Moorthy, B. Omeprazole attenuates hyperoxic injury in H441 cells via the aryl hydrocarbon receptor. Free Radic. Biol. Med. 2011, 51, 1910–1917. [Google Scholar] [CrossRef] [PubMed]
- Menon, R.T.; Shrestha, A.K.; Barrios, R.; Shivanna, B. Hyperoxia Disrupts Extracellular Signal-Regulated Kinases 1/2-Induced Angiogenesis in the Developing Lungs. Int. J. Mol. Sci. 2018, 19, 1525. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, A.K.; Bettini, M.L.; Menon, R.T.; Gopal, V.Y.N.; Huang, S.; Edwards, D.P.; Pammi, M.; Barrios, R.; Shivanna, B. Consequences of Early Postnatal Lipopolysaccharide Exposure on Developing Lungs in Mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 316, L229–L244. [Google Scholar] [CrossRef] [PubMed]
- Arnaoutova, I.; Kleinman, H.K. In vitro angiogenesis: Endothelial cell tube formation on gelled basement membrane extract. Nat. Protoc. 2010, 5, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Guidolin, D.; Conconi, M.T.; Nico, B.; Baiguera, S.; Parnigotto, P.P.; Vacca, A.; Nussdorfer, G.G. Vinblastine inhibits the angiogenic response induced by adrenomedullin in vitro and in vivo. Oncogene 2003, 22, 6458–6461. [Google Scholar] [CrossRef] [PubMed]
- Menon, R.T.; Shrestha, A.K.; Barrios, R.; Reynolds, C.; Shivanna, B. Tie-2 Cre-Mediated Deficiency of Extracellular Signal-Regulated Kinase 2 Potentiates Experimental Bronchopulmonary Dysplasia-Associated Pulmonary Hypertension in Neonatal Mice. Int. J. Mol. Sci. 2020, 21, 2408. [Google Scholar] [CrossRef]
- de Visser, Y.P.; Walther, F.J.; Laghmani el, H.; Boersma, H.; van der Laarse, A.; Wagenaar, G.T. Sildenafil attenuates pulmonary inflammation and fibrin deposition, mortality and right ventricular hypertrophy in neonatal hyperoxic lung injury. Respir. Res. 2009, 10, 30. [Google Scholar] [CrossRef]
- Wagenaar, G.T.; ter Horst, S.A.; van Gastelen, M.A.; Leijser, L.M.; Mauad, T.; van der Velden, P.A.; de Heer, E.; Hiemstra, P.S.; Poorthuis, B.J.; Walther, F.J. Gene expression profile and histopathology of experimental bronchopulmonary dysplasia induced by prolonged oxidative stress. Free Radic. Biol. Med. 2004, 36, 782–801. [Google Scholar] [CrossRef]
- ter Horst, S.A.; Walther, F.J.; Poorthuis, B.J.; Hiemstra, P.S.; Wagenaar, G.T. Inhaled nitric oxide attenuates pulmonary inflammation and fibrin deposition and prolongs survival in neonatal hyperoxic lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L35–L44. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.C.; Zhu, Y.; Lu, H.Y.; Ju, H.M.; Xu, S.Q.; Qiao, Y.; Wei, S.J. Type 2 innate lymphoid cell-derived amphiregulin regulates type II alveolar epithelial cell transdifferentiation in a mouse model of bronchopulmonary dysplasia. Int. Immunopharmacol. 2023, 122, 110672. [Google Scholar] [CrossRef] [PubMed]
- Polosa, R.; Prosperini, G.; Leir, S.H.; Holgate, S.T.; Lackie, P.M.; Davies, D.E. Expression of c-erbB receptors and ligands in human bronchial mucosa. Am. J. Respir. Cell Mol. Biol. 1999, 20, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Aida, S.; Tamai, S.; Sekiguchi, S.; Shimizu, N. Distribution of epidermal growth factor and epidermal growth factor receptor in human lung: Immunohistochemical and immunoelectron-microscopic studies. Respiration 1994, 61, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Vermeer, P.D.; Panko, L.; Karp, P.; Lee, J.H.; Zabner, J. Differentiation of human airway epithelia is dependent on erbB2. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 291, L175–L180. [Google Scholar] [CrossRef] [PubMed]
