

Cystathionine Gamma Lyase Is Regulated by Flow and Controls Smooth Muscle Migration in Human Saphenous Vein

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Vessels Culture
2.2. Cell Culture
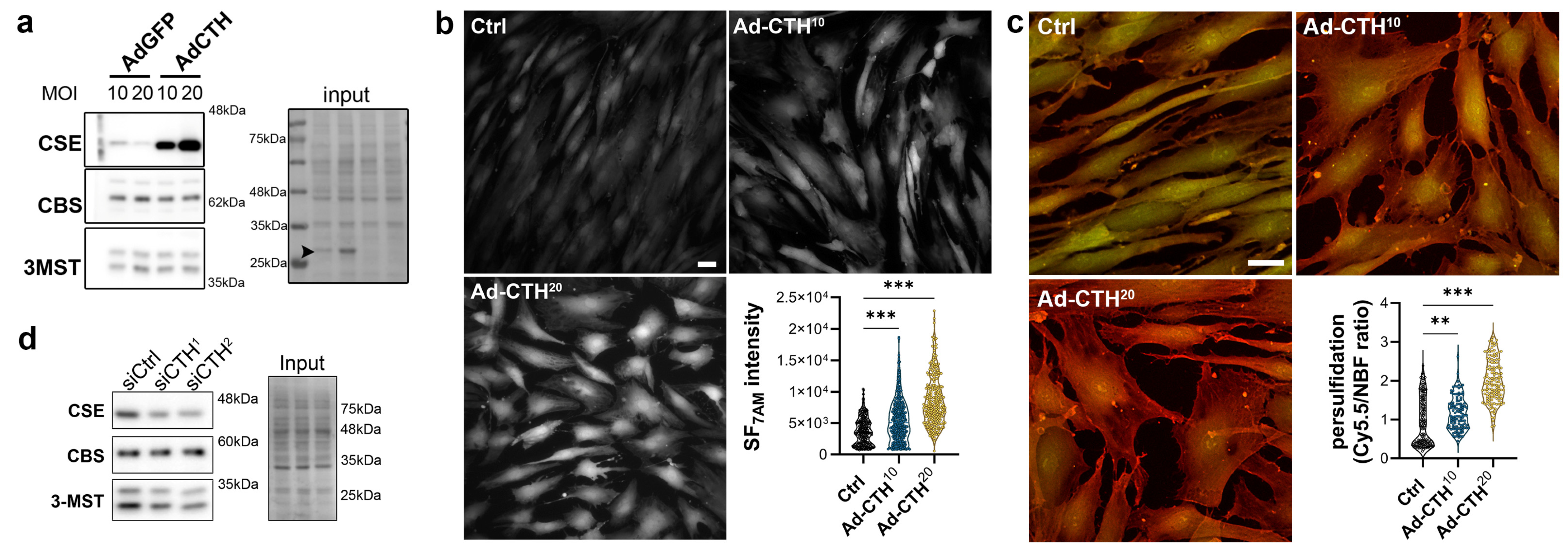
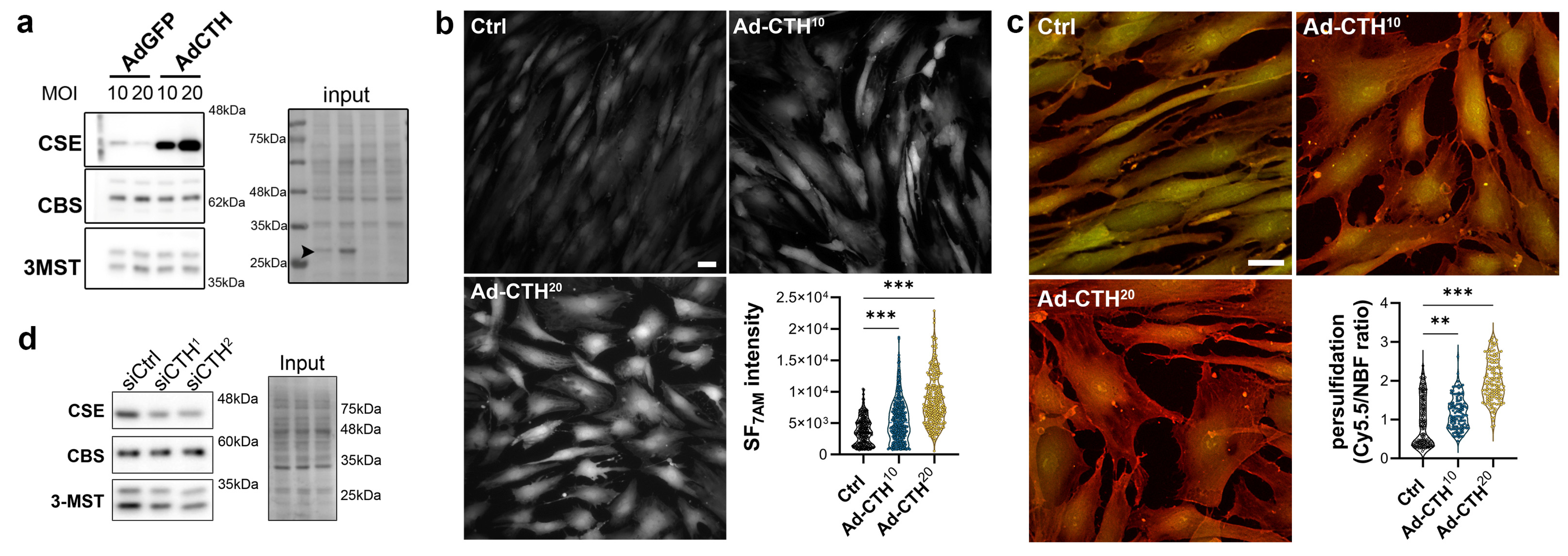
2.3. siRNA-Mediated Knock-Down and Adenoviral-Mediated Overexpression
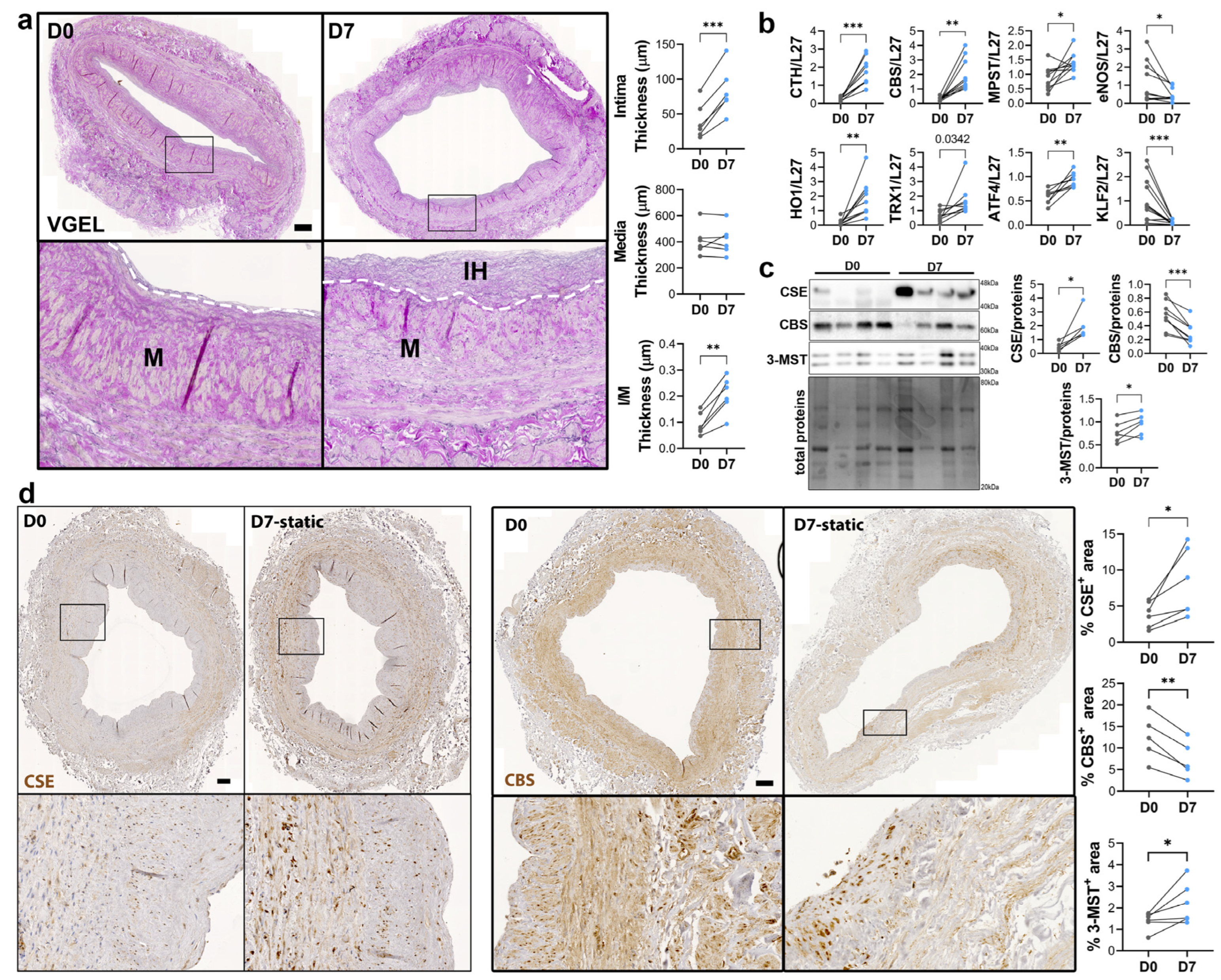
2.4. Histology
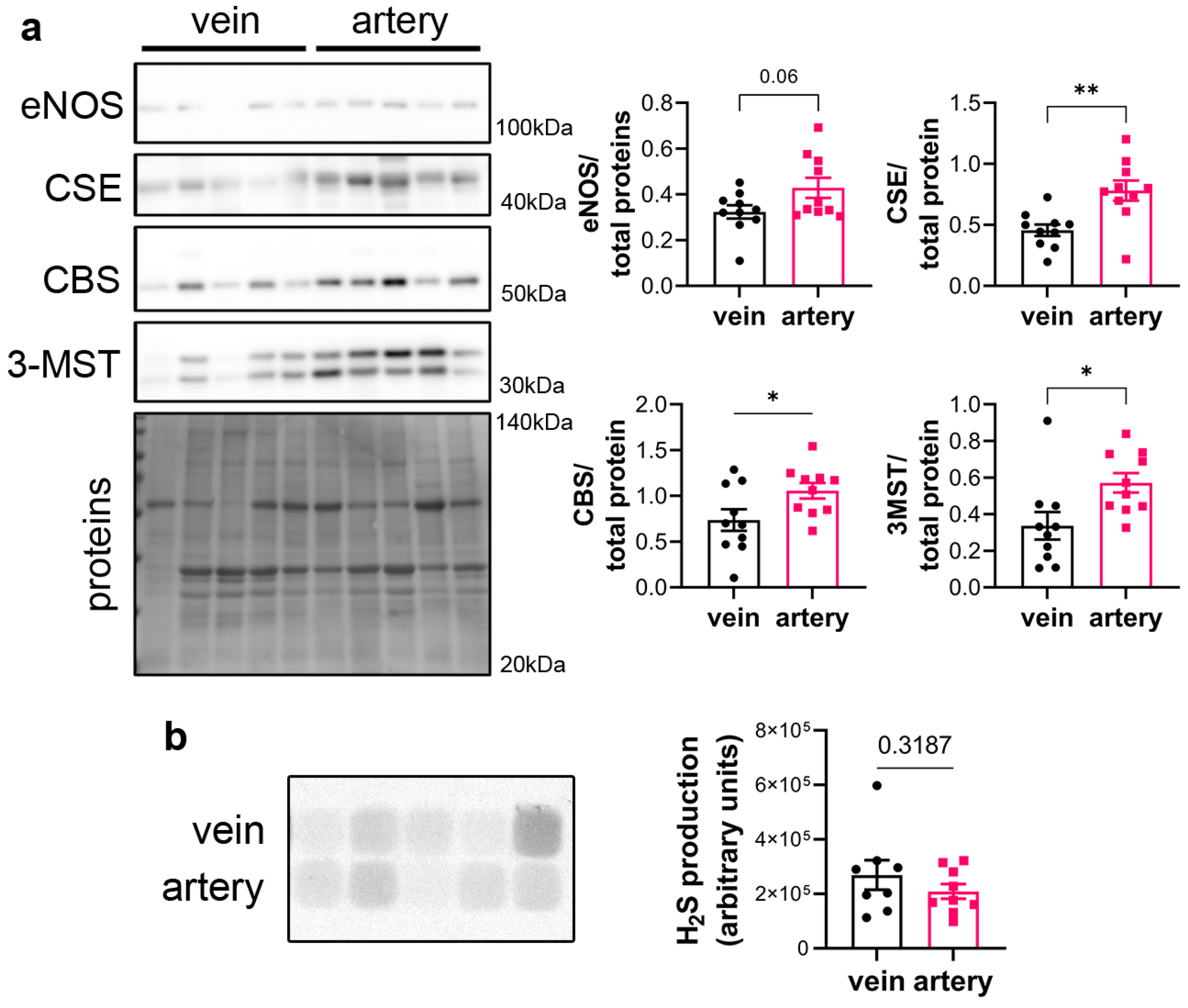
2.5. Western Blotting
2.6. Reverse Transcription and Quantitative Polymerase Chain Reaction (RT-qPCR)
2.7. Lead Acetate (CSE/CBS Activity Assay)
2.8. H2S and Persulfidation Measurement
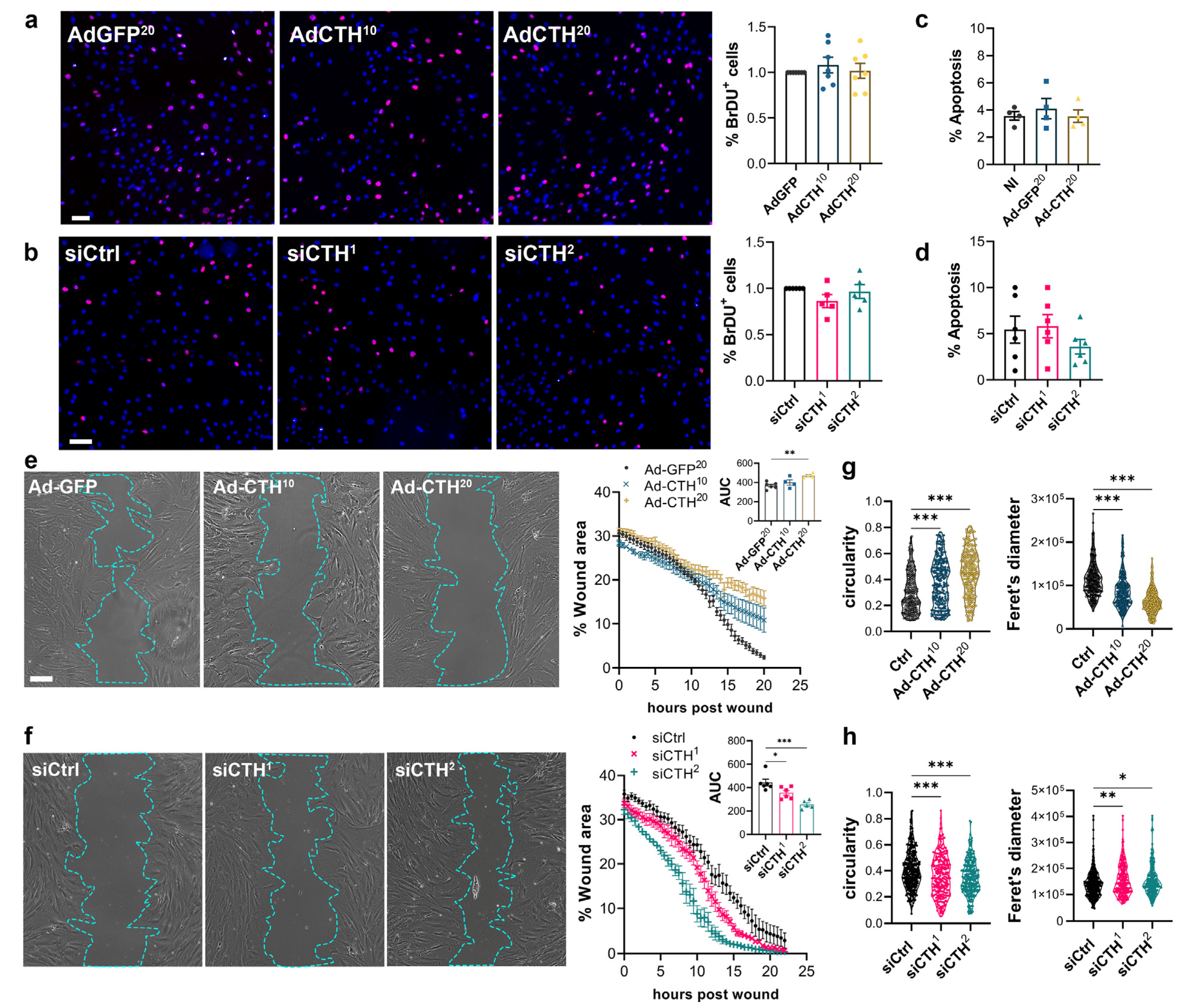
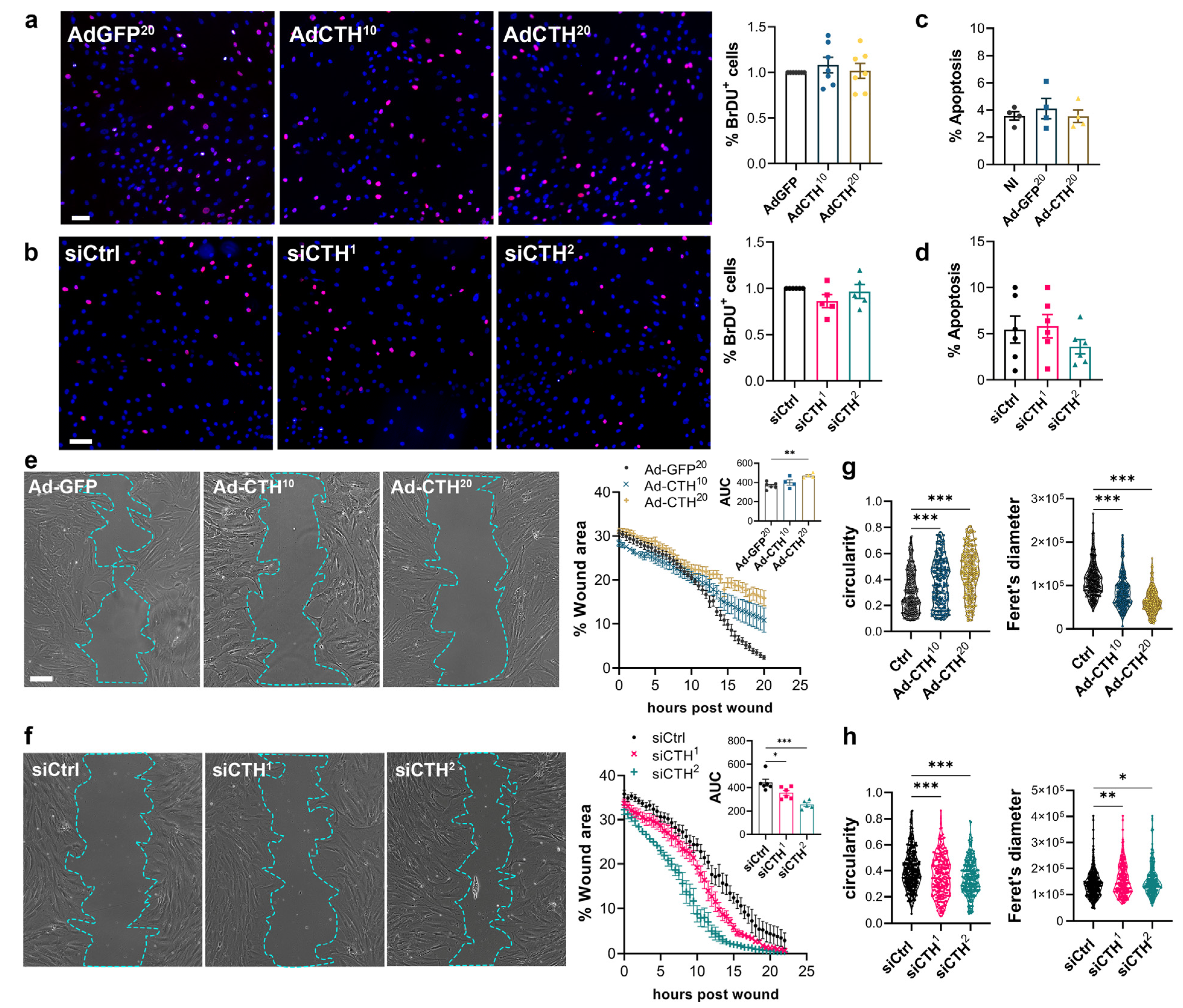
2.9. BrdU Assay
2.10. Wound Healing Assay
2.11. Statistical Analyses
2.12. Ethics Statement
3. Results
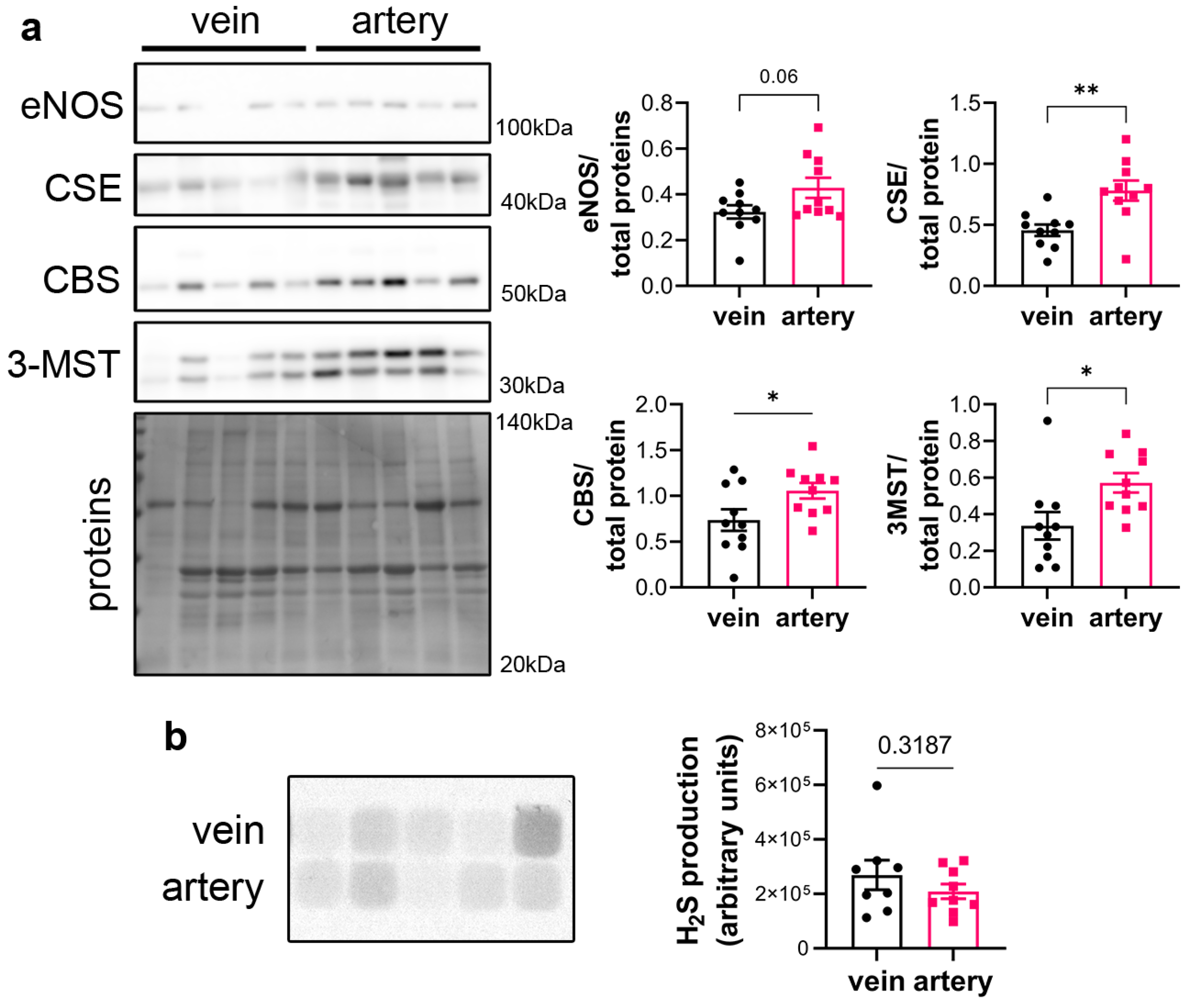
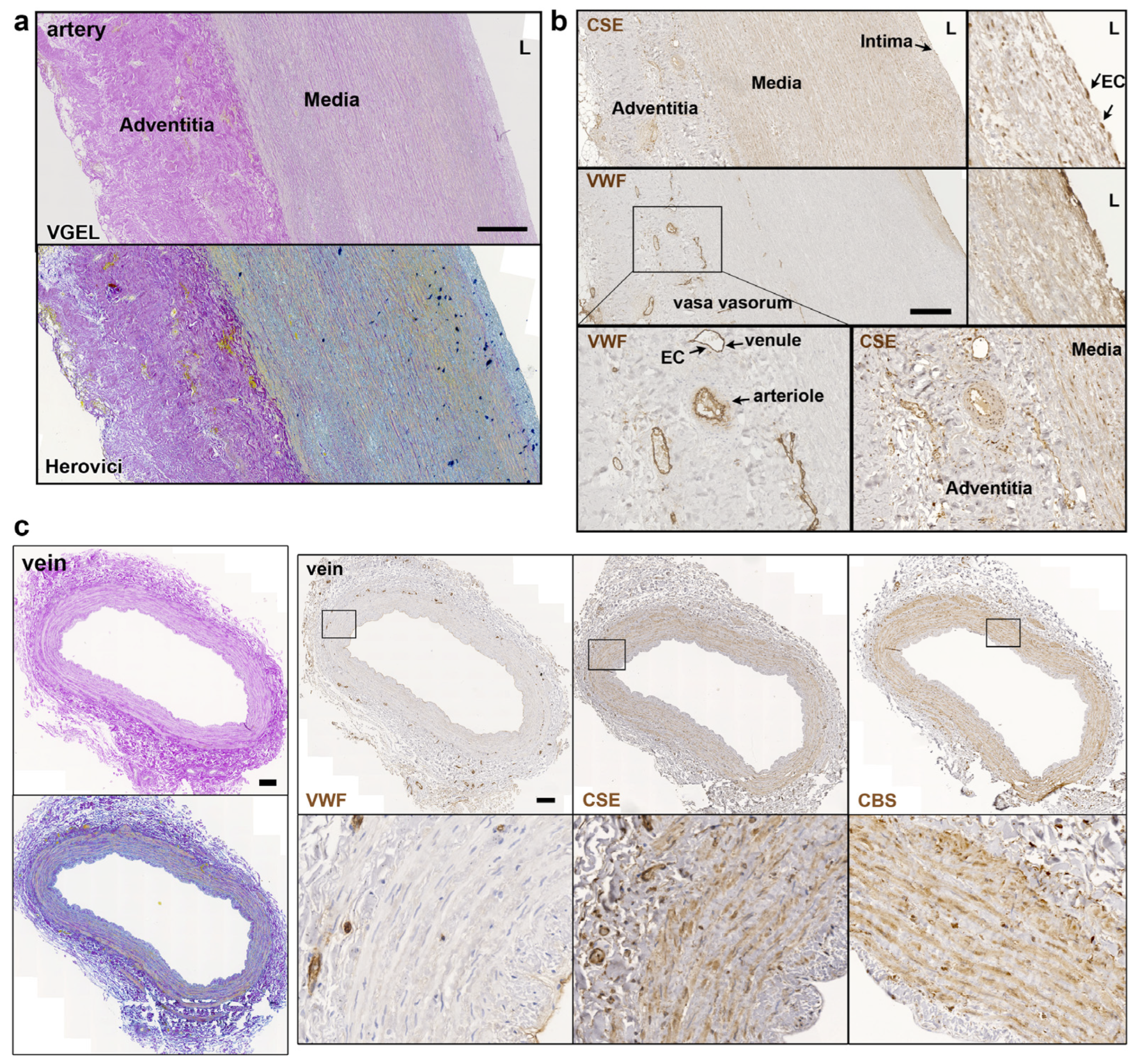
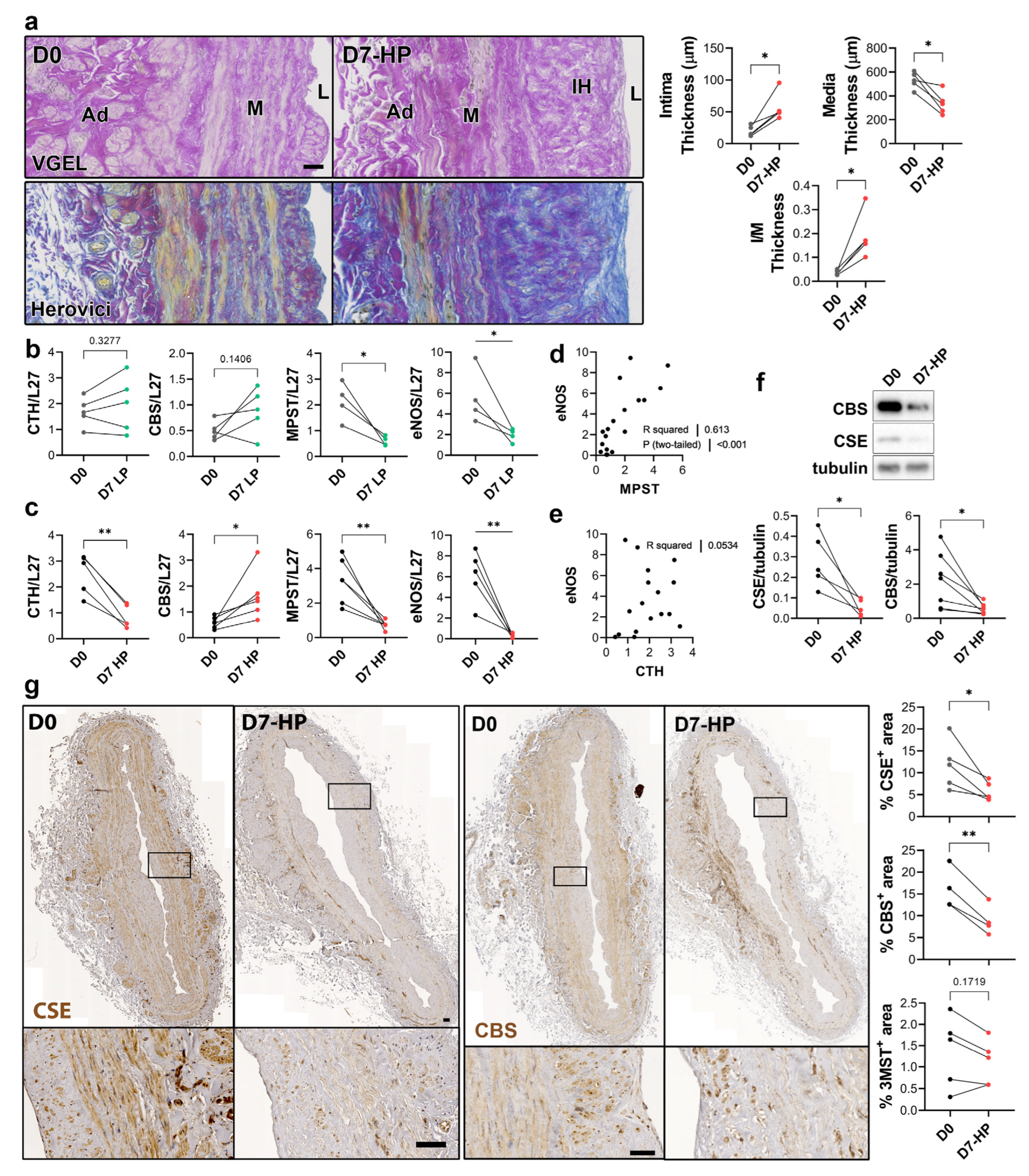
3.1. CSE Is Expressed in the Media and Intima of Human Saphenous Vein and Artery Segments
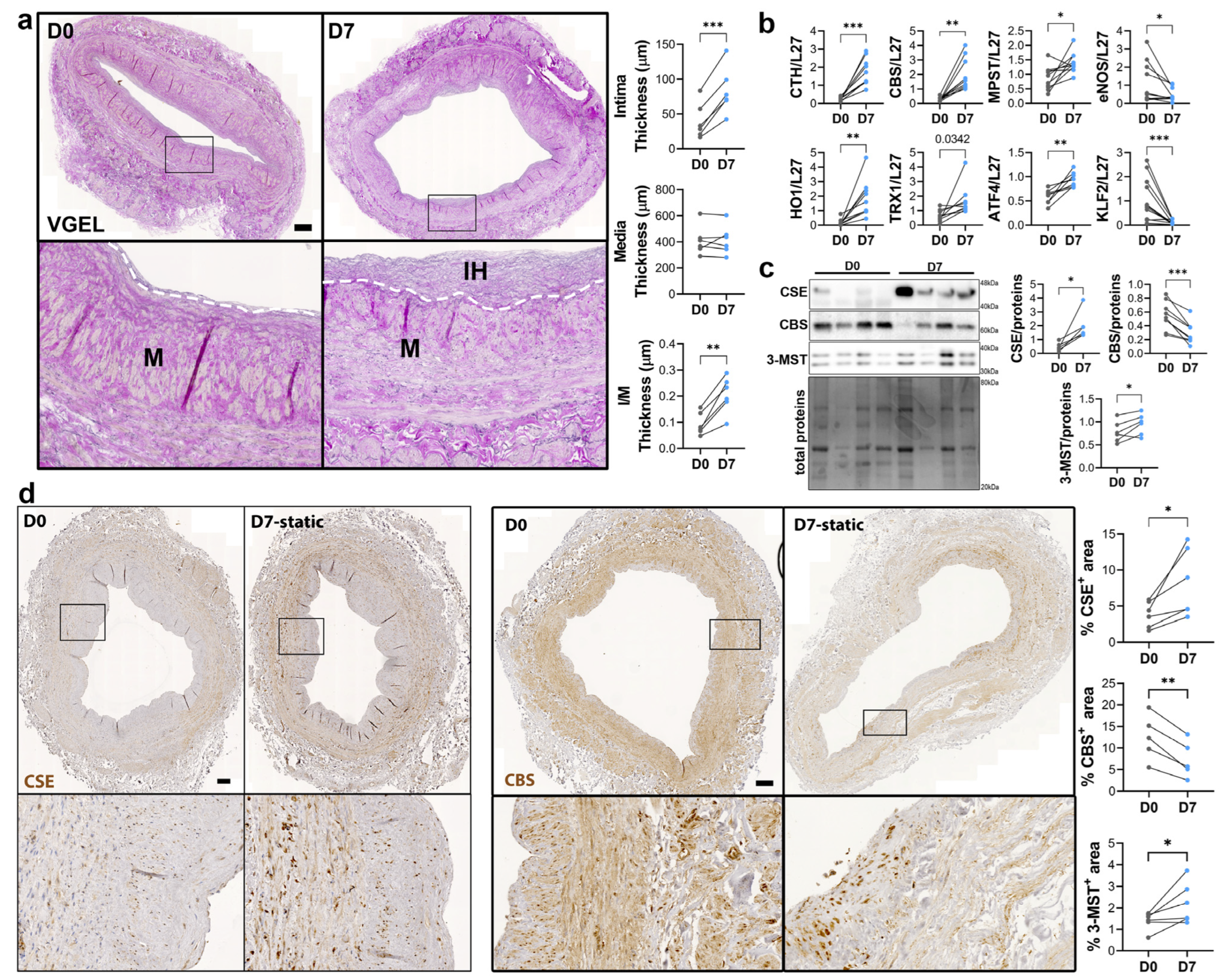
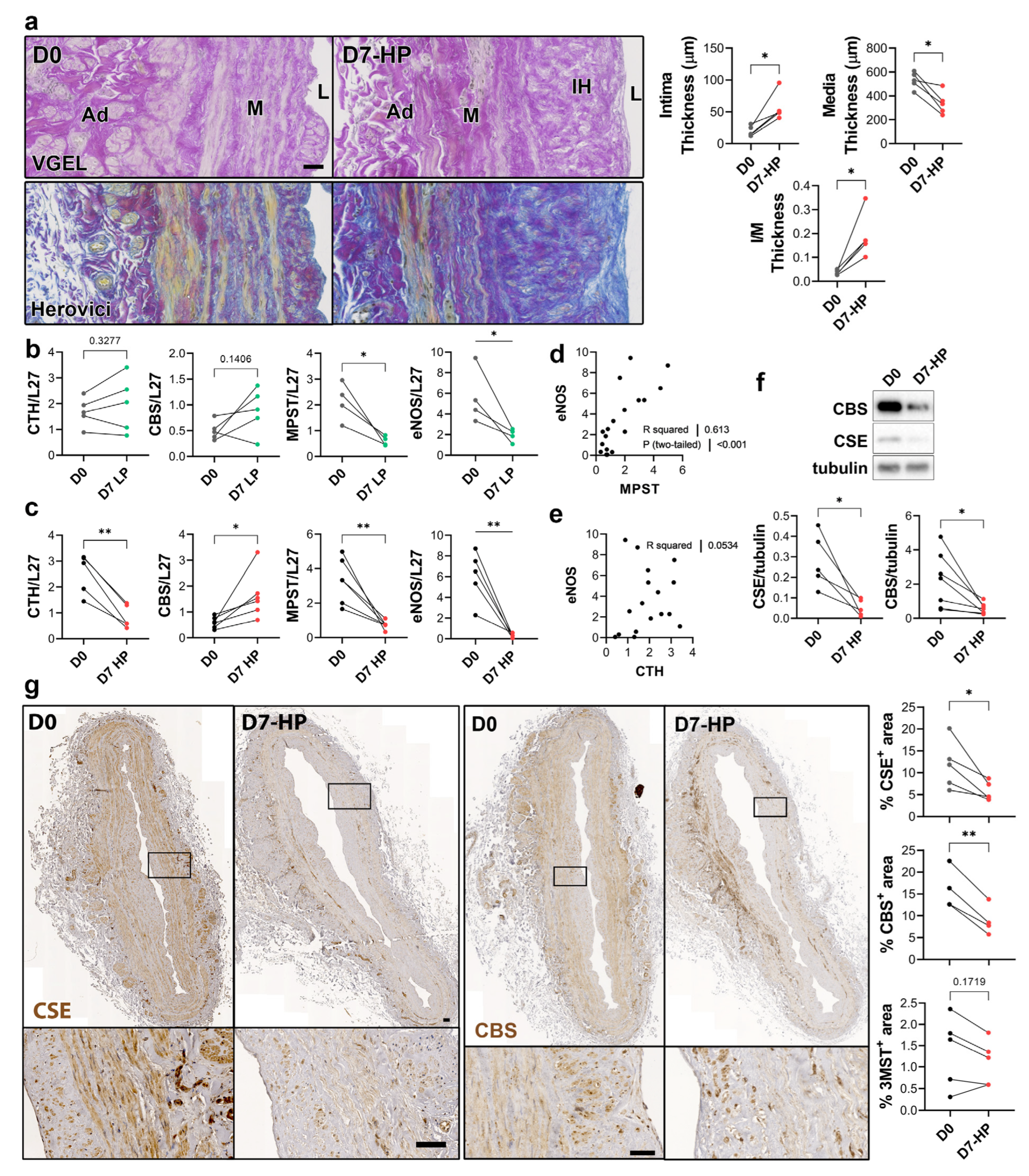
3.2. CSE Expression Is Regulated by Flow
3.3. CSE Regulates Human VSMC Migration
4. Discussion
4.1. Limitations
4.2. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Melnik, T.; Jordan, O.; Corpataux, J.M.; Delie, F.; Saucy, F. Pharmacological Prevention of Intimal Hyperplasia: A State-of-the-Art Review. Pharmacol. Ther. 2022, 235, 108157. [Google Scholar] [CrossRef]
- Simpson, E.L.; Kearns, B.; Stevenson, M.D.; Cantrell, A.J.; Littlewood, C.; Michaels, J.A. Enhancements to angioplasty for peripheral arterial occlusive disease: Systematic review, cost-effectiveness assessment and expected value of information analysis. Health Technol. Assess. 2014, 18, 1–252. [Google Scholar] [CrossRef]
- Cirino, G.; Szabo, C.; Papapetropoulos, A. Physiological roles of hydrogen sulfide in mammalian cells, tissues and organs. Physiol. Rev. 2022, 103, 31–276. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.K.; Bull, R.; Rains, J.L.; Bass, P.F.; Levine, S.N.; Reddy, S.; McVie, R.; Bocchini, J.A.; Achari, A.E.; Born, J.C.V.D.; et al. Low Levels of Hydrogen Sulfide in the Blood of Diabetes Patients and Streptozotocin-Treated Rats Causes Vascular Inflammation? Antioxid. Redox Signal 2010, 12, 1333–1337. [Google Scholar] [CrossRef] [PubMed]
