Abstract
Butein (BU) and homobutein (HB) are bioactive polyhydroxylated chalcones widespread in dietary plants, whose antioxidant properties require mechanistic definition. They were investigated by inhibited autoxidation kinetic studies of methyl linoleate in Triton™ X-100 micelles at pH 7.4, 37 °C. Butein had kinh = (3.0 ± 0.9) × 104 M−1s−1 showing a chain-breaking mechanism with higher antioxidant activity than reference α-tocopherol (kinh = (2.2 ± 0.6) × 104 M−1s−1), particularly concerning the stoichiometry or peroxyl radical trapping n = 3.7 ± 1.1 vs. 2.0 for tocopherol. Homobutein had kinh = (2.8 ± 0.9) × 103 M−1s−1, pairing the relative BDEOH measured by radical equilibration EPR as 78.4 ± 0.2 kcal/mol for BU and estimated as 82.6 kcal/mol for HB. The inhibition of mushroom tyrosinase (mTYR) by HB and BU was also investigated. BU gives a reversible uncompetitive inhibition of monophenolase reaction with KI′ = 9.95 ± 2.69 µM and mixed-type diphenolase inhibition with KI = 3.30 ± 0.75 µM and KI′ = 18.75 ± 5.15 µM, while HB was nearly competitive toward both mono- and diphenolase with respective KI of 2.76 ± 0.70 µM and 2.50 ± 1.56 µM. IC50 values (monophenolase/diphenolase at 1 mM substrate) were 10.88 ± 2.19 µM/15.20 ± 1.25 µM, 14.78 ± 1.05 µM/12.36 ± 2.00 µM, and 33.14 ± 5.03 µM/18.27 ± 3.42 µM, respectively, for BU, HB, and reference kojic acid. Molecular docking studies confirmed the mechanism. Results indicate very potent antioxidant activity for BU and potent anti-tyrosinase activity for both chalcones, which is discussed in relation to bioactivity toward protection from skin disorders and food oxidative spoilage.
Keywords:
skin whitening; food safety; chalcones; polyphenols; peroxyl radicals; melanin; kinetics; mechanism; molecular docking 1. Introduction
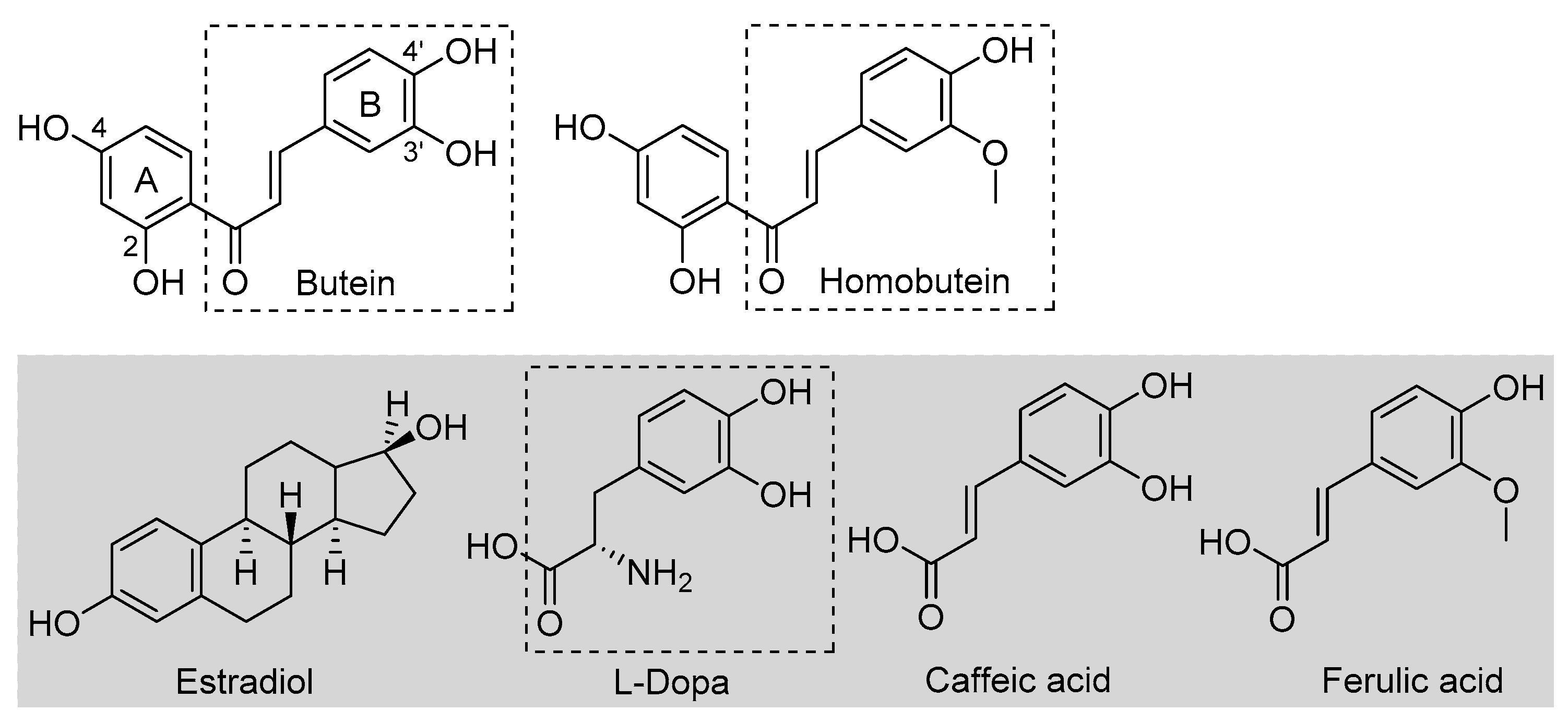
Butein is a natural polyphenolic chalcone (Figure 1) found in a very large variety of botanical sources belonging to different families, such as Asteraceae (e.g., Coreopsis lanceolata L., Dahlia variabilis Desf.), Asparagaceae (e.g., Sansevieria liberica Ger.), Anacardiaceae (e.g., Semecarpus anacardium L.), Fabaceae (e.g., Butea frondosa Roxb., Butea monosperma Taub., Acacia pycnatha Benth.), Pinaceae (e.g., Abies pindrow Royle.), Solanaceae (e.g., Solanum lycopersicum Lam.), and others [1]. As such, it is a very important dietary polyphenol that is specifically considered a nutraceutical owing to its many beneficial properties, which include protection against some forms of cancer and anti-angiogenic, anti-inflammatory, antidiabetic, neuroprotective, hepatoprotective, nephroprotective, and anti-hypertensive properties [1,2,3,4,5]. Plant extracts rich in butein (e.g., D. variabilis, B. monosperma, etc.) have a long tradition of use in folk medicine, particularly in China, Korea, and Japan [1,2]. Some identified mechanisms as the basis of its bioactivity include the up- or downregulation of enzymes, such as protein kinases, and interference with the NF-kB signaling pathway [1,2,3,4]. Structural similarity to estrogens (Figure 1) is attributed a role in its bioactivity [2]. However, bioactivity has also been associated with its antioxidant activity [6]. This has been the subject of different studies, highlighting an indirect antioxidant behavior via the activation of endogenous cellular antioxidant defenses [7]. It was recently demonstrated that a distinct mechanism to explain such antioxidant activity by butein is via the activation of the NRf2 signaling pathway [8].
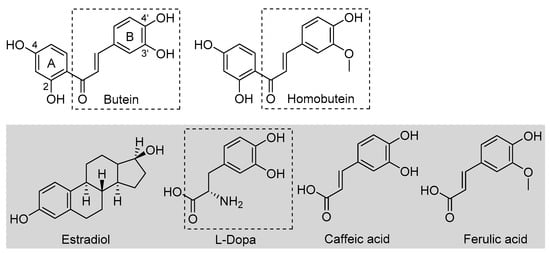
Figure 1.
Structures of butein and homobutein, along with related reference compounds.
An early study also indicated that butein has a direct antioxidant action: it can trap peroxyl radicals in water but not in hexane, and it was able to reduce the markers of oxidative damage (TBARS) in metal-catalyzed oxidation of LDL—an activity that was attributed to its ability in chelating transition metal ions, iron, and copper, preventing peroxidation initiated by Fenton reaction [9]. This would be a preventive albeit direct mechanism. Indeed, a recent computational study also pointed toward iron chelation to explain the activity of butein, which was surpassed by its analogue homobutein [10], differing only for methylation of the -OH in 3′ position in B-ring (Figure 1). Not much else is known about the antioxidant activity of homobutein, which also showed anti-cancer and anti-inflammatory activity [11], except it was found ineffective, at variance with butein, in reducing the production of reactive oxygen species (ROS) in breast cancer cells [6].
Butein and homobutein bear structural similarities to the well-established antioxidants caffeic acid and ferulic acid (Figure 1), which are known to inhibit lipid peroxidation by the direct chain-breaking mechanism by trapping chain-carrying alkylperoxyl radicals [12,13]. We hypothesized that the overlooked chain-breaking mechanism might also be prevalent in explaining the direct antioxidant behavior of butein and homobutein, which we investigated in detail on kinetic grounds by electron paramagnetic resonance (EPR) and by oxygen uptake kinetic studies in the inhibited peroxidation of methyl linoleate micelles.
