
PTC596-Induced BMI-1 Inhibition Fights Neuroblastoma Multidrug Resistance by Inducing Ferroptosis

 ,
,  , , ,
, , ,  and
and 
Abstract
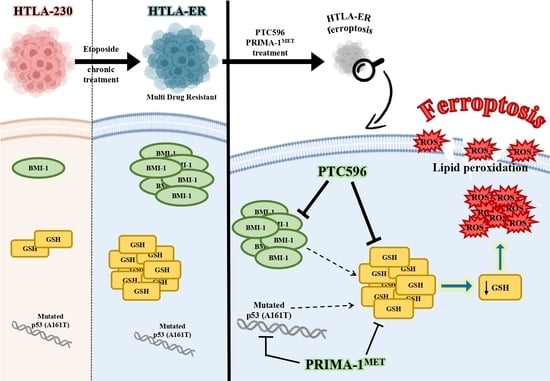
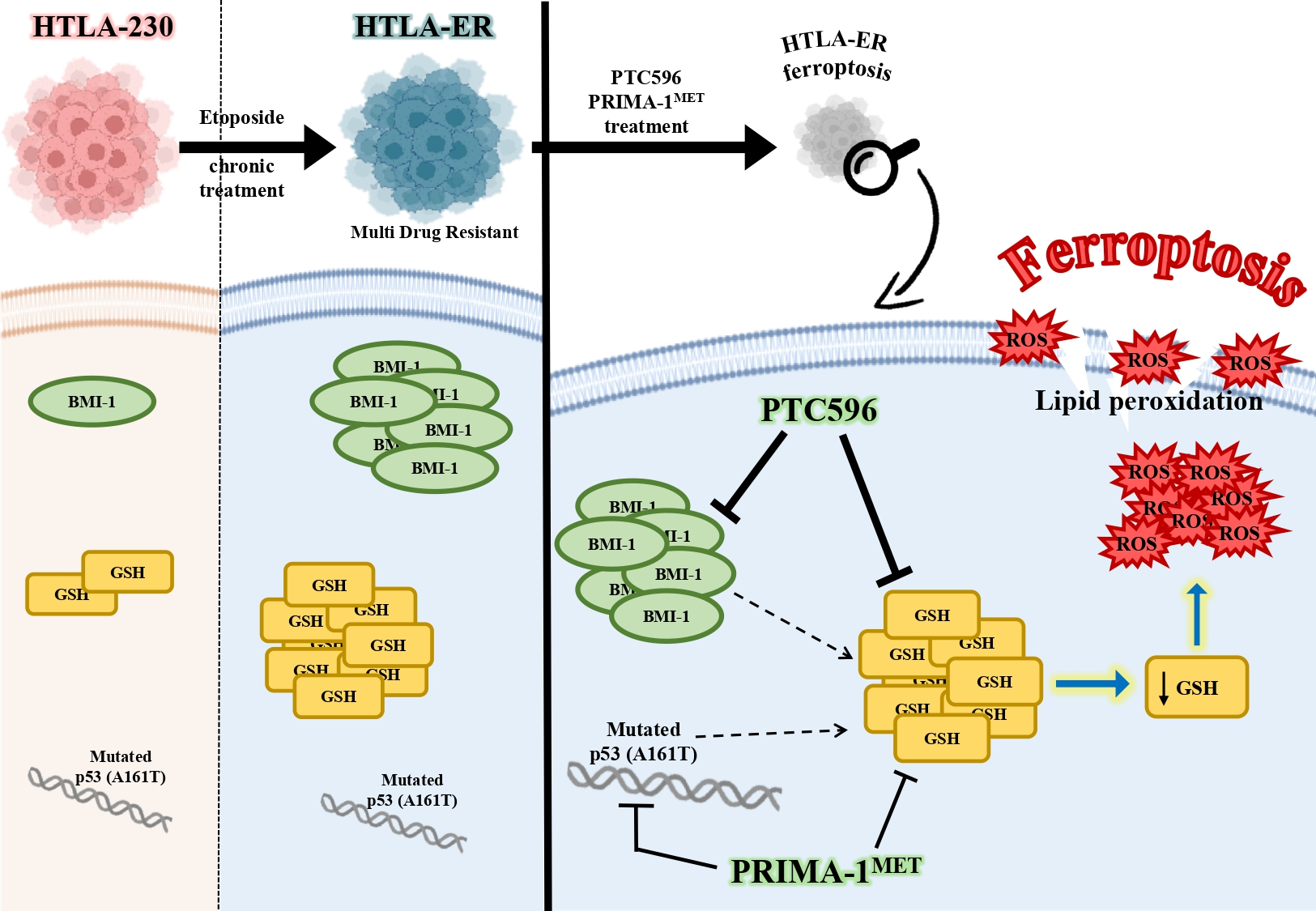
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Cultures
2.3. Treatments
2.4. Cell Viability Assay (MTS)
2.5. Total Protein Extraction and Quantification
2.6. Western Blot Analysis
2.7. Clonogenic Assay
2.8. Cancer Stem Cell Formation Assessment
2.9. H2O2 Level Determination
2.10. HPLC Analysis of Intracellular GSH Levels
2.11. Lipid Peroxidation Assay
2.12. GPX4 Activity
2.13. Statistical Analysis
3. Results
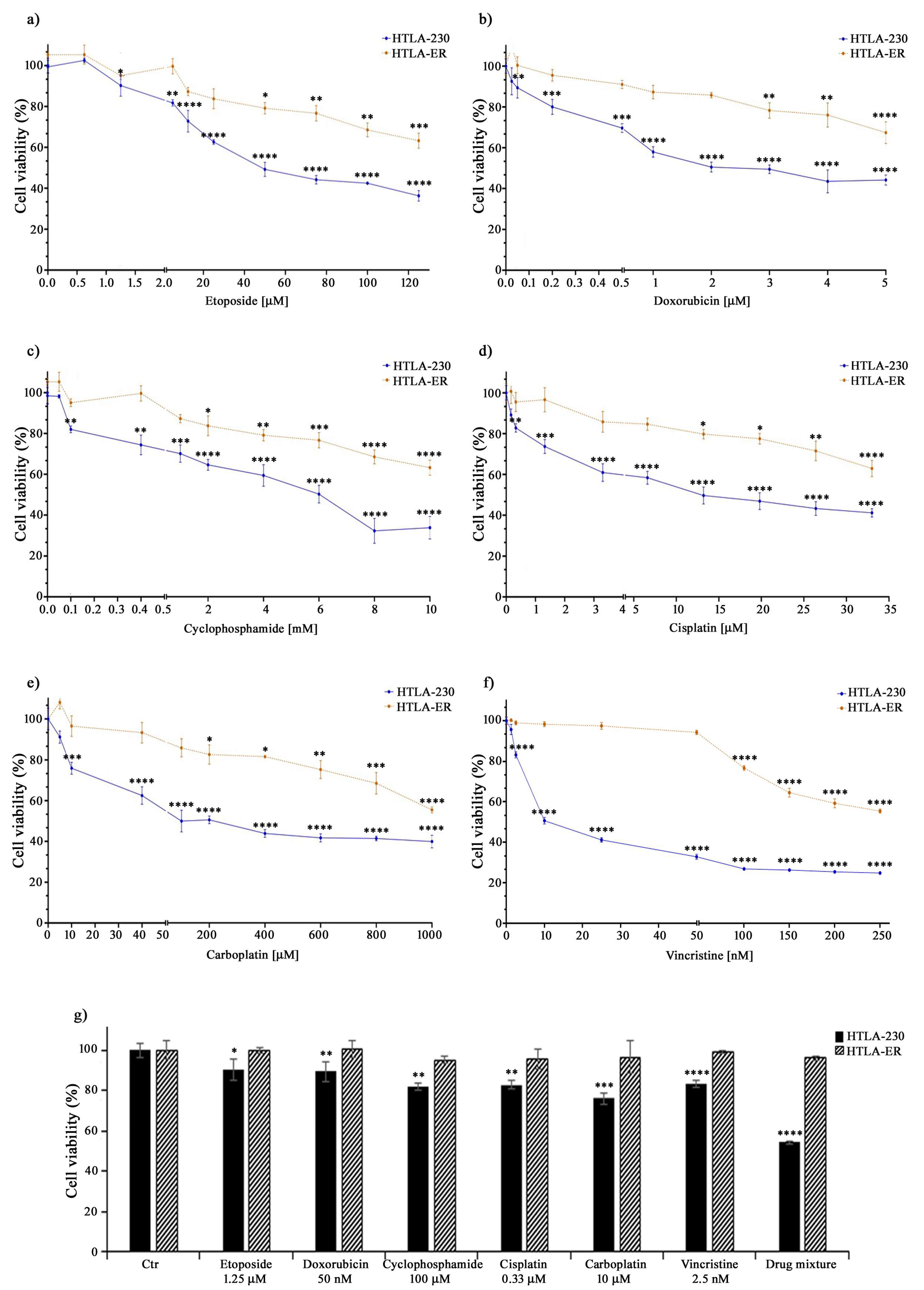
3.1. HTLA-ER Developed a Multidrug-Resistant Phenotype
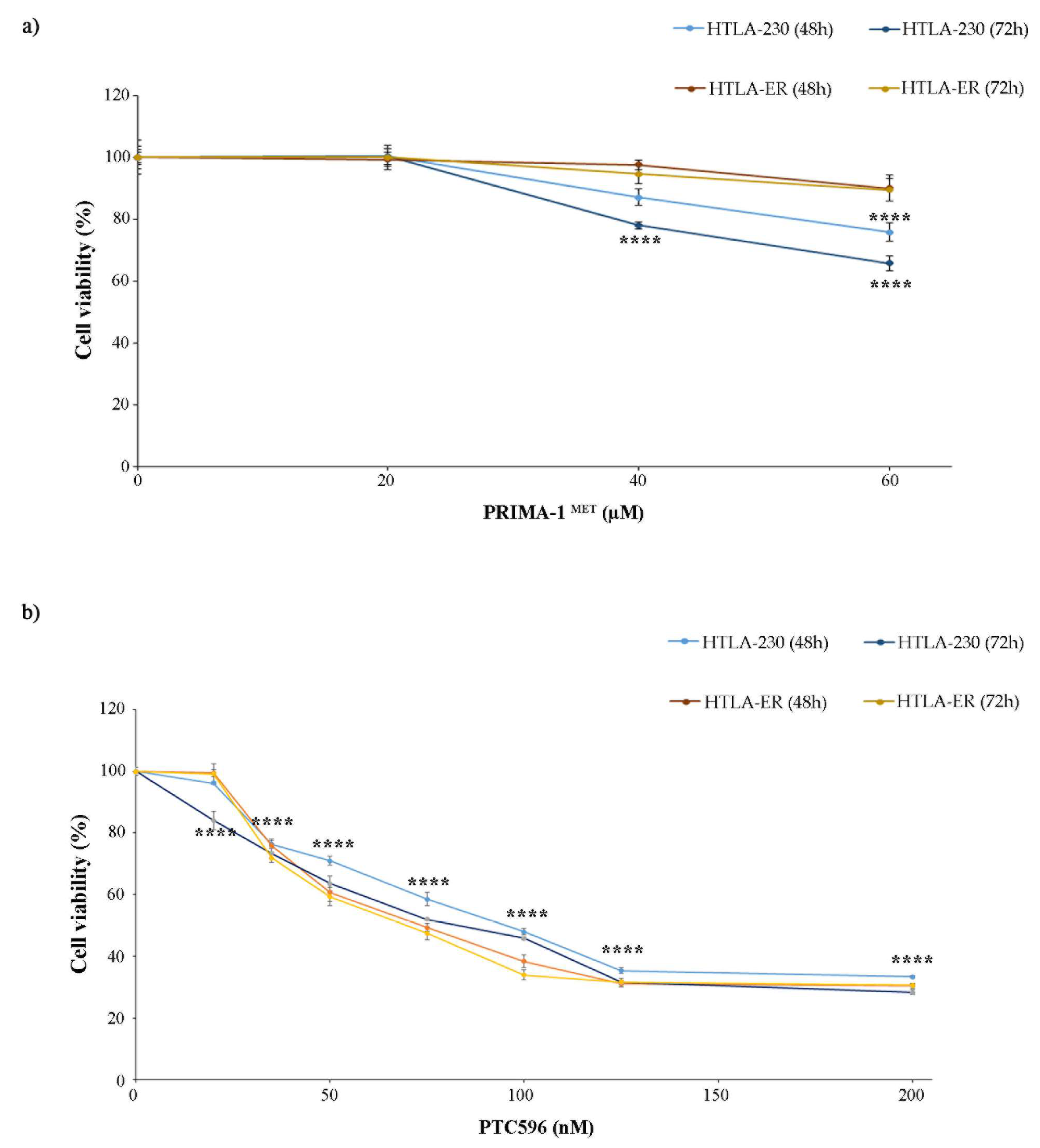
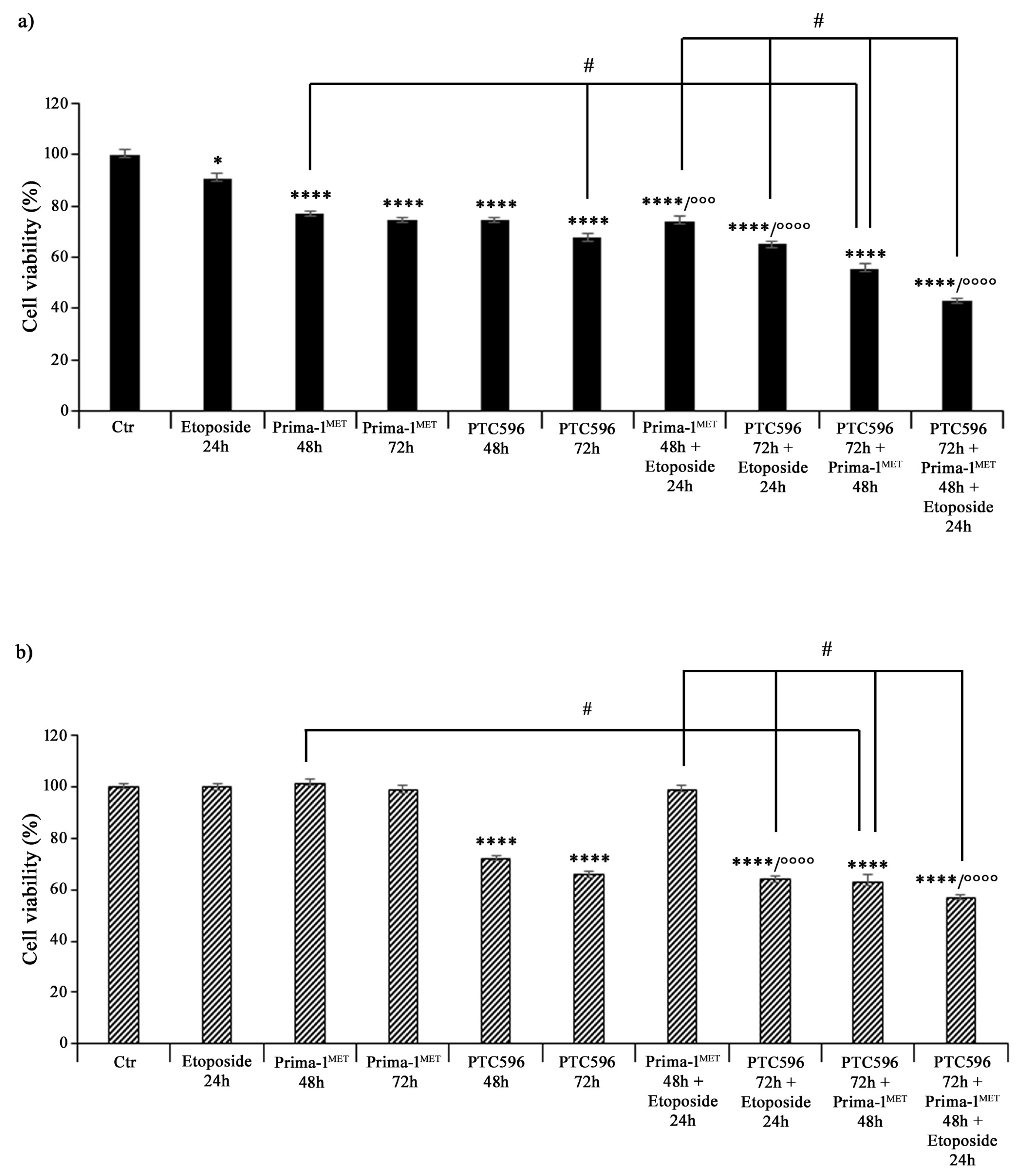
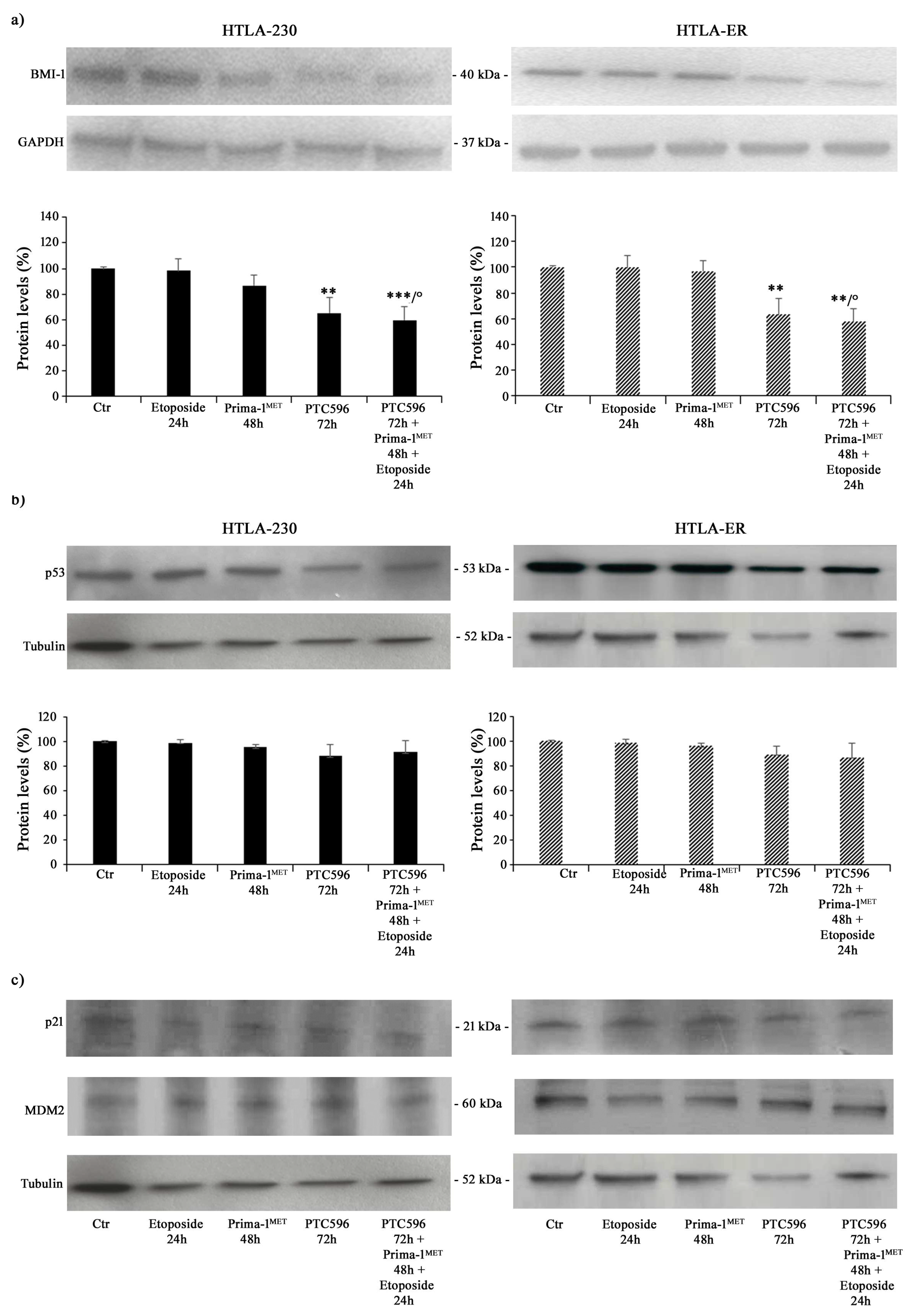