- Islam, K.N.; Polhemus, D.J.; Donnarumma, E.; Brewster, L.P.; Lefer, D.J. Hydrogen Sulfide Levels and Nuclear Factor-Erythroid 2-Related Factor 2 (NRF2) Activity Are Attenuated in the Setting of Critical Limb Ischemia (CLI). J. Am. Heart Assoc. 2015, 4, e001986. [Google Scholar] [CrossRef] [PubMed]
- Beard, R.S., Jr.; Bearden, S.E. Vascular complications of cystathionine beta-synthase deficiency: Future directions for homocysteine-to-hydrogen sulfide research. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H13–H26. [Google Scholar] [CrossRef]
- Longchamp, A.; MacArthur, M.R.; Trocha, K.; Ganahl, J.; Mann, C.G.; Kip, P.; King, W.W.; Sharma, G.; Tao, M.; Mitchell, S.J.; et al. Plasma Hydrogen Sulfide Is Positively Associated With Post-operative Survival in Patients Undergoing Surgical Revascularization. Front. Cardiovasc. Med. 2021, 8, 750926. [Google Scholar] [CrossRef]
- Yang, G.; Li, H.; Tang, G.; Wu, L.; Zhao, K.; Cao, Q.; Xu, C.; Wang, R. Increased neointimal formation in cystathionine gamma-lyase deficient mice: Role of hydrogen sulfide in alpha5beta1-integrin and matrix metalloproteinase-2 expression in smooth muscle cells. J. Mol. Cell. Cardiol. 2012, 52, 677–688. [Google Scholar] [CrossRef]
- Macabrey, D.; Longchamp, A.; MacArthur, M.R.; Lambelet, M.; Urfer, S.; Deglise, S.; Allagnat, F. Sodium thiosulfate acts as a hydrogen sulfide mimetic to prevent intimal hyperplasia via inhibition of tubulin polymerisation. EBioMedicine 2022, 78, 103954. [Google Scholar] [CrossRef]
- Trocha, K.M.; Kip, P.; Tao, M.; MacArthur, M.R.; Trevino-Villarreal, J.H.; Longchamp, A.; Toussaint, W.; Lambrecht, B.N.; de Vries, M.R.; Quax, P.H.A.; et al. Short-term preoperative protein restriction attenuates vein graft disease via induction of cystathionine gamma-lyase. Cardiovasc. Res. 2020, 116, 416–428. [Google Scholar] [CrossRef]