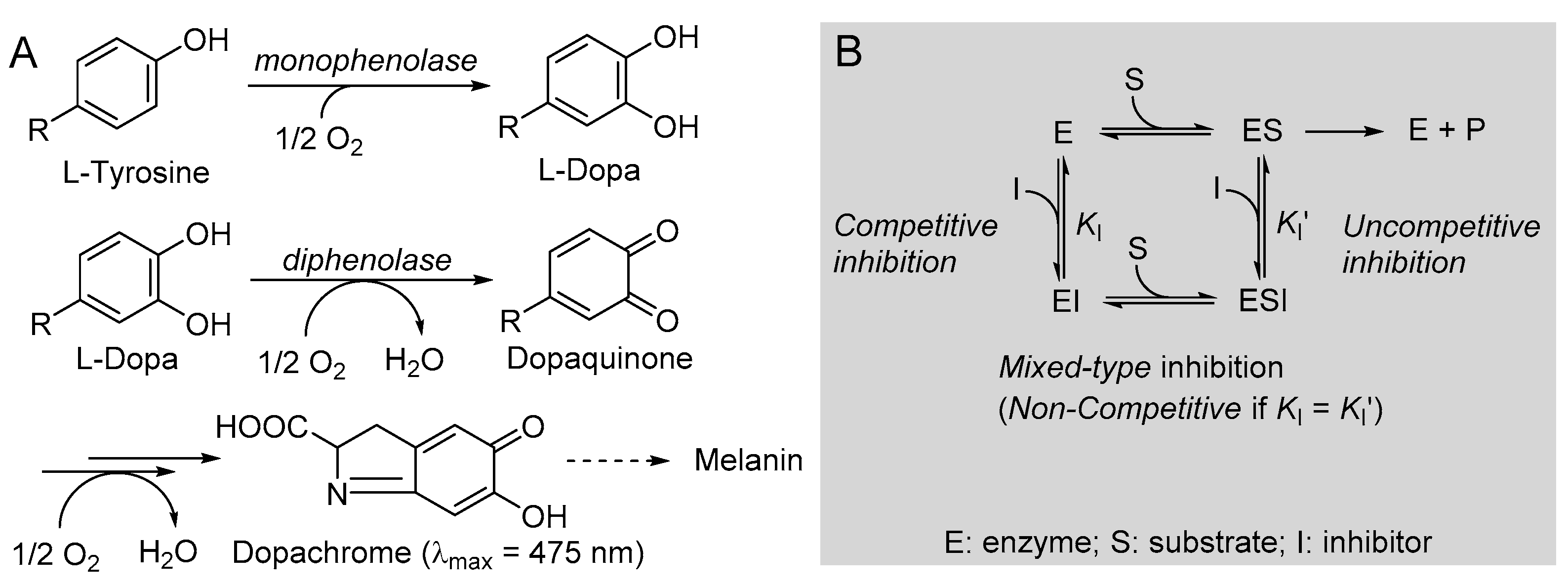
One additional distinctive antioxidant action is via the inhibition of tyrosinase. Tyrosinase (EC 1.14.18.1) is a polyphenol oxidase enzyme, highly conserved in most living organisms, which regulates the biosynthesis of melanin, catalyzing two consecutive reactions: the monophenolase, consisting of the hydroxylation of a phenolic substrate (e.g., l-tyrosine) to the catechol, and the diphenolase, consisting in the oxidation of a catechol (e.g., l-dopa) to the ortho-quinone, which will then undergo further spontaneous oxidation and polymerization to afford the melanic pigments.
Inhibition of tyrosinase has major roles and applications, e.g., in biomedicine, to contrast skin pigmentation disorders like melasma [14,15,16,17] and in food safety, to prevent enzymatic food oxidative spoilage on storage [16,17,18,19].
The structural similarities of butein and homobutein with natural tyrosinase substrate l-dopa (Figure 1) suggest their possible bioactivity as inhibitors. Indeed, polyhydroxylated chalcones are known to inhibit tyrosinase [20,21,22], and some components of this class, such as morachalcone A found in Morus alba, are among the most potent natural tyrosinase inhibitors known to date [23]. No data are available in this regard for homobutein, while butein was reported ineffective toward diphenolase reaction, and data on activity toward monophenolase reaction appear conflicting [24]. Given their importance, we performed a detailed kinetic investigation on the inhibition of both tyrosinase reactions by butein and homobutein and used molecular docking computations to help rationalize the mechanism.
We anticipate that butein is an excellent chain-breaking antioxidant that largely outperforms homobutein and, beyond expectations, it outperforms even reference α-tocopherol (α-TOH) with an unusual mechanism. On the other hand, both butein and homobutein are potent inhibitors of both monophenolase and diphenolase tyrosinase reactions, with homobutein slightly outperforming butein while showing a partly different mechanism despite the similar structure.
2. Materials and Methods
2.1. Materials
Butein ((E)-1-(2,4-dihydroxyphenyl)-3-(3,4-dihydroxyphenyl)prop-2-en-1-one or 2′,3,4,4′-tetrahydroxychalcone) and homobutein ((E)-1-(2,4-dihydroxyphenyl)-3-(4-hydroxy-3-methoxyphenyl)prop-2-en-1-one or 2′,4,4′-trihydroxy-3-methoxychalcone) were purchased from Cymit Quimica (Barcelona, Spain). AAPH (2,2′-azobis(2-methylpropionamidine) dihydrochloride), methyl linoleate (MeLin; ≥98%) and Triton™ X-100, (R,R,R)-α-tocopherol (α-TOH), l-tyrosine (≥98%), l-dopa (3,4-dihydroxy-l-phenylalanine; ≥98%), kojic acid (5-hydroxy-2-hydroxymethyl-4H-4-pyranone; ≥98.5%), and mushroom tyrosinase (mTYR; EC 1.14.18.1, activity = 3410 units/mg) were obtained from Sigma-Aldrich (Milan, Italy) and used as received. Fresh mTYR stock solutions were prepared every second day and stored at 4 °C. Before use, tyrosinase activity was analyzed spectrophotometrically to define the necessary dilution to fixed tyrosinase Sigma units for consistent results. One Sigma unit corresponds to an increase in absorbance at 280 nm of 0.001 per minute at pH 6.8 in a 3 mL reaction mixture containing l-tyrosine. One Sigma unit corresponds to 1.65 × 10−4 international units (I.U.) for monophenolase activity and to 2.24 × 10−2 I.U. for diphenolase activity [25]. Di-tert-butylperoxide (Sigma-Aldrich) was percolated twice through activated basic alumina, and 2,4,6-tri-tert-butylphenol (TBP, 98%) was recrystallized from hexane. Stock solutions of AAPH phosphate buffer (pH 7.4) were prepared every day and stored at 4 °C between subsequent uses to avoid loss of title. Solvents and other chemicals were of the highest available grade (Sigma-Aldrich, Merck, VWR; Milan, Italy) and were used as received.
2.2. Inhibited Autoxidation Studies in Micelles
Autoxidation studies were performed as previously described [19,26], with slight modifications. In a typical experiment, 2.5 mL of air-saturated buffered (PBS, 50 mM, pH 7.4) aqueous dispersion of MeLin (final concentration 2.74 mM) in Triton™ X-100 (final concentration 16 mM) micelles were prepared by vortex mixing. A fresh stock solution of AAPH was added (final concentration 2.5 mM), immediately followed by addition (5–30 μL) of a stock solution of the antioxidant in acetonitrile (final concentration of 1–20 μM) and by additional vortex mixing (5 s). The mixture was sealed in a 2 mL glass vial provided with a PTFE-coated stirring bar and capped with the O2 sensor. The sample was equilibrated at 37 °C in a thermostatted bath equipped with a sealed magnetic stirrer, and oxygen concentration was monitored with time as previously described [19]. Oxygen consumption in the absence of antioxidants was compared with that recorded in the presence of butein, homobutein, or α-tocopherol (α-TOH) as the reference antioxidant [19]. The inhibition rate constant kinh was obtained from oxygen consumption plots by Equation (1) for AH = butein or α-TOH and by Equation (2) for homobutein, where R0 and Rinh are the rates of O2 consumption in the absence and presence of the antioxidant [27,28,29], using kp = 36 M−1s−1 and 2kt = 3.52 × 105 M−1s−1 for MeLin in micelles [30]. The stoichiometric factor n was determined from the length of the inhibited period τ by Equation (3) [29]. The rate of initiation Ri was determined in preliminary experiments using α-TOH as the inhibitor (n = 2) by Equation (3) [29].
2.3. Electron Paramagnetic Resonance (EPR) Spectroscopy
Deoxygenated acetonitrile solutions containing the phenols (0.01–0.001 M) and di-tert-butyl peroxide (10% v/v) were sealed under nitrogen in a 2 mm ID suprasil quartz EPR tube. The sample was inserted in the thermostatted (30 °C) cavity of an X-band EPR spectrometer and photolyzed with a mercury–xenon lamp (240–400 nm, max 4500 mW/cm2). Spectra were recorded with the following settings: modulation amplitude 0.2–1 Gauss, sweep width 30–60 Gauss, modulation frequency 100 kHz, frequency 9.76 GHz, sweep time 60 s, and microwave power 0.1–1 mW. Measured g-factors were corrected with respect to that of TEMPO (g = 2.0064) [31] and of DPPH radical [32]. In ReqEPR experiments, mixtures of TBP and butein were analyzed to obtain the molar ratio of the two equilibrating radicals by comparison of the digitized experimental spectra with computer-simulated ones, as previously described [33,34]. This afforded the equilibrium constant, Keq [34]. Different irradiation power levels (20% to 100%) and different ratios of the two phenols were tested to guarantee that the two species were at equilibrium [33].
2.4. Tyrosnase Inbibition
The kinetics of tyrosinase reaction with or without inhibitor was studied by UV–Vis spectrophotometry, similar to previous methods [35,36,37], following our recent protocol [18]. Measurements were performed at 30 °C in phosphate buffer (50 mM, pH 6.8) in polystyrene low-volume cuvettes (1.5 mL, l = 1 cm) with a double-beam spectrophotometer. l-Tyrosine and l-dopa (5 levels, 0.05–1 mM) were used as the substrate of mushroom tyrosinase (mTYR, 5.0 U/mL and 2.5 U/mL, respectively, for mono- and diphenolase reactions) for monophenolase and diphenolase reactions, respectively. Butein or homobutein (0 to 12 μM) and kojic acid (0 to 50 μM) were comparatively tested as inhibitors. The concentrations 1.4 µM, 2.8 µM, 5.6 µM, and 11.2 µM were tested for butein (mono- and diphenolase inhibition) and for homobutein monophenolase inhibition, while the concentrations 0.175 µM, 0.7 µM, 1.4 µM, and 5.6 µM were used for diphenolase inhibition by homobutein. The reaction was monitored at 475 nm for up to 60 min, following dopachrome formation. Initial velocity (V = ΔA/Δmin) was converted in μM/min according to Lambert–Beer law using the molar extinction coefficient ε = 3700 M−1cm−1 for dopachrome at λmax = 475 nm. Michaelis–Menten parameters (Km e Vmax) were obtained by processing initial velocity vs. substrate concentration data by non-linear fitting to M-M Equation (4), using Sigmaplot 11.0 (Systat Software Inc., San Jose, CA, USA) [18]. Linearized Lineweaver–Burk Equation (5) was used to identify the inhibition mode [35]. In both equations, V indicates the measured initial rate of reaction, [S] is the initial substrate concentration, while Vmax and Km are, respectively, the maximum reaction rate (at saturating substrate concentration) and the M-M constant, with the substrate concentration yielding half-maximum rate [35].
2.5. Stability of Inhibitors in the Presence of Oxygen
To evaluate whether butein and homobutein can be a substrate for mTYR and their stability toward oxidation under the experimental conditions of this study, they were incubated at 30 °C in air-saturated buffer (PBS, 50 mM, pH 6.8) solution at a concentration of 0.05 M and 0.1 M (by dilution of a concentrated stock in acetonitrile) in the presence or absence of mTYR (5 U/mL and 7.5 U/mL) and monitored over 60–90 min by recording the full UV–Vis spectrum (200–800 nm), and by recording the oxygen consumption in the O2 uptake apparatus described in Section 2.2 [18]. The kinetics of spectral variation or O2 consumption were analyzed using Sigmaplot 11.0.