- Chokki, M.; Yamamura, S.; Eguchi, H.; Masegi, T.; Horiuchi, H.; Tanabe, H.; Kamimura, T.; Yasuoka, S. Human airway trypsin-like protease increases mucin gene expression in airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2004, 30, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, S.; Ramgolam, K.; Baulig, A.; Marano, F.; Baeza-Squiban, A. Fine particulate matter induces amphiregulin secretion by bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 2004, 30, 421–427. [Google Scholar] [CrossRef]
- Dial, C.F.; Tune, M.K.; Doerschuk, C.M.; Mock, J.R. Foxp3(+) Regulatory T Cell Expression of Keratinocyte Growth Factor Enhances Lung Epithelial Proliferation. Am. J. Respir. Cell Mol. Biol. 2017, 57, 162–173. [Google Scholar] [CrossRef]
- Bhatt, A.J.; Pryhuber, G.S.; Huyck, H.; Watkins, R.H.; Metlay, L.A.; Maniscalco, W.M. Disrupted pulmonary vasculature and decreased vascular endothelial growth factor, Flt-1, and TIE-2 in human infants dying with bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2001, 164, 1971–1980. [Google Scholar] [CrossRef]
- Hasan, J.; Beharry, K.D.; Valencia, A.M.; Strauss, A.; Modanlou, H.D. Soluble vascular endothelial growth factor receptor 1 in tracheal aspirate fluid of preterm neonates at birth may be predictive of bronchopulmonary dysplasia/chronic lung disease. Pediatrics 2009, 123, 1541–1547. [Google Scholar] [CrossRef]
- Thébaud, B.; Ladha, F.; Michelakis, E.D.; Sawicka, M.; Thurston, G.; Eaton, F.; Hashimoto, K.; Harry, G.; Haromy, A.; Korbutt, G.; et al. Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia-induced lung injury: Evidence that angiogenesis participates in alveolarization. Circulation 2005, 112, 2477–2486. [Google Scholar] [CrossRef] [PubMed]
- Syed, M.; Das, P.; Pawar, A.; Aghai, Z.H.; Kaskinen, A.; Zhuang, Z.W.; Ambalavanan, N.; Pryhuber, G.; Andersson, S.; Bhandari, V. Hyperoxia causes miR-34a-mediated injury via angiopoietin-1 in neonatal lungs. Nat. Commun. 2017, 8, 1173. [Google Scholar] [CrossRef] [PubMed]
- Maniscalco, W.M.; Watkins, R.H.; Pryhuber, G.S.; Bhatt, A.; Shea, C.; Huyck, H. Angiogenic factors and alveolar vasculature: Development and alterations by injury in very premature baboons. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 282, L811–L823. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, A.; Gwini, S.M.; Menahem, S.; Allison, B.J.; Miller, S.L.; Polglase, G.R. Preterm growth restriction and bronchopulmonary dysplasia: The vascular hypothesis and related physiology. J. Physiol. 2019, 597, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Wallace, B.; Peisl, A.; Seedorf, G.; Nowlin, T.; Kim, C.; Bosco, J.; Kenniston, J.; Keefe, D.; Abman, S.H. Anti-sFlt-1 Therapy Preserves Lung Alveolar and Vascular Growth in Antenatal Models of Bronchopulmonary Dysplasia. Am. J. Respir. Crit. Care Med. 2018, 197, 776–787. [Google Scholar] [CrossRef]
- Tang, J.R.; Markham, N.E.; Lin, Y.J.; McMurtry, I.F.; Maxey, A.; Kinsella, J.P.; Abman, S.H. Inhaled nitric oxide attenuates pulmonary hypertension and improves lung growth in infant rats after neonatal treatment with a VEGF receptor inhibitor. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L344–L351. [Google Scholar] [CrossRef]