3.2. PTC596 Affects the Viability of Parental and HTLA-ER Cells and Reduces BMI-1 Expression Levels
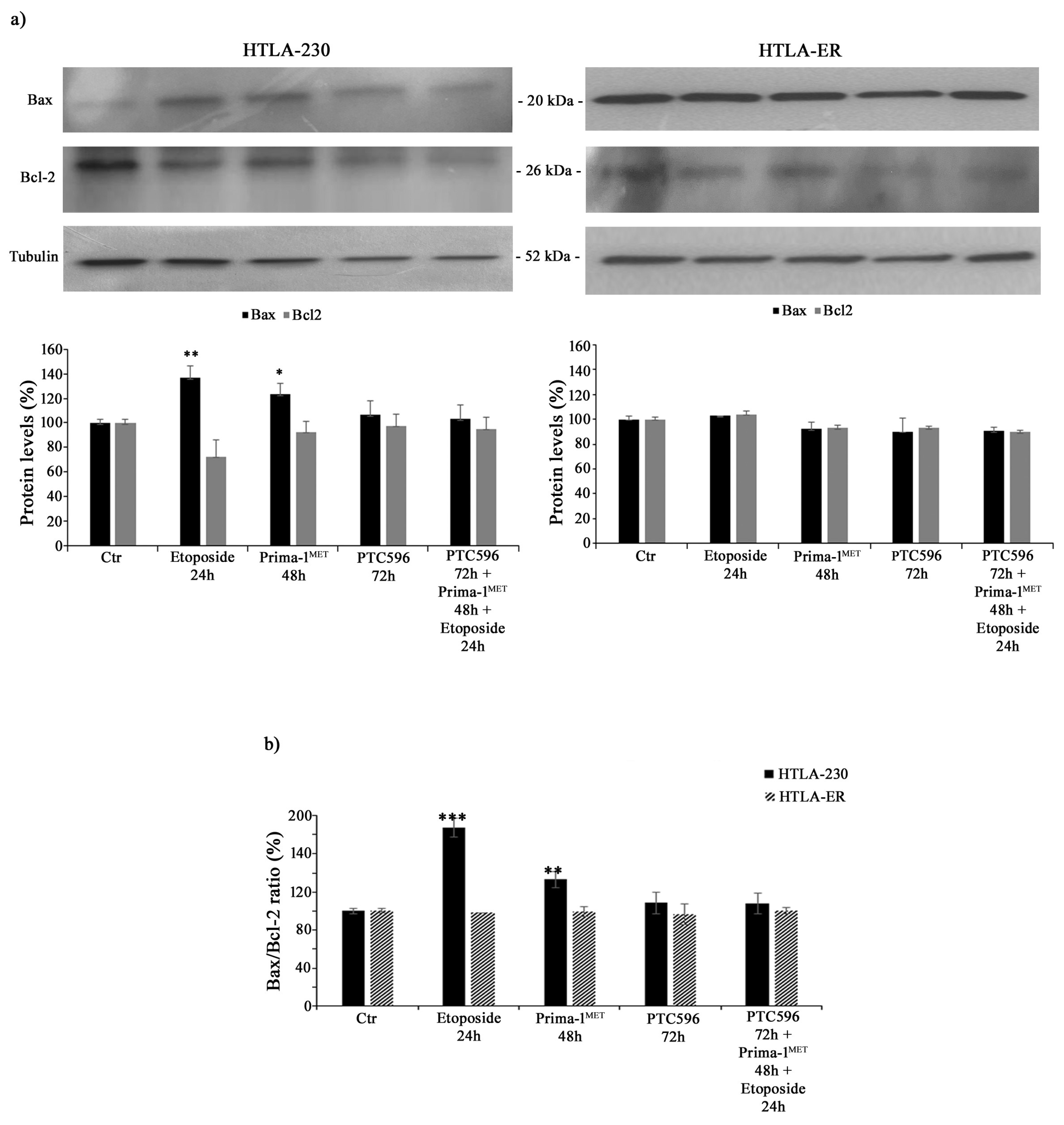
3.3. PTC596 Does Not Alter the Expression of Apoptotic Proteins in Parental and HTLA-ER Cells
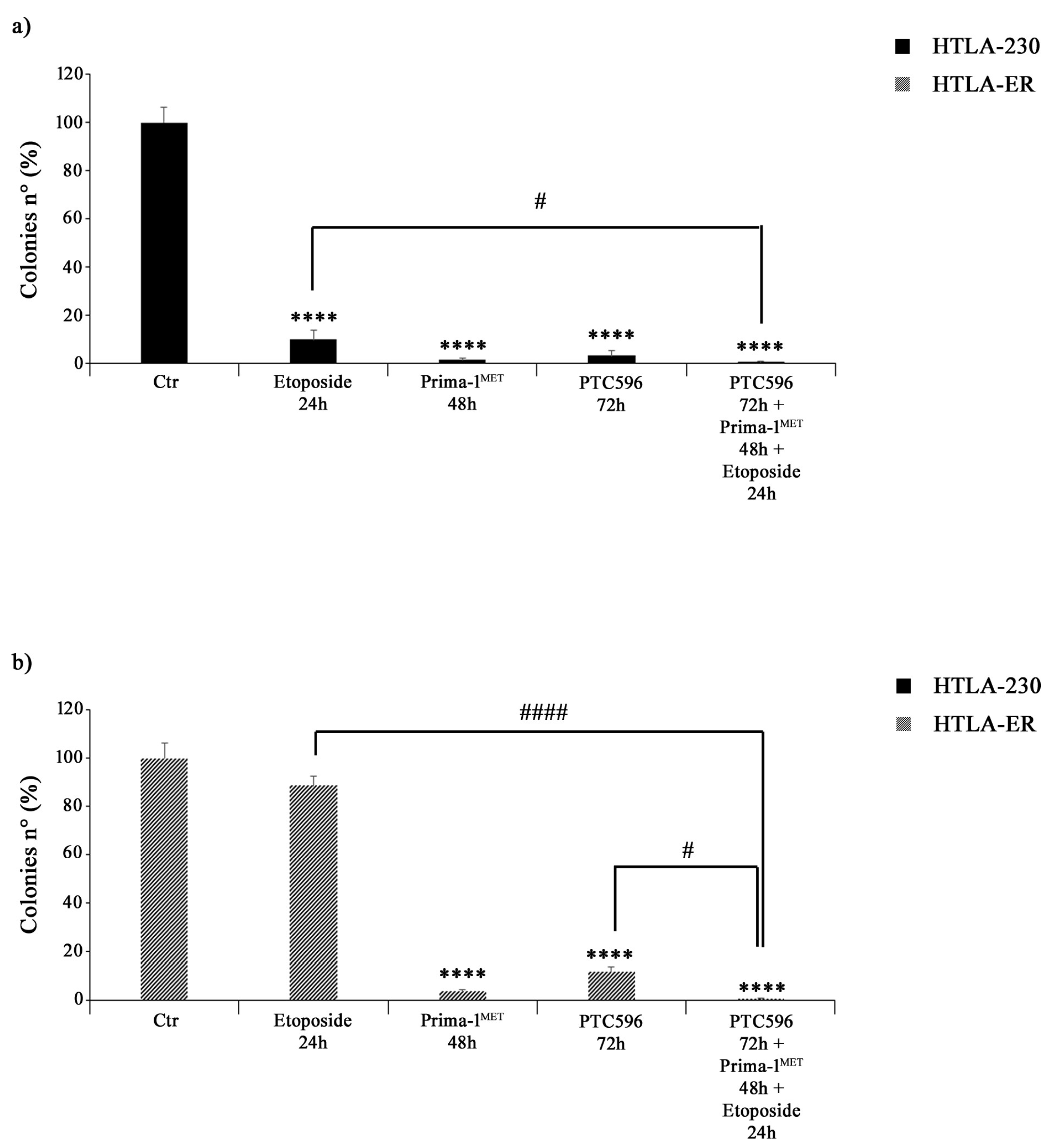
3.4. PRIMA-1MET and PTC596 Alone or in Combination Strongly Reduce the Clonogenic Potential of Parental and HTLA-ER Cells
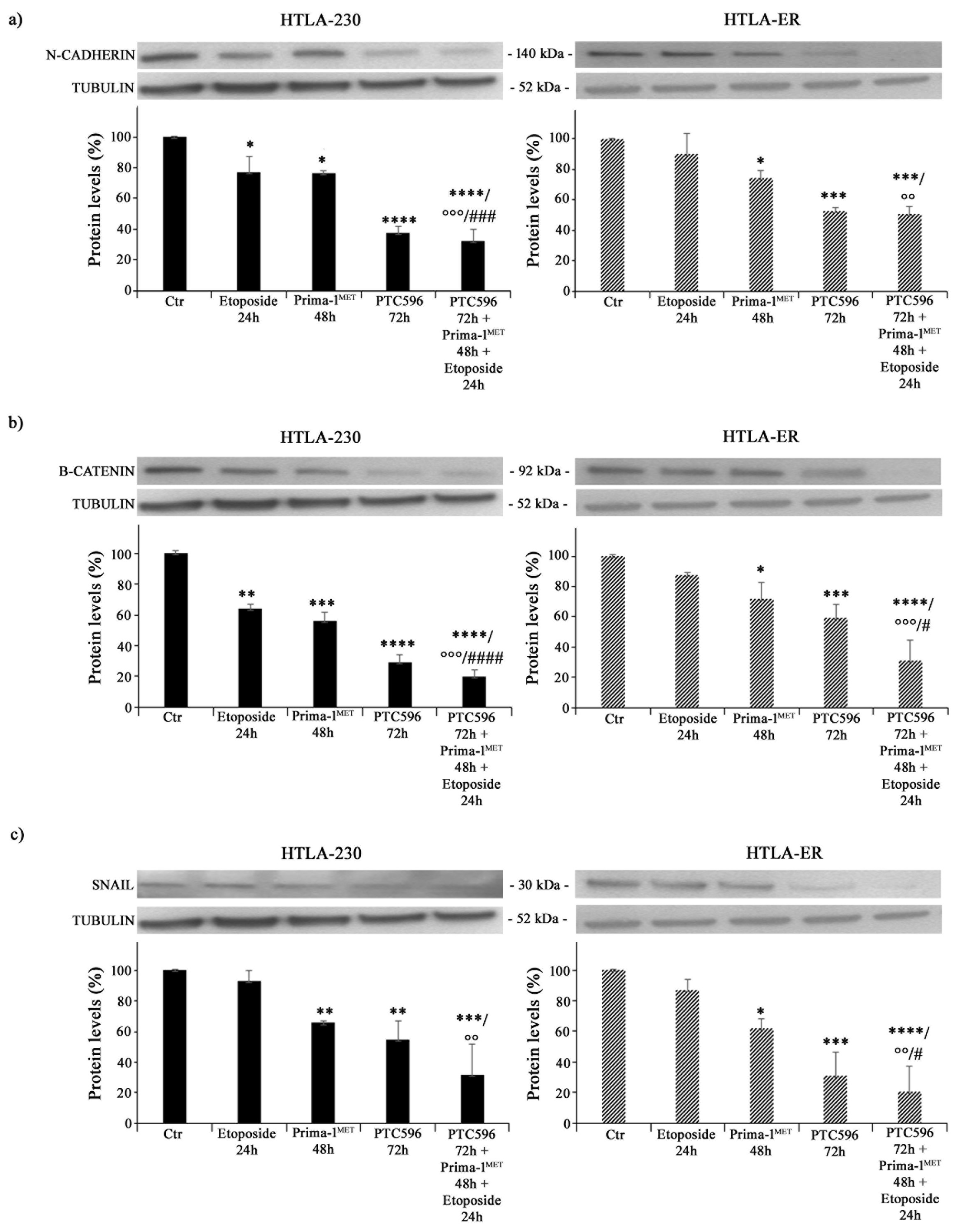
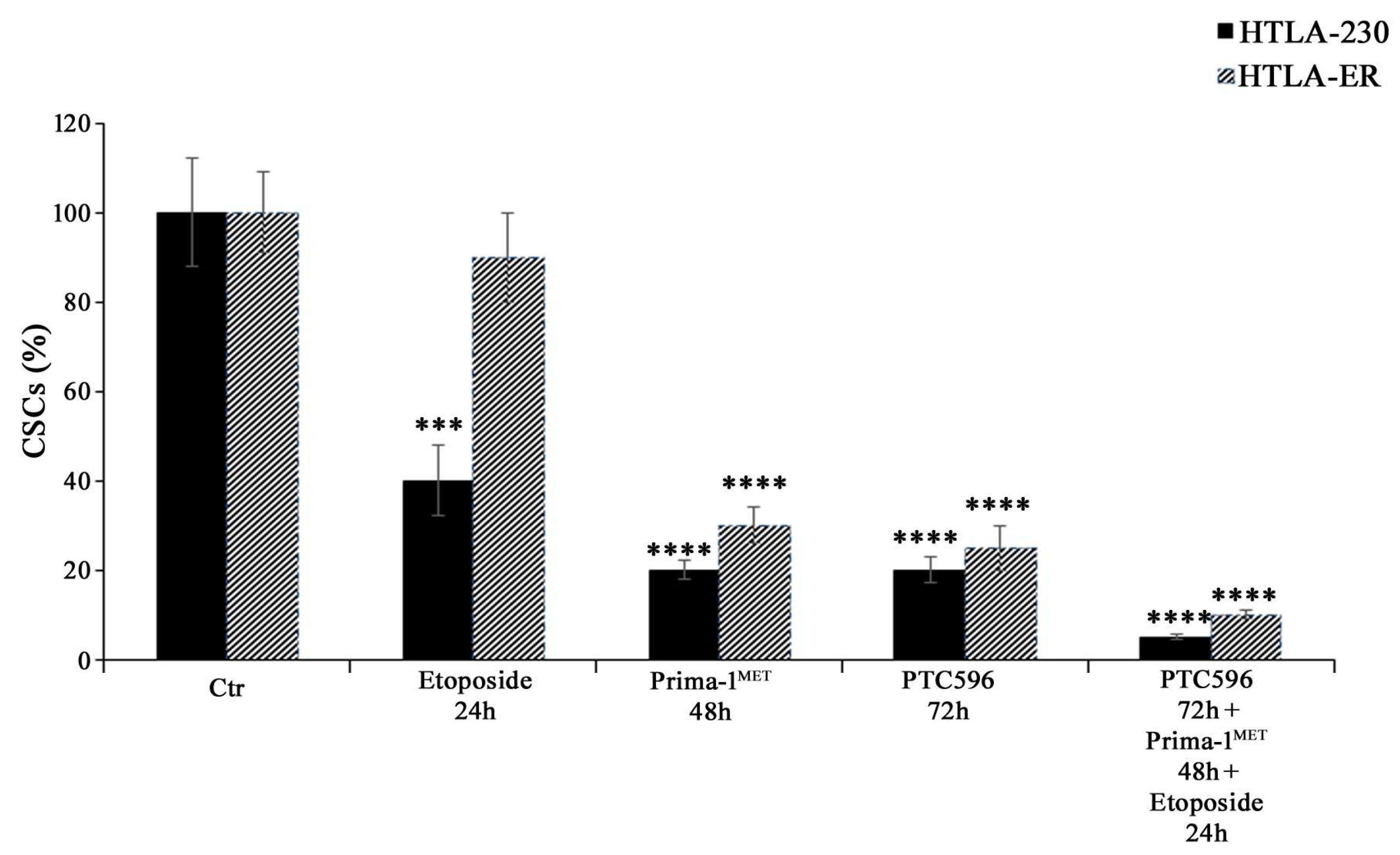
3.5. PRIMA-1MET and PTC596, Alone or in Combination, Reduce the Expression of Crucial EMT-Promoting Proteins and Inhibit CSC Formation
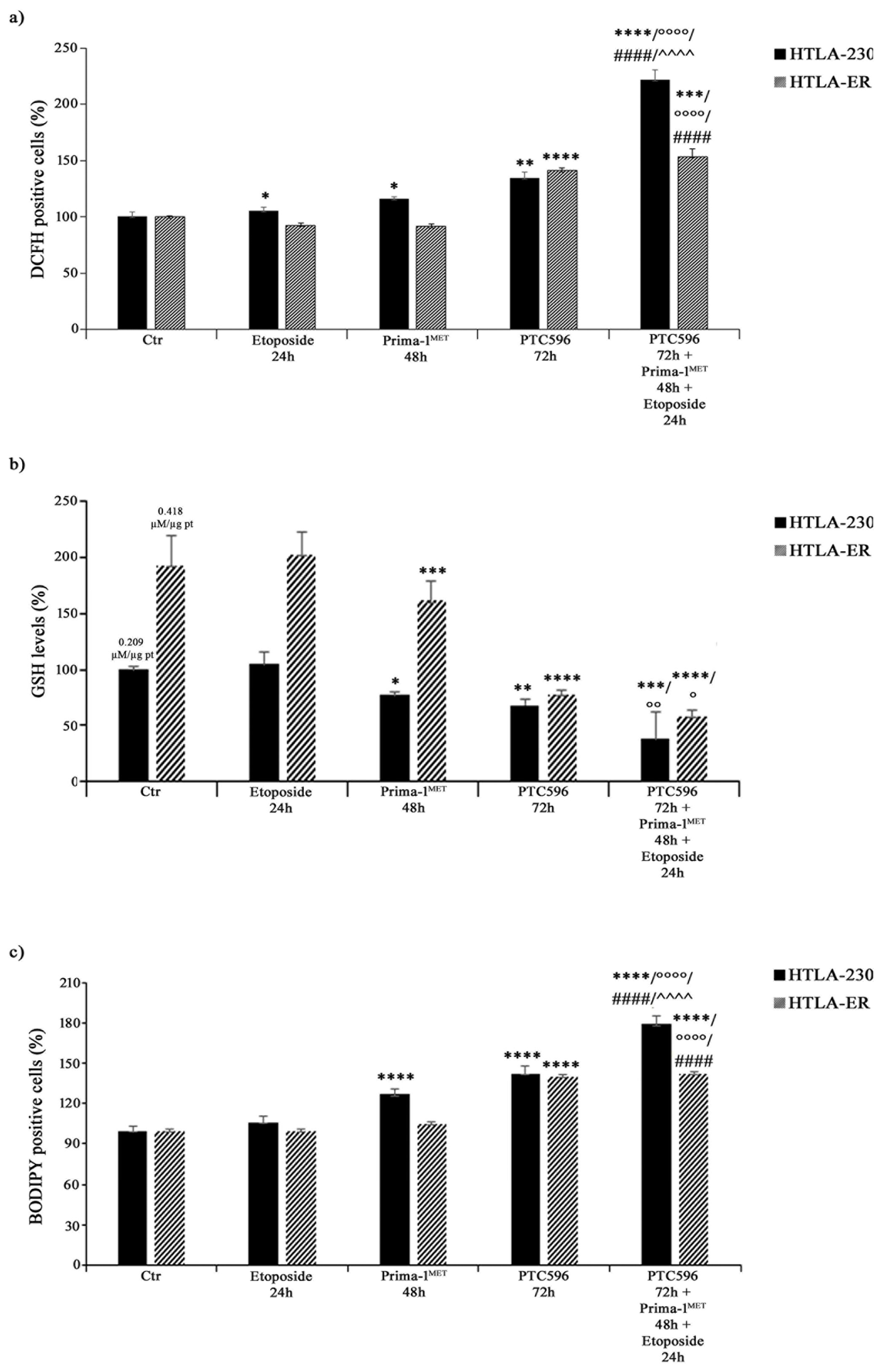
3.6. PRIMA-1MET and PTC596, Alone or in Combination, Enhance H2O2 Production, Deplete Intracellular GSH, and Induce Lipid Peroxidation
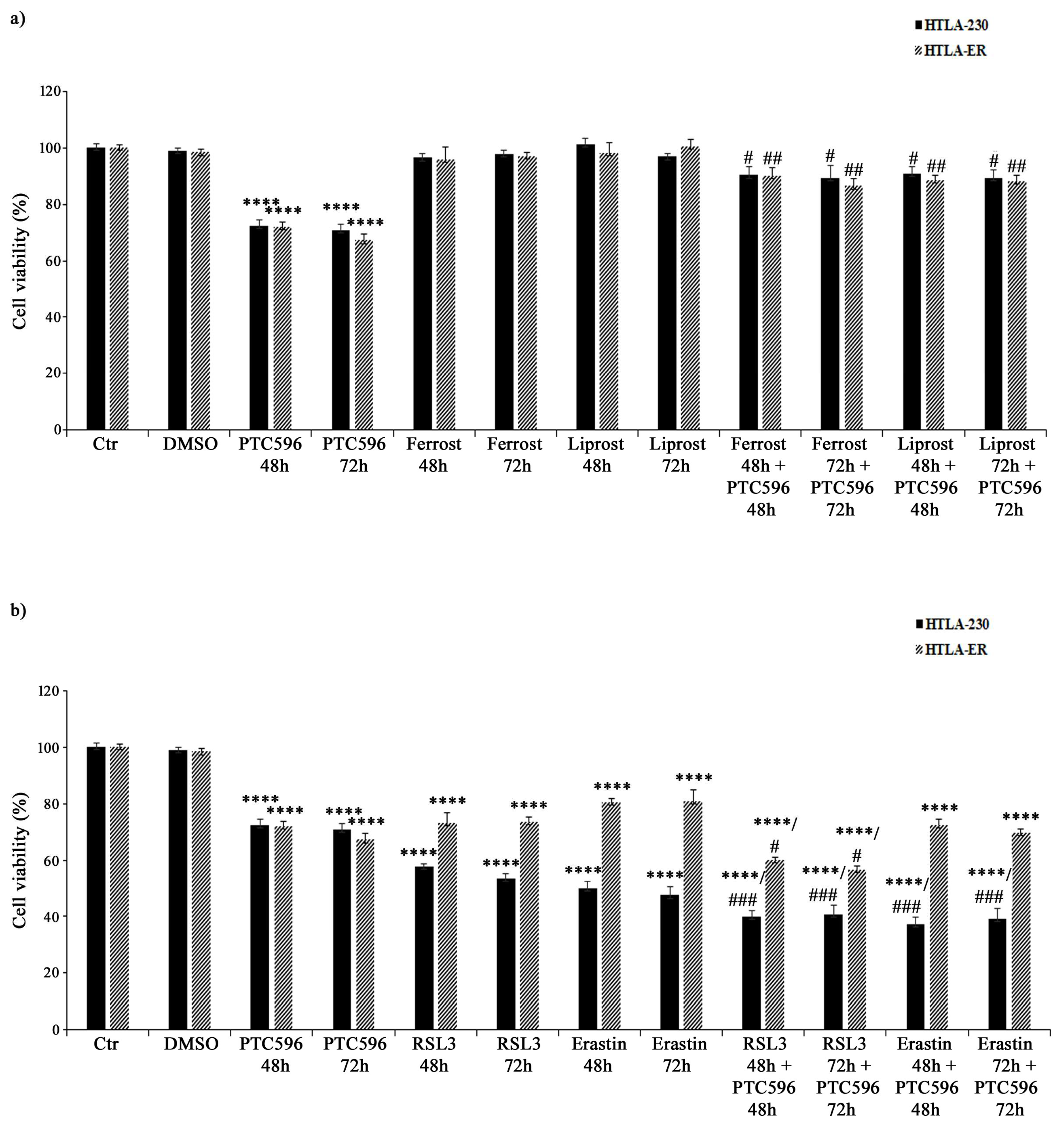
3.7. PTC596 Exerts Its Cytotoxic Effect by Inducing Ferroptosis
4. Discussion