2.6. Molecular Docking
Molecular docking calculations of the reversible inhibitors (butein and homobutein) and the mushroom tyrosinase structure were performed by using Autodock Vina 1.1.2 [36], Autodock v4.2.6 [37], and Autodock GPU [38]. The three-dimensional (3D) structure of the Agaricus bisporus tyrosinase (PDB ID: 2Y9X, Chain A) was downloaded from RCSB Protein Data Bank (https://www.rcsb.org (accessed on 3 February 2023)) as a deoxy form tyrosinase. The 2D structures of the compounds were drawn by ChemDraw Pro. 20.0 and converted to 3D structure by Chem3D Ultra 20.0 software. The AutoDockTools 1.5.6 [37] package was employed to generate the docking input files. All bound water and ligands of the protein were eliminated, and the polar hydrogen was added and optimized. The ligands were prepared by merging non-polar hydrogen atoms and defining rotatable bonds. All atoms within 3 Å from the center of mass of Histine in complex with the crystal structure were defined as a docking pocket using VMD 1.9.3 software. The search grid of the key site of tyrosinase was identified as center x: −7.645, center y: −25.444, and center z: −38.149 with dimensions size x: 25, size y: 25, and size z: 25. Considering the docking score for ligand-to-receptor binding, which involves electrostatic, van der Waals, and solvation energies, we chose the amino acid residues (VAL283, GLY281, ASN260, ARG268) as the flexible side chains to run the flexible docking [39]. In order to increase the docking accuracy, the value of exhaustiveness was set to 32, and the default parameters were used if it was not mentioned. Cu formal charge in the active pocket was set to +1. The best-scoring pose, as judged by the Vina docking score, was chosen and visually analyzed using PyMoL 2.5.0 software (http://www.pymol.org/, accessed on 5 December 2022). The root-mean-square deviation (RMSD) was validated between the co-crystallized heavy atoms coordinates of the inhibitors and the theoretical poses determined in the calculations using PyMoL 2.5.0 software. The interaction energies between the enzyme and inhibitors were then calculated using Autodock Vina 1.1.2, Autodock v4.2.6, and Autodock GPU.
2.7. Statistical Analysis
Each measurement was performed at least in triplicate and reported as average ± standard error. In autoxidation studies, 3–5 different concentrations were tested for each antioxidant. In tyrosinase kinetics, Vmax and Vmaxapp and Km and Kmapp in the absence and presence of inhibitors were determined from non-linear regression of M-M plots based on 5–14 concentrations of the substrate, each with 4 concentrations of the inhibitor, which were analyzed by Shapiro–Wilk test with significance set at p ≤ 0.05.
3. Results and Discussion
3.1. Antioxidant Activity in the Inhibited Autoxidation of Methyl Linoleate Micelles
To study the antioxidant activity of butein and homobutein on quantitative grounds, we performed inhibited autoxidation studies using biomimetic neutral micelles of methyl linoleate (MeLin) in aqueous Triton™ X-100 (with 50 mM PBS, pH 7.4, 37 °C), since it is a well-validated model we and others used in previous studies [19,26]. The reaction kinetics was followed by monitoring oxygen consumption via miniaturized NIR-fluorescence-quenching O2 probes, according to a previously validated protocol [19], both in the absence and in the presence of different concentrations of butein or homobutein. The reaction was initiated thermally in the aqueous phase by the controlled decomposition of the azo compound AAPH so as to clearly distinguish the chain-breaking mechanism from other efficacies based on interference with the initiation process (e.g., the Fenton reaction by metal chelation) and α-TOH was used as the reference antioxidant.
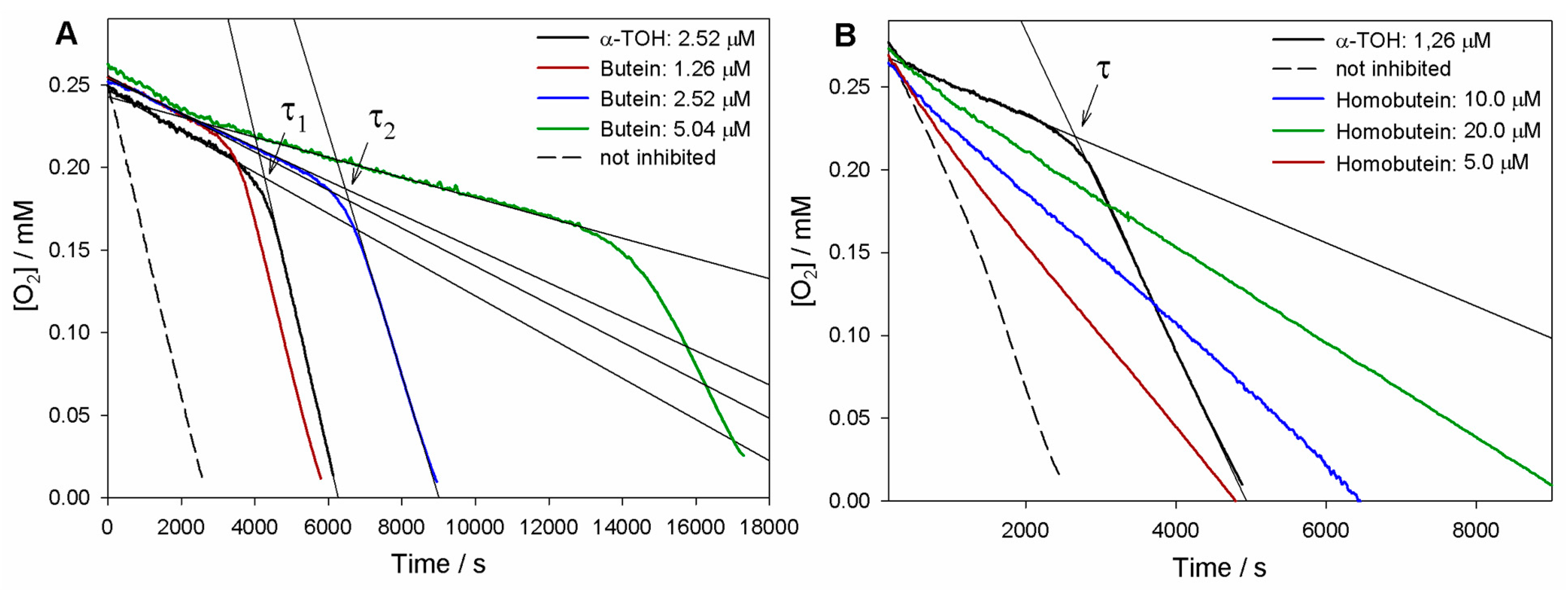
As can be seen in Figure 2, both butein and homobutein effectively inhibited MeLin autoxidation, albeit with significantly different kinetic behavior. Reference α-TOH produced a marked inhibition of the autoxidation for a duration τ until it was completely consumed, then the autoxidation restarted at an uninhibited rate. The length of the inhibited period τ depends on the concentration of the antioxidant and on the stoichiometric factor n of peroxyl radical trapping (Equation (3)), i.e., the number of radicals trapped by one antioxidant molecule. Compared to α-TOH, homobutein was unable to produce a neat inhibited period; however, it slowed down autoxidation in a marked and dose-dependent fashion already in the low micromolar concentration range. This implies a substantial kinh value, albeit significantly lower than α-TOH, which was determined from the slope of oxygen consumption plots via Equation (2) as (2.8 ± 0.9) × 103 M−1s−1 (Table 1). The corresponding kinh value for α-TOH was determined as (2.2 ± 0.6) × 104 M−1s−1, in good agreement with the previous literature in the same model system [19,26]; therefore, the value for homobutein was about one order of magnitude lower. However, the kinh value for homobutein was similar to or higher than that previously reported for other well-established antioxidants in the same model systems, e.g., bakuchiol [19] and resveratrol [26], which stands for its relevance as an antioxidant.
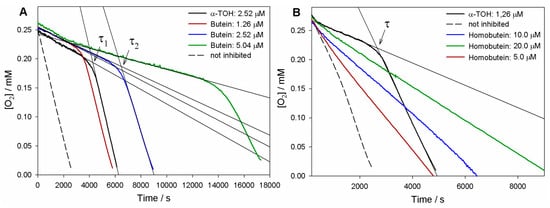
Figure 2.
Oxygen uptake plots during the autoxidation of MeLin (2.74 mM) and Triton™ X-100 (16 mM) micelles in 50 mM PBS (pH 7.4), initiated by 2.5 mM AAPH at 37 °C and inhibited by (A) Butein at different concentrations or (B) Homobutein at different concentrations vs. α-TOH. In (A), τ1 and τ2 indicate, respectively, the inhibition time of 2.5 µM α-TOH and 2.5 µM butein.

Table 1.
Inhibition rate constant, stoichiometric factor, and bond dissociation enthalpy of the reactive OH for the investigated chalcones vs. α-TOH in the AAPH (2.5 mM) initiated autoxidation of MeLin/Triton™ X-100 micelles at 37 °C, pH 7.4.
Instead, the antioxidant activity recorded for butein was surprisingly high (Figure 2). Not only was a neat inhibition period produced, but this was markedly more extended than that produced by α-TOH at the same concentration. The resulting stoichiometric factor n showed some variability from experiment to experiment, but this was consistently and significantly higher than the canonical n = 2 of α-TOH and the vast majority of phenolic antioxidants, averaging at 3.7 peroxyl radicals trapped by one molecule of butein (Table 1).
In addition, the rate of oxygen consumption during the inhibited period was slightly lower than with α-TOH, implying a faster trapping of peroxyl radicals (Equation (1)). Indeed, the measured kinh = (3.0 ± 0.9) × 104 M−1s−1 was slightly higher than that of α-TOH, completing the picture that indicates an overall higher antioxidant performance of butein.
3.2. EPR Spectroscopy
The reactivity of (phenolic) chain-breaking antioxidants toward formal hydrogen atom transfer (HAT) to a radical such as peroxyl radicals is dictated by the strength of the phenolic O-H bond being broken, i.e., by its bond dissociation enthalpy (BDEOH), and by the steric hindrance in ortho position to the reactive OH [40,41]. Indeed, there are well-established Evans–Polanyi linear free-energy correlations between the BDEOH and the inhibition rate constant kinh for phenolic antioxidants [19,40]. One very accurate method to measure the BDEOH is via radical equilibration experiments using electron paramagnetic resonance spectroscopy, the ReqEPR technique [40]. It consists of photolyzing the “unknown” phenol to be studied (UPhOH) in a mixture with a reference phenol (RPhOH) in the cavity of the EPR spectrometer with the addition of a peroxide (e.g., di-tert-butylperoxide) as a photochemical initiator. Analysis and deconvolution of the EPR spectrum containing both equilibrating radical species affords the equilibrium constant Keq (see Figure 3), which in turn allows determining the ∆G° of equilibration. Since it has been shown that ∆S° is negligible for such an equilibrium [40], this affords the BDEOH of UPhOH if the value is known for RPhOH (Figure 3A).

