
Brain Iron Metabolism, Redox Balance and Neurological Diseases



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
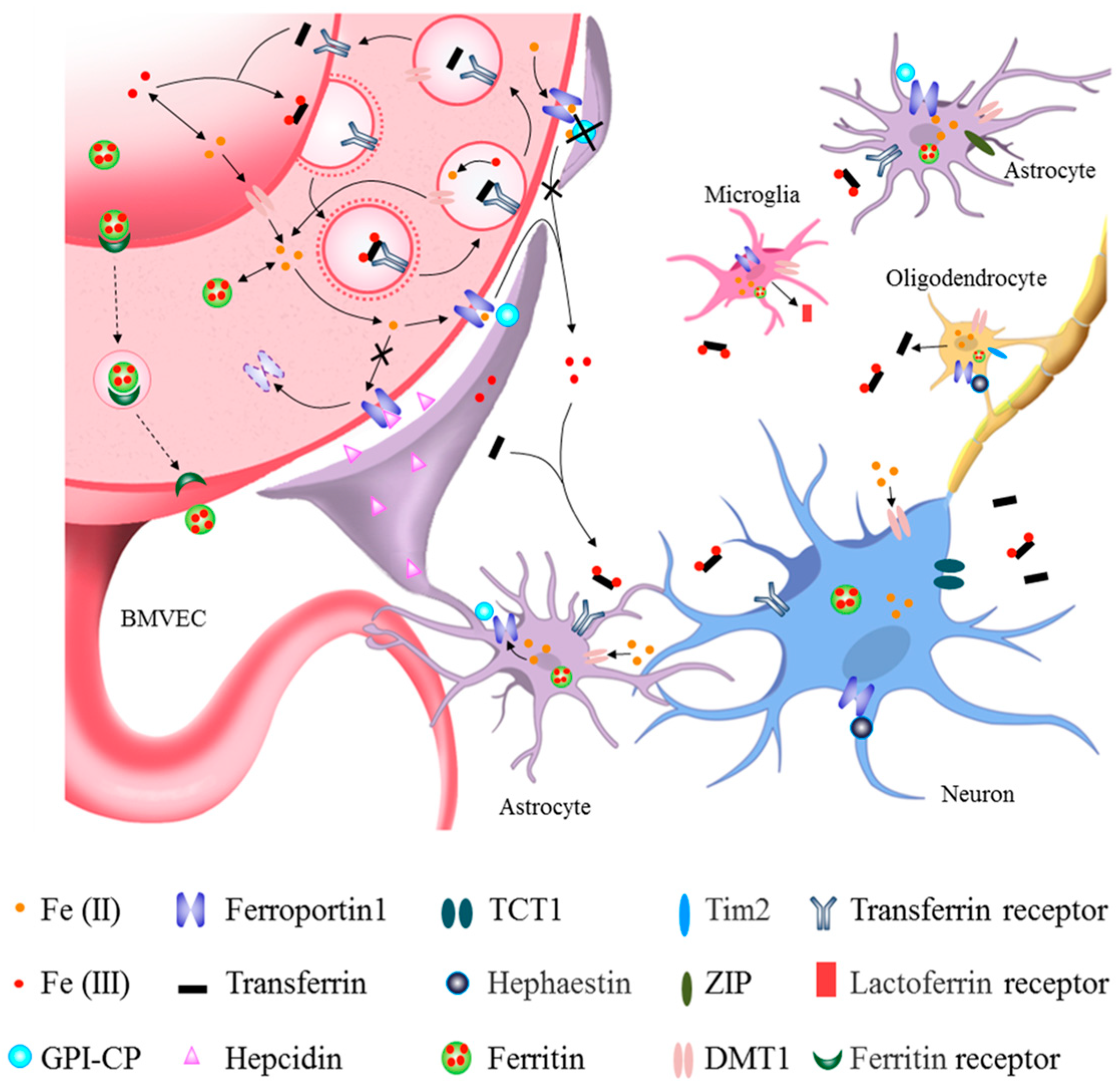
2. Brain Iron Metabolism
2.1. Iron Uptake into Brain Parenchyma from Circulatory Iron across the BBB
2.2. Iron Uptake and Metabolism in Neurons and Glia
3. Iron, Redox Balance and Oxidative Damage
3.1. Iron Dysregulation Induces ROS Generation
3.2. Iron-Induced Neuronal Death
4. Role of Iron in Neurological Diseases
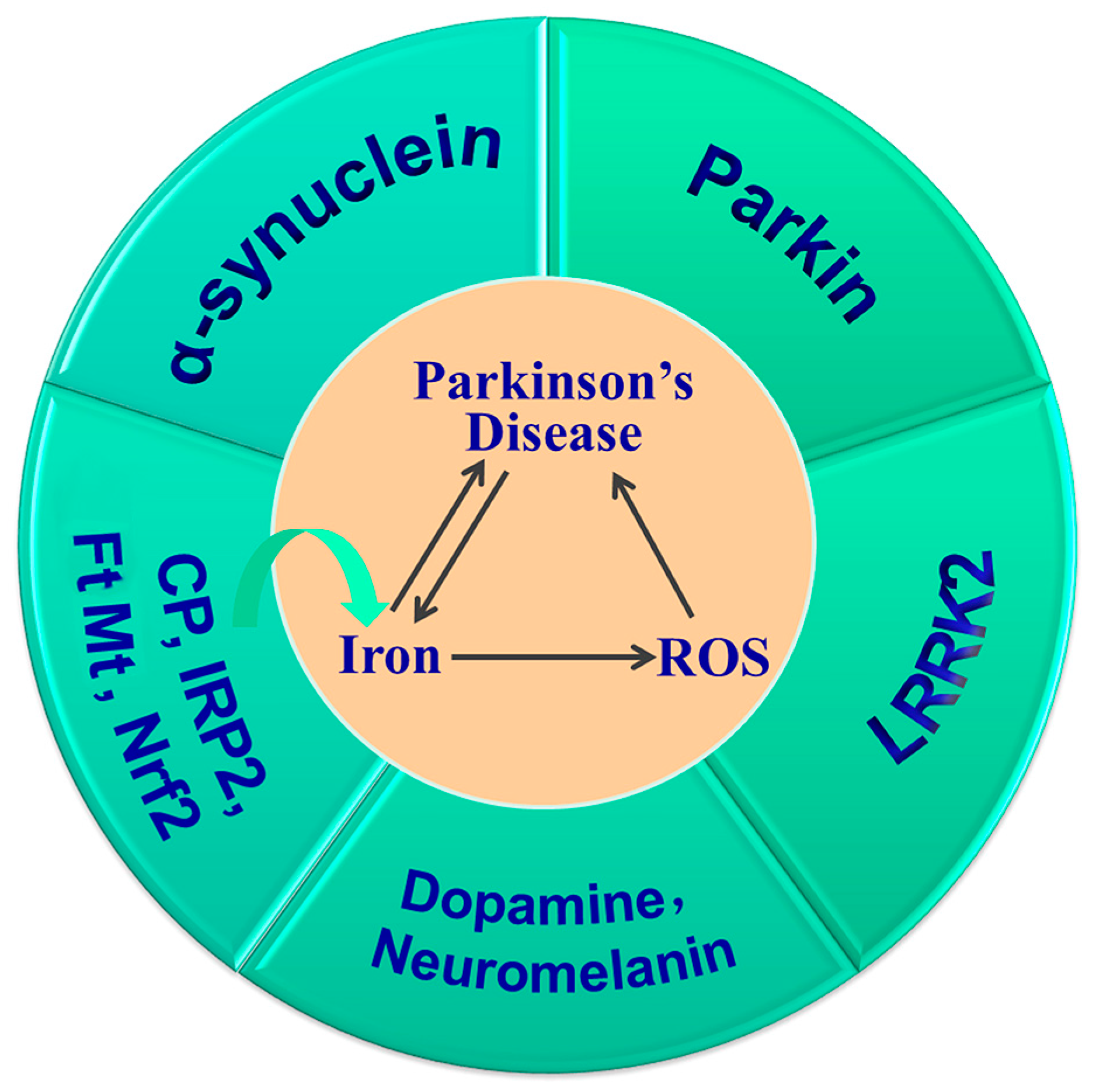
4.1. Iron and Parkinson’s Disease
4.1.1. Iron Dysregulation in PD
4.1.2. Iron Overload Exacerbates PD and Related Mechanisms
Iron and α-Synuclein
Iron, Dopamine and Neuromelanin
Iron and Parkin
Iron and Leucine-Rich Repeat Kinase 2
4.2. Iron and Alzheimer’s Disease
4.2.1. Iron Dysregulation in AD
4.2.2. Iron in Aβ and Tau Pathology
4.2.3. Mechanism of Iron and Oxidative Stress in AD Pathogenesis
4.3. Iron and Stroke
4.3.1. Iron Regulation in Ischemic Stroke
4.3.2. Mechanism Underlying Cell Death Induced by Iron Dysregulation
4.4. Iron and Neuropsychiatric Disorders
4.5. Iron and Abnormal Neurodevelopment
4.5.1. Iron Deficiency Affects Neurodevelopment and Function
4.5.2. Iron Deficiency Affects Development of Oligodendrocytes and Myelination
4.5.3. Mechanisms Underlying ID and Hippocampal Development
5. Targeting Iron Metabolism in the Treatment of Neurological Diseases
5.1. Iron Chelators
5.1.1. Iron Chelators in AD
5.1.2. Iron Chelators in PD
5.1.3. Iron Chelators in Stroke
5.2. Iron Chelators in New Administration Forms
5.3. Key Molecules of Brain Iron Metabolism as Targets
5.4. Iron Supplements
5.5. Antioxidants and Anti-Inflammatory Reagents Regulate Iron Metabolism as Targets
6. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef]
- Faucheux, B.A.; Nillesse, N.; Damier, P.; Spik, G.; Mouatt-Prigent, A.; Pierce, A.; Leveugle, B.; Kubis, N.; Hauw, J.J.; Agid, Y.; et al. Expression of lactoferrin receptors is increased in the mesencephalon of patients with Parkinson disease. Proc. Natl. Acad. Sci. USA 1995, 92, 9603–9607. [Google Scholar] [CrossRef]
- Peters, D.G.; Connor, J.R.; Meadowcroft, M.D. The relationship between iron dyshomeostasis and amyloidogenesis in Alzheimer’s disease: Two sides of the same coin. Neurobiol. Dis. 2015, 81, 49–65. [Google Scholar] [CrossRef]
- Riggins, T.; Miller, N.C.; Bauer, P.J.; Georgieff, M.K.; Nelson, C.A. Consequences of low neonatal iron status due to maternal diabetes mellitus on explicit memory performance in childhood. Dev. Neuropsychol. 2009, 34, 762–779. [Google Scholar] [CrossRef] [PubMed]
- Lozoff, B.; Beard, J.; Connor, J.; Barbara, F.; Georgieff, M.; Schallert, T. Long-lasting neural and behavioral effects of iron deficiency in infancy. Nutr. Rev. 2006, 64, S34–S43. discussion S72-91. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.R.; Menzies, S.L. Relationship of iron to oligodendrocytes and myelination. Glia 1996, 17, 83–93. [Google Scholar] [CrossRef]
- Wu, Q.; Hao, Q.; Li, H.; Wang, B.; Wang, P.; Jin, X.; Yu, P.; Gao, G.; Chang, Y.Z. Brain iron deficiency and affected contextual fear memory in mice with conditional Ferroportin1 ablation in the brain. FASEB J. 2021, 35, e21174. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Wang, P.; Han, Y.; Ma, Q.; Liu, Z.; Zhong, S.; Lu, Y.; Chen, R.; Sun, L.; Wu, Q.; et al. Ferrodifferentiation regulates neurodevelopment via ROS generation. Sci. China Life Sci. 2023. online ahead of print. [Google Scholar] [CrossRef]
- Ke, Y.; Ming Qian, Z. Iron misregulation in the brain: A primary cause of neurodegenerative disorders. Lancet Neurol. 2003, 2, 246–253. [Google Scholar] [CrossRef]
- Oshiro, S.; Morioka, M.S.; Kikuchi, M. Dysregulation of iron metabolism in Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Adv. Pharmacol. Sci. 2011, 2011, 378278. [Google Scholar] [CrossRef]
- You, L.; Yu, P.P.; Dong, T.; Guo, W.; Chang, S.; Zheng, B.; Ci, Y.; Wang, F.; Yu, P.; Gao, G.; et al. Astrocyte-derived hepcidin controls iron traffic at the blood–brain-barrier via regulating ferroportin 1 of microvascular endothelial cells. Cell Death Dis. 2022, 13, 667. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.M.; Fu, L.J.; Duan, X.L.; Crooks, D.R.; Yu, P.; Qian, Z.M.; Di, X.J.; Li, J.; Rouault, T.A.; Chang, Y.Z. Role of hepcidin in murine brain iron metabolism. Cell. Mol. Life Sci. 2010, 67, 123–133. [Google Scholar] [CrossRef]
- Jeong, S.Y.; David, S. Glycosylphosphatidylinositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. J. Biol. Chem. 2003, 278, 27144–27148. [Google Scholar] [CrossRef]
- Li, Z.D.; Li, H.; Kang, S.; Cui, Y.G.; Zheng, H.; Wang, P.; Han, K.; Yu, P.; Chang, Y.Z. The divergent effects of astrocyte ceruloplasmin on learning and memory function in young and old mice. Cell Death Dis. 2022, 13, 1006. [Google Scholar] [CrossRef]
- Levi, S.; Tiranti, V. Neurodegeneration with Brain Iron Accumulation Disorders: Valuable Models Aimed at Understanding the Pathogenesis of Iron Deposition. Pharmaceuticals 2019, 12, 27. [Google Scholar] [CrossRef]
- Wise, R.M.; Wagener, A.; Fietzek, U.M.; Klopstock, T.; Mosharov, E.V.; Zucca, F.A.; Sulzer, D.; Zecca, L.; Burbulla, L.F. Interactions of dopamine, iron, and alpha-synuclein linked to dopaminergic neuron vulnerability in Parkinson’s disease and Neurodegeneration with Brain Iron Accumulation disorders. Neurobiol. Dis. 2022, 175, 105920. [Google Scholar] [CrossRef]
- Crapper McLachlan, D.R.; Dalton, A.J.; Kruck, T.P.; Bell, M.Y.; Smith, W.L.; Kalow, W.; Andrews, D.F. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 1991, 337, 1304–1308. [Google Scholar] [CrossRef]
- Kupershmidt, L.; Amit, T.; Bar-Am, O.; Youdim, M.B.; Weinreb, O. The novel multi-target iron chelating-radical scavenging compound M30 possesses beneficial effects on major hallmarks of Alzheimer’s disease. Antioxid. Redox Signal. 2012, 17, 860–877. [Google Scholar] [CrossRef]
- Zeng, X.; An, H.; Yu, F.; Wang, K.; Zheng, L.; Zhou, W.; Bao, Y.; Yang, J.; Shen, N.; Huang, D. Benefits of Iron Chelators in the Treatment of Parkinson’s Disease. Neurochem. Res. 2021, 46, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Stephenson, G.; Ben Shachar, D. Ironing iron out in Parkinson’s disease and other neurodegenerative diseases with iron chelators: A lesson from 6-hydroxydopamine and iron chelators, desferal and VK-28. Ann. N. Y. Acad. Sci. 2004, 1012, 306–325. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.; Roberts, R.L.; Bero, C. Deferoxamine posttreatment reduces ischemic brain injury in neonatal rats. Stroke 1994, 25, 1039–1045. [Google Scholar] [CrossRef]
- Patt, A.; Horesh, I.R.; Berger, E.M.; Harken, A.H.; Repine, J.E. Iron depletion or chelation reduces ischemia/reperfusion-induced edema in gerbil brains. J. Pediatr. Surg. 1990, 25, 224–227; discussion 227–228. [Google Scholar] [CrossRef]
- Moos, T.; Morgan, E.H. Transferrin and transferrin receptor function in brain barrier systems. Cell. Mol. Neurobiol. 2000, 20, 77–95. [Google Scholar] [CrossRef]
- Yang, W.M.; Jung, K.J.; Lee, M.O.; Lee, Y.S.; Lee, Y.H.; Nakagawa, S.; Niwa, M.; Cho, S.S.; Kim, D.W. Transient expression of iron transport proteins in the capillary of the developing rat brain. Cell. Mol. Neurobiol. 2011, 31, 93–99. [Google Scholar] [CrossRef]
- Zheng, H.; Guo, X.; Kang, S.; Li, Z.; Tian, T.; Li, J.; Wang, F.; Yu, P.; Chang, S.; Chang, Y.Z. Cdh5-mediated Fpn1 deletion exerts neuroprotective effects during the acute phase and inhibitory effects during the recovery phase of ischemic stroke. Cell Death Dis. 2023, 14, 161. [Google Scholar] [CrossRef]
- Klomp, L.W.; Farhangrazi, Z.S.; Dugan, L.L.; Gitlin, J.D. Ceruloplasmin gene expression in the murine central nervous system. J. Clin. Investig. 1996, 98, 207–215. [Google Scholar] [CrossRef]
- Patel, B.N.; Dunn, R.J.; David, S. Alternative RNA splicing generates a glycosylphosphatidylinositol-anchored form of ceruloplasmin in mammalian brain. J. Biol. Chem. 2000, 275, 4305–4310. [Google Scholar] [CrossRef]
- Wang, L.; Liu, X.; You, L.H.; Ci, Y.Z.; Chang, S.; Yu, P.; Gao, G.; Chang, Y.Z. Hepcidin and iron regulatory proteins coordinately regulate ferroportin 1 expression in the brain of mice. J. Cell. Physiol. 2019, 234, 7600–7607. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Qian, Z.M.; Duan, X.L.; Chang, Y.Z. The structure of DMT1 and its regulation. Chin. J. Neuroanat. 2004, 20, 205–209. [Google Scholar]
- Yu, P.; Chang, Y.Z. Brain Iron Metabolism and Regulation. Adv. Exp. Med. Biol. 2019, 1173, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Zheng, W.; Zlokovic, B.V. Brain capillary endothelium and choroid plexus epithelium regulate transport of transferrin-bound and free iron into the rat brain. J. Neurochem. 2004, 88, 813–820. [Google Scholar] [CrossRef]
- Thirupathi, A.; Chang, Y.Z. Brain Iron Metabolism and CNS Diseases. Adv. Exp. Med. Biol. 2019, 1173, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.; Devraj, K.; Ingram, J.; Slagle-Webb, B.; Madhankumar, A.B.; Liu, X.; Klinger, M.; Simpson, I.A.; Connor, J.R. Ferritin: A novel mechanism for delivery of iron to the brain and other organs. Am. J. Physiol. Cell Physiol. 2007, 293, C641–C649. [Google Scholar] [CrossRef]