- Meng, Q.H.; Yang, G.; Yang, W.; Jiang, B.; Wu, L.; Wang, R. Protective effect of hydrogen sulfide on balloon injury-induced neointima hyperplasia in rat carotid arteries. Am. J. Pathol. 2007, 170, 1406–1414. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Liang, G.; Zhang, F.; Chen, Y.; Zhang, H. Effect of hydrogen sulfide on restenosis of peripheral arteries after angioplasty. Mol. Med. Rep. 2012, 5, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Macabrey, D.; Deslarzes-Dubuis, C.; Longchamp, A.; Lambelet, M.; Ozaki, C.K.; Corpataux, J.M.; Allagnat, F.; Deglise, S. Hydrogen Sulphide Release via the Angiotensin Converting Enzyme Inhibitor Zofenopril Prevents Intimal Hyperplasia in Human Vein Segments and in a Mouse Model of Carotid Artery Stenosis. Eur. J. Vasc. Endovasc. Surg. 2022, 63, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Longchamp, A.; Kaur, K.; Macabrey, D.; Dubuis, C.; Corpataux, J.M.; Deglise, S.; Matson, J.B.; Allagnat, F. Hydrogen sulfide-releasing peptide hydrogel limits the development of intimal hyperplasia in human vein segments. Acta Biomater. 2019, 97, 374–384. [Google Scholar] [CrossRef]
- Cheng, Y.; Ndisang, J.F.; Tang, G.; Cao, K.; Wang, R. Hydrogen sulfide-induced relaxation of resistance mesenteric artery beds of rats. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2316–H2323. [Google Scholar] [CrossRef]
- Mustafa, A.K.; Sikka, G.; Gazi, S.K.; Steppan, J.; Jung, S.M.; Bhunia, A.K.; Barodka, V.M.; Gazi, F.K.; Barrow, R.K.; Wang, R.; et al. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ. Res. 2011, 109, 1259–1268. [Google Scholar] [CrossRef]
- Kiesworo, K.; MacArthur, M.R.; Kip, P.; Agius, T.; Macabrey, D.; Lambelet, M.; Hamard, L.; Ozaki, C.K.; Mitchell, J.R.; Deglise, S.; et al. Cystathionine-gamma-lyase overexpression modulates oxidized nicotinamide adenine dinucleotide biosynthesis and enhances neovascularization. JVS Vasc. Sci. 2023, 4, 100095. [Google Scholar] [CrossRef]
- Yang, G.; Wu, L.; Bryan, S.; Khaper, N.; Mani, S.; Wang, R. Cystathionine gamma-lyase deficiency and overproliferation of smooth muscle cells. Cardiovasc. Res. 2010, 86, 487–495. [Google Scholar] [CrossRef]