Figure 3.
(A) ReqEPR equilibration study to measure the BDE of phenolic antioxidants by EPR spectroscopy and (B) influence of the solvent on the kinetics and thermodynamics of the HAT reaction.
Using well-established reference 2,4,6-tri-tert-butylphenol (TBP, BDEOH = 80.1 kcal/mol [40]), we performed the equilibration studies with butein in acetonitrile, owing to its insufficient solubility in apolar solvents, which are normally needed in these types of experiments to afford BDE values equivalent to the “gas-phase” [40]. The EPR spectra indicated that the phenoxyl radical of butein forms by HAT from the catechol B-ring (see Supplementary Materials) affording Keq = 1.64 ± 0.63 and BDEOH = 79.8 ± 0.2 kcal/mol (in acetonitrile at 298 K). To convert this value into a gas-phase equivalent, it is necessary to correct for the effect of the solvent, which increases the apparent BDE value by H-bonding to the phenols (see Figure 3B). Such solvent effects can be accounted for quantitatively, as detailed in Figure 3B, by using Abraham’s solvatochromic parameters α2H and β2H, describing, respectively, the H-bond donating ability of the phenol and the H-bond accepting ability of the solvent [42,43]. Considering that the solvent effect is negligible for TBP due to steric hindrance by the t-butyl groups in ortho [40], that α2H for catechol is 0.73 and β2H for acetonitrile is 0.39 [42], the DBE measured in acetonitrile must be downscaled by −1.4 kcal/mol to afford BDEOH = 78.4 ± 0.2 kcal/mol for butein in apolar solvent/gas-phase.
When we turned to homobutein, unfortunately, we were unable to obtain EPR spectra of sufficient quality to determine the BDE by ReqEPR, owing to the much lower persistency of the corresponding phenoxyl radical. However, its value can be estimated from that of butein considering the additive contribution of ring substituents on the BDE of phenols [40], i.e., −6 kcal/mol for ortho-OH and −1.8 kcal/mol for ortho-OCH3. This affords BDEOH ~ 82.6 kcal/mol for homobutein.
The much lower BDEOH of butein compared to homobutein justifies its much better antioxidant performance. Indeed, the BDEOH of butein is lower than that of other catechol antioxidants such as hydroxytyrosol (80.8 kcal/mol [44]) and similar to well-established 3,5-di-tert-butylcatechol (78.2 kcal/mol [45]), which speaks for its excellent potential as a chain-breaking antioxidant.
3.3. Explaining the Excellent Antioxidant Activity of Butein in MeLin Micelles
While both butein and homobutein were effective antioxidants in the protection of methyl linoleate micelles, the performance of butein was truly exceptional and difficult to explain on the basis of the factors normally governing the reactivity of phenolic antioxidants. The value of n > 2 is hardly justified by the presence of other phenolic groups in the A ring as they are in relative meta-position and both conjugated with the electron-withdrawing carbonyl group, along with the occurrence of an intramolecular H-bond between the carbonyl and the OH in 2. This suggests a BDEOH value > 86 kcal/mol for any OH group in A ring, which rules out their contribution in quenching peroxyl radicals [40]. While it cannot be excluded that the large n value arises from subsequent reactions of butein semiquinone radical to form dimeric structures endowed with radical trapping ability, as it was proposed for resveratrol [26], the concomitant very large kinh value prompts a different explanation. Indeed, kinh exceeds that of α-TOH despite butein having BDEOH higher by 1.3 kcal/mol compared to α-TOH (see Section 3.2), which suggests the involvement of a different antioxidant mechanism. Likely, the catechol nature of butein allows its recycling during the autoxidation via the reduction of the semiquinone radical (QH•) and the quinone exhaust product (Q) by hydroperoxyl radicals (HOO•) released as a side reaction during the autoxidation of methyl linoleate [45,46,47], as depicted in Equations (6) and (7).
Q + HOO• → QH• + O2
QH• + HOO• → QH2 + O2
Reactions (6) and (7) have been found to be faster than the reaction of a catechol (QH2) with chain-carrying peroxyl radicals [48], which might help explain the higher reactivity of butein. This mechanism, based on the hydroperoxyl radical as a sacrificial reducing agent, has recently been demonstrated as key in explaining the antioxidant activity of melanin biopolymers (similarly based on the catechol/quinone redox chemistry) [48], and it is at the basis of the synergic antioxidant activity of catechols with terpenes like γ-terpinene [47]; furthermore, it explains the exceptional antioxidant activity of nitroxides in lipophilic environments such as in biological membranes [49]. We suggest it would have a role in the excellent antioxidant behavior of butein.
3.4. Kinetics of Inhibition of Mushroom Tyrosinase (mTYR)
The kinetics of monophenolase and diphenolase reactions of mTYR were investigated using, respectively, l-tyrosine and l-dopa as the natural substrates, in both cases monitoring the formation of dopachrome at λmax = 475 nm, as depicted in Figure 4.
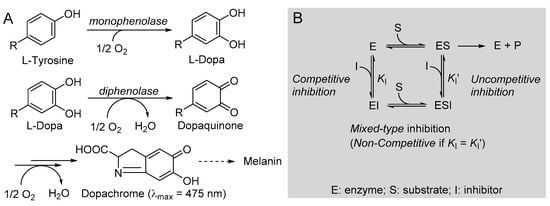
Figure 4.
(A) Reactions involved in tyrosinase-catalyzed biosynthesis of melanin and (B) schematic representation of the possible reversible inhibition types.
Non-linear regression of Michaelis–Menten (M-M) plot of the initial rate vs. substrate concentration (Equation (4)) afforded the parameters Vmax = 3.85 ± 0.04 µM/min and the M-M constant Km = 0.19 ± 0.01 mM for the monophenolase reaction and Vmax = 11.69 ± 0.12 µM/min, Km = 0.24 ± 0.01 mM for the diphenolase reaction, which are in good agreement with previous work from our group [18,19] and from others [38].
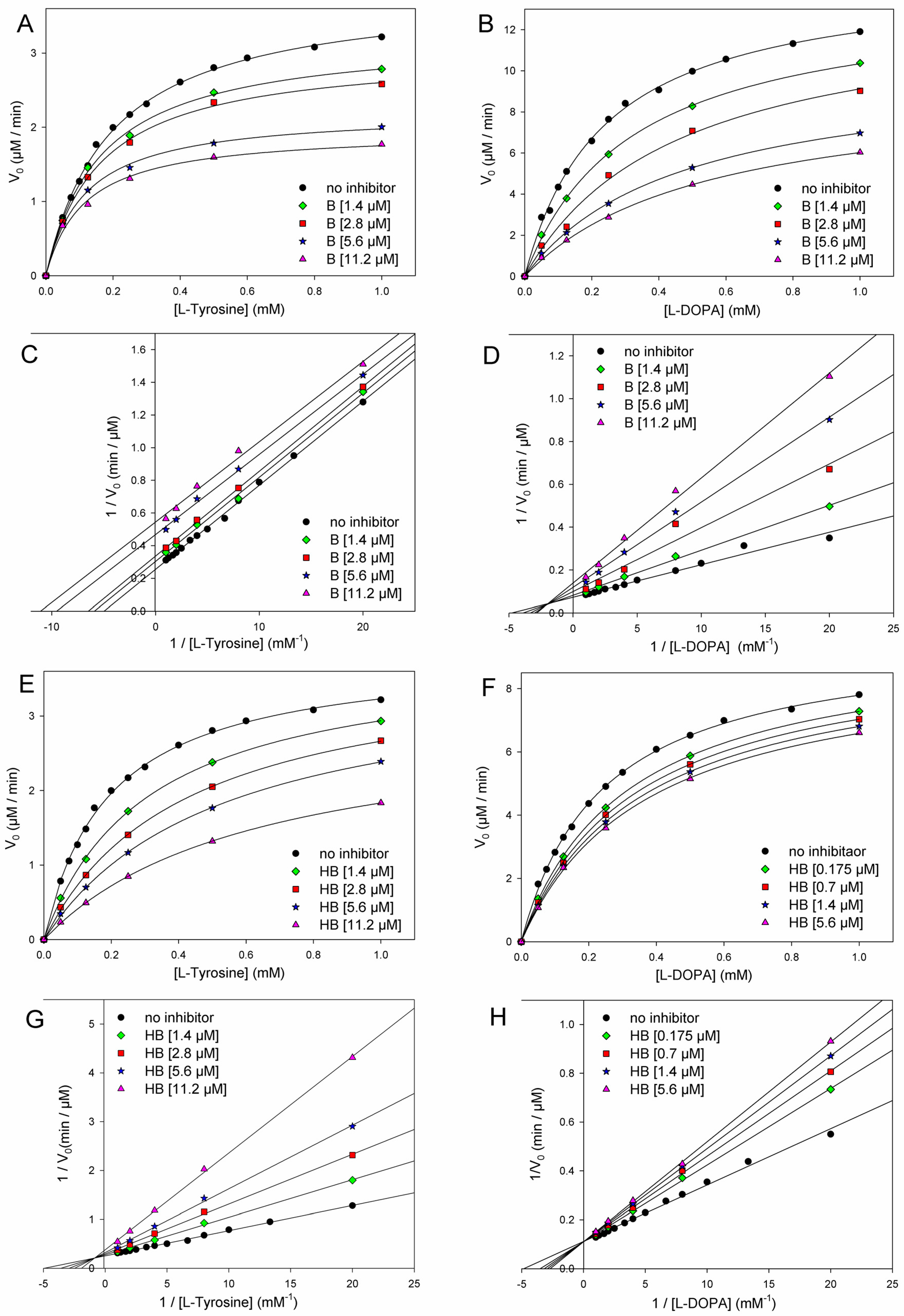
The addition of butein at micromolar levels significantly reduced the rate of dopachrome formation (Figure 5). In the monophenolase reaction (substrate = l-tyrosine), non-linear regression of the M-M plot showed a decrease of both the apparent Vmax and Km (Vmaxapp and Kmapp) in the presence of increasing concentrations of the inhibitor (see Table 2), so that their ratio remained approximately constant. This infrequent kinetic behavior is typical of uncompetitive inhibitors, which act by binding the enzyme–substrate (E-S) complex rather than the free enzyme [35], as exemplified in Figure 4. Indeed, the Lineweaver–Burk (L-B) plot in Figure 5 showed parallel lines for the reaction without inhibitor or with the growing concentration of butein [35]. Inhibition potency can be accurately quantified by the M-M inhibition constant KI′, which represents the dissociation constant of the E-S-I complex—lower values indicate higher potency—that can be obtained by comparing Vmax in the presence and absence of the inhibitor (Equation (8)) or Km in the presence and absence of the inhibitor (Equation (9)) [35]. Results collected in Table 2 show good agreement between the two calculation methods, affording an averaged KI′ = 9.95 ± 2.69 µM, which indicates the high inhibition potency of butein toward the monophenolase reaction.