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.M. Defining Why Cancer Develops in Children. N. Engl. J. Med. 2015, 373, 2373–2375. [Google Scholar] [CrossRef] [PubMed]
- Pinto, N.R.; Applebaum, M.A.; Volchenboum, S.L.; Matthay, K.K.; London, W.B.; Ambros, P.F.; Nakagawara, A.; Berthold, F.; Schleiermacher, G.; Park, J.R.; et al. Advances in Risk Classification and Treatment Strategies for Neuroblastoma. J. Clin. Oncol. 2015, 33, 3008–3017. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.; Sadeghi, S.; Tabatabaeian, H. Battling Chemoresistance in Cancer: Root Causes and Strategies to Uproot Them. Int. J. Mol. Sci. 2021, 22, 9451. [Google Scholar] [CrossRef]
- Marengo, B.; Nitti, M.; Furfaro, A.L.; Colla, R.; Ciucis, C.D.; Marinari, U.M.; Pronzato, M.A.; Traverso, N.; Domenicotti, C. Redox Homeostasis and Cellular Antioxidant Systems: Crucial Players in Cancer Growth and Therapy. Oxid. Med. Cell Longev. 2016, 2016, 6235641. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Duffy, M.J.; Synnott, N.C.; Crown, J. Mutant p53 as a target for cancer treatment. Eur. J. Cancer 2017, 83, 258–265. [Google Scholar] [CrossRef]
- Bykov, V.J.; Zache, N.; Stridh, H.; Westman, J.; Bergman, J.; Selivanova, G.; Wiman, K.G. PRIMA-1(MET) synergizes with Cisplatin to induce tumor cell apoptosis. Oncogene 2005, 24, 3484–3491. [Google Scholar] [CrossRef] [PubMed]
- Zandi, R.; Selivanova, G.; Christensen, C.L.; Gerds, T.A.; Willumsen, B.M.; Poulsen, H.S. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin. Cancer Res. 2011, 17, 2830–2841. [Google Scholar] [CrossRef] [PubMed]
- Demir, S.; Boldrin, E.; Sun, Q.; Hampp, S.; Tausch, E.; Eckert, C.; Ebinger, M.; Handgretinger, R.; Kronnie, G.T.; Wiesmüller, L.; et al. Therapeutic targeting of mutant p53 in pediatric acute lymphoblastic leukemia. Haematologica 2020, 105, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Elayapillai, S.; Ramraj, S.; Benbrook, D.M.; Bieniasz, M.; Wang, L.; Pathuri, G.; Isingizwe, Z.R.; Kennedy, A.L.; Zhao, Y.D.; Lightfoot, S.; et al. Potential and mechanism of mebendazole for treatment and maintenance of ovarian cancer. Gynecol. Oncol. 2021, 160, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Deneberg, S.; Cherif, H.; Lazarevic, V.; Andersson, P.O.; von Euler, M.; Juliusson, G.; Lehmann, S. An open-label phase I dose-finding study of APR-246 in hematological malignancies. Blood Cancer J. 2016, 6, e447. [Google Scholar] [CrossRef] [PubMed]
- Safety Study of APR-246 in Patients With Refractory Hematologic Cancer or Prostate Cancer, NCT00900614. 2010. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT00900614 (accessed on 4 June 2023).