- Shibuya, N.; Mikami, Y.; Kimura, Y.; Nagahara, N.; Kimura, H. Vascular endothelium expresses 3-mercaptopyruvate sulfurtransferase and produces hydrogen sulfide. J. Biochem. 2009, 146, 623–626. [Google Scholar] [CrossRef]
- Bibli, S.I.; Hu, J.; Leisegang, M.S.; Wittig, J.; Zukunft, S.; Kapasakalidi, A.; Fisslthaler, B.; Tsilimigras, D.; Zografos, G.; Filis, K.; et al. Shear stress regulates cystathionine gamma lyase expression to preserve endothelial redox balance and reduce membrane lipid peroxidation. Redox Biol. 2020, 28, 101379. [Google Scholar] [CrossRef]
- Yuan, S.; Yurdagul, A., Jr.; Peretik, J.M.; Alfaidi, M.; Al Yafeai, Z.; Pardue, S.; Kevil, C.G.; Orr, A.W. Cystathionine gamma-Lyase Modulates Flow-Dependent Vascular Remodeling. Arter. Thromb. Vasc. Biol. 2018, 38, 2126–2136. [Google Scholar] [CrossRef]
- Macabrey, D.; Joniova, J.; Gasser, Q.; Bechelli, C.; Longchamp, A.; Urfer, S.; Lambelet, M.; Fu, C.Y.; Schwarz, G.; Wagnieres, G.; et al. Sodium thiosulfate, a source of hydrogen sulfide, stimulates endothelial cell proliferation and neovascularization. Front. Cardiovasc. Med. 2022, 9, 965965. [Google Scholar] [CrossRef] [PubMed]
- Saucy, F.; Probst, H.; Alonso, F.; Berard, X.; Deglise, S.; Dunoyer-Geindre, S.; Mazzolai, L.; Kruithof, E.; Haefliger, J.A.; Corpataux, J.M. Ex vivo pulsatile perfusion of human saphenous veins induces intimal hyperplasia and increased levels of the plasminogen activator inhibitor 1. Eur. Surg. Res. Eur. Chir. Forsch. Rech. Chir. Eur. 2010, 45, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Berard, X.; Deglise, S.; Alonso, F.; Saucy, F.; Meda, P.; Bordenave, L.; Corpataux, J.M.; Haefliger, J.A. Role of hemodynamic forces in the ex vivo arterialization of human saphenous veins. J. Vasc. Surg. 2013, 57, 1371–1382. [Google Scholar] [CrossRef]
- Longchamp, A.; Allagnat, F.; Berard, X.; Alonso, F.; Haefliger, J.A.; Deglise, S.; Corpataux, J.M. Procedure for human saphenous veins ex vivo perfusion and external reinforcement. J. Vis. Exp. 2014, e52079. [Google Scholar] [CrossRef]