- Qian, Z.M.; Chang, Y.Z.; Zhu, L.; Yang, L.; Du, J.R.; Ho, K.P.; Wang, Q.; Li, L.Z.; Wang, C.Y.; Ge, X.; et al. Development and iron-dependent expression of hephaestin in different brain regions of rats. J. Cell. Biochem. 2007, 102, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A.; Zhang, D.L.; Jeong, S.Y. Brain iron homeostasis, the choroid plexus, and localization of iron transport proteins. Metab. Brain Dis. 2009, 24, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, J.M. Iron and oxygen radicals in brain. Ann. Neurol. 1992, 32, S16–S21. [Google Scholar] [CrossRef]
- Gałazka-Friedman, J.; Friedman, A. Controversies about iron in parkinsonian and control substantia nigra. Acta Neurobiol. Exp. (Wars) 1997, 57, 217–225. [Google Scholar] [PubMed]
- Lu, L.N.; Qian, Z.M.; Wu, K.C.; Yung, W.H.; Ke, Y. Expression of Iron Transporters and Pathological Hallmarks of Parkinson’s and Alzheimer’s Diseases in the Brain of Young, Adult, and Aged Rats. Mol. Neurobiol. 2017, 54, 5213–5224. [Google Scholar] [CrossRef]
- Wang, Z.; Zeng, Y.N.; Yang, P.; Jin, L.Q.; Xiong, W.C.; Zhu, M.Z.; Zhang, J.Z.; He, X.; Zhu, X.H. Axonal iron transport in the brain modulates anxiety-related behaviors. Nat. Chem. Biol. 2019, 15, 1214–1222. [Google Scholar] [CrossRef]
- Moos, T. Brain iron homeostasis. Dan. Med. Bull. 2002, 49, 279–301. [Google Scholar]
- Xu, J.Q.; Liu, Q.Q.; Huang, S.Y.; Duan, C.Y.; Lu, H.B.; Cao, Y.; Hu, J.Z. The lymphatic system: A therapeutic target for central nervous system disorders. Neural Regen. Res. 2023, 18, 1249–1256. [Google Scholar] [CrossRef]
- Hahn, P.; Qian, Y.; Dentchev, T.; Chen, L.; Beard, J.; Harris, Z.L.; Dunaief, J.L. Disruption of ceruloplasmin and hephaestin in mice causes retinal iron overload and retinal degeneration with features of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2004, 101, 13850–13855. [Google Scholar] [CrossRef]
- Ke, Y.; Qian, Z.M. Brain iron metabolism: Neurobiology and neurochemistry. Prog. Neurobiol. 2007, 83, 149–173. [Google Scholar] [CrossRef]
- Attieh, Z.K.; Mukhopadhyay, C.K.; Seshadri, V.; Tripoulas, N.A.; Fox, P.L. Ceruloplasmin ferroxidase activity stimulates cellular iron uptake by a trivalent cation-specific transport mechanism. J. Biol. Chem. 1999, 274, 1116–1123. [Google Scholar] [CrossRef]
- Andersen, H.H.; Johnsen, K.B.; Moos, T. Iron deposits in the chronically inflamed central nervous system and contributes to neurodegeneration. Cell. Mol. Life Sci. 2014, 71, 1607–1622. [Google Scholar] [CrossRef]
- Lane, D.J.; Robinson, S.R.; Czerwinska, H.; Bishop, G.M.; Lawen, A. Two routes of iron accumulation in astrocytes: Ascorbate-dependent ferrous iron uptake via the divalent metal transporter (DMT1) plus an independent route for ferric iron. Biochem. J. 2010, 432, 123–132. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Y.; Song, N.; Wang, J.; Jiang, H.; Xie, J. New Progress on the Role of Glia in Iron Metabolism and Iron-Induced Degeneration of Dopamine Neurons in Parkinson’s Disease. Front. Mol. Neurosci. 2017, 10, 455. [Google Scholar] [CrossRef] [PubMed]
- Oshiro, S.; Kawahara, M.; Kuroda, Y.; Zhang, C.; Cai, Y.; Kitajima, S.; Shirao, M. Glial cells contribute more to iron and aluminum accumulation but are more resistant to oxidative stress than neuronal cells. Biochim. Biophys. Acta 2000, 1502, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Todorich, B.; Zhang, X.; Slagle-Webb, B.; Seaman, W.E.; Connor, J.R. Tim-2 is the receptor for H-ferritin on oligodendrocytes. J. Neurochem. 2008, 107, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Bishop, G.M.; Dang, T.N.; Dringen, R.; Robinson, S.R. Accumulation of non-transferrin-bound iron by neurons, astrocytes, and microglia. Neurotox. Res. 2011, 19, 443–451. [Google Scholar] [CrossRef]
- Fillebeen, C.; Ruchoux, M.M.; Mitchell, V.; Vincent, S.; Benaïssa, M.; Pierce, A. Lactoferrin is synthesized by activated microglia in the human substantia nigra and its synthesis by the human microglial CHME cell line is upregulated by tumor necrosis factor alpha or 1-methyl-4-phenylpyridinium treatment. Brain Res. Mol. Brain Res. 2001, 96, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Urrutia, P.; Aguirre, P.; Esparza, A.; Tapia, V.; Mena, N.P.; Arredondo, M.; González-Billault, C.; Núñez, M.T. Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J. Neurochem. 2013, 126, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; You, L.; Chang, Y. Iron metabolism in Parkinson’s disease. In Oxidative Stress and Redox Signalling in Parkinson’s Disease; The Royal Society of Chemistry: London, UK, 2017; pp. 255–276. [Google Scholar]
- Bradbury, M.W. Transport of iron in the blood–brain-cerebrospinal fluid system. J. Neurochem. 1997, 69, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Siddappa, A.J.; Rao, R.B.; Wobken, J.D.; Casperson, K.; Leibold, E.A.; Connor, J.R.; Georgieff, M.K. Iron deficiency alters iron regulatory protein and iron transport protein expression in the perinatal rat brain. Pediatr. Res. 2003, 53, 800–807. [Google Scholar] [CrossRef]
- Pantopoulos, K. Iron metabolism and the IRE/IRP regulatory system: An update. Ann. N. Y. Acad. Sci. 2004, 1012, 1–13. [Google Scholar] [CrossRef]
- Muckenthaler, M.U.; Galy, B.; Hentze, M.W. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu. Rev. Nutr. 2008, 28, 197–213. [Google Scholar] [CrossRef]
- You, L.H.; Yan, C.Z.; Zheng, B.J.; Ci, Y.Z.; Chang, S.Y.; Yu, P.; Gao, G.F.; Li, H.Y.; Dong, T.Y.; Chang, Y.Z. Astrocyte hepcidin is a key factor in LPS-induced neuronal apoptosis. Cell Death Dis. 2017, 8, e2676. [Google Scholar] [CrossRef]
- Kautz, L.; Jung, G.; Du, X.; Gabayan, V.; Chapman, J.; Nasoff, M.; Nemeth, E.; Ganz, T. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of β-thalassemia. Blood 2015, 126, 2031–2037. [Google Scholar] [CrossRef]
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, E.; Ganz, T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 2014, 46, 678–684. [Google Scholar] [CrossRef]
- Ashraf, A.; Michaelides, C.; Walker, T.A.; Ekonomou, A.; Suessmilch, M.; Sriskanthanathan, A.; Abraha, S.; Parkes, A.; Parkes, H.G.; Geraki, K.; et al. Regional Distributions of Iron, Copper and Zinc and Their Relationships with Glia in a Normal Aging Mouse Model. Front. Aging Neurosci. 2019, 11, 351. [Google Scholar] [CrossRef]
- Merkofer, M.; Kissner, R.; Hider, R.C.; Brunk, U.T.; Koppenol, W.H. Fenton chemistry and iron chelation under physiologically relevant conditions: Electrochemistry and kinetics. Chem. Res. Toxicol. 2006, 19, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Cabantchik, Z.I. Labile iron in cells and body fluids: Physiology, pathology, and pharmacology. Front. Pharmacol. 2014, 5, 45. [Google Scholar] [CrossRef]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef] [PubMed]
- von der Mark, C.; Ivanov, R.; Eutebach, M.; Maurino, V.G.; Bauer, P.; Brumbarova, T. Reactive oxygen species coordinate the transcriptional responses to iron availability in Arabidopsis. J. Exp. Bot. 2021, 72, 2181–2195. [Google Scholar] [CrossRef]
- Koskenkorva-Frank, T.S.; Weiss, G.; Koppenol, W.H.; Burckhardt, S. The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: Insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free Radic. Biol. Med. 2013, 65, 1174–1194. [Google Scholar] [CrossRef]
- Tewari, R.K.; Hadacek, F.; Sassmann, S.; Lang, I. Iron deprivation-induced reactive oxygen species generation leads to non-autolytic PCD in Brassica napus leaves. Environ. Exp. Bot. 2013, 91, 74–83. [Google Scholar] [CrossRef]
- Bush, A.I. Metals and neuroscience. Curr. Opin. Chem. Biol. 2000, 4, 184–191. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Y.; Zhang, J.H.; Han, K.; Zhang, X.; Bai, X.; You, L.H.; Yu, P.; Shi, Z.; Chang, Y.Z.; et al. Astrocyte hepcidin ameliorates neuronal loss through attenuating brain iron deposition and oxidative stress in APP/PS1 mice. Free Radic. Biol. Med. 2020, 158, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wang, J.; Rogers, J.; Xie, J.X. Brain Iron Metabolism Dysfunction in Parkinson’s Disease. Mol. Neurobiol. 2017, 54, 3078–3101. [Google Scholar] [CrossRef]
- Masaldan, S.; Bush, A.I.; Devos, D.; Rolland, A.S.; Moreau, C. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radic. Biol. Med. 2019, 133, 221–233. [Google Scholar] [CrossRef]
- Zhang, Y.; Bai, X.; Zhang, Y.; Yao, S.; Cui, Y.; You, L.H.; Yu, P.; Chang, Y.Z.; Gao, G. Hippocampal Iron Accumulation Impairs Synapses and Memory via Suppressing Furin Expression and Downregulating BDNF Maturation. Mol. Neurobiol. 2022, 59, 5574–5590. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.H.; Kwon, K.C.; Hwang, D.J.; Koo, J.H.; Um, H.S.; Song, H.S.; Kim, J.S.; Jang, Y.; Cho, J.Y. Treadmill Exercise Alleviates Brain Iron Dyshomeostasis Accelerating Neuronal Amyloid-β Production, Neuronal Cell Death, and Cognitive Impairment in Transgenic Mice Model of Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 3208–3223. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Yang, K.S.; Kang, S.W.; Woo, H.A.; Chang, T.S. Controlled elimination of intracellular H(2)O(2): Regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxid. Redox Signal. 2005, 7, 619–626. [Google Scholar] [CrossRef]
- Lillig, C.H.; Berndt, C.; Holmgren, A. Glutaredoxin systems. Biochim. Biophys. Acta 2008, 1780, 1304–1317. [Google Scholar] [CrossRef]