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef]
- Tessoulin, B.; Descamps, G.; Moreau, P.; Maïga, S.; Lodé, L.; Godon, C.; Marionneau-Lambot, S.; Oullier, T.; Le Gouill, S.; Amiot, M.; et al. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood 2014, 124, 1626–1636. [Google Scholar] [CrossRef]
- Yoshikawa, N.; Kajiyama, H.; Nakamura, K.; Utsumi, F.; Niimi, K.; Mitsui, H.; Sekiya, R.; Suzuki, S.; Shibata, K.; Callen, D.; et al. PRIMA-1MET induces apoptosis through accumulation of intracellular reactive oxygen species irrespective of p53 status and chemo-sensitivity in epithelial ovarian cancer cells. Oncol. Rep. 2016, 35, 2543–2552. [Google Scholar] [CrossRef]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xC-/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef]
- Wang, E.; Bhattacharyya, S.; Szabolcs, A.; Rodriguez-Aguayo, C.; Jennings, N.B.; Lopez-Berestein, G.; Mukherjee, P.; Sood, A.K.; Bhattacharya, R. Enhancing chemotherapy response with Bmi-1 silencing in ovarian cancer. PLoS ONE 2011, 6, e17918. [Google Scholar] [CrossRef]
- Calao, M.; Sekyere, E.O.; Cui, H.J.; Cheung, B.B.; Thomas, W.D.; Keating, J.; Chen, J.B.; Raif, A.; Jankowski, K.; Davies, N.P.; et al. Direct effects of Bmi1 on p53 protein stability inactivates oncoprotein stress responses in embryonal cancer precursor cells at tumor initiation. Oncogene 2013, 32, 3616–3626. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Li, J.; Song, L. Bmi-1, stem cells and cancer. Acta Biochim. Biophys. Sin. 2009, 41, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Honig, A.; Weidler, C.; Häusler, S.; Krockenberger, M.; Buchholz, S.; Köster, F.; Segerer, S.E.; Dietl, J.; Engel, J.B. Overexpression of polycomb protein BMI-1 in human specimens of breast, ovarian, endometrial and cervical cancer. Anticancer Res. 2010, 30, 1559–1564. [Google Scholar] [PubMed]
- Zhang, X.; Wang, C.X.; Zhu, C.B.; Zhang, J.; Kan, S.F.; Du, L.T.; Li, W.; Wang, L.L.; Wang, S. Overexpression of Bmi-1 in uterine cervical cancer: Correlation with clinicopathology and prognosis. Int. J. Gynecol. Cancer 2010, 20, 1597–1603. [Google Scholar] [PubMed]
- Wang, X.; Wang, C.; Zhang, X.; Hua, R.; Gan, L.; Huang, M.; Zhao, L.; Ni, S.; Guo, W. Bmi-1 regulates stem cell-like properties of gastric cancer cells via modulating miRNAs. J. Hematol. Oncol. 2016, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, Q.; Bu, X.; Zhang, C.; Chen, H.; Sha, W.; Liu, W. Overexpression of Bmi 1 promotes epithelial mesenchymal transition in CD133+Hep G2 cells. Mol. Med. Rep. 2017, 16, 6156–6161. [Google Scholar] [CrossRef]
- Marengo, B.; Monti, P.; Miele, M.; Menichini, P.; Ottaggio, L.; Foggetti, G.; Pulliero, A.; Izzotti, A.; Speciale, A.; Garbarino, O.; et al. Etoposide-resistance in a neuroblastoma model cell line is associated with 13q14.3 mono-allelic deletion and miRNA-15a/16-1 down-regulation. Sci. Rep. 2018, 8, 13762. [Google Scholar] [CrossRef]
- Li, Y.; Tian, Z.; Tan, Y.; Lian, G.; Chen, S.; Chen, S.; Li, J.; Li, X.; Huang, K.; Chen, Y. Bmi-1-induced miR-27a and miR-155 promote tumor metastasis and chemoresistance by targeting RKIP in gastric cancer. Mol. Cancer 2020, 19, 109. [Google Scholar] [CrossRef]
- Yu, J.; Chen, L.; Bao, Z.; Liu, Y.; Liu, G.; Li, F.; Li, L. BMI 1 promotes invasion and metastasis in endometrial adenocarcinoma and is a poor prognostic factor. Oncol. Rep. 2020, 43, 1630–1640. [Google Scholar] [CrossRef]
- Yin, T.; Wei, H.; Leng, Z.; Yang, Z.; Gou, S.; Wu, H.; Zhao, G.; Hu, X.; Wang, C. Bmi-1 promotes the chemoresistance, invasion and tumorigenesis of pancreatic cancer cells. Chemotherapy 2011, 57, 488–496. [Google Scholar] [CrossRef]
- Chen, Y.; Lian, G.; Ou, G.; Yang, K.; Chen, J.; Li, H.; Chen, S.; Li, J.; Zeng, L.; Huang, K. Inverse association between Bmi-1 and RKIP affecting clinical outcome of gastric cancer and revealing the potential molecular mechanisms underlying tumor metastasis and chemotherapy resistance. Gastric Cancer 2016, 19, 392–402. [Google Scholar] [CrossRef]
- Wang, M.C.; Li, C.L.; Cui, J.; Jiao, M.; Wu, T.; Jing, L.I.; Nan, K.J. BMI-1, a promising therapeutic target for human cancer. Oncol. Lett. 2015, 10, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C.; Wagner, A.; Loeffelberger, M.; Bruckner, D.; Jakab, M.; Berr, F.; Di Fazio, P.; Ocker, M.; Neureiter, D.; Pichler, M.; et al. The BMI1 inhibitor PTC-209 is a potential compound to halt cellular growth in biliary tract cancer cells. Oncotarget 2016, 7, 745–758. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Liu, L.; Sharma, S.; Liu, H.; Yang, W.; Sun, X.; Dong, Q. Bmi-1 confers adaptive radioresistance to KYSE-150R esophageal carcinoma cells. Biochem. Biophys. Res. Commun. 2012, 425, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Zhang, W.; Wang, C.Y. BMI1 Inhibition Eliminates Residual Cancer Stem Cells after PD1 Blockade and Activates Antitumor Immunity to Prevent Metastasis and Relapse. Cell Stem Cell 2020, 27, 238–253.e6. [Google Scholar] [CrossRef]
- Herzog, A.E.; Warner, K.A.; Zhang, Z.; Bellile, E.; Bhagat, M.A.; Castilho, R.M.; Wolf, G.T.; Polverini, P.J.; Pearson, A.T.; Nör, J.E. The IL-6R and Bmi-1 axis controls self-renewal and chemoresistance of head and neck cancer stem cells. Cell Death Dis. 2021, 12, 988. [Google Scholar] [CrossRef]
- Nishida, Y.; Maeda, A.; Kim, M.J.; Cao, L.; Kubota, Y.; Ishizawa, J.; AlRawi, A.; Kato, Y.; Iwama, A.; Fujisawa, M.; et al. The novel BMI-1 inhibitor PTC596 downregulates MCL-1 and induces p53-independent mitochondrial apoptosis in acute myeloid leukemia progenitor cells. Blood Cancer J. 2017, 7, e527. [Google Scholar] [CrossRef]
- Maeda, A.; Nishida, Y.; Weetall, M.; Cao, L.; Branstrom, A.; Ishizawa, J.; Nii, T.; Schober, W.D.; Abe, Y.; Matsue, K.; et al. Targeting of BMI-1 expression by the novel small molecule PTC596 in mantle cell lymphoma. Oncotarget 2018, 9, 28547–28560. [Google Scholar] [CrossRef]
- Bolomsky, A.; Muller, J.; Stangelberger, K.; Lejeune, M.; Duray, E.; Breid, H.; Vrancken, L.; Pfeiffer, C.; Hübl, W.; Willheim, M.; et al. The anti-mitotic agents PTC-028 and PTC596 display potent activity in pre-clinical models of multiple myeloma but challenge the role of BMI-1 as an essential tumour gene. Br. J. Haematol. 2020, 190, 877–890. [Google Scholar] [CrossRef]
- Wu, J.H.; Batist, G. Glutathione and glutathione analogues; therapeutic potentials. Biochim. Biophys. Acta 2013, 1830, 3350–3353. [Google Scholar] [CrossRef]