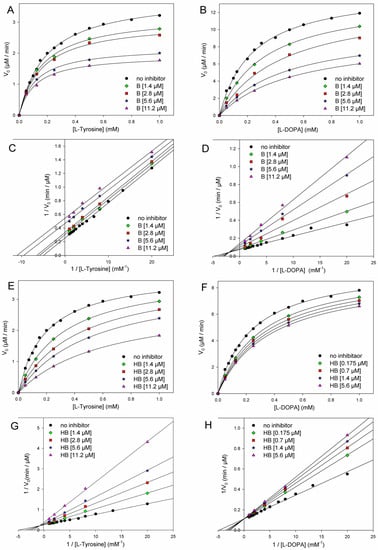
Figure 5.
Kinetics of mTYR inhibition at 30 °C (pH = 6.8) by butein (A–D) and homobutein (E–H) showing the following: Michaelis–Menten plot of monophenolase activity (A,E) and of diphenolase activity (B,F) and the corresponding Lineweaver–Burk plots for monophenolase (C,G) and diphenolase (D,H) inhibition at the concentrations indicated in the legends. Enzyme concentrations for substrate l-dopa and l-tyrosine were 2.5 U/mL and 5.0 U/mL, respectively.
Table 2.
Kinetic parameters for the inhibition of mTYR by butein at 30 °C (pH 6.8).
The behavior of butein toward the diphenolase reaction (substrate = l-dopa) was kinetically different, as indicated by the L-B plot in Figure 5, which shows the crossing of the regression lines for inhibited and uninhibited reactions in the second quadrant of the Cartesian plane. This is indicative of a mixed-type inhibition (competitive + uncompetitive) and is confirmed by data in Table 2, showing a decrease of Vmax and a significant increase of Km on increasing the inhibitor concentration [18,35]. It implies that butein is able to bind both the free enzyme E and the E-S complex with different affinities (Figure 2), as quantified by the respective dissociation constants KI and KI′, which can conveniently be determined by Equations (10) and (11) [18,35].
where
Results collected in Table 2 indicate that the competitive mechanism is largely prevailing since KI = 3.30 ± 0.75 µM (competitive) is about 5-fold lower than KI′ (uncompetitive). Overall, kinetic data prove a high inhibition potency of butein both toward monophenolase and, particularly, toward diphenolase reactions of mTYR, at variance with previous knowledge (vide infra) [24].
Homobutein showed even higher inhibition efficacy toward both monophenolase and diphenolase reactions. Concerning the diphenolase reaction, clear inhibition was already detectable at a concentration as low as 175 nM (Figure 3). For both reactions, linearized L-B plots showed regression lines crossing close to the vertical axis, which would be indicative of competitive inhibition [18,19,35]. Nearly competitive inhibition is also confirmed by analysis of Vmaxapp and Kmapp data collected in Table 2: while Km grows significantly on increasing the concentration of the inhibitor, Vmax remains nearly constant. However, since the crossing points in L-B plots are slightly off-axis (in the II quarter), and some decrease in Vmax is observed, at least for the highest inhibitor’s concentrations, we, more rigorously, treated both inhibitions as being of mixed-type and determined both M-M inhibition constants KI and KI′ according to Equations (8) and (9). Results in Table 3 indicate that the competitive mechanism is largely prevailing as KI values are one and two orders of magnitude lower than the corresponding KI′ for diphenolase and monophenolase reactions, respectively. This indicates that KI′ is poorly contributing to explaining the inhibiting behavior of homobutein. In other words, homobutein behaves as a nearly competitive inhibitor, i.e., it acts by competing with the natural substrate for interaction with the active site of the enzyme. Of interest, the low values of KI, both for monophenolase and diphenolase reactions, as 2.75 ± 0.70 µM and 2.50 ± 1.56 µM, are indicative of high potency, surpassing both butein and reference kojic acid [18].
Table 3.
Kinetic parameters for the inhibition of mTYR by homobutein at 30 °C (pH 6.8).
In a previous study from our group [18], kojic acid was found to give nearly competitive mixed-type inhibition toward the monophenolase reaction with KI = 10.91 μM, while inhibition was mixed-type toward the diphenolase reaction, with KI and KI′ values of 9.91 μM and 20.97 μM, respectively. These values are significantly higher than those found here for butein and particularly for homobutein, indicating substantially higher inhibition by the chalcones. Current values are at variance with a previous report on butein, which indicated competitive inhibition for the monophenolase reaction with KI of 1.41 mM, i.e., orders of magnitude higher than found here and with a different mechanism [24]. The reasons for such diverse results are currently unknown.
3.5. Determination of IC50 against mTYR
Determination of M-M inhibition constants KI and KI′, along with assignment of the inhibition mode, is the most reliable method to accurately quantify the potency of enzyme inhibitors, as it is independent of the experimental settings, like substrate and enzyme concentrations [18,35]. Nonetheless, it is most common in the scientific literature to find enzyme inhibitors characterized by their IC50; the concentration of the inhibitor is able to decrease the rate of the enzyme-catalyzed reaction, i.e., the apparent enzyme activity, by 50% under the same settings. Therefore, to allow comparison with literature data, we determined the IC50 both toward mono- and diphenolase reactions of mTYR for butein and homobutein, along with kojic acid chosen as reference inhibitor, using Langmuir isotherm Equation (12) [35], where at each substrate concentration, V0 and VI indicate, respectively, the rate of reaction in the absence or in the presence of the inhibitor at concentration [I]. Results in Table 4 show a clear dependence on the substrate concentration, as expected [35]. While IC50 of butein decreases on increasing the concentration of l-tyrosine for monophenolase inhibition, owing to the uncompetitive mechanism that requires pre-formation of the E-S complex, values variably increase with the concentration of substrate for butein diphenolase inhibition and for homobutein and kojic acid, all showing a mixed-type inhibition with the (variable) prevalence of competitive behavior. This further highlights the limits of IC50 in quantifying inhibitors’ performance.
Table 4.
Values of IC50 for butein, homobutein, and kojic acid in the inhibition of mTYR (30 °C, pH 6.8) at each concentration of substrate (l-Tyrosine or l-Dopa) for mono- and diphenolase reactions.
Nonetheless, potencies can be compared, taking [substrate] = 1 mM as “standard” settings [18]. Results for reference kojic acid are in good agreement with the previous literature [18,19,22,39], and IC50 for butein and homobutein, against both mono- and diphenolase reactions, were sensibly lower, confirming their higher inhibiting potency. Concerning butein, our values are lower but compatible with those of Khatib et al. [22] for monophenolase inhibition. However, they differ for diphenolase inhibition since butein was reported to be poorly effective even at concentrations > 100 µM [22]. While we do not have an explanation for this divergent outcome, we note that our IC50 values are fully consistent with the KI values from kinetic studies.
3.6. Molecular Docking
To help rationalize the inhibiting mechanism of butein and homobutein, molecular docking simulations were performed against mTYR (PDBID: 2Y9X) to determine binding structures and affinities [16,50,51] using the AutoDock suite [39]. Validation of the docking settings was obtained with a 2Y9X crystal structure bearing co-crystallized tropolone in the active pocket. The tropolone inhibitor was removed and successfully re-docked in the original position.
Computations were first performed by individually docking butein, homobutein, and substrates l-tyrosine and l-dopa; setting flexibility in the ligand and rigid structure in the protein; and were repeated using AutoDock 4.2, AutoDock GPU, and AutoDock Vina to identify consistent binding poses and the protein residues which are mainly involved in interaction with the ligands. Results guided the setting of the flexible residues in AutoDock Vina, which was used for re-docking all the ligands so as to obtain optimized interaction at a low computational cost [37]. This removed unfavorable interactions and produced a significant decrease (i.e., numerical increase in the negative value) in the calculated binding energy.