- Wilson, D.F. Oxidative phosphorylation: Regulation and role in cellular and tissue metabolism. J. Physiol. 2017, 595, 7023–7038. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Wu, J.R.; Tuo, Q.Z.; Lei, P. Ferroptosis, a Recent Defined Form of Critical Cell Death in Neurological Disorders. J. Mol. Neurosci. 2018, 66, 197–206. [Google Scholar] [CrossRef]
- Zuo, Y.; Xie, J.; Li, X.; Li, Y.; Thirupathi, A.; Zhang, J.; Yu, P.; Gao, G.; Chang, Y.; Shi, Z. Ferritinophagy-Mediated Ferroptosis Involved in Paraquat-Induced Neurotoxicity of Dopaminergic Neurons: Implication for Neurotoxicity in PD. Oxid. Med. Cell. Longev. 2021, 2021, 9961628. [Google Scholar] [CrossRef]
- Parkinson, J. An Essay on the Shaking Palsy; Whittingham & Rowland, Neely & Jones: London, UK, 1817. [Google Scholar]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Przedborski, S. The two-century journey of Parkinson disease research. Nat. Rev. Neurosci. 2017, 18, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Sofic, E.; Riederer, P.; Heinsen, H.; Beckmann, H.; Reynolds, G.P.; Hebenstreit, G.; Youdim, M.B. Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain. J. Neural Transm. 1988, 74, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Wells, F.R.; Agid, F.; Agid, Y.; Lees, A.J.; Jenner, P.; Marsden, C.D. Increased nigral iron content in postmortem parkinsonian brain. Lancet 1987, 2, 1219–1220. [Google Scholar] [CrossRef]
- He, Y.; Thong, P.S.; Lee, T.; Leong, S.K.; Mao, B.Y.; Dong, F.; Watt, F. Dopaminergic cell death precedes iron elevation in MPTP-injected monkeys. Free. Radic. Biol. Med. 2003, 35, 540–547. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Lee, T.; Leong, S.K. Time course of dopaminergic cell death and changes in iron, ferritin and transferrin levels in the rat substantia nigra after 6-hydroxydopamine (6-OHDA) lesioning. Free Radic. Res. 1999, 31, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Ndayisaba, A.; Kaindlstorfer, C.; Wenning, G.K. Iron in Neurodegeneration—Cause or Consequence? Front. Neurosci. 2019, 13, 180. [Google Scholar] [CrossRef]
- Biondetti, E.; Santin, M.D.; Valabrègue, R.; Mangone, G.; Gaurav, R.; Pyatigorskaya, N.; Hutchison, M.; Yahia-Cherif, L.; Villain, N.; Habert, M.O.; et al. The spatiotemporal changes in dopamine, neuromelanin and iron characterizing Parkinson’s disease. Brain 2021, 144, 3114–3125. [Google Scholar] [CrossRef]
- You, L.H.; Li, F.; Wang, L.; Zhao, S.E.; Wang, S.M.; Zhang, L.L.; Zhang, L.H.; Duan, X.L.; Yu, P.; Chang, Y.Z. Brain iron accumulation exacerbates the pathogenesis of MPTP-induced Parkinson’s disease. Neuroscience 2015, 284, 234–246. [Google Scholar] [CrossRef]
- Ci, Y.Z.; Li, H.; You, L.H.; Jin, Y.; Zhou, R.; Gao, G.; Hoi, M.P.M.; Wang, C.; Chang, Y.Z.; Yu, P. Iron overload induced by IRP2 gene knockout aggravates symptoms of Parkinson’s disease. Neurochem. Int. 2020, 134, 104657. [Google Scholar] [CrossRef]
- You, L.; Wang, J.; Liu, T.; Zhang, Y.; Han, X.; Wang, T.; Guo, S.; Dong, T.; Xu, J.; Anderson, G.J.; et al. Targeted Brain Delivery of Rabies Virus Glycoprotein 29-Modified Deferoxamine-Loaded Nanoparticles Reverses Functional Deficits in Parkinsonian Mice. ACS Nano 2018, 12, 4123–4139. [Google Scholar] [CrossRef]
- Weinreb, O.; Mandel, S.; Youdim, M.B.H.; Amit, T. Targeting dysregulation of brain iron homeostasis in Parkinson’s disease by iron chelators. Free. Radic. Biol. Med. 2013, 62, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Kaur, D.; Yantiri, F.; Rajagopalan, S.; Kumar, J.; Mo, J.Q.; Boonplueang, R.; Viswanath, V.; Jacobs, R.; Yang, L.; Beal, M.F.; et al. Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: A novel therapy for Parkinson’s disease. Neuron 2003, 37, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Loréal, O.; Turlin, B.; Pigeon, C.; Moisan, A.; Ropert, M.; Morice, P.; Gandon, Y.; Jouanolle, A.M.; Vérin, M.; Hider, R.C.; et al. Aceruloplasminemia: New clinical, pathophysiological and therapeutic insights. J. Hepatol. 2002, 36, 851–856. [Google Scholar] [CrossRef]
- Harris, Z.L.; Klomp, L.W.; Gitlin, J.D. Aceruloplasminemia: An inherited neurodegenerative disease with impairment of iron homeostasis. Am. J. Clin. Nutr. 1998, 67, 972s–977s. [Google Scholar] [CrossRef]
- Hellman, N.E.; Gitlin, J.D. Ceruloplasmin metabolism and function. Annu. Rev. Nutr. 2002, 22, 439–458. [Google Scholar] [CrossRef]
- Jeong, S.Y.; David, S. Age-related changes in iron homeostasis and cell death in the cerebellum of ceruloplasmin-deficient mice. J. Neurosci. 2006, 26, 9810–9819. [Google Scholar] [CrossRef]
- Patel, B.N.; Dunn, R.J.; Jeong, S.Y.; Zhu, Q.; Julien, J.P.; David, S. Ceruloplasmin regulates iron levels in the CNS and prevents free radical injury. J. Neurosci. 2002, 22, 6578–6586. [Google Scholar] [CrossRef]
- Ayton, S.; Lei, P.; Duce, J.A.; Wong, B.X.; Sedjahtera, A.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I. Ceruloplasmin dysfunction and therapeutic potential for Parkinson disease. Ann. Neurol. 2013, 73, 554–559. [Google Scholar] [CrossRef]
- Ayton, S.; Lei, P.; Adlard, P.A.; Volitakis, I.; Cherny, R.A.; Bush, A.I.; Finkelstein, D.I. Iron accumulation confers neurotoxicity to a vulnerable population of nigral neurons: Implications for Parkinson’s disease. Mol. Neurodegener. 2014, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- De Domenico, I.; McVey Ward, D.; Kaplan, J. Regulation of iron acquisition and storage: Consequences for iron-linked disorders. Nat. Rev. Mol. Cell Biol. 2008, 9, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Meyron-Holtz, E.G.; Ghosh, M.C.; Iwai, K.; LaVaute, T.; Brazzolotto, X.; Berger, U.V.; Land, W.; Ollivierre-Wilson, H.; Grinberg, A.; Love, P.; et al. Genetic ablations of iron regulatory proteins 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. EMBO J. 2004, 23, 386–395. [Google Scholar] [CrossRef]
- LaVaute, T.; Smith, S.; Cooperman, S.; Iwai, K.; Land, W.; Meyron-Holtz, E.; Drake, S.K.; Miller, G.; Abu-Asab, M.; Tsokos, M.; et al. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat. Genet. 2001, 27, 209–214. [Google Scholar] [CrossRef]
- Ghosh, M.C.; Zhang, D.L.; Rouault, T.A. Iron misregulation and neurodegenerative disease in mouse models that lack iron regulatory proteins. Neurobiol. Dis. 2015, 81, 66–75. [Google Scholar] [CrossRef]
- Jia, F.; Li, H.; Jiao, Q.; Li, C.; Fu, L.; Cui, C.; Jiang, H.; Zhang, L. Deubiquitylase OTUD3 prevents Parkinson’s disease through stabilizing iron regulatory protein 2. Cell Death Dis. 2022, 13, 418. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.H.; Nie, G.; Duan, X.L.; Rouault, T.; Wu, W.S.; Ning, B.; Zhang, N.; Chang, Y.Z.; Zhao, B.L. Neuroprotective mechanism of mitochondrial ferritin on 6-hydroxydopamine-induced dopaminergic cell damage: Implication for neuroprotection in Parkinson’s disease. Antioxid. Redox Signal. 2010, 13, 783–796. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Cui, Y.; Ren, Q.; Yan, B.; Zhao, Y.; Yu, P.; Gao, G.; Shi, H.; Chang, S.; Chang, Y.Z. Mitochondrial ferritin attenuates cerebral ischaemia/reperfusion injury by inhibiting ferroptosis. Cell Death Dis. 2021, 12, 447. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Cui, Y.; Liu, Y.; Li, Z.; Bai, H.; Zhao, Y.; Chang, Y.Z. Mitochondrial ferritin alleviates apoptosis by enhancing mitochondrial bioenergetics and stimulating glucose metabolism in cerebral ischemia reperfusion. Redox Biol. 2022, 57, 102475. [Google Scholar] [CrossRef] [PubMed]
- Holze, C.; Michaudel, C.; Mackowiak, C.; Haas, D.A.; Benda, C.; Hubel, P.; Pennemann, F.L.; Schnepf, D.; Wettmarshausen, J.; Braun, M.; et al. Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat. Immunol. 2018, 19, 130–140. [Google Scholar] [CrossRef]
- Han, K.; Jin, X.; Guo, X.; Cao, G.; Tian, S.; Song, Y.; Zuo, Y.; Yu, P.; Gao, G.; Chang, Y.Z. Nrf2 knockout altered brain iron deposition and mitigated age-related motor dysfunction in aging mice. Free Radic. Biol. Med. 2021, 162, 592–602. [Google Scholar] [CrossRef]
- Wang, Z.L.; Yuan, L.; Li, W.; Li, J.Y. Ferroptosis in Parkinson’s disease: Glia-neuron crosstalk. Trends Mol. Med. 2022, 28, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Hare, D.J.; Double, K.L. Iron and dopamine: A toxic couple. Brain 2016, 139, 1026–1035. [Google Scholar] [CrossRef]
- Perfeito, R.; Lázaro, D.F.; Outeiro, T.F.; Rego, A.C. Linking alpha-synuclein phosphorylation to reactive oxygen species formation and mitochondrial dysfunction in SH-SY5Y cells. Mol. Cell. Neurosci. 2014, 62, 51–59. [Google Scholar] [CrossRef]
- Lu, Y.; Prudent, M.; Fauvet, B.; Lashuel, H.A.; Girault, H.H. Phosphorylation of α-Synuclein at Y125 and S129 alters its metal binding properties: Implications for understanding the role of α-Synuclein in the pathogenesis of Parkinson’s Disease and related disorders. ACS Chem. Neurosci. 2011, 2, 667–675. [Google Scholar] [CrossRef]
- Zucca, F.A.; Segura-Aguilar, J.; Ferrari, E.; Muñoz, P.; Paris, I.; Sulzer, D.; Sarna, T.; Casella, L.; Zecca, L. Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson’s disease. Prog. Neurobiol. 2017, 155, 96–119. [Google Scholar] [CrossRef]
- Roth, J.A.; Singleton, S.; Feng, J.; Garrick, M.; Paradkar, P.N. Parkin regulates metal transport via proteasomal degradation of the 1B isoforms of divalent metal transporter 1. J. Neurochem. 2010, 113, 454–464. [Google Scholar] [CrossRef]