- Lewis-Wambi, J.S.; Swaby, R.; Kim, H.; Jordan, V.C. Potential of l-buthionine sulfoximine to enhance the apoptotic action of estradiol to reverse acquired antihormonal resistance in metastatic breast cancer. J. Steroid Biochem. Mol. Biol. 2009, 114, 33–39. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Colla, R.; Izzotti, A.; De Ciucis, C.; Fenoglio, D.; Ravera, S.; Speciale, A.; Ricciarelli, R.; Furfaro, A.L.; Pulliero, A.; Passalacqua, M.; et al. Glutathione-mediated antioxidant response and aerobic metabolism: Two crucial factors involved in determining the multi-drug resistance of high-risk neuroblastoma. Oncotarget 2016, 7, 70715–70737. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, J.; Ora, I.; Porn-Ares, I.; Pahlman, S. Arsenic trioxide-induced death of neuroblastoma cells involves activation of Bax and does not require p53. Clin. Cancer Res. 2004, 10, 3179–3188. [Google Scholar] [CrossRef] [PubMed]
- Hadjidaniel, M.D.; Reynolds, C.P. Antagonism of cytotoxic chemotherapy in neuroblastoma cell lines by 13-cis-retinoic acid is mediated by the antiapoptotic Bcl-2 family proteins. Mol. Cancer Ther. 2010, 9, 3164–3174. [Google Scholar] [CrossRef] [PubMed]
- Estlin, E.J.; Veal, G.J. Clinical and cellular pharmacology in relation to solid tumours of childhood. Cancer Treat. Rev. 2003, 29, 253–273. [Google Scholar] [CrossRef]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, L.; Speciale, A.; Valenti, G.E.; Traverso, N.; Ravera, S.; Garbarino, O.; Leardi, R.; Farinini, E.; Roveri, A.; Ursini, F.; et al. PKCα Inhibition as a Strategy to Sensitize Neuroblastoma Stem Cells to Etoposide by Stimulating Ferroptosis. Antioxidants 2021, 10, 691. [Google Scholar] [CrossRef]
- Alfei, S.; Marengo, B.; Valenti, G.E.; Domenicotti, C. Synthesis of Polystyrene-Based Cationic Nanomaterials with Pro-Oxidant Cytotoxic Activity on Etoposide-Resistant Neuroblastoma Cells. Nanomaterials 2021, 11, 977. [Google Scholar] [CrossRef]
- Fariss, M.W.; Reed, D.J. High-performance liquid chromatography of thiols and disulfides: Dinitrophenol derivatives. Methods Enzymol. 1987, 143, 101–109. [Google Scholar] [CrossRef]
- Asensi, M.; Sastre, J.; Pallardo, F.V.; Lloret, A.; Lehner, M.; Garcia-de-la Asuncion, J.; Viña, J. Ratio of reduced to oxidized glutathione as indicator of oxidative stress status and DNA damage. Methods Enzymol. 1999, 299, 267–276. [Google Scholar] [CrossRef]
- Balbis, E.; Patriarca, S.; Furfaro, A.L.; Millanta, S.; Sukkar, S.G.; Marinari, U.M.; Pronzato, M.A.; Cottalasso, D.; Traverso, N. Whey proteins influence hepatic glutathione after CCl4 intoxication. Toxicol. Ind. Health 2009, 25, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Roveri, A.; Maiorino, M.; Ursini, F. Enzymatic and immunological measurements of soluble and membrane-bound phospholipidhydroperoxide glutathione peroxidase. Methods Enzymol. 1994, 233, 202–212. [Google Scholar] [PubMed]
- Zahreddine, H.; Borden, K.L. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Qiu, B.; Matthay, K.K. Advancing therapy for neuroblastoma. Nat. Rev. Clin. Oncol. 2022, 19, 515–533. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Xu, X.; Tang, Y.; Zhang, C.; Li, J.; Ouyang, Y.; Ju, J.; Bie, P.; Wang, H. miR-15a inhibits cell proliferation and epithelial to mesenchymal transition in pancreatic ductal adenocarcinoma by down-regulating Bmi-1 expression. Cancer Lett. 2014, 344, 40–46. [Google Scholar] [CrossRef]
- Menichini, P.; Monti, P.; Speciale, A.; Cutrona, G.; Matis, S.; Fais, F.; Taiana, E.; Neri, A.; Bomben, R.; Gentile, M.; et al. Antitumor Effects of PRIMA-1 and PRIMA-1Met(APR246) in Hematological Malignancies: Still a Mutant P53-Dependent Affair? Cells 2021, 10, 98. [Google Scholar] [CrossRef]
- Shapiro, G.I.; O’Mara, E.; Laskin, O.L.; Gao, L.; Baird, J.D.; Spiegel, R.J.; Kaushik, D.; Weetall, M.; Colacino, J.; O’Keefe, K.; et al. Pharmacokinetics and Safety of PTC596, a Novel Tubulin-Binding Agent, in Subjects With Advanced Solid Tumors. Clin. Pharmacol. Drug Dev. 2021, 10, 940–949. [Google Scholar] [CrossRef]
- Marine, J.C. p53 stabilization: The importance of nuclear import. Cell Death Differ. 2010, 17, 191–192. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef]
- Gupta, P.B.; Fillmore, C.M.; Jiang, G.; Shapira, S.D.; Tao, K.; Kuperwasser, C.; Lander, E.S. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 2011, 146, 633–644. [Google Scholar] [CrossRef]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–73. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Morel, A.P.; Lièvre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Haim, N.; Roman, J.; Nemec, J.; Sinha, B.K. Peroxidative free radical formation and O-demethylation of Etoposide(VP-16) and teniposide(VM-26). Biochem. Biophys. Res. Commun. 1986, 135, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Smolyarova, D.D.; Podgorny, O.V.; Bilan, D.S.; Belousov, V.V. A guide to genetically encoded tools for the study of H2O2. FEBS J. 2022, 289, 5382–5395. [Google Scholar] [CrossRef]
- Gamcsik, M.P.; Kasibhatla, M.S.; Teeter, S.D.; Colvin, O.M. Glutathione levels in human tumors. Biomarkers 2012, 17, 671–691. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893.e13. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.A.; Gay, C.M.; Xi, Y.; Sivajothi, S.; Sivakamasundari, V.; Fujimoto, J.; Bolisetty, M.; Hartsfield, P.M.; Balasubramaniyan, V.; Chalishazar, M.D.; et al. Single-cell analyses reveal increased intratumoral heterogeneity after the onset of therapy resistance in small-cell lung cancer. Nat. Cancer 2020, 1, 423–436. [Google Scholar] [CrossRef] [PubMed]
- El-Sayes, N.; Vito, A.; Mossman, K. Tumor Heterogeneity: A Great Barrier in the Age of Cancer Immunotherapy. Cancers 2021, 13, 806. [Google Scholar] [CrossRef] [PubMed]
- Garbarino, O.; Valenti, G.E.; Monteleone, L.; Pietra, G.; Mingari, M.C.; Benzi, A.; Bruzzone, S.; Ravera, S.; Leardi, R.; Farinini, E.; et al. PLAX4032 resistance of patient-derived melanoma cells: Crucial role of oxidative metabolism. Front. Oncol. 2023, 13, 1210130. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Ahluwalia, P.; Wang, L.; Howard, J.; Kolhe, R.; Rojiani, A.M.; Rojiani, M.V. TIMP-1 Dependent Modulation of Metabolic Profiles Impacts Chemoresistance in NSCLC. Cells 2022, 11, 3036. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhou, Y.; Duan, W.; Song, J.; Wei, S.; Xia, S.; Wang, Y.; Du, X.; Li, E.; Ren, C.; et al. Glutathione peroxidase 4-dependent glutathione high-consumption drives acquired platinum chemoresistance in lung cancer-derived brain metastasis. Clin. Transl. Med. 2021, 11, e517. [Google Scholar] [CrossRef] [PubMed]
- Jagust, P.; Alcalá, S.; Sainz, B., Jr.; Heeschen, C.; Sancho, P. Glutathione metabolism is essential for self-renewal and chemoresistance of pancreatic cancer stem cells. World J. Stem Cells 2020, 12, 1410–1428. [Google Scholar] [CrossRef]