- Kunig, A.M.; Balasubramaniam, V.; Markham, N.E.; Seedorf, G.; Gien, J.; Abman, S.H. Recombinant human VEGF treatment transiently increases lung edema but enhances lung structure after neonatal hyperoxia. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 291, L1068–L1078. [Google Scholar] [CrossRef]
- Appuhn, S.V.; Siebert, S.; Myti, D.; Wrede, C.; Surate Solaligue, D.E.; Pérez-Bravo, D.; Brandenberger, C.; Schipke, J.; Morty, R.E.; Grothausmann, R.; et al. Capillary Changes Precede Disordered Alveolarization in a Mouse Model of Bronchopulmonary Dysplasia. Am. J. Respir. Cell Mol. Biol. 2021, 65, 81–91. [Google Scholar] [CrossRef]
- Menon, R.T.; Shrestha, A.K.; Reynolds, C.L.; Barrios, R.; Caron, K.M.; Shivanna, B. Adrenomedullin Is Necessary to Resolve Hyperoxia-Induced Experimental Bronchopulmonary Dysplasia and Pulmonary Hypertension in Mice. Am. J. Pathol. 2020, 190, 711–722. [Google Scholar] [CrossRef]
- Ren, X.; Ustiyan, V.; Pradhan, A.; Cai, Y.; Havrilak, J.A.; Bolte, C.S.; Shannon, J.M.; Kalin, T.V.; Kalinichenko, V.V. FOXF1 transcription factor is required for formation of embryonic vasculature by regulating VEGF signaling in endothelial cells. Circ. Res. 2014, 115, 709–720. [Google Scholar] [CrossRef]
- Wang, G.; Wen, B.; Deng, Z.; Zhang, Y.; Kolesnichenko, O.A.; Ustiyan, V.; Pradhan, A.; Kalin, T.V.; Kalinichenko, V.V. Endothelial progenitor cells stimulate neonatal lung angiogenesis through FOXF1-mediated activation of BMP9/ACVRL1 signaling. Nat. Commun. 2022, 13, 2080. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.L.; Gong, J.; Rizal, S.; Peterson, A.L.; Chang, J.; Yao, C.; Dennery, P.A.; Yao, H. Upregulating carnitine palmitoyltransferase 1 attenuates hyperoxia-induced endothelial cell dysfunction and persistent lung injury. Respir. Res. 2022, 23, 205. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhu, M.; Wang, L.; Chen, C.; Song, Y. Amphiregulin potentiates airway inflammation and mucus hypersecretion induced by urban particulate matter via the EGFR-PI3Kα-AKT/ERK pathway. Cell Signal 2019, 53, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Pu, J.; He, F.; Liao, B.; Hao, B.; Hong, W.; Ye, X.; Chen, J.; Zhao, J.; Liu, S.; et al. Positive feedback of the amphiregulin-EGFR-ERK pathway mediates PM2.5 from wood smoke-induced MUC5AC expression in epithelial cells. Sci. Rep. 2017, 7, 11084. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, J.; Harada, C.; Kawaguchi, T.; Suetsugu, S.; Maeyama, T.; Inoshima, I.; Hamada, N.; Kuwano, K.; Nakanishi, Y. Amphiregulin attenuates bleomycin-induced pneumopathy in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 298, L131–L138. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.S.; Lee, H.J.; Nishida, M.; Lee, M.S.; Tamura, R.; Yamashita, S.; Matsuzawa, Y.; Lee, I.K.; Koh, G.Y. Betacellulin and amphiregulin induce upregulation of cyclin D1 and DNA synthesis activity through differential signaling pathways in vascular smooth muscle cells. Circ. Res. 2003, 93, 302–310. [Google Scholar] [CrossRef]
- Qin, L.; Tamasi, J.; Raggatt, L.; Li, X.; Feyen, J.H.; Lee, D.C.; Dicicco-Bloom, E.; Partridge, N.C. Amphiregulin is a novel growth factor involved in normal bone development and in the cellular response to parathyroid hormone stimulation. J. Biol. Chem. 2005, 280, 3974–3981. [Google Scholar] [CrossRef]