- Mamais, A.; Kluss, J.H.; Bonet-Ponce, L.; Landeck, N.; Langston, R.G.; Smith, N.; Beilina, A.; Kaganovich, A.; Ghosh, M.C.; Pellegrini, L.; et al. Mutations in LRRK2 linked to Parkinson disease sequester Rab8a to damaged lysosomes and regulate transferrin-mediated iron uptake in microglia. PLoS Biol. 2021, 19, e3001480. [Google Scholar] [CrossRef]
- Castellani, R.; Siedlak, S.; Perry, G.; Smith, M. Sequestration of iron by Lewy bodies in Parkinson’s disease. Acta Neuropathol. 2000, 100, 111–114. [Google Scholar] [CrossRef]
- Golts, N.; Snyder, H.; Frasier, M.; Theisler, C.; Choi, P.; Wolozin, B. Magnesium inhibits spontaneous and iron-induced aggregation of alpha-synuclein. J. Biol. Chem. 2002, 277, 16116–16123. [Google Scholar] [CrossRef] [PubMed]
- Friedlich, A.L.; Tanzi, R.E.; Rogers, J.T. The 5′-untranslated region of Parkinson’s disease alpha-synuclein messengerRNA contains a predicted iron responsive element. Mol. Psychiatry 2007, 12, 222–223. [Google Scholar] [CrossRef] [PubMed]
- McDowall, J.S.; Ntai, I.; Honeychurch, K.C.; Hart, J.P.; Colin, P.; Schneider, B.L.; Brown, D.R. Alpha-synuclein ferrireductase activity is detectible in vivo, is altered in Parkinson’s disease and increases the neurotoxicity of DOPAL. Mol. Cell. Neurosci. 2017, 85, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wen, X.; Jiang, H.; Wang, J.; Song, N.; Xie, J. Interactions between iron and α-synuclein pathology in Parkinson’s disease. Free. Radic. Biol. Med. 2019, 141, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Jia, F.; Song, N.; Wang, W.; Du, X.; Chi, Y.; Jiang, H. High Dietary Iron Supplement Induces the Nigrostriatal Dopaminergic Neurons Lesion in Transgenic Mice Expressing Mutant A53T Human Alpha-Synuclein. Front. Aging Neurosci. 2018, 10, 97. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef]
- Reynolds, M.R.; Berry, R.W.; Binder, L.I. Nitration in neurodegeneration: Deciphering the “Hows” “nYs”. Biochemistry 2007, 46, 7325–7336. [Google Scholar] [CrossRef] [PubMed]
- Hodara, R.; Norris, E.H.; Giasson, B.I.; Mishizen-Eberz, A.J.; Lynch, D.R.; Lee, V.M.; Ischiropoulos, H. Functional consequences of alpha-synuclein tyrosine nitration: Diminished binding to lipid vesicles and increased fibril formation. J. Biol. Chem. 2004, 279, 47746–47753. [Google Scholar] [CrossRef]
- Burai, R.; Ait-Bouziad, N.; Chiki, A.; Lashuel, H.A. Elucidating the Role of Site-Specific Nitration of α-Synuclein in the Pathogenesis of Parkinson’s Disease via Protein Semisynthesis and Mutagenesis. J. Am. Chem. Soc. 2015, 137, 5041–5052. [Google Scholar] [CrossRef]
- Kleinknecht, A.; Popova, B.; Lázaro, D.F.; Pinho, R.; Valerius, O.; Outeiro, T.F.; Braus, G.H. C-Terminal Tyrosine Residue Modifications Modulate the Protective Phosphorylation of Serine 129 of α-Synuclein in a Yeast Model of Parkinson’s Disease. PLoS Genet. 2016, 12, e1006098. [Google Scholar] [CrossRef]
- Shavali, S.; Combs, C.K.; Ebadi, M. Reactive macrophages increase oxidative stress and alpha-synuclein nitration during death of dopaminergic neuronal cells in co-culture: Relevance to Parkinson’s disease. Neurochem. Res. 2006, 31, 85–94. [Google Scholar] [CrossRef]
- Reynolds, A.D.; Glanzer, J.G.; Kadiu, I.; Ricardo-Dukelow, M.; Chaudhuri, A.; Ciborowski, P.; Cerny, R.; Gelman, B.; Thomas, M.P.; Mosley, R.L.; et al. Nitrated alpha-synuclein-activated microglial profiling for Parkinson’s disease. J. Neurochem. 2008, 104, 1504–1525. [Google Scholar] [CrossRef]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Feany, M.B. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 2005, 8, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Wu, S.; Wang, Z.; Ge, L.; Rizak, J.D.; Wu, J.; Li, J.; Xu, L.; Lv, L.; Yin, Y.; et al. Phosphorylated α-Synuclein Accumulations and Lewy Body-like Pathology Distributed in Parkinson’s Disease-Related Brain Areas of Aged Rhesus Monkeys Treated with MPTP. Neuroscience 2018, 379, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Ko, L.W.; Kulathingal, J.; Jiang, P.; Sevlever, D.; Yen, S.H. Oxidative stress-induced phosphorylation, degradation and aggregation of alpha-synuclein are linked to upregulated CK2 and cathepsin D. Eur. J. Neurosci. 2007, 26, 863–874. [Google Scholar] [CrossRef]
- Xu, J.; Kao, S.Y.; Lee, F.J.; Song, W.; Jin, L.W.; Yankner, B.A. Dopamine-dependent neurotoxicity of alpha-synuclein: A mechanism for selective neurodegeneration in Parkinson disease. Nat. Med. 2002, 8, 600–606. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Park. Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Holemans, S.; Javoy-Agid, F.; Agid, Y.; De Paermentier, F.; Laterre, E.C.; Maloteaux, J.M. Sulfonylurea binding sites in normal human brain and in Parkinson’s disease, progressive supranuclear palsy and Huntington’s disease. Brain Res. 1994, 642, 327–333. [Google Scholar] [CrossRef]
- Takanashi, M.; Mochizuki, H.; Yokomizo, K.; Hattori, N.; Mori, H.; Yamamura, Y.; Mizuno, Y. Iron accumulation in the substantia nigra of autosomal recessive juvenile parkinsonism (ARJP). Park. Relat. Disord. 2001, 7, 311–314. [Google Scholar] [CrossRef]
- Zhang, C.W.; Tai, Y.K.; Chai, B.H.; Chew, K.C.M.; Ang, E.T.; Tsang, F.; Tan, B.W.Q.; Hong, E.T.E.; Asad, A.B.A.; Chuang, K.H.; et al. Transgenic Mice Overexpressing the Divalent Metal Transporter 1 Exhibit Iron Accumulation and Enhanced Parkin Expression in the Brain. Neuromolecular Med. 2017, 19, 375–386. [Google Scholar] [CrossRef]
- Kluss, J.H.; Mamais, A.; Cookson, M.R. LRRK2 links genetic and sporadic Parkinson’s disease. Biochem. Soc. Trans. 2019, 47, 651–661. [Google Scholar] [CrossRef]
- Barrett, J.C.; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; Barmada, M.M.; et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef]
- Pyatigorskaya, N.; Sharman, M.; Corvol, J.C.; Valabregue, R.; Yahia-Cherif, L.; Poupon, F.; Cormier-Dequaire, F.; Siebner, H.; Klebe, S.; Vidailhet, M.; et al. High nigral iron deposition in LRRK2 and Parkin mutation carriers using R2* relaxometry. Mov. Disord. 2015, 30, 1077–1084. [Google Scholar] [CrossRef]
- Gillardon, F.; Schmid, R.; Draheim, H. Parkinson’s disease-linked leucine-rich repeat kinase 2(R1441G) mutation increases proinflammatory cytokine release from activated primary microglial cells and resultant neurotoxicity. Neuroscience 2012, 208, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Moehle, M.S.; Webber, P.J.; Tse, T.; Sukar, N.; Standaert, D.G.; DeSilva, T.M.; Cowell, R.M.; West, A.B. LRRK2 inhibition attenuates microglial inflammatory responses. J. Neurosci. 2012, 32, 1602–1611. [Google Scholar] [CrossRef] [PubMed]
- Hattula, K.; Furuhjelm, J.; Tikkanen, J.; Tanhuanpää, K.; Laakkonen, P.; Peränen, J. Characterization of the Rab8-specific membrane traffic route linked to protrusion formation. J. Cell Sci. 2006, 119, 4866–4877. [Google Scholar] [CrossRef]
- Rivero-Ríos, P.; Romo-Lozano, M.; Madero-Pérez, J.; Thomas, A.P.; Biosa, A.; Greggio, E.; Hilfiker, S. The G2019S variant of leucine-rich repeat kinase 2 (LRRK2) alters endolysosomal trafficking by impairing the function of the GTPase RAB8A. J. Biol. Chem. 2019, 294, 4738–4758. [Google Scholar] [CrossRef]
- Jia, R.; Liu, Y.; Shuai, K.; Zhou, C.; Chen, L.; Zhu, L.; Wu, X.M. The Relationship between Iron and LRRK2 in a 6-OHDA-Induced Parkinson’s Disease Model. Int. J. Mol. Sci. 2023, 24, 3709. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Amyloid beta-protein and the genetics of Alzheimer’s disease. J. Biol. Chem. 1996, 271, 18295–18298. [Google Scholar] [CrossRef]
- Lane, D.J.R.; Ayton, S.; Bush, A.I. Iron and Alzheimer’s Disease: An Update on Emerging Mechanisms. J. Alzheimers Dis. 2018, 64, S379–S395. [Google Scholar] [CrossRef]
- Jan, A.T.; Azam, M.; Rahman, S.; Almigeiti, A.M.S.; Choi, D.H.; Lee, E.J.; Haq, Q.M.R.; Choi, I. Perspective Insights into Disease Progression, Diagnostics, and Therapeutic Approaches in Alzheimer’s Disease: A Judicious Update. Front. Aging Neurosci. 2017, 9, 356. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.R.; Menzies, S.L.; St Martin, S.M.; Mufson, E.J. A histochemical study of iron, transferrin, and ferritin in Alzheimer’s diseased brains. J. Neurosci. Res. 1992, 31, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Meadowcroft, M.D.; Connor, J.R.; Smith, M.B.; Yang, Q.X. MRI and Histological Analysis of Beta-Amyloid Plaques in Both Human Alzheimer’s Disease and APP/PS1 Transgenic Mice. J. Magn. Reson. Imaging 2009, 29, 997–1007. [Google Scholar] [CrossRef]
- Chen, L.L.; Fan, Y.G.; Zhao, L.X.; Zhang, Q.; Wang, Z.Y. The metal ion hypothesis of Alzheimer’s disease and the anti-neuroinflammatory effect of metal chelators. Bioorg. Chem. 2023, 131, 106301. [Google Scholar] [CrossRef]
- Ge, X.; Zhang, Y.; Zuo, Y.; Israr, M.; Li, B.; Yu, P.; Gao, G.; Chang, Y.Z.; Shi, Z. Transcriptomic analysis reveals the molecular mechanism of Alzheimer-related neuropathology induced by sevoflurane in mice. J. Cell. Biochem. 2019, 120, 17555–17565. [Google Scholar] [CrossRef]