- Forte, D.; García-Fernández, M.; Sánchez-Aguilera, A.; Stavropoulou, V.; Fielding, C.; Martín-Pérez, D.; López, J.A.; Costa, A.S.H.; Tronci, L.; Nikitopoulou, E.; et al. Bone Marrow Mesenchymal Stem Cells Support Acute Myeloid Leukemia Bioenergetics and Enhance Antioxidant Defense and Escape from Chemotherapy. Cell Metab. 2020, 32, 829–843.e9. [Google Scholar] [CrossRef]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef]
- Huber, G.F.; Albinger-Hegyi, A.; Soltermann, A.; Roessle, M.; Graf, N.; Haerle, S.K.; Holzmann, D.; Moch, H.; Hegyi, I. Expression patterns of Bmi-1 and p16 significantly correlate with overall, disease-specific, and recurrence-free survival in oropharyngeal squamous cell carcinoma. Cancer 2011, 117, 4659–4670. [Google Scholar] [CrossRef]
- Yu, D.; Liu, Y.; Yang, J.; Jin, C.; Zhao, X.; Cheng, J.; Liu, X.; Qi, X. Clinical implications of BMI-1 in cancer stem cells of laryngeal carcinoma. Cell Biochem. Biophys. 2015, 71, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; He, G.; Li, J.; Li, J.; Guo, X.; Gao, J. Hypoxic exposure activates the B cell-specific Moloney murine leukaemia virus integration site 1/PI3K/Akt axis and promotes EMT in leukaemia stem cells. Oncol. Lett. 2021, 21, 98. [Google Scholar] [CrossRef] [PubMed]
- Patyka, M.; Sharifi, Z.; Petrecca, K.; Mansure, J.; Jean-Claude, B.; Sabri, S. Sensitivity to PRIMA-1MET is associated with decreased MGMT in human glioblastoma cells and glioblastoma stem cells irrespective of p53 status. Oncotarget 2016, 7, 60245–60269. [Google Scholar] [CrossRef] [PubMed]
- Jagani, Z.; Wiederschain, D.; Loo, A.; He, D.; Mosher, R.; Fordjour, P.; Monahan, J.; Morrissey, M.; Yao, Y.M.; Lengauer, C.; et al. The Polycomb group protein Bmi-1 is essential for the growth of multiple myeloma cells. Cancer Res. 2010, 70, 5528–5538. [Google Scholar] [CrossRef] [PubMed]
- Semenov, O.; Daks, A.; Fedorova, O.; Shuvalov, O.; Barlev, N.A. Opposing Roles of Wild-type and Mutant p53 in the Process of Epithelial to Mesenchymal Transition. Front. Mol. Biosci. 2022, 9, 928399. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Woo, S.M.; Seo, S.U.; Kwon, T.K. Inhibition of BMI-1 Induces Apoptosis through Downregulation of DUB3-Mediated Mcl-1 Stabilization. Int. J. Mol. Sci. 2021, 22, 10107. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Sengupta, S.; Zhu, X.; Mishra, D.K.; Phoenix, T.; Dyer, L.; Fuller, C.; Stevenson, C.B.; DeWire, M.; Fouladi, M.; et al. Diffuse Intrinsic Pontine Glioma Cells Are Vulnerable to Mitotic Abnormalities Associated with BMI-1 Modulation. Mol. Cancer Res. 2020, 18, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Zhang, M.Q.; Conserva, F.; Hosny, G.; Selivanova, G.; Bykov, V.J.; Arnér, E.S.; Wiman, K.G. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2013, 4, e881. [Google Scholar] [CrossRef]
- Mohell, N.; Alfredsson, J.; Fransson, Å.; Uustalu, M.; Byström, S.; Gullbo, J.; Hallberg, A.; Bykov, V.J.; Björklund, U.; Wiman, K.G. APR-246 overcomes resistance to Cisplatin and Doxorubicin in ovarian cancer cells. Cell Death Dis. 2015, 6, e1794. [Google Scholar] [CrossRef]
- Haffo, L.; Lu, J.; Bykov, V.J.N.; Martin, S.S.; Ren, X.; Coppo, L.; Wiman, K.G.; Holmgren, A. Inhibition of the glutaredoxin and thioredoxin systems and ribonucleotide reductase by mutant p53-targeting compound APR-246. Sci. Rep. 2018, 8, 12671. [Google Scholar] [CrossRef]
- Hang, W.; Yin, Z.X.; Liu, G.; Zeng, Q.; Shen, X.F.; Sun, Q.H.; Li, D.D.; Jian, Y.P.; Zhang, Y.H.; Wang, Y.S.; et al. Piperlongumine and p53-reactivator APR-246 selectively induce cell death in HNSCC by targeting GSTP1. Oncogene 2018, 37, 3384–3398. [Google Scholar] [CrossRef] [PubMed]
- Mlakar, V.; Jurkovic Mlakar, S.; Lesne, L.; Marino, D.; Rathi, K.S.; Maris, J.M.; Ansari, M.; Gumy-Pause, F. PRIMA-1MET-induced neuroblastoma cell death is modulated by p53 and mycn through glutathione level. J. Exp. Clin. Cancer Res. 2019, 38, 69. [Google Scholar] [CrossRef] [PubMed]
- Ceder, S.; Eriksson, S.E.; Cheteh, E.H.; Dawar, S.; Corrales Benitez, M.; Bykov, V.J.N.; Fujihara, K.M.; Grandin, M.; Li, X.; Ramm, S.; et al. A thiol-bound drug reservoir enhances APR-246-induced mutant p53 tumor cell death. EMBO Mol. Med. 2021, 13, e10852. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, R.L. Iron accumulation, glutathione depletion, and lipid peroxidation must occur simultaneously during ferroptosis and are mutually amplifying events. Med. Hypotheses 2017, 101, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Floros, K.V.; Cai, J.; Jacob, S.; Kurupi, R.; Fairchild, C.K.; Shende, M.; Coon, C.M.; Powell, K.M.; Belvin, B.R.; Hu, B.; et al. MYCN-Amplified Neuroblastoma Is Addicted to Iron and Vulnerable to Inhibition of the System Xc-/Glutathione Axis. Cancer Res. 2021, 81, 1896–1908. [Google Scholar] [CrossRef]
- Alborzinia, H.; Flórez, A.F.; Kreth, S.; Brückner, L.M.; Yildiz, U.; Gartlgruber, M.; Odoni, D.I.; Poschet, G.; Garbowicz, K.; Shao, C.; et al. MYCN mediates cysteine addiction and sensitizes neuroblastoma to ferroptosis. Nat. Cancer 2022, 3, 471–485. [Google Scholar] [CrossRef]
- Lu, Y.; Yang, Q.; Su, Y.; Ji, Y.; Li, G.; Yang, X.; Xu, L.; Lu, Z.; Dong, J.; Wu, Y.; et al. MYCN mediates TFRC-dependent ferroptosis and reveals vulnerabilities in neuroblastoma. Cell Death Dis. 2021, 12, 511. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Dose Compared with That Clinically Used | HTLA-230 Cells’ IC50 | HTLA-ER Cells’ IC50 |
|---|---|---|---|
| Etoposide (μM) | 1.25 | 69.79 | 221.4 |
| Doxorubicin (μM) | 0.05 | 3.21 | 8.88 |
| Cyclophosphamide (mM) | 0.1 | 5.30 | 14.86 |
| Cisplatin (μM) | 0.33 | 19.78 | 54.58 |
| Carboplatin (μM) | 10 | 509.3 | 1345.02 |
| Vincristine (nM) | 2.5 | 51.6 | 234.66 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valenti, G.E.; Roveri, A.; Venerando, R.; Menichini, P.; Monti, P.; Tasso, B.; Traverso, N.; Domenicotti, C.; Marengo, B. PTC596-Induced BMI-1 Inhibition Fights Neuroblastoma Multidrug Resistance by Inducing Ferroptosis. Antioxidants 2024, 13, 3. https://doi.org/10.3390/antiox13010003
Valenti GE, Roveri A, Venerando R, Menichini P, Monti P, Tasso B, Traverso N, Domenicotti C, Marengo B. PTC596-Induced BMI-1 Inhibition Fights Neuroblastoma Multidrug Resistance by Inducing Ferroptosis. Antioxidants. 2024; 13(1):3. https://doi.org/10.3390/antiox13010003
Chicago/Turabian StyleValenti, Giulia Elda, Antonella Roveri, Rina Venerando, Paola Menichini, Paola Monti, Bruno Tasso, Nicola Traverso, Cinzia Domenicotti, and Barbara Marengo. 2024. "PTC596-Induced BMI-1 Inhibition Fights Neuroblastoma Multidrug Resistance by Inducing Ferroptosis" Antioxidants 13, no. 1: 3. https://doi.org/10.3390/antiox13010003
APA StyleValenti, G. E., Roveri, A., Venerando, R., Menichini, P., Monti, P., Tasso, B., Traverso, N., Domenicotti, C., & Marengo, B. (2024). PTC596-Induced BMI-1 Inhibition Fights Neuroblastoma Multidrug Resistance by Inducing Ferroptosis. Antioxidants, 13(1), 3. https://doi.org/10.3390/antiox13010003