

Therapeutic Potential of Heterocyclic Compounds Targeting Mitochondrial Calcium Homeostasis and Signaling in Alzheimer’s Disease and Parkinson’s Disease

 ,
, 
 ,
,
Abstract
1. Introduction
2. Mitochondrial Deficiencies and Oxidative Stress as Close Partners in Alzheimer’s Disease
3. Ca2+ Dysregulation and Downstream Effects in Alzheimer’s Disease
4. Impaired Mitochondrial Function and Associated Oxidative Damage in Parkinson’s Disease
5. Altered Ca2+ Homeostasis and Concomitant Neurotoxicity in Parkinson’s Disease
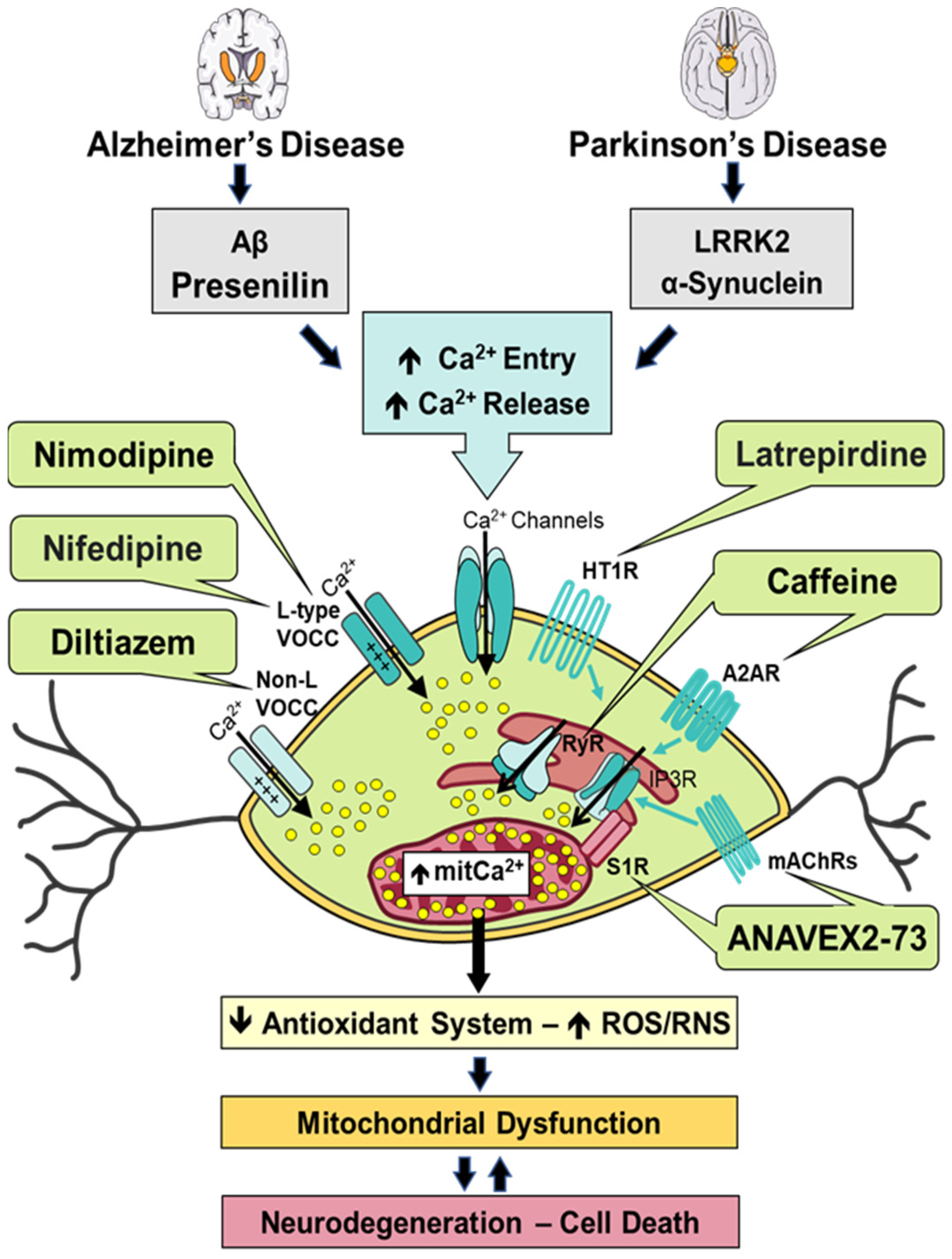
6. Modulation of Calcium Signaling and Homeostasis by Heterocyclic Compounds in Alzheimer’s Disease and Parkinson’s Disease
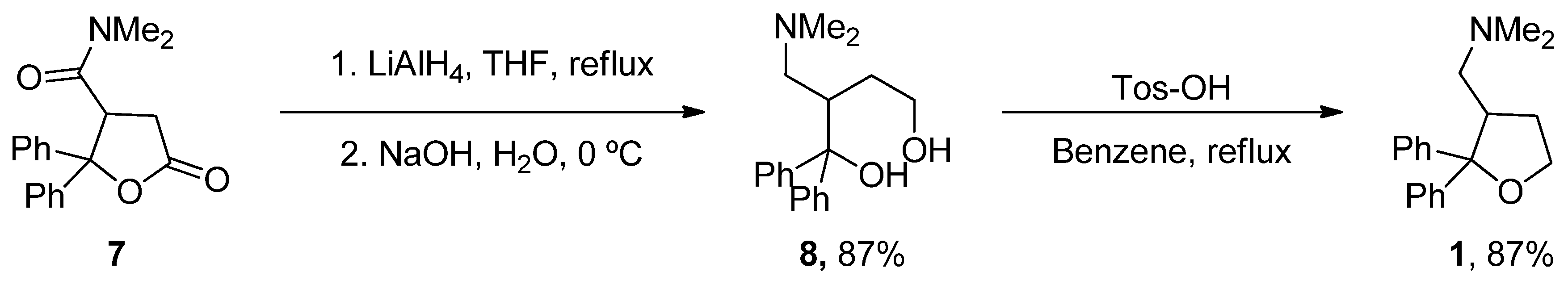
6.1. ANAVEX2-73
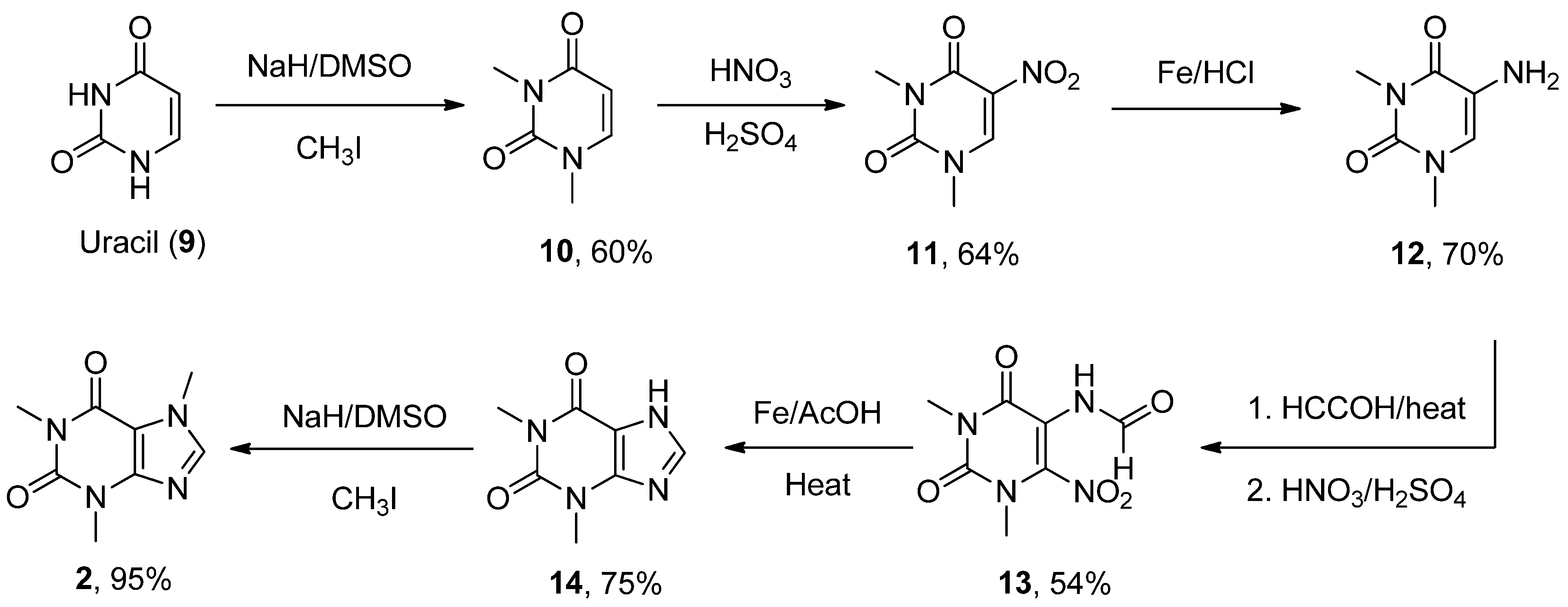
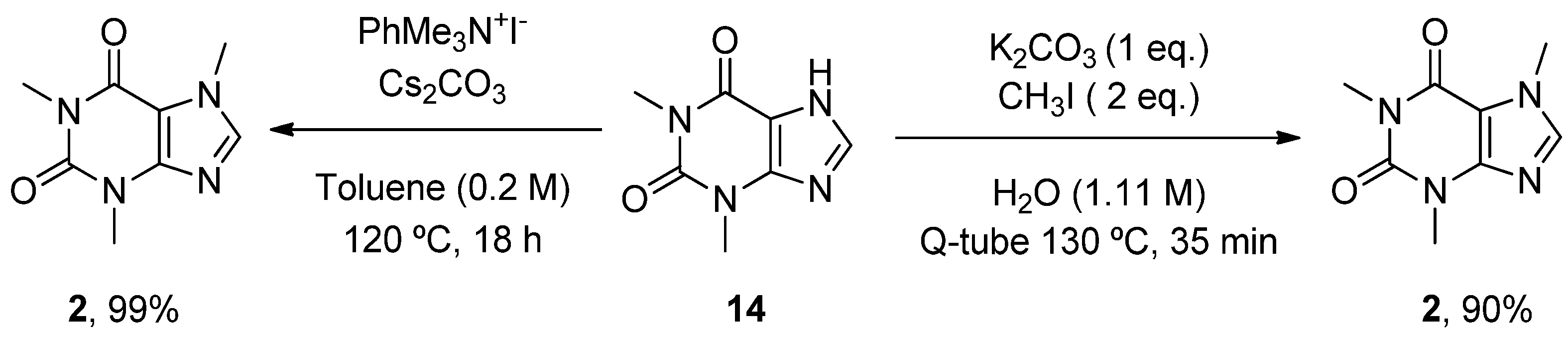
6.2. Caffeine
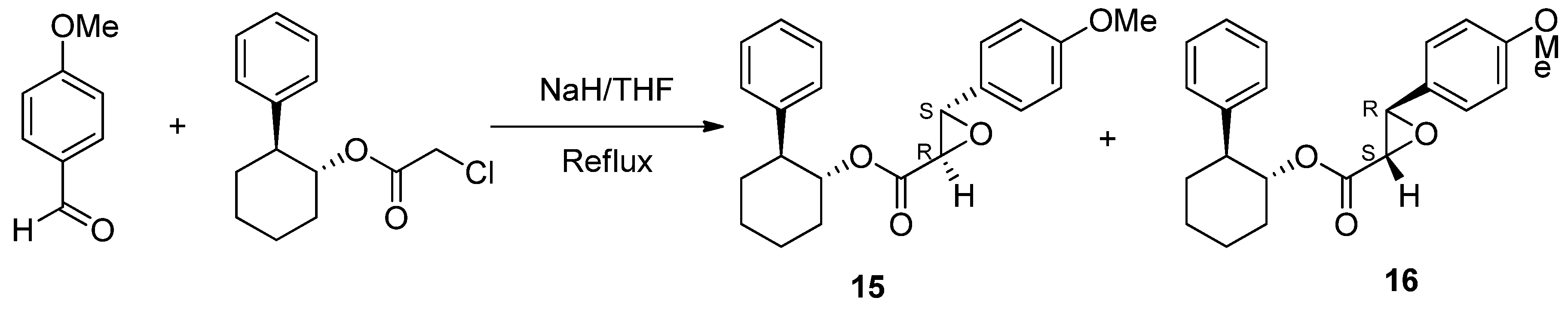
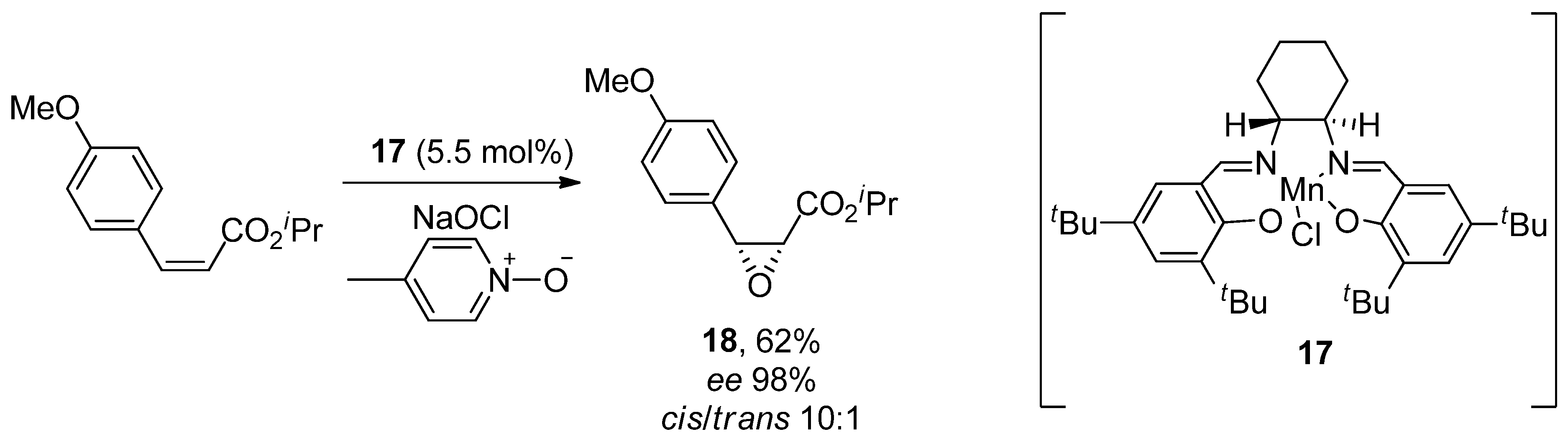