- Zhao, Y.S.; Zhang, L.H.; Yu, P.P.; Gou, Y.J.; Zhao, J.; You, L.H.; Wang, Z.Y.; Zheng, X.; Yan, L.J.; Yu, P.; et al. Ceruloplasmin, a Potential Therapeutic Agent for Alzheimer’s Disease. Antioxid. Redox Signal. 2018, 28, 1323–1337. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gou, Y.J.; Zhang, Y.; Li, J.; Han, K.; Xu, Y.; Li, H.; You, L.H.; Yu, P.; Chang, Y.Z.; et al. Hepcidin overexpression in astrocytes alters brain iron metabolism and protects against amyloid-β induced brain damage in mice. Cell Death Discov. 2020, 6, 113. [Google Scholar] [CrossRef]
- Kuiper, M.A.; Mulder, C.; van Kamp, G.J.; Scheltens, P.; Wolters, E.C. Cerebrospinal fluid ferritin levels of patients with Parkinson’s disease, Alzheimer’s disease, and multiple system atrophy. J. Neural Transm. Park. Dis. Dement. Sect. 1994, 7, 109–114. [Google Scholar] [CrossRef]
- Gao, G.; Chang, Y.Z. Mitochondrial ferritin in the regulation of brain iron homeostasis and neurodegenerative diseases. Front. Pharmacol. 2014, 5, 19. [Google Scholar] [CrossRef]
- Wu, Q.; Wu, W.S.; Su, L.; Zheng, X.; Wu, W.Y.; Santambrogio, P.; Gou, Y.J.; Hao, Q.; Wang, P.N.; Li, Y.R.; et al. Mitochondrial Ferritin Is a Hypoxia-Inducible Factor 1alpha-Inducible Gene That Protects from Hypoxia-Induced Cell Death in Brain. Antioxid. Redox Signal. 2019, 30, 198–212. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.S.; Zhao, Y.S.; Shi, Z.H.; Chang, S.Y.; Nie, G.J.; Duan, X.L.; Zhao, S.M.; Wu, Q.; Yang, Z.L.; Zhao, B.L.; et al. Mitochondrial ferritin attenuates β-amyloid-induced neurotoxicity: Reduction in oxidative damage through the Erk/P38 mitogen-activated protein kinase pathways. Antioxid. Redox Signal. 2013, 18, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.D.; Ma, J.K.; Luo, Z.Y.; Tai, Q.X.; Wang, P.; Guan, P.P. Transferrin is responsible for mediating the effects of iron ions on the regulation of anterior pharynx-defective-1alpha/beta and Presenilin 1 expression via PGE(2) and PGD(2) at the early stage of Alzheimer’s Disease. Aging 2018, 10, 3117–3135. [Google Scholar] [CrossRef]
- Dong, X.; Gao, W.; Shao, T.; Xie, H.; Bai, J.; Zhao, J.; Chai, X. Age-related changes of brain iron load changes in the frontal cortex in APPswe/PS1DeltaE9 transgenic mouse model of Alzheimer’s disease. J. Trace Elem. Med. Biol. 2015, 30, 118–123. [Google Scholar] [CrossRef]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Rowan, M.J.; Selkoe, D.J. Amyloid-beta oligomers: Their production, toxicity and therapeutic inhibition. Biochem. Soc. Trans. 2002, 30, 552–557. [Google Scholar] [CrossRef]
- Guillemot, J.; Canuel, M.; Essalmani, R.; Prat, A.; Seidah, N.G. Implication of the proprotein convertases in iron homeostasis: Proprotein convertase 7 sheds human transferrin receptor 1 and furin activates hepcidin. Hepatology 2013, 57, 2514–2524. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, X.; Bai, X.; Yao, S.; Chang, Y.Z.; Gao, G. The emerging role of furin in neurodegenerative and neuropsychiatric diseases. Transl. Neurodegener. 2022, 11, 39. [Google Scholar] [CrossRef]
- Silvestri, L.; Camaschella, C. A potential pathogenetic role of iron in Alzheimer’s disease. J. Cell. Mol. Med. 2008, 12, 1548–1550. [Google Scholar] [CrossRef]
- Rogers, J.T.; Randall, J.D.; Cahill, C.M.; Eder, P.S.; Huang, X.; Gunshin, H.; Leiter, L.; McPhee, J.; Sarang, S.S.; Utsuki, T.; et al. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 2002, 277, 45518–45528. [Google Scholar] [CrossRef]
- Gamblin, T.C.; King, M.E.; Kuret, J.; Berry, R.W.; Binder, L.I. Oxidative regulation of fatty acid-induced tau polymerization. Biochemistry 2000, 39, 14203–14210. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zheng, J.; Liu, G.; Zeng, C.; Xu, E.; Zhu, W.; Anderson, G.J.; Chen, H. High Dietary Iron Disrupts Iron Homeostasis and Induces Amyloid-β and Phospho-τ Expression in the Hippocampus of Adult Wild-Type and APP/PS1 Transgenic Mice. J. Nutr. 2019, 149, 2247–2254. [Google Scholar] [CrossRef]
- Multhaup, G.; Huber, O.; Buee, L.; Galas, M.C. Amyloid Precursor Protein (APP) Metabolites APP Intracellular Fragment (AICD), Abeta42, and Tau in Nuclear Roles. J. Biol. Chem. 2015, 290, 23515–23522. [Google Scholar] [CrossRef]
- Duce, J.A.; Tsatsanis, A.; Cater, M.A.; James, S.A.; Robb, E.; Wikhe, K.; Leong, S.L.; Perez, K.; Johanssen, T.; Greenough, M.A.; et al. Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 2010, 142, 857–867. [Google Scholar] [CrossRef]
- Onukwufor, J.O.; Dirksen, R.T.; Wojtovich, A.P. Iron Dysregulation in Mitochondrial Dysfunction and Alzheimer’s Disease. Antioxidants 2022, 11, 692. [Google Scholar] [CrossRef] [PubMed]
- Duan, G.; Li, J.; Duan, Y.; Zheng, C.; Guo, Q.; Li, F.; Zheng, J.; Yu, J.; Zhang, P.; Wan, M.; et al. Mitochondrial Iron Metabolism: The Crucial Actors in Diseases. Molecules 2022, 28, 29. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Dexter, D.T.; Crichton, R.R. Ageing, neuroinflammation and neurodegeneration. Front. Biosci. (Schol. Ed.) 2015, 7, 189–204. [Google Scholar] [CrossRef]
- Wang, P.; Wu, Q.; Wu, W.; Li, H.; Guo, Y.; Yu, P.; Gao, G.; Shi, Z.; Zhao, B.; Chang, Y.Z. Mitochondrial Ferritin Deletion Exacerbates β-Amyloid-Induced Neurotoxicity in Mice. Oxid. Med. Cell. Longev. 2017, 2017, 1020357. [Google Scholar] [CrossRef] [PubMed]
- Salami, A.; Papenberg, G.; Sitnikov, R.; Laukka, E.J.; Persson, J.; Kalpouzos, G. Elevated neuroinflammation contributes to the deleterious impact of iron overload on brain function in aging. Neuroimage 2021, 230, 117792. [Google Scholar] [CrossRef] [PubMed]
- Nnah, I.C.; Lee, C.H.; Wessling-Resnick, M. Iron potentiates microglial interleukin-1beta secretion induced by amyloid-beta. J. Neurochem. 2020, 154, 177–189. [Google Scholar] [CrossRef]
- Nakamura, K.; Kawakami, T.; Yamamoto, N.; Tomizawa, M.; Fujiwara, T.; Ishii, T.; Harigae, H.; Ogasawara, K. Activation of the NLRP3 inflammasome by cellular labile iron. Exp. Hematol. 2016, 44, 116–124. [Google Scholar] [CrossRef]
- Zhou, S.; Du, X.; Xie, J.; Wang, J. Interleukin-6 regulates iron-related proteins through c-Jun N-terminal kinase activation in BV2 microglial cell lines. PLoS ONE 2017, 12, e0180464. [Google Scholar] [CrossRef] [PubMed]
- Rathore, K.I.; Redensek, A.; David, S. Iron homeostasis in astrocytes and microglia is differentially regulated by TNF-alpha and TGF-beta1. Glia 2012, 60, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Bolognin, S.; Drago, D.; Messori, L.; Zatta, P. Chelation therapy for neurodegenerative diseases. Med. Res. Rev. 2009, 29, 547–570. [Google Scholar] [CrossRef]
- Radi, E.; Formichi, P.; Battisti, C.; Federico, A. Apoptosis and oxidative stress in neurodegenerative diseases. J. Alzheimers Dis. 2014, 42 (Suppl. S3), S125–S152. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.C.V.; Khatri, P. Stroke. Lancet 2020, 396, 129–142. [Google Scholar] [CrossRef]
- Ding, H.; Yan, C.Z.; Shi, H.; Zhao, Y.S.; Chang, S.Y.; Yu, P.; Wu, W.S.; Zhao, C.Y.; Chang, Y.Z.; Duan, X.L. Hepcidin is involved in iron regulation in the ischemic brain. PLoS ONE 2011, 6, e25324. [Google Scholar] [CrossRef]
- Wang, P.; Ren, Q.; Shi, M.; Liu, Y.; Bai, H.; Chang, Y.Z. Overexpression of Mitochondrial Ferritin Enhances Blood–brain Barrier Integrity Following Ischemic Stroke in Mice by Maintaining Iron Homeostasis in Endothelial Cells. Antioxidants 2022, 11, 1257. [Google Scholar] [CrossRef]
- Zhang, D.-L.; Rouault, T.A. How does hepcidin hinder ferroportin activity? Blood 2018, 131, 839–840. [Google Scholar] [CrossRef]
- Yang, L.; Wang, D.; Wang, X.T.; Lu, Y.P.; Zhu, L. The roles of hypoxia-inducible Factor-1 and iron regulatory protein 1 in iron uptake induced by acute hypoxia. Biochem. Biophys. Res. Commun. 2018, 507, 128–135. [Google Scholar] [CrossRef]
- Cheah, J.H.; Kim, S.F.; Hester, L.D.; Clancy, K.W.; Patterson, S.E., 3rd; Papadopoulos, V.; Snyder, S.H. NMDA receptor-nitric oxide transmission mediates neuronal iron homeostasis via the GTPase Dexras1. Neuron 2006, 51, 431–440. [Google Scholar] [CrossRef]
- Riedl, S.J.; Salvesen, G.S. The apoptosome: Signalling platform of cell death. Nat. Rev. Mol. Cell Biol. 2007, 8, 405–413. [Google Scholar] [CrossRef]
- Ott, M.; Robertson, J.D.; Gogvadze, V.; Zhivotovsky, B.; Orrenius, S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. USA 2002, 99, 1259–1263. [Google Scholar] [CrossRef]
- Mantzaris, M.D.; Bellou, S.; Skiada, V.; Kitsati, N.; Fotsis, T.; Galaris, D. Intracellular labile iron determines H2O2-induced apoptotic signaling via sustained activation of ASK1/JNK-p38 axis. Free Radic. Biol. Med. 2016, 97, 454–465. [Google Scholar] [CrossRef]
- Ichijo, H.; Nishida, E. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997, 275, 90–94. [Google Scholar] [CrossRef]
- Saitoh, M.; Nishitoh, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, H.; Humeniuk, L.; Kozlov, N.; Roigas, S.; Adel, S.; Heydeck, D. The evolutionary hypothesis of reaction specificity of mammalian ALOX15 orthologs. Prog. Lipid Res. 2018, 72, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.X.; Ding, S.J.; Xiao, L.; Guo, W.; Zhan, Q. Desferoxamine preconditioning protects against cerebral ischemia in rats by inducing expressions of hypoxia inducible factor 1 alpha and erythropoietin. Neurosci. Bull. 2008, 24, 89–95. [Google Scholar] [CrossRef]