- Longchamp, A.; Alonso, F.; Dubuis, C.; Allagnat, F.; Berard, X.; Meda, P.; Saucy, F.; Corpataux, J.M.; Deglise, S.; Haefliger, J.A. The use of external mesh reinforcement to reduce intimal hyperplasia and preserve the structure of human saphenous veins. Biomaterials 2014, 35, 2588–2599. [Google Scholar] [CrossRef] [PubMed]
- Allagnat, F.; Dubuis, C.; Lambelet, M.; Le Gal, L.; Alonso, F.; Corpataux, J.M.; Deglise, S.; Haefliger, J.A. Connexin37 reduces smooth muscle cell proliferation and intimal hyperplasia in a mouse model of carotid artery ligation. Cardiovasc. Res. 2017, 113, 805–816. [Google Scholar] [CrossRef]
- Teuscher, A.C.; Statzer, C.; Pantasis, S.; Bordoli, M.R.; Ewald, C.Y. Assessing Collagen Deposition During Aging in Mammalian Tissue and in Caenorhabditis elegans. Methods Mol. Biol. 2019, 1944, 169–188. [Google Scholar] [CrossRef]
- Hine, C.; Harputlugil, E.; Zhang, Y.; Ruckenstuhl, C.; Lee, B.C.; Brace, L.; Longchamp, A.; Trevino-Villarreal, J.H.; Mejia, P.; Ozaki, C.K.; et al. Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell 2015, 160, 132–144. [Google Scholar] [CrossRef]
- Lin, V.S.; Lippert, A.R.; Chang, C.J. Cell-trappable fluorescent probes for endogenous hydrogen sulfide signaling and imaging H2O2-dependent H2S production. Proc. Natl. Acad. Sci. USA 2013, 110, 7131–7135. [Google Scholar] [CrossRef]
- Suarez-Arnedo, A.; Torres Figueroa, F.; Clavijo, C.; Arbelaez, P.; Cruz, J.C.; Munoz-Camargo, C. An image J plugin for the high throughput image analysis of in vitro scratch wound healing assays. PLoS ONE 2020, 15, e0232565. [Google Scholar] [CrossRef] [PubMed]
- Corsello, T.; Komaravelli, N.; Casola, A. Role of Hydrogen Sulfide in NRF2- and Sirtuin-Dependent Maintenance of Cellular Redox Balance. Antioxidants 2018, 7, 129. [Google Scholar] [CrossRef]