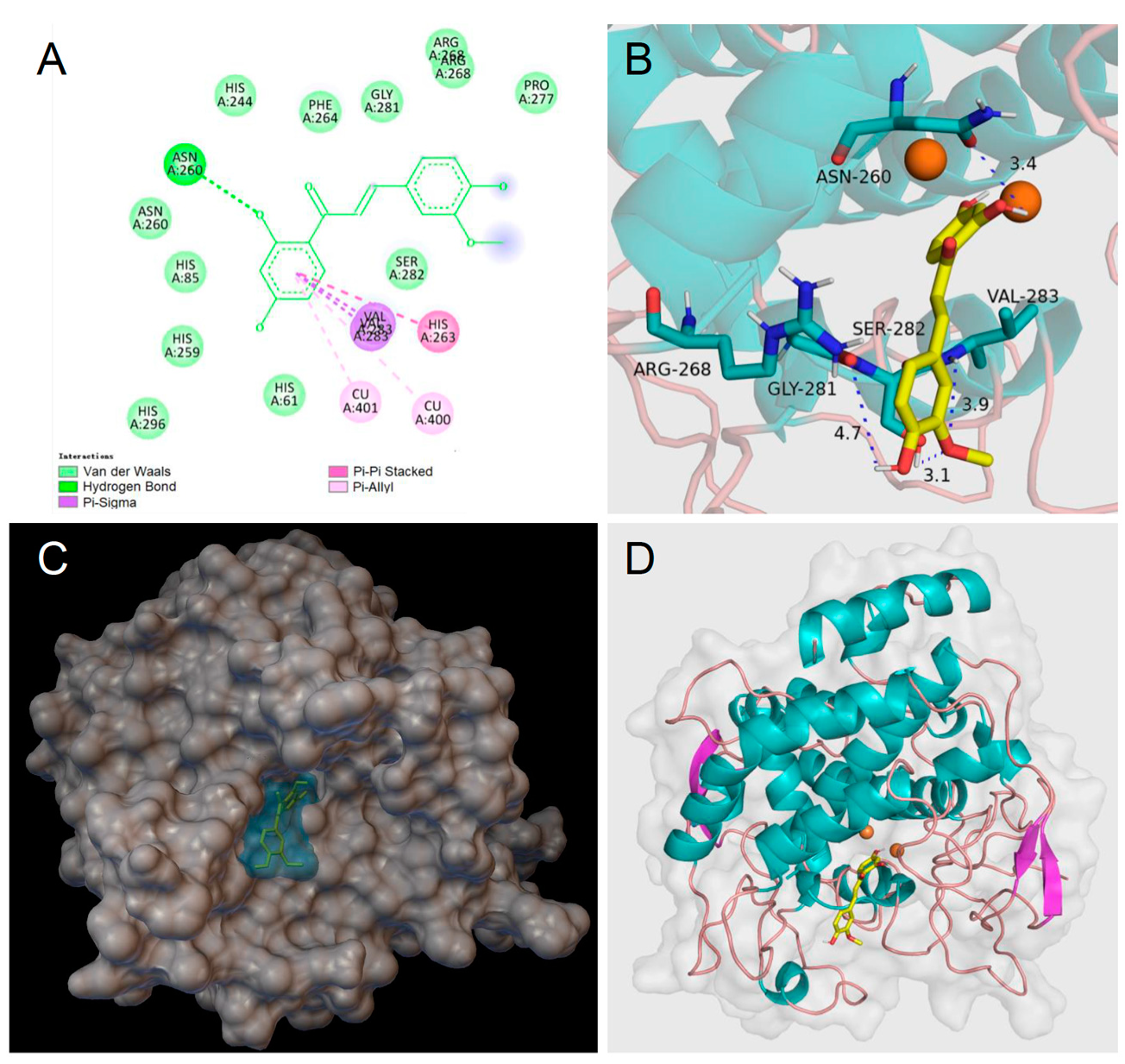
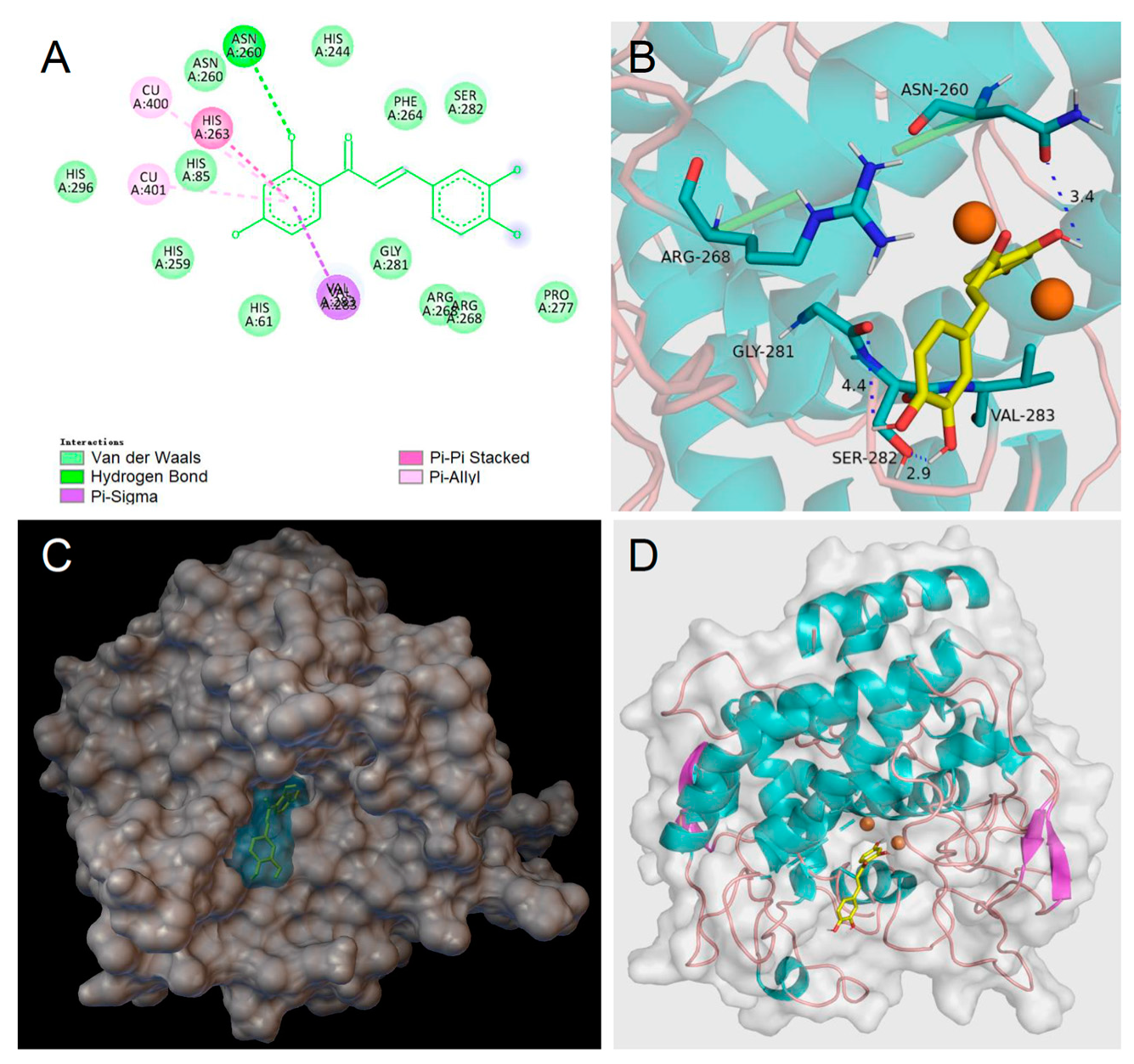
Homobutein’s stabilizing interactions were mainly with ASN260, VAL283, VAL263, and HIS85 (Figure 6), and the binding energy was—7.14 kcal/mol, just slightly lower than calculated for butein (−6.86 Kcal/mol) stabilized by ARG268, ASN260 VAL283, and ALA286 (Figure 7). The lower binding energy for homobutein compared to butein matches with the lower KI value for homobutein compared to butein measured for diphenolase inhibition (see Table 2 and Table 3), although calculated binding energies should not be overinterpreted, and differences in the order of 0.5 kcal/mol or lower should be regarded as merely indicative [51].
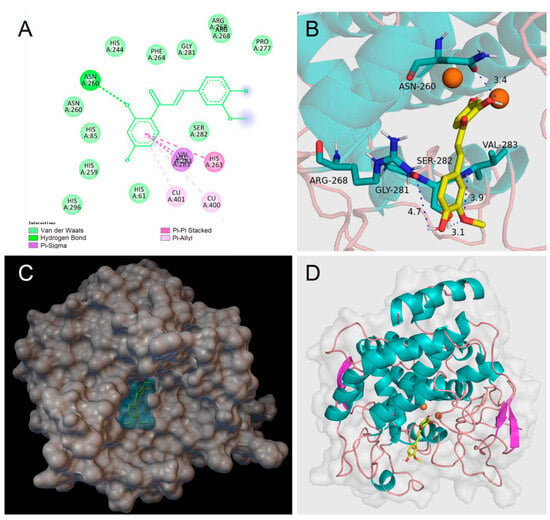
Figure 6.
Molecular docking for homobutein (HB) to mTYR: (A) 2D interaction diagram of 2Y9X-HB complex, (B) detail of the active pocket with copper centers in orange, (C) molecular surface structure with the binding area in cyan, and (D) full view of 3D interaction of HB (in yellow) with 2Y9X.
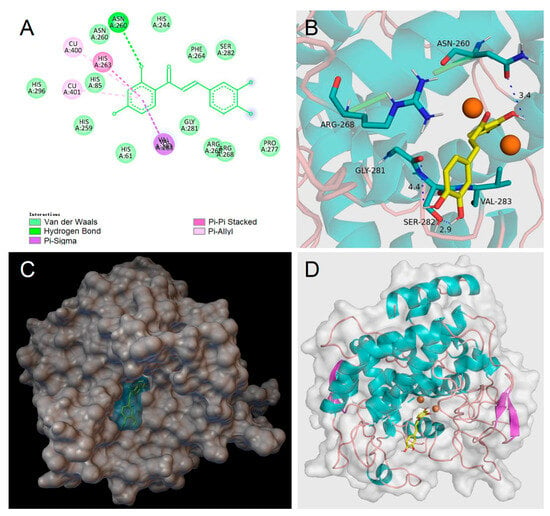
Figure 7.
Molecular docking for butein (BU) to mTYR: (A) 2D interaction diagram of 2Y9X-BU complex, (B) detail of the active pocket with copper centers in orange, (C) molecular surface structure with the binding area in cyan, and (D) full view of 3D interaction of BU (in yellow) with 2Y9X.
Of interest, both inhibitors had lower energies than the ligands, i.e., l-dopa, −6.31 kcal/mol, and l-tyrosine, −5.86 kcal/mol, which would justify their displacement and competitive inhibition. Further studies would be needed to help rationalize the uncompetitive inhibition of butein when the substrate is l-tyrosine, which we plan to pursue in further work. Meanwhile, current results lend support to our kinetic measurements, indicating prevailing competitive, reversible inhibition of mTYR by both butein and homobutein.
3.7. Stability of Butein and Homobutein toward Air and mTYR
Both butein and homobutein are polyphenols, mimicking the structure of natural l-tyrosine and l-dopa substrates; therefore, besides being potent inhibitors, they might also behave as alternative substrates of mushroom tyrosinase, being transformed by the enzyme during the kinetic studies, which would affect the measured kinetics. This has been previously reported for some phenolic inhibitors like caffeic acid and ferulic acid [52]. To investigate this aspect, both butein and homobutein were tested as substrates for mTYR, i.e., they were incubated in PBS at pH 6.8 (30 °C) in the presence and absence of (7 U/mL) mTYR at a much higher concentration than when used as inhibitors (0.05 mM and 0.1 mM, similar to those used for substrates l-tyrosine and l-dopa), in the absence of other substrates, i.e., without l-tyrosine and l-dopa. Their full UV–Vis spectrum was monitored for 60 min. Results showed a rapid spectral variation attributable to the complexation with the enzyme for butein, followed by a very slow decay of its concentration, which was identical in the presence or absence of the enzyme (see Supplementary Materials). Homobutein instead showed no initial spectral variation but a very slow decay of its concentration, which was identical in the presence and absence of the enzyme. This indicates that neither butein nor homobutein behave as substrates of mTYR. To confirm our judgment, we repeated the experiments by monitoring the oxygen consumption during the reactions, according to our previously validated protocol [18,19], since O2 is the obliged oxidant during the tyrosinase reaction and its time-course would parallel the consumption of substrates [18]. Both butein and homobutein showed negligible oxygen consumption with a rate of 2–4 nM/s (120–480 nM/min), i.e., orders of magnitude slower than that found for natural substrates l-tyrosine and l-dopa under similar settings [18,19]. Additionally, oxygen consumption was nearly identical in the absence and presence of the enzyme. Since this kinetic study does not rely on the formation of any specific oxidation product with a detectable spectrum and it is not affected by interference from other species absorbing in the same spectral window, it rules out that butein or homobutein are significant substrates for mTYR (see Supplementary Materials). Both UV–Vis and O2 uptake kinetics converge, showing some spontaneous slow oxidation of the chalcones in solution; however, the measured rates imply a negligible consumption during the typical experiment under our settings, ruling out any significant interference in the measured kinetics.