- Guo, X.; Jin, X.; Han, K.; Kang, S.; Tian, S.; Lv, X.; Feng, M.; Zheng, H.; Zuo, Y.; Xu, G.; et al. Iron promotes neurological function recovery in mice with ischemic stroke through endogenous repair mechanisms. Free Radic. Biol. Med. 2022, 182, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Tuo, Q.Z.; Lei, P.; Jackman, K.A.; Li, X.L.; Xiong, H.; Li, X.L.; Liuyang, Z.Y.; Roisman, L.; Zhang, S.T.; Ayton, S.; et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry 2017, 22, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- DeGregorio-Rocasolano, N.; Marti-Sistac, O.; Ponce, J.; Castello-Ruiz, M.; Millan, M.; Guirao, V.; Garcia-Yebenes, I.; Salom, J.B.; Ramos-Cabrer, P.; Alborch, E.; et al. Iron-loaded transferrin (Tf) is detrimental whereas iron-free Tf confers protection against brain ischemia by modifying blood Tf saturation and subsequent neuronal damage. Redox Biol. 2018, 15, 143–158. [Google Scholar] [CrossRef]
- Zhao, Y.; Xin, Z.; Li, N.; Chang, S.; Chen, Y.; Geng, L.; Chang, H.; Shi, H.; Chang, Y.Z. Nano-liposomes of lycopene reduces ischemic brain damage in rodents by regulating iron metabolism. Free Radic. Biol. Med. 2018, 124, 1–11. [Google Scholar] [CrossRef]
- Bandelow, B.; Michaelis, S. Epidemiology of anxiety disorders in the 21st century. Dialogues Clin. Neurosci. 2015, 17, 327–335. [Google Scholar] [CrossRef]
- Shah, H.E.; Bhawnani, N.; Ethirajulu, A.; Alkasabera, A.; Onyali, C.B.; Anim-Koranteng, C.; Mostafa, J.A. Iron Deficiency-Induced Changes in the Hippocampus, Corpus Striatum, and Monoamines Levels That Lead to Anxiety, Depression, Sleep Disorders, and Psychotic Disorders. Cureus 2021, 13, e18138. [Google Scholar] [CrossRef]
- Li, Z.; Wang, W.; Xin, X.; Song, X.; Zhang, D. Association of total zinc, iron, copper and selenium intakes with depression in the US adults. J. Affect. Disord. 2018, 228, 68–74. [Google Scholar] [CrossRef]
- Stewart, R.; Hirani, V. Relationship between depressive symptoms, anemia, and iron status in older residents from a national survey population. Psychosom. Med. 2012, 74, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Beard, J.L.; Connor, J.R. Iron status and neural functioning. Annu. Rev. Nutr. 2003, 23, 41–58. [Google Scholar] [CrossRef]
- Sobotka, T.J.; Whittaker, P.; Sobotka, J.M.; Brodie, R.E.; Quander, D.Y.; Robl, M.; Bryant, M.; Barton, C.N. Neurobehavioral dysfunctions associated with dietary iron overload. Physiol. Behav. 1996, 59, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Philibert, R.A.; Sandhu, H.; Hollenbeck, N.; Gunter, T.; Adams, W.; Madan, A. The relationship of 5HTT (SLC6A4) methylation and genotype on mRNA expression and liability to major depression and alcohol dependence in subjects from the Iowa Adoption Studies. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2008, 147, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Wessling-Resnick, M. Iron and mechanisms of emotional behavior. J. Nutr. Biochem. 2014, 25, 1101–1107. [Google Scholar] [CrossRef]
- Bastian, T.W.; von Hohenberg, W.C.; Mickelson, D.J.; Lanier, L.M.; Georgieff, M.K. Iron Deficiency Impairs Developing Hippocampal Neuron Gene Expression, Energy Metabolism, and Dendrite Complexity. Dev. Neurosci. 2016, 38, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Denny, C.A.; Kheirbek, M.A.; Alba, E.L.; Tanaka, K.F.; Brachman, R.A.; Laughman, K.B.; Tomm, N.K.; Turi, G.F.; Losonczy, A.; Hen, R. Hippocampal memory traces are differentially modulated by experience, time, and adult neurogenesis. Neuron 2014, 83, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ramirez, S.; Pang, P.T.; Puryear, C.B.; Govindarajan, A.; Deisseroth, K.; Tonegawa, S. Optogenetic stimulation of a hippocampal engram activates fear memory recall. Nature 2012, 484, 381–385. [Google Scholar] [CrossRef]
- McEchron, M.D.; Cheng, A.Y.; Liu, H.; Connor, J.R.; Gilmartin, M.R. Perinatal nutritional iron deficiency permanently impairs hippocampus-dependent trace fear conditioning in rats. Nutr. Neurosci. 2005, 8, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Georgieff, M.K. Iron deficiency in pregnancy. Am. J. Obstet. Gynecol. 2020, 223, 516–524. [Google Scholar] [CrossRef]
- Rice, D.; Barone, S., Jr. Critical periods of vulnerability for the developing nervous system: Evidence from humans and animal models. Environ. Health Perspect. 2000, 108 (Suppl. S3), 511–533. [Google Scholar] [CrossRef]
- Di Bella, D.J.; Habibi, E.; Stickels, R.R.; Scalia, G.; Brown, J.; Yadollahpour, P.; Yang, S.M.; Abbate, C.; Biancalani, T.; Macosko, E.Z.; et al. Molecular logic of cellular diversification in the mouse cerebral cortex. Nature 2021, 595, 554–559. [Google Scholar] [CrossRef]
- Evsyukova, I.; Plestant, C.; Anton, E. Integrative mechanisms of oriented neuronal migration in the developing brain. Annu. Rev. Cell Dev. Biol. 2013, 29, 299–353. [Google Scholar] [CrossRef]
- Gilmore, E.C.; Walsh, C.A. Genetic causes of microcephaly and lessons for neuronal development. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 461–478. [Google Scholar] [CrossRef]
- Gleeson, J.G.; Allen, K.M.; Fox, J.W.; Lamperti, E.D.; Berkovic, S.; Scheffer, I.; Cooper, E.C.; Dobyns, W.B.; Minnerath, S.R.; Ross, M.E. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 1998, 92, 63–72. [Google Scholar] [CrossRef]
- Juric-Sekhar, G.; Hevner, R.F. Malformations of cerebral cortex development: Molecules and mechanisms. Annu. Rev. Pathol. 2019, 14, 293–318. [Google Scholar] [CrossRef]
- Kahn, R.S.; Sommer, I.E.; Murray, R.M.; Meyer-Lindenberg, A.; Weinberger, D.R.; Cannon, T.D.; O’Donovan, M.; Correll, C.U.; Kane, J.M.; van Os, J.; et al. Schizophrenia. Nat. Rev. Dis. Prim. 2015, 1, 15067. [Google Scholar] [CrossRef]
- Hazlett, H.C.; Gu, H.; Munsell, B.C.; Kim, S.H.; Styner, M.; Wolff, J.J.; Elison, J.T.; Swanson, M.R.; Zhu, H.; Botteron, K.N.; et al. Early brain development in infants at high risk for autism spectrum disorder. Nature 2017, 542, 348–351. [Google Scholar] [CrossRef]
- McCann, S.; Perapoch Amado, M.; Moore, S.E. The Role of Iron in Brain Development: A Systematic Review. Nutrients 2020, 12, 2001. [Google Scholar] [CrossRef]
- McLean, E.; Cogswell, M.; Egli, I.; Wojdyla, D.; de Benoist, B. Worldwide prevalence of anaemia, WHO Vitamin and Mineral Nutrition Information System, 1993–2005. Public. Health Nutr. 2009, 12, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Fretham, S.J.; Carlson, E.S.; Georgieff, M.K. The role of iron in learning and memory. Adv. Nutr. 2011, 2, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.C.; Jacobson, J.L.; Burden, M.J.; Armony-Sivan, R.; Dodge, N.C.; Angelilli, M.L.; Lozoff, B.; Jacobson, S.W. Iron deficiency anemia and cognitive function in infancy. Pediatrics 2010, 126, e427–e434. [Google Scholar] [CrossRef]
- Lozoff, B. Iron deficiency and child development. Food Nutr. Bull. 2007, 28, S560–S571. [Google Scholar] [CrossRef]
- Basu, S.; Kumar, D.; Anupurba, S.; Verma, A.; Kumar, A. Effect of maternal iron deficiency anemia on fetal neural development. J. Perinatol. 2018, 38, 233–239. [Google Scholar] [CrossRef]
- Berglund, S.K.; Torres-Espínola, F.J.; García-Valdés, L.; Segura, M.T.; Martínez-Zaldívar, C.; Padilla, C.; Rueda, R.; Pérez García, M.; McArdle, H.J.; Campoy, C. The impacts of maternal iron deficiency and being overweight during pregnancy on neurodevelopment of the offspring. Br. J. Nutr. 2017, 118, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kim, J.; Buckett, P.D.; Böhlke, M.; Maher, T.J.; Wessling-Resnick, M. Severe postnatal iron deficiency alters emotional behavior and dopamine levels in the prefrontal cortex of young male rats. J. Nutr. 2011, 141, 2133–2138. [Google Scholar] [CrossRef]
- Beard, J. Recent evidence from human and animal studies regarding iron status and infant development. J. Nutr. 2007, 137, 524s–530s. [Google Scholar] [CrossRef]
- de los Monteros, A.E.; Korsak, R.A.; Tran, T.; Vu, D.; de Vellis, J.; Edmond, J. Dietary iron and the integrity of the developing rat brain: A study with the artificially-reared rat pup. Cell. Mol. Biol. 2000, 46, 501–515. [Google Scholar]
- Beard, J.L.; Wiesinger, J.A.; Connor, J.R. Pre- and postweaning iron deficiency alters myelination in Sprague-Dawley rats. Dev. Neurosci. 2003, 25, 308–315. [Google Scholar] [CrossRef]
- Ortiz, E.; Pasquini, J.M.; Thompson, K.; Felt, B.; Butkus, G.; Beard, J.; Connor, J.R. Effect of manipulation of iron storage, transport, or availability on myelin composition and brain iron content in three different animal models. J. Neurosci. Res. 2004, 77, 681–689. [Google Scholar] [CrossRef]
- Kwik-Uribe, C.L.; Golub, M.S.; Keen, C.L. Chronic marginal iron intakes during early development in mice alter brain iron concentrations and behavior despite postnatal iron supplementation. J. Nutr. 2000, 130, 2040–2048. [Google Scholar] [CrossRef] [PubMed]
- Markova, V.; Holm, C.; Pinborg, A.B.; Thomsen, L.L.; Moos, T. Impairment of the Developing Human Brain in Iron Deficiency: Correlations to Findings in Experimental Animals and Prospects for Early Intervention Therapy. Pharmaceuticals 2019, 12, 120. [Google Scholar] [CrossRef] [PubMed]
- Beard, J. Iron deficiency alters brain development and functioning. J. Nutr. 2003, 133, 1468s–1472s. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Chen, H.; Zhao, R.; Zhu, M.; Nie, G. Nanomedicine targets iron metabolism for cancer therapy. Cancer Sci. 2022, 113, 828. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; He, Y.; Jin, X.; Xie, J.; Yu, P.; Gao, G.; Chang, S.; Zhang, J.; Chang, Y.Z. CHIR99021 Maintenance of the Cell Stemness by Regulating Cellular Iron Metabolism. Antioxidants 2023, 12, 377. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.V.; Carlson, E.S.; Fretham, S.J.; Georgieff, M.K. Early-life iron deficiency anemia alters neurotrophic factor expression and hippocampal neuron differentiation in male rats. J. Nutr. 2008, 138, 2495–2501. [Google Scholar] [CrossRef]