- Nicholson, C.K.; Lambert, J.P.; Molkentin, J.D.; Sadoshima, J.; Calvert, J.W. Thioredoxin 1 is essential for sodium sulfide-mediated cardioprotection in the setting of heart failure. Arter. Thromb. Vasc. Biol. 2013, 33, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.; Dong, J.; Jin, S.; Teng, X.; Wu, Y. Endogenous hydrogen sulfide-mediated MAPK inhibition preserves endothelial function through TXNIP signaling. Free Radic. Biol. Med. 2017, 110, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wu, L.; Wang, R. Pro-apoptotic effect of endogenous H2S on human aorta smooth muscle cells. FASEB J. 2006, 20, 553–555. [Google Scholar] [CrossRef]
- Bibli, S.I.; Hu, J.; Sigala, F.; Wittig, I.; Heidler, J.; Zukunft, S.; Tsilimigras, D.I.; Randriamboavonjy, V.; Wittig, J.; Kojonazarov, B.; et al. Cystathionine gamma Lyase Sulfhydrates the RNA Binding Protein Human Antigen R to Preserve Endothelial Cell Function and Delay Atherogenesis. Circulation 2019, 139, 101–114. [Google Scholar] [CrossRef]
- Bucci, M.; Papapetropoulos, A.; Vellecco, V.; Zhou, Z.; Zaid, A.; Giannogonas, P.; Cantalupo, A.; Dhayade, S.; Karalis, K.P.; Wang, R.; et al. cGMP-dependent protein kinase contributes to hydrogen sulfide-stimulated vasorelaxation. PLoS ONE 2012, 7, e53319. [Google Scholar] [CrossRef]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef]
- Chakraborty, R.; Chatterjee, P.; Dave, J.M.; Ostriker, A.C.; Greif, D.M.; Rzucidlo, E.M.; Martin, K.A. Targeting smooth muscle cell phenotypic switching in vascular disease. JVS Vasc. Sci. 2021, 2, 79–94. [Google Scholar] [CrossRef]
- Chappell, J.; Harman, J.L.; Narasimhan, V.M.; Yu, H.; Foote, K.; Simons, B.D.; Bennett, M.R.; Jorgensen, H.F. Extensive Proliferation of a Subset of Differentiated, yet Plastic, Medial Vascular Smooth Muscle Cells Contributes to Neointimal Formation in Mouse Injury and Atherosclerosis Models. Circ. Res. 2016, 119, 1313–1323. [Google Scholar] [CrossRef]
- Worssam, M.D.; Lambert, J.; Oc, S.; Taylor, J.C.; Taylor, A.L.; Dobnikar, L.; Chappell, J.; Harman, J.L.; Figg, N.L.; Finigan, A.; et al. Cellular mechanisms of oligoclonal vascular smooth muscle cell expansion in cardiovascular disease. Cardiovasc. Res. 2022, 119, 1279–1294. [Google Scholar] [CrossRef]