4. Conclusions
Both butein and homobutein are effective antioxidants working with a chain-breaking mechanism consisting of the fast trapping of chain-carrying peroxyl radicals (kinh 3.0 × 104 and 2.8 × 103 M−1s−1, respectively). The lower BDEOH of catechol butein (78.4 kcal/mol) compared to homobutein (82.6 kcal/mol) justifies its higher reactivity. However, it explains the outstanding antioxidant performance in the protection of methyl linoleate micelles only in part. Indeed, butein was able to outperform nature’s premiere antioxidant, α-tocopherol, despite its BDEOH being higher by 1.3 kcal/mol, and, most interestingly, butein was able to trap almost twice as many peroxyl radicals per molecule of antioxidant compared to α-tocopherol. This behavior is reminiscent of the behavior previously found for catechol/quinone antioxidant systems [47] and, most notably, by catechol-based polydopamine [48] in the presence of a source of hydroperoxyl (HOO•) radicals. It is based on the very fast reduction by HOO• of the semiquinone and quinone from the antioxidant to re-generate fresh catechol ready to carry on the inhibition. However, it was observed here without the deliberate addition of a source of hydroperoxyl radicals. We suggest that butein is able to exploit the HOO• radical formed as a side event during the autoxidation of methyl linoleate [45,46,47]. This mechanism is likely of major relevance in biological systems, where O2-•/HOO• is produced under both physiologic and pathologic conditions, which lends support to the redox bioactivity reported for butein in previous studies [6,7], complementing other mechanisms such as iron chelation [10] and induction of the NRf2 signaling system [8]. Indeed, catechol-type antioxidants have been shown to possess major potential in handling redox-related pathologies like neurological disorders [53].
Along with their antioxidant activity, both butein and homobutein have major efficacy in the inhibition of tyrosinase, showing a prevailing competitive mechanism when l-dopa is the substrate (diphenolase reaction), with KI of 3.30 and 2.50 µM, respectively, both outperforming the reference kojic acid. This mechanism, which is supported by molecular docking studies, is investigated for the first time for homobutein and is at variance from previous knowledge for butein. Both chalcones were also active against the l-tyrosine substrate (monophenolase), albeit with a different mechanism, which was uncompetitive for butein (KI′ = 9.95 µM), and it was nearly competitive for homobutein (KI = 2.76 µM). While the mechanism of homobutein is nicely supported by molecular docking, that of butein appears to require further efforts for full rationalization.
Of interest, the combined antioxidant and anti-tyrosinase activities open previously unexplored applications for the two chalcones, particularly in dermatology, to contrast photoaging, melasma, and age-related pigmentation disorders [14] and in food safety, where they would protect from oxidative food spoilage, both directly caused by oxygen and enzyme-mediated [19]. Clearly, such potential would require further studies, e.g., in cells or in food samples, for full validation and possible exploitation.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/antiox12091763/s1. Figures S1 and S2: EPR spectra of butein and TPB mixtures; Figures S3–S10: Uv–Vis spectra of butein and homobutein incubated with mTYR and kinetic analysis; Figures S11 and S12: plots of O2 uptake during incubation of butein and homobutein with mTYR.
Author Contributions
Conceptualization, L.V.; methodology, A.C. and M.M.; software, V.B.; validation, A.C. and V.B.; formal analysis, L.V., W.P. and I.G.; investigation, W.P., I.G. and T.S.; resources, L.V., writing—original draft preparation, L.V.; supervision, L.V., M.M., A.C. and V.B.; project administration, L.V. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by MUR (Rome, Italy), project FEROX, grant number 20227XZKBY, PRIN2022, and by the University of Bologna, grant RFO2022.
Data Availability Statement
Data are available within the article and Supplementary Materials.
Acknowledgments
We thank Anastasia Grigoryeva for assistance with O2 uptake kinetics and Marco Lucarini for access to the EPR simulation software.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Semwal, R.B.; Semwal, D.K.; Combrinck, S.; Viljoen, A. Butein: From ancient traditional remedy to modern nutraceutical. Phytochem. Lett. 2015, 11, 188–201. [Google Scholar] [CrossRef]
- Padmavathi, G.; Roy, N.K.; Bordoloi, D.; Arfuso, F.; Mishra, S.; Sethi, G.; Bishayee, A.; Kunnumakkara, A.B. Butein in health and disease: A comprehensive review. Phytomedicine 2017, 25, 118–127. [Google Scholar] [CrossRef]
- Lee, S.H.; Seo, G.S.; Sohn, D.H. Inhibition of lipopolysaccharide-induced expression of inducible nitric oxide synthase by butein in RAW264.7 cells. Biochem. Biophys. Res. Commun. 2004, 323, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.R.; Prasad, S.; Sung, B.; Aggarwal, B.B. The role of chalcones in suppression of NF-kappaB-mediated inflammation and cancer. Int. Immunopharmacol. 2011, 11, 295–309. [Google Scholar] [CrossRef]
- Lee, S.-H.; Choi, W.-C.; Kim, K.-S.; Park, J.-W.; Lee, S.H.; Yoon, S.-W. Shrinkage of Gastric Cancer in an elderly patient who received Rhus verniciflua Stokes extract. J. Altern. Complement. Med. 2010, 16, 497–500. [Google Scholar] [CrossRef]
- Cho, S.-G.; Woo, S.-M.; Ko, S.-G. Butein suppresses breast cancer growth by reducing a production of intracellular reactive oxygen species. J. Exp. Clin. Cancer Res. 2014, 33, 51. [Google Scholar] [CrossRef]
- Szuster-Ciesielska, A.; Mizerska-Dudka, M.; Daniluk, J.; Kandefer-Szerszen, M. Butein inhibits ethanol-induced activation of liver stellate cells through TGF-beta, NFkappaB, p38, and JNK signaling pathways and inhibition of oxidative stress. J. Gas-troenterol. 2013, 48, 222–237. [Google Scholar] [CrossRef]
- Tungalag, T.; Park, K.W.; Yang, D.K. Butein Ameliorates Oxidative Stress in H9c2 Cardiomyoblasts through Activation of the NRF2 Signaling Pathway. Antioxidants 2022, 11, 1430. [Google Scholar] [CrossRef]
- Cheng, Z.-J.; Kuo, S.-C.; Chan, S.-C.; Ko, F.-N.; Teng, C.-M. Antioxidant properties of butein isolated from Dalbergia odorifera. Biochim. Biophys. Acta Lipids Lipid Metab. 1998, 1392, 291–299. [Google Scholar] [CrossRef]
- Serobatse, K.R.N.; Kabanda, M.M. Antioxidant and antimalarial properties of butein and homobutein based on their ability to chelate iron (II and III) cations: A DFT study in vacuo and in solution. Eur. Food Res. Technol. 2016, 242, 71–90. [Google Scholar] [CrossRef]
- Orlikova, B.; Schnekenburger, M.; Zloh, M.; Golais, F.; Diederich, M.; Tasdemir, D. Natural chalcones as dual inhibitors of HDACs and NF-κB. Oncol. Rep. 2012, 28, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Amorati, R.; Pedulli, G.F.; Cabrini, L.; Zambonin, L.; Landi, L. Solvent and pH Effects on the Antioxidant Activity of Caffeic and Other Phenolic Acids. J. Agric. Food Chem. 2006, 54, 2932–2937. [Google Scholar] [CrossRef]
- Matera, R.; Gabbanini, S.; Berretti, S.; Amorati, R.; De Nicola, G.R.; Iori, R.; Valgimigli, L. Acylated anthocyanins from sprouts of Raphanus sativus cv. Sango: Isolation, structure elucidation and antioxidant activity. Food Chem. 2015, 166, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Panzella, L.; Napolitano, A. Natural and Bioinspired Phenolic Compounds as Tyrosinase Inhibitors for the Treatment of Skin Hyperpigmentation: Recent Advances. Cosmetics 2019, 6, 57. [Google Scholar] [CrossRef]
- Nicolaidou, E.; Katsambas, A.D. Pigmentation disorders: Hyperpigmentation and hypopigmentation. Clin. Dermatol. 2014, 32, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Hering, A.; Stefanowicz-Hajduk, J.; Dziomba, S.; Halasa, R.; Krzemieniecki, R.; Sappati, S.; Baginski, M.; Ochocka, J.R. Mangiferin Affects Melanin Synthesis by an Influence on Tyrosinase: Inhibition, Mechanism of Action and Molecular Docking Studies. Antioxidants 2023, 12, 1016. [Google Scholar] [CrossRef]