- Jorgenson, L.A.; Sun, M.; O’Connor, M.; Georgieff, M.K. Fetal iron deficiency disrupts the maturation of synaptic function and efficacy in area CA1 of the developing rat hippocampus. Hippocampus 2005, 15, 1094–1102. [Google Scholar] [CrossRef]
- Brunette, K.E.; Tran, P.V.; Wobken, J.D. Gestational and neonatal iron deficiency alters apical dendrite structure of CA1 pyramidal neurons in adult rat hippocampus. Dev. Neurosci. 2010, 32, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Callahan, L.S.; Thibert, K.A.; Wobken, J.D.; Georgieff, M.K. Early-life iron deficiency anemia alters the development and long-term expression of parvalbumin and perineuronal nets in the rat hippocampus. Dev. Neurosci. 2013, 35, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Greminger, A.R.; Lee, D.L.; Shrager, P.; Mayer-Pröschel, M. Gestational iron deficiency differentially alters the structure and function of white and gray matter brain regions of developing rats. J. Nutr. 2014, 144, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Felt, B.T.; Beard, J.L.; Schallert, T.; Shao, J.; Aldridge, J.W.; Connor, J.R.; Georgieff, M.K.; Lozoff, B. Persistent neurochemical and behavioral abnormalities in adulthood despite early iron supplementation for perinatal iron deficiency anemia in rats. Behav. Brain Res. 2006, 171, 261–270. [Google Scholar] [CrossRef]
- Unger, E.L.; Paul, T.; Murray-Kolb, L.E.; Felt, B.; Jones, B.C.; Beard, J.L. Early iron deficiency alters sensorimotor development and brain monoamines in rats. J. Nutr. 2007, 137, 118–124. [Google Scholar] [CrossRef]
- Mihaila, C.; Schramm, J.; Strathmann, F.G.; Lee, D.L.; Gelein, R.M.; Luebke, A.E.; Mayer-Pröschel, M. Identifying a window of vulnerability during fetal development in a maternal iron restriction model. PLoS ONE 2011, 6, e17483. [Google Scholar] [CrossRef]
- Greminger, A.R.; Mayer-Pröschel, M. Identifying the threshold of iron deficiency in the central nervous system of the rat by the auditory brainstem response. ASN Neuro 2015, 7, 1759091415569911. [Google Scholar] [CrossRef]
- Fretham, S.J.; Carlson, E.S.; Georgieff, M.K. Neuronal-specific iron deficiency dysregulates mammalian target of rapamycin signaling during hippocampal development in nonanemic genetic mouse models. J. Nutr. 2013, 143, 260–266. [Google Scholar] [CrossRef]
- Benkovic, S.A.; Connor, J.R. Ferritin, transferrin, and iron in selected regions of the adult and aged rat brain. J. Comp. Neurol. 1993, 338, 97–113. [Google Scholar] [CrossRef]
- Connor, J.R.; Pavlick, G.; Karli, D.; Menzies, S.L.; Palmer, C. A histochemical study of iron-positive cells in the developing rat brain. J. Comp. Neurol. 1995, 355, 111–123. [Google Scholar] [CrossRef]
- Horiquini-Barbosa, E.; Gibb, R.; Kolb, B.; Bray, D.; Lachat, J.J. Tactile stimulation partially prevents neurodevelopmental changes in visual tract caused by early iron deficiency. Brain Res. 2017, 1657, 130–139. [Google Scholar] [CrossRef]
- Rosato-Siri, M.V.; Marziali, L.; Guitart, M.E.; Badaracco, M.E.; Puntel, M.; Pitossi, F.; Correale, J.; Pasquini, J.M. Iron Availability Compromises Not Only Oligodendrocytes But Also Astrocytes and Microglial Cells. Mol. Neurobiol. 2018, 55, 1068–1081. [Google Scholar] [CrossRef]
- Guitart, M.E.; Vence, M.; Correale, J.; Pasquini, J.M.; Rosato-Siri, M.V. Ontogenetic oligodendrocyte maturation through gestational iron deprivation: The road not taken. Glia 2019, 67, 1760–1774. [Google Scholar] [CrossRef]
- Rao, R.; Ennis, K.; Oz, G.; Lubach, G.R.; Georgieff, M.K.; Coe, C.L. Metabolomic analysis of cerebrospinal fluid indicates iron deficiency compromises cerebral energy metabolism in the infant monkey. Neurochem. Res. 2013, 38, 573–580. [Google Scholar] [CrossRef]
- Mudd, A.T.; Fil, J.E.; Knight, L.C.; Dilger, R.N. Dietary Iron Repletion following Early-Life Dietary Iron Deficiency Does Not Correct Regional Volumetric or Diffusion Tensor Changes in the Developing Pig Brain. Front. Neurol. 2017, 8, 735. [Google Scholar] [CrossRef] [PubMed]
- Felt, B.T.; Lozoff, B. Brain iron and behavior of rats are not normalized by treatment of iron deficiency anemia during early development. J. Nutr. 1996, 126, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Lozoff, B.; Jimenez, E.; Hagen, J.; Mollen, E.; Wolf, A.W. Poorer behavioral and developmental outcome more than 10 years after treatment for iron deficiency in infancy. Pediatrics 2000, 105, E51. [Google Scholar] [CrossRef] [PubMed]
- Uecker, A.; Nadel, L. Spatial locations gone awry: Object and spatial memory deficits in children with fetal alcohol syndrome. Neuropsychologia 1996, 34, 209–223. [Google Scholar] [CrossRef]
- Carlson, E.S.; Stead, J.D.H.; Neal, C.R. Perinatal iron deficiency results in altered developmental expression of genes mediating energy metabolism and neuronal morphogenesis in hippocampus. Hippocampus 2007, 17, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Bastian, T.W.; von Hohenberg, W.C.; Georgieff, M.K.; Lanier, L.M. Chronic Energy Depletion due to Iron Deficiency Impairs Dendritic Mitochondrial Motility during Hippocampal Neuron Development. J. Neurosci. 2019, 39, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Bastian, T.W.; Rao, R.; Tran, P.V.; Georgieff, M.K. The Effects of Early-Life Iron Deficiency on Brain Energy Metabolism. Neurosci. Insights 2020, 15, 2633105520935104. [Google Scholar] [CrossRef] [PubMed]
- Perng, V.; Li, C.; Klocke, C.R.; Navazesh, S.E.; Pinneles, D.K.; Lein, P.J.; Ji, P. Iron Deficiency and Iron Excess Differently Affect Dendritic Architecture of Pyramidal Neurons in the Hippocampus of Piglets. J. Nutr. 2021, 151, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Lien, Y.C.; Condon, D.E.; Georgieff, M.K.; Simmons, R.A.; Tran, P.V. Dysregulation of Neuronal Genes by Fetal-Neonatal Iron Deficiency Anemia Is Associated with Altered DNA Methylation in the Rat Hippocampus. Nutrients 2019, 11, 1191. [Google Scholar] [CrossRef] [PubMed]
- Erber, L.N.; Luo, A.; Gong, Y.; Beeson, M.; Tu, M.; Tran, P.; Chen, Y. Iron Deficiency Reprograms Phosphorylation Signaling and Reduces O-GlcNAc Pathways in Neuronal Cells. Nutrients 2021, 13, 179. [Google Scholar] [CrossRef]
- Erber, L.; Liu, S.; Gong, Y.; Tran, P.; Chen, Y. Quantitative Proteome and Transcriptome Dynamics Analysis Reveals Iron Deficiency Response Networks and Signature in Neuronal Cells. Molecules 2022, 27, 484. [Google Scholar] [CrossRef]
- DeMaman, A.S.; Melo, P.; Homem, J.M.; Tavares, M.A.; Lachat, J.J. Effectiveness of iron repletion in the diet for the optic nerve development of anaemic rats. Eye 2010, 24, 901–908. [Google Scholar] [CrossRef]
- Unger, E.L.; Hurst, A.R.; Georgieff, M.K.; Schallert, T.; Rao, R.; Connor, J.R.; Kaciroti, N.; Lozoff, B.; Felt, B. Behavior and monoamine deficits in prenatal and perinatal iron deficiency are not corrected by early postnatal moderate-iron or high-iron diets in rats. J. Nutr. 2012, 142, 2040–2049. [Google Scholar] [CrossRef]
- Georgieff, M.K. Long-term brain and behavioral consequences of early iron deficiency. Nutr. Rev. 2011, 69 (Suppl. S1), S43–S48. [Google Scholar] [CrossRef]
- Mudd, A.T.; Fil, J.E.; Knight, L.C.; Lam, F.; Liang, Z.P.; Dilger, R.N. Early-Life Iron Deficiency Reduces Brain Iron Content and Alters Brain Tissue Composition Despite Iron Repletion: A Neuroimaging Assessment. Nutrients 2018, 10, 135. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Bush, A.I. Metals and Alzheimer’s Disease: How Far Have We Come in the Clinic? J. Alzheimers Dis. 2018, 62, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; He, M.L. Deferoxamine enhances alternative activation of microglia and inhibits amyloid beta deposits in APP/PS1 mice. Brain Res. 2017, 1677, 86–92. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, J.; Song, N.; Xie, J.; Jiang, H. Up-regulation of divalent metal transporter 1 is involved in 1-methyl-4-phenylpyridinium (MPP(+))-induced apoptosis in MES23.5 cells. Neurobiol. Aging 2009, 30, 1466–1476. [Google Scholar] [CrossRef] [PubMed]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.C.; Spino, M.; Connelly, J.; Tricta, F.; et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 2017, 7, 1398. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.J.; Chen, S.D.; Lin, K.L.; Liou, C.W.; Lan, M.Y.; Chuang, Y.C.; Wang, P.W.; Lee, J.J.; Wang, F.S.; Lin, H.Y.; et al. Iron Brain Menace: The Involvement of Ferroptosis in Parkinson Disease. Cells 2022, 11, 3829. [Google Scholar] [CrossRef] [PubMed]
- Devos, D.; Labreuche, J.; Rascol, O.; Corvol, J.C.; Duhamel, A.; Guyon Delannoy, P.; Poewe, W.; Compta, Y.; Pavese, N.; Růžička, E.; et al. Trial of Deferiprone in Parkinson’s Disease. N. Engl. J. Med. 2022, 387, 2045–2055. [Google Scholar] [CrossRef]
- Castellanos, M.; Puig, N.; Carbonell, T.; Castillo, J.; Martinez, J.; Rama, R.; Davalos, A. Iron intake increases infarct volume after permanent middle cerebral artery occlusion in rats. Brain Res. 2002, 952, 1–6. [Google Scholar] [CrossRef]