- Wang, Y.; Nanda, V.; Direnzo, D.; Ye, J.; Xiao, S.; Kojima, Y.; Howe, K.L.; Jarr, K.U.; Flores, A.M.; Tsantilas, P.; et al. Clonally expanding smooth muscle cells promote atherosclerosis by escaping efferocytosis and activating the complement cascade. Proc. Natl. Acad. Sci. USA 2020, 117, 15818–15826. [Google Scholar] [CrossRef]
- Longchamp, A.; Allagnat, F.; Alonso, F.; Kuppler, C.; Dubuis, C.; Ozaki, C.K.; Mitchell, J.R.; Berceli, S.; Corpataux, J.M.; Deglise, S.; et al. Connexin43 Inhibition Prevents Human Vein Grafts Intimal Hyperplasia. PLoS ONE 2015, 10, e0138847. [Google Scholar] [CrossRef] [PubMed]
- Peleli, M.; Bibli, S.I.; Li, Z.; Chatzianastasiou, A.; Varela, A.; Katsouda, A.; Zukunft, S.; Bucci, M.; Vellecco, V.; Davos, C.H.; et al. Cardiovascular phenotype of mice lacking 3-mercaptopyruvate sulfurtransferase. Biochem. Pharmacol. 2020, 176, 113833. [Google Scholar] [CrossRef]
- D’Onofrio, N.; Prattichizzo, F.; Martino, E.; Anastasio, C.; Mele, L.; La Grotta, R.; Sardu, C.; Ceriello, A.; Marfella, R.; Paolisso, G.; et al. MiR-27b attenuates mitochondrial oxidative stress and inflammation in endothelial cells. Redox Biol. 2023, 62, 102681. [Google Scholar] [CrossRef] [PubMed]
- Demolli, S.; Doddaballapur, A.; Devraj, K.; Stark, K.; Manavski, Y.; Eckart, A.; Zehendner, C.M.; Lucas, T.; Korff, T.; Hecker, M.; et al. Shear stress-regulated miR-27b controls pericyte recruitment by repressing SEMA6A and SEMA6D. Cardiovasc. Res. 2017, 113, 681–691. [Google Scholar] [CrossRef]
- Ngowi, E.E.; Afzal, A.; Sarfraz, M.; Khattak, S.; Zaman, S.U.; Khan, N.H.; Li, T.; Jiang, Q.Y.; Zhang, X.; Duan, S.F.; et al. Role of hydrogen sulfide donors in cancer development and progression. Int. J. Biol. Sci. 2021, 17, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Bibli, S.I.; Hu, J.; Looso, M.; Weigert, A.; Ratiu, C.; Wittig, J.; Drekolia, M.K.; Tombor, L.; Randriamboavonjy, V.; Leisegang, M.S.; et al. Mapping the Endothelial Cell S-Sulfhydrome Highlights the Crucial Role of Integrin Sulfhydration in Vascular Function. Circulation 2021, 143, 935–948. [Google Scholar] [CrossRef] [PubMed]