- Choi, J.; Neupane, T.; Baral, R.; Jee, J.-G. Hydroxamic Acid as a Potent Metal-Binding Group for Inhibiting Tyrosinase. Antioxidants 2022, 11, 280. [Google Scholar] [CrossRef]
- Guo, Y.; Cariola, A.; Matera, R.; Gabbanini, S.; Valgimigli, L. Real-time oxygen sensing as a powerful tool to investigate tyrosinase kinetics allows revising mechanism and activity of inhibition by glabridin. Food Chem. 2022, 393, 133423. [Google Scholar] [CrossRef]
- Cariola, A.; El Chami, M.; Granatieri, J.; Valgimigli, L. Anti-tyrosinase and antioxidant activity of meroterpene bakuchiol from Psoralea corylifolia (L.). Food Chem. 2023, 405, 134953. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Namasivayam, V.; Manickam, M.; Jung, S.-H. Inhibitors of Melanogenesis: An Updated Review. J. Med. Chem. 2018, 61, 7395–7418. [Google Scholar] [CrossRef]
- Zolghadri, S.; Bahrami, A.; Khan, M.T.H.; Munoz-Munoz, J.; Garcia-Molina, F.; Garcia-Canovas, F.; Saboury, A.A. A comprehensive review on tyrosinase inhibitors. J. Enz. Inhib. Med. Chem. 2019, 34, 279–309. [Google Scholar] [CrossRef]
- Khatib, S.; Nerya, O.; Musa, R.; Shmuel, M.; Tamira, S.; Vaya, J. Chalcones as potent tyrosinase inhibitors: The importance of a 2,4-substituted resorcinol moiety. Bioorg. Med. Chem. 2005, 13, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tao, G.; Chen, J.; Zheng, Z.-P. Characterization of a New Flavone and Tyrosinase Inhibition Constituents from the Twigs of Morus alba L. Molecules 2016, 21, 1130. [Google Scholar] [CrossRef]
- Nerya, O.; Musa, R.; Khatib, S.; Tamir, S.; Vaya, J. Chalcones as potent tyrosinase inhibitors: The effect of hydroxyl positions and numbers. Phytochemistry 2004, 65, 1389–1395. [Google Scholar] [CrossRef]
- Fenoll, L.G.; Rodriguez-Lopez, J.N.; Garcia-Molina, F.; Garcìa-Canovas, F.; Tudela, J. Unification for the Expression of the Monophenolase and Diphenolase Activities of Tyrosinase. IUBMB Life 2002, 54, 137–141. [Google Scholar] [CrossRef]
- Konopko, A.; Litwinienko, G. Unexpected Role of pH and Microenvironment on the Antioxidant and Synergistic Activity of Resveratrol in Model Micellar and Liposomal Systems. J. Org. Chem. 2022, 87, 1698–1709. [Google Scholar] [CrossRef]
- Amorati, R.; Baschieri, A.; Morroni, G.; Gambino, R.; Valgimigli, L. Peroxyl Radical Reactions in Water Solution: A Gym for Proton-Coupled Electron-Transfer Theories. Chem. Eur. J. 2016, 22, 7924–7934. [Google Scholar] [CrossRef]
- Johansson, H.; Shanks, D.; Engman, L.; Amorati, R.; Pedulli, G.F.; Valgimigli, L. Long-lasting antioxidant protection: A re-generable BHA analogue. J. Org. Chem. 2010, 75, 7535–7541. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.W.; Doba, T.; Gabe, E.; Hughes, L.; Lee, F.L.; Prasad, L.; Ingold, K.U. Autoxidation of biological molecules. 4. Maximizing the antioxidant activity of phenols. J. Am. Chem. Soc. 1985, 107, 7053–7065. [Google Scholar] [CrossRef]
- Barclay, L.R.C.; Baskin, K.A.; Locke, S.J.; Schaffer, T.D. Benzophenone-photosensitized autoxidation of linoleate in solution and sodium dodecyl sulfate micelles. Can. J. Chem. 1987, 65, 2229–2240. [Google Scholar] [CrossRef]
- Valgimigli, L.; Valgimigli, M.; Gaiani, S.; Pedulli, G.F.; Bolondi, L. Measurement of oxidative stress in human liver by EPR spin-probe technique. Free Radic. Res. 2000, 33, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Valgimigli, L.; Ingold, K.U.; Lusztyk, J. Solvent effects on the reactivity and free spin distribution of 2,2-diphenyl-1-picrylhydrazyl radicals. J. Org. Chem. 1996, 61, 7947–7950. [Google Scholar] [CrossRef]
- Brigati, G.; Lucarini, M.; Mugnaini, V.; Pedulli, G.F. Determination of the Substituent Effect on the O−H Bond Dissociation Enthalpies of Phenolic Antioxidants by the EPR Radical Equilibration Technique. J. Org. Chem. 2002, 67, 4828–4832. [Google Scholar] [CrossRef] [PubMed]
- Lucarini, M.; Pedulli, G.F.; Valgimigli, L.; Amorati, R.; Minisci, F. Thermochemical and kinetic studies of a bisphenol anti-oxidant. J. Org. Chem. 2001, 66, 5456–5462. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A. Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis, 2nd ed.; Wiley-VCH: New York, NY, USA, 2000. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Computat. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef]
- Santos-Martins, D.; Solis-Vasquez, L.; Tillack, A.F.; Sanner, M.F.; Koch, A.; Forli, S. Accelerating AutoDock4 with GPUs and Gradient-Based Local Search. J. Chem. Theory Comput. 2021, 17, 1060–1073. [Google Scholar] [CrossRef]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef]
- Lucarini, M.; Pedulli, G.F. Free radical intermediates in the inhibition of the autoxidation reaction. Chem. Soc. Rev. 2010, 39, 2106–2119. [Google Scholar] [CrossRef]
- Foti, M.C. Antioxidant properties of phenols. J. Pharm. Pharmacol. 2007, 59, 1673–1685. [Google Scholar] [CrossRef]
- Litwinienko, G.; Ingold, K.U. Solvent Effects on the Rates and Mechanisms of Reaction of Phenols with Free Radicals. Acc. Chem. Res. 2007, 40, 222–230. [Google Scholar] [CrossRef]
- Lucarini, M.; Pedulli, G.F.; Guerra, M. A Critical Evaluation of the Factors Determining the Effect of Intramolecular Hydrogen Bonding on the O-H Bond Dissociation Enthalpy of Catechol and of Flavonoid Antioxidants. Chem. Eur. J. 2004, 10, 933–939. [Google Scholar] [CrossRef]
- Amorati, R.; Valgimigli, L.; Panzella, L.; Napolitano, A.; D’Ischia, M. 5-S-lipoylhydroxytyrosol, a multidefense antioxidant featuring a solvent-tunable peroxyl radical-scavenging 3-thio-1,2-dihydroxybenzene motif. J. Org. Chem. 2013, 78, 9857–9864. [Google Scholar] [CrossRef] [PubMed]
- Baschieri, A.; Jin, Z.; Amorati, R. Hydroperoxyl radical (HOO•) as a reducing agent: Unexpected synergy with antioxidants. A review. Free Radic. Res. 2023, 57, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Harrison, K.A.; Haidasz, E.A.; Griesser, M.; Pratt, D.A. Inhibition of hydrocarbon autoxidation by nitroxide-catalyzed cross-dismutation of hydroperoxyl and alkylperoxyl radicals. Chem. Sci. 2018, 9, 6068–6079. [Google Scholar] [CrossRef]
- Guo, Y.; Amorati, R. The Underrecognized Role of the Hydroperoxyl (HOO•) Radical in Chain Propagation of Lipids and its Implication in Antioxidant Activity. In Lipid Oxidation in Food and Biological Systems; Bravo-Diaz, C., Ed.; Springer: Cham, Switzerland, 2022. [Google Scholar] [CrossRef]
- Guo, Y.; Baschieri, A.; Mollica, F.; Valgimigli, L.; Cedrowski, J.; Litwinienko, G.; Amorati, R. Hydrogen Atom Transfer from HOO• to ortho-Quinones Explains the Antioxidant Activity of Polydopamine. Angew. Chem. Int. Ed. 2021, 60, 15220–15224. [Google Scholar] [CrossRef] [PubMed]
- Mollica, F.; Lucernati, R.; Amorati, R. Expanding the spectrum of polydopamine antioxidant activity by nitroxide conjugation. J. Mater. Chem. B 2021, 9, 9980–9988. [Google Scholar] [CrossRef]
- Song, X.; Ni, M.; Zhang, Y.; Zhang, G.; Pan, J.; Gong, D. Comparing the inhibitory abilities of epigallocatechin-3-gallate and gallocatechin gallate against tyrosinase and their combined effects with kojic acid. Food Chem. 2021, 349, 129172. [Google Scholar] [CrossRef]
- Deri, B.; Kanteev, M.; Goldfeder, M.; Lecina, D.; Guallar, V.; Adir, N.; Fishman, A. The unravelling of the complex pattern of tyrosinase inhibition. Sci. Rep. 2016, 6, 34993. [Google Scholar] [CrossRef]
- Mayr, F.; Sturm, S.; Ganzera, M.; Waltenberger, B.; Martens, S.; Schwaiger, S.; Schuster, D.; Stuppner, H. Mushroom Tyrosinase-Based Enzyme Inhibition Assays Are Not Suitable for Bioactivity-Guided Fractionation of Extracts. J. Nat. Prod. 2019, 82, 136–147. [Google Scholar] [CrossRef]
- Cichon, N.; Saluk-Bijak, J.; Gorniak, L.; Przyslo, L.; Bijak, M. Flavonoids as a Natural Enhancer of Neuroplasticity—An Overview of the Mechanism of Neurorestorative Action. Antioxidants 2020, 9, 1035. [Google Scholar] [CrossRef] [PubMed]
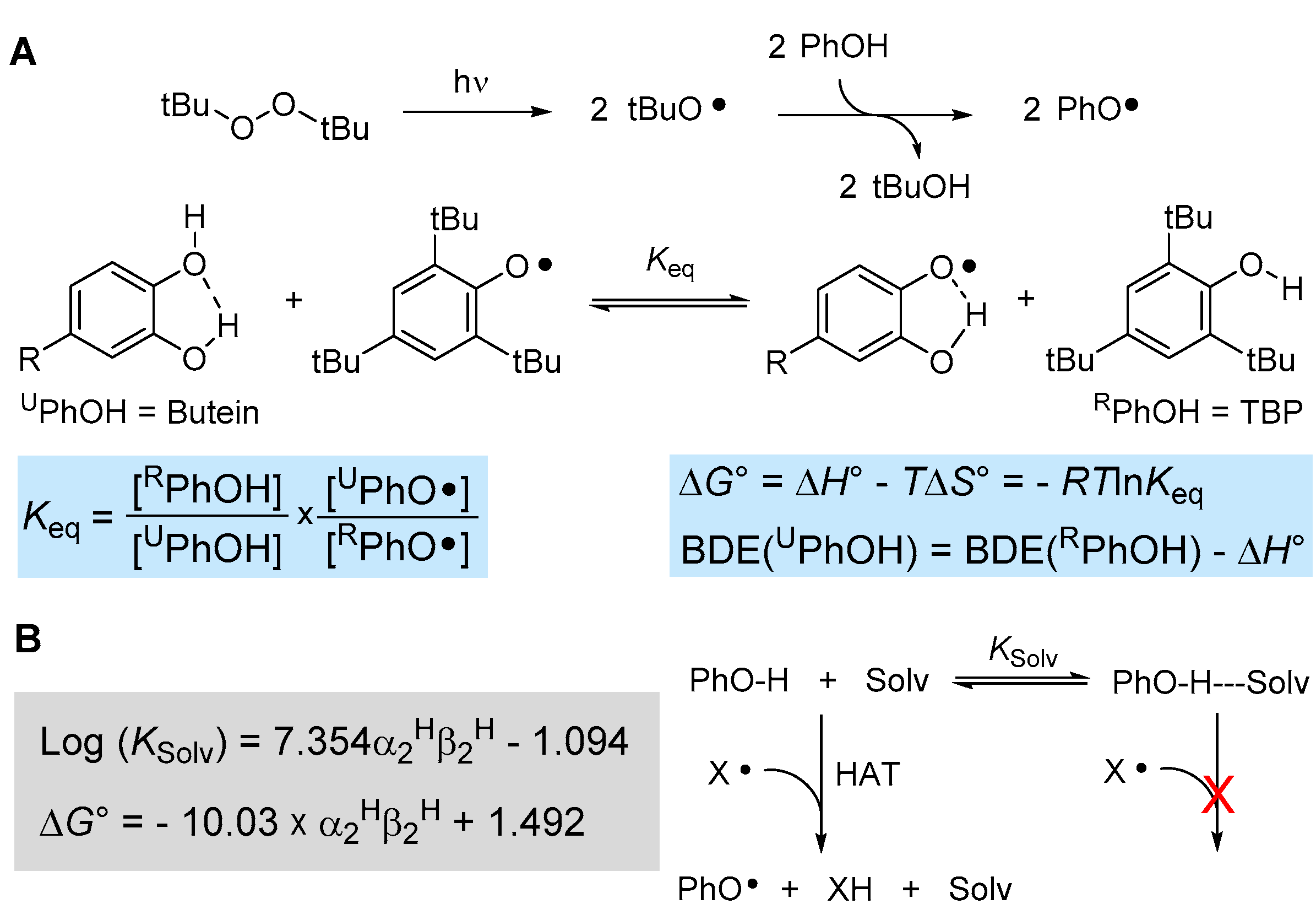
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).