- Shadid, M.; Buonocore, G.; Groenendaal, F.; Moison, R.; Ferrali, M.; Berger, H.M.; van Bel, F. Effect of deferoxamine and allopurinol on non-protein-bound iron concentrations in plasma and cortical brain tissue of newborn lambs following hypoxia-ischemia. Neurosci. Lett. 1998, 248, 5–8. [Google Scholar] [CrossRef]
- Sarco, D.P.; Becker, J.; Palmer, C.; Sheldon, R.A.; Ferriero, D.M. The neuroprotective effect of deferoxamine in the hypoxic-ischemic immature mouse brain. Neurosci. Lett. 2000, 282, 113–116. [Google Scholar] [CrossRef]
- Papazisis, G.; Pourzitaki, C.; Sardeli, C.; Lallas, A.; Amaniti, E.; Kouvelas, D. Deferoxamine decreases the excitatory amino acid levels and improves the histological outcome in the hippocampus of neonatal rats after hypoxia-ischemia. Pharmacol. Res. 2008, 57, 73–78. [Google Scholar] [CrossRef]
- Selim, M.; Foster, L.D.; Moy, C.S.; Xi, G.; Hill, M.D.; Morgenstern, L.B.; Greenberg, S.M.; James, M.L.; Singh, V.; Clark, W.M.; et al. Deferoxamine mesylate in patients with intracerebral haemorrhage (i-DEF): A multicentre, randomised, placebo-controlled, double-blind phase 2 trial. Lancet Neurol. 2019, 18, 428–438. [Google Scholar] [CrossRef]
- Foster, L.; Robinson, L.; Yeatts, S.D.; Conwit, R.A.; Shehadah, A.; Lioutas, V.; Selim, M. Effect of Deferoxamine on Trajectory of Recovery After Intracerebral Hemorrhage: A Post Hoc Analysis of the i-DEF Trial. Stroke 2022, 53, 2204–2210. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Wang, J.; Foster, L.D.; Yeatts, S.D.; Moy, C.; Mocco, J.; Selim, M. Effect of Deferoxamine on Outcome According to Baseline Hematoma Volume: A Post Hoc Analysis of the i-DEF Trial. Stroke 2022, 53, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.J. Oral chelators deferasirox and deferiprone for transfusional iron overload in thalassemia major: New data, new questions. Blood 2006, 107, 3436–3441. [Google Scholar] [CrossRef] [PubMed]
- Jakaria, M.; Belaidi, A.A.; Bush, A.I.; Ayton, S. Ferroptosis as a mechanism of neurodegeneration in Alzheimer’s disease. J. Neurochem. 2021, 159, 804–825. [Google Scholar] [CrossRef]
- Vlachodimitropoulou, E.; Chen, Y.L.; Garbowski, M.; Koonyosying, P.; Psaila, B.; Sola-Visner, M.; Cooper, N.; Hider, R.; Porter, J. Eltrombopag: A powerful chelator of cellular or extracellular iron(III) alone or combined with a second chelator. Blood 2017, 130, 1923–1933. [Google Scholar] [CrossRef]
- Boddaert, N.; Le Quan Sang, K.H.; Rotig, A.; Leroy-Willig, A.; Gallet, S.; Brunelle, F.; Sidi, D.; Thalabard, J.C.; Munnich, A.; Cabantchik, Z.I. Selective iron chelation in Friedreich ataxia: Biologic and clinical implications. Blood 2007, 110, 401–408. [Google Scholar] [CrossRef]
- Sohn, Y.S.; Breuer, W.; Munnich, A.; Cabantchik, Z.I. Redistribution of accumulated cell iron: A modality of chelation with therapeutic implications. Blood 2008, 111, 1690–1699. [Google Scholar] [CrossRef]
- Kosyakovsky, J.; Fine, J.M.; Frey, W.H., 2nd; Hanson, L.R. Mechanisms of Intranasal Deferoxamine in Neurodegenerative and Neurovascular Disease. Pharmaceuticals 2021, 14, 95. [Google Scholar] [CrossRef]
- Guo, C.; Wang, P.; Zhong, M.L.; Wang, T.; Huang, X.S.; Li, J.Y.; Wang, Z.Y. Deferoxamine inhibits iron induced hippocampal tau phosphorylation in the Alzheimer transgenic mouse brain. Neurochem. Int. 2013, 62, 165–172. [Google Scholar] [CrossRef]
- Guo, C.; Zhang, Y.X.; Wang, T.; Zhong, M.L.; Yang, Z.H.; Hao, L.J.; Chai, R.; Zhang, S. Intranasal deferoxamine attenuates synapse loss via up-regulating the P38/HIF-1alpha pathway on the brain of APP/PS1 transgenic mice. Front. Aging Neurosci. 2015, 7, 104. [Google Scholar] [CrossRef]
- Fine, J.M.; Stroebel, B.M.; Faltesek, K.A.; Terai, K.; Haase, L.; Knutzen, K.E.; Kosyakovsky, J.; Bowe, T.J.; Fuller, A.K.; Frey, W.H.; et al. Intranasal delivery of low-dose insulin ameliorates motor dysfunction and dopaminergic cell death in a 6-OHDA rat model of Parkinson’s Disease. Neurosci. Lett. 2020, 714, 134567. [Google Scholar] [CrossRef]
- Fine, J.M.; Forsberg, A.C.; Renner, D.B.; Faltesek, K.A.; Mohan, K.G.; Wong, J.C.; Arneson, L.C.; Crow, J.M.; Frey, W.H., 2nd; Hanson, L.R. Intranasally-administered deferoxamine mitigates toxicity of 6-OHDA in a rat model of Parkinson׳s disease. Brain Res. 2014, 1574, 96–104. [Google Scholar] [CrossRef]
- Hanson, L.R.; Roeytenberg, A.; Martinez, P.M.; Coppes, V.G.; Sweet, D.C.; Rao, R.J.; Marti, D.L.; Hoekman, J.D.; Matthews, R.B.; Frey, W.H., 2nd; et al. Intranasal deferoxamine provides increased brain exposure and significant protection in rat ischemic stroke. J. Pharmacol. Exp. Ther. 2009, 330, 679–686. [Google Scholar] [CrossRef]
- Zhao, Y.P.; Tian, S.Y.; Zhang, J.; Cheng, X.; Huang, W.P.; Cao, G.L.; Chang, Y.Z.; Wang, H.; Nie, G.J.; Qiu, W. Regulation of neuroinflammation with GLP-1 receptor targeting nanostructures to alleviate Alzheimer’s symptoms in the disease models. Nano Today 2022, 44, 101457. [Google Scholar] [CrossRef]
- Zheng, W.; Xin, N.; Chi, Z.H.; Zhao, B.L.; Zhang, J.; Li, J.Y.; Wang, Z.Y. Divalent metal transporter 1 is involved in amyloid precursor protein processing and Abeta generation. FASEB J. 2009, 23, 4207–4217. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Lu, J.; Hao, X.; Li, H.; Zhang, G.; Liu, X.; Li, X.; Zhao, C.; Kuang, W.; Chen, D.; et al. FTH1 Inhibits Ferroptosis Through Ferritinophagy in the 6-OHDA Model of Parkinson’s Disease. Neurotherapeutics 2020, 17, 1796–1812. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Chang, S.Y.; Wu, Q.; Gou, Y.J.; Jia, L.; Cui, Y.M.; Yu, P.; Shi, Z.H.; Wu, W.S.; Gao, G.; et al. The Protective Role of Mitochondrial Ferritin on Erastin-Induced Ferroptosis. Front. Aging Neurosci. 2016, 8, 308. [Google Scholar] [CrossRef] [PubMed]
- You, L.H.; Li, Z.; Duan, X.L.; Zhao, B.L.; Chang, Y.Z.; Shi, Z.H. Mitochondrial ferritin suppresses MPTP-induced cell damage by regulating iron metabolism and attenuating oxidative stress. Brain Res. 2016, 1642, 33–42. [Google Scholar] [CrossRef]
- Guo, X.; Zheng, H.; Guo, Y.; Wang, Y.; Anderson, G.J.; Ci, Y.; Yu, P.; Geng, L.; Chang, Y.Z. Nasal delivery of nanoliposome-encapsulated ferric ammonium citrate can increase the iron content of rat brain. J. Nanobiotechnology 2017, 15, 42. [Google Scholar] [CrossRef]
- Villalón-García, I.; Álvarez-Córdoba, M.; Povea-Cabello, S.; Talaverón-Rey, M.; Villanueva-Paz, M.; Luzón-Hidalgo, R.; Suárez-Rivero, J.M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Salas, J.J.; et al. Vitamin E prevents lipid peroxidation and iron accumulation in PLA2G6-Associated Neurodegeneration. Neurobiol. Dis. 2022, 165, 105649. [Google Scholar] [CrossRef]
- Romero, A.; Ramos, E.; de Los Ríos, C.; Egea, J.; Del Pino, J.; Reiter, R.J. A review of metal-catalyzed molecular damage: Protection by melatonin. J. Pineal Res. 2014, 56, 343–370. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, M.S.; Schumacher-Schuh, A.; Cardoso, A.M.; Bochi, G.V.; Baldissarelli, J.; Kegler, A.; Santana, D.; Chaves, C.M.; Schetinger, M.R.; Moresco, R.N.; et al. Iron and Oxidative Stress in Parkinson’s Disease: An Observational Study of Injury Biomarkers. PLoS ONE 2016, 11, e0146129. [Google Scholar] [CrossRef]
- Santoro, M.M. The Antioxidant Role of Non-mitochondrial CoQ10: Mystery Solved! Cell Metab. 2020, 31, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Shults, C.W.; Oakes, D.; Kieburtz, K.; Beal, M.F.; Haas, R.; Plumb, S.; Juncos, J.L.; Nutt, J.; Shoulson, I.; Carter, J.; et al. Effects of coenzyme Q10 in early Parkinson disease: Evidence of slowing of the functional decline. Arch. Neurol. 2002, 59, 1541–1550. [Google Scholar] [CrossRef]
- Mandel, S.; Amit, T.; Reznichenko, L.; Weinreb, O.; Youdim, M.B. Green tea catechins as brain-permeable, natural iron chelators-antioxidants for the treatment of neurodegenerative disorders. Mol. Nutr. Food Res. 2006, 50, 229–234. [Google Scholar] [CrossRef]
- Mandel, S.A.; Avramovich-Tirosh, Y.; Reznichenko, L.; Zheng, H.; Weinreb, O.; Amit, T.; Youdim, M.B. Multifunctional activities of green tea catechins in neuroprotection. Modulation of cell survival genes, iron-dependent oxidative stress and PKC signaling pathway. Neurosignals 2005, 14, 46–60. [Google Scholar] [CrossRef]
- Morgan, L.A.; Grundmann, O. Preclinical and Potential Applications of Common Western Herbal Supplements as Complementary Treatment in Parkinson’s Disease. J. Diet. Suppl. 2017, 14, 453–466. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, G.; You, L.; Zhang, J.; Chang, Y.-Z.; Yu, P. Brain Iron Metabolism, Redox Balance and Neurological Diseases. Antioxidants 2023, 12, 1289. https://doi.org/10.3390/antiox12061289
Gao G, You L, Zhang J, Chang Y-Z, Yu P. Brain Iron Metabolism, Redox Balance and Neurological Diseases. Antioxidants. 2023; 12(6):1289. https://doi.org/10.3390/antiox12061289
Chicago/Turabian StyleGao, Guofen, Linhao You, Jianhua Zhang, Yan-Zhong Chang, and Peng Yu. 2023. "Brain Iron Metabolism, Redox Balance and Neurological Diseases" Antioxidants 12, no. 6: 1289. https://doi.org/10.3390/antiox12061289
APA StyleGao, G., You, L., Zhang, J., Chang, Y.-Z., & Yu, P. (2023). Brain Iron Metabolism, Redox Balance and Neurological Diseases. Antioxidants, 12(6), 1289. https://doi.org/10.3390/antiox12061289