- Misra, A.; Sheikh, A.Q.; Kumar, A.; Luo, J.; Zhang, J.; Hinton, R.B.; Smoot, L.; Kaplan, P.; Urban, Z.; Qyang, Y.; et al. Integrin beta3 inhibition is a therapeutic strategy for supravalvular aortic stenosis. J. Exp. Med. 2016, 213, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Slepian, M.J.; Massia, S.P.; Dehdashti, B.; Fritz, A.; Whitesell, L. Beta3-integrins rather than beta1-integrins dominate integrin-matrix interactions involved in postinjury smooth muscle cell migration. Circulation 1998, 97, 1818–1827. [Google Scholar] [CrossRef]
- Hu, J.; Leisegang, M.S.; Looso, M.; Dumbovic, G.; Wittig, J.; Drekolia, M.-K.; Guenther, S.; John, D.; Siragusa, M.; Zukunft, S.; et al. A novel role for cystathionine γ lyase in the control of p53: Impact on endothelial senescence and metabolic reprograming. bioRxiv 2022. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Li, Y.; Li, L.; Xu, S.; Feng, X. Hydrogen Sulfide (H2s)-Releasing Compounds: Therapeutic Potential in Cardiovascular Diseases. Front. Pharmacol. 2018, 9, 1066. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, S.; Deslarzes-Dubuis, C.; Urfer, S.; Lambelet, M.; Déglise, S.; Allagnat, F. Cystathionine Gamma Lyase Is Regulated by Flow and Controls Smooth Muscle Migration in Human Saphenous Vein. Antioxidants 2023, 12, 1731. https://doi.org/10.3390/antiox12091731
Zhao S, Deslarzes-Dubuis C, Urfer S, Lambelet M, Déglise S, Allagnat F. Cystathionine Gamma Lyase Is Regulated by Flow and Controls Smooth Muscle Migration in Human Saphenous Vein. Antioxidants. 2023; 12(9):1731. https://doi.org/10.3390/antiox12091731
Chicago/Turabian StyleZhao, Shuang, Céline Deslarzes-Dubuis, Severine Urfer, Martine Lambelet, Sébastien Déglise, and Florent Allagnat. 2023. "Cystathionine Gamma Lyase Is Regulated by Flow and Controls Smooth Muscle Migration in Human Saphenous Vein" Antioxidants 12, no. 9: 1731. https://doi.org/10.3390/antiox12091731
APA StyleZhao, S., Deslarzes-Dubuis, C., Urfer, S., Lambelet, M., Déglise, S., & Allagnat, F. (2023). Cystathionine Gamma Lyase Is Regulated by Flow and Controls Smooth Muscle Migration in Human Saphenous Vein. Antioxidants, 12(9), 1731. https://doi.org/10.3390/antiox12091731