- Kansra, S.; Stoll, S.W.; Johnson, J.L.; Elder, J.T. Autocrine extracellular signal-regulated kinase (ERK) activation in normal human keratinocytes: Metalloproteinase-mediated release of amphiregulin triggers signaling from ErbB1 to ERK. Mol. Biol. Cell 2004, 15, 4299–4309. [Google Scholar] [CrossRef][Green Version]
- Young, C.D.; Zimmerman, L.J.; Hoshino, D.; Formisano, L.; Hanker, A.B.; Gatza, M.L.; Morrison, M.M.; Moore, P.D.; Whitwell, C.A.; Dave, B.; et al. Activating PIK3CA Mutations Induce an Epidermal Growth Factor Receptor (EGFR)/Extracellular Signal-regulated Kinase (ERK) Paracrine Signaling Axis in Basal-like Breast Cancer. Mol. Cell Proteom. 2015, 14, 1959–1976. [Google Scholar] [CrossRef]
- Yotsumoto, F.; Fukami, T.; Yagi, H.; Funakoshi, A.; Yoshizato, T.; Kuroki, M.; Miyamoto, S. Amphiregulin regulates the activation of ERK and Akt through epidermal growth factor receptor and HER3 signals involved in the progression of pancreatic cancer. Cancer Sci. 2010, 101, 2351–2360. [Google Scholar] [CrossRef]
- Li Calzi, S.; Shaw, L.C.; Moldovan, L.; Shelley, W.C.; Qi, X.; Racette, L.; Quigley, J.L.; Fortmann, S.D.; Boulton, M.E.; Yoder, M.C.; et al. Progenitor cell combination normalizes retinal vascular development in the oxygen-induced retinopathy (OIR) model. JCI Insight 2019, 4, e129224. [Google Scholar] [CrossRef] [PubMed]
- Stzepourginski, I.; Nigro, G.; Jacob, J.M.; Dulauroy, S.; Sansonetti, P.J.; Eberl, G.; Peduto, L. CD34+ mesenchymal cells are a major component of the intestinal stem cells niche at homeostasis and after injury. Proc. Natl. Acad. Sci. USA 2017, 114, E506–E513. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhong, Y.; Li, X.; Huang, X.; Du, L. Anti-placental growth factor antibody ameliorates hyperoxia-mediated impairment of lung development in neonatal rats. Braz. J. Med. Biol. Res. 2020, 53, e8917. [Google Scholar] [CrossRef] [PubMed]
- Perveen, S.; Patel, H.; Arif, A.; Younis, S.; Codipilly, C.N.; Ahmed, M. Role of EC-SOD overexpression in preserving pulmonary angiogenesis inhibited by oxidative stress. PLoS ONE 2012, 7, e51945. [Google Scholar] [CrossRef]
- Wang, C.Q.; Huang, Y.W.; Wang, S.W.; Huang, Y.L.; Tsai, C.H.; Zhao, Y.M.; Huang, B.F.; Xu, G.H.; Fong, Y.C.; Tang, C.H. Amphiregulin enhances VEGF-A production in human chondrosarcoma cells and promotes angiogenesis by inhibiting miR-206 via FAK/c-Src/PKCδ pathway. Cancer Lett. 2017, 385, 261–270. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thapa, S.; Shankar, N.; Shrestha, A.K.; Civunigunta, M.; Gaikwad, A.S.; Shivanna, B. Amphiregulin Exerts Proangiogenic Effects in Developing Murine Lungs. Antioxidants 2024, 13, 78. https://doi.org/10.3390/antiox13010078
Thapa S, Shankar N, Shrestha AK, Civunigunta M, Gaikwad AS, Shivanna B. Amphiregulin Exerts Proangiogenic Effects in Developing Murine Lungs. Antioxidants. 2024; 13(1):78. https://doi.org/10.3390/antiox13010078
Chicago/Turabian StyleThapa, Shyam, Nithyapriya Shankar, Amrit Kumar Shrestha, Monish Civunigunta, Amos S. Gaikwad, and Binoy Shivanna. 2024. "Amphiregulin Exerts Proangiogenic Effects in Developing Murine Lungs" Antioxidants 13, no. 1: 78. https://doi.org/10.3390/antiox13010078
APA StyleThapa, S., Shankar, N., Shrestha, A. K., Civunigunta, M., Gaikwad, A. S., & Shivanna, B. (2024). Amphiregulin Exerts Proangiogenic Effects in Developing Murine Lungs. Antioxidants, 13(1), 78. https://doi.org/10.3390/antiox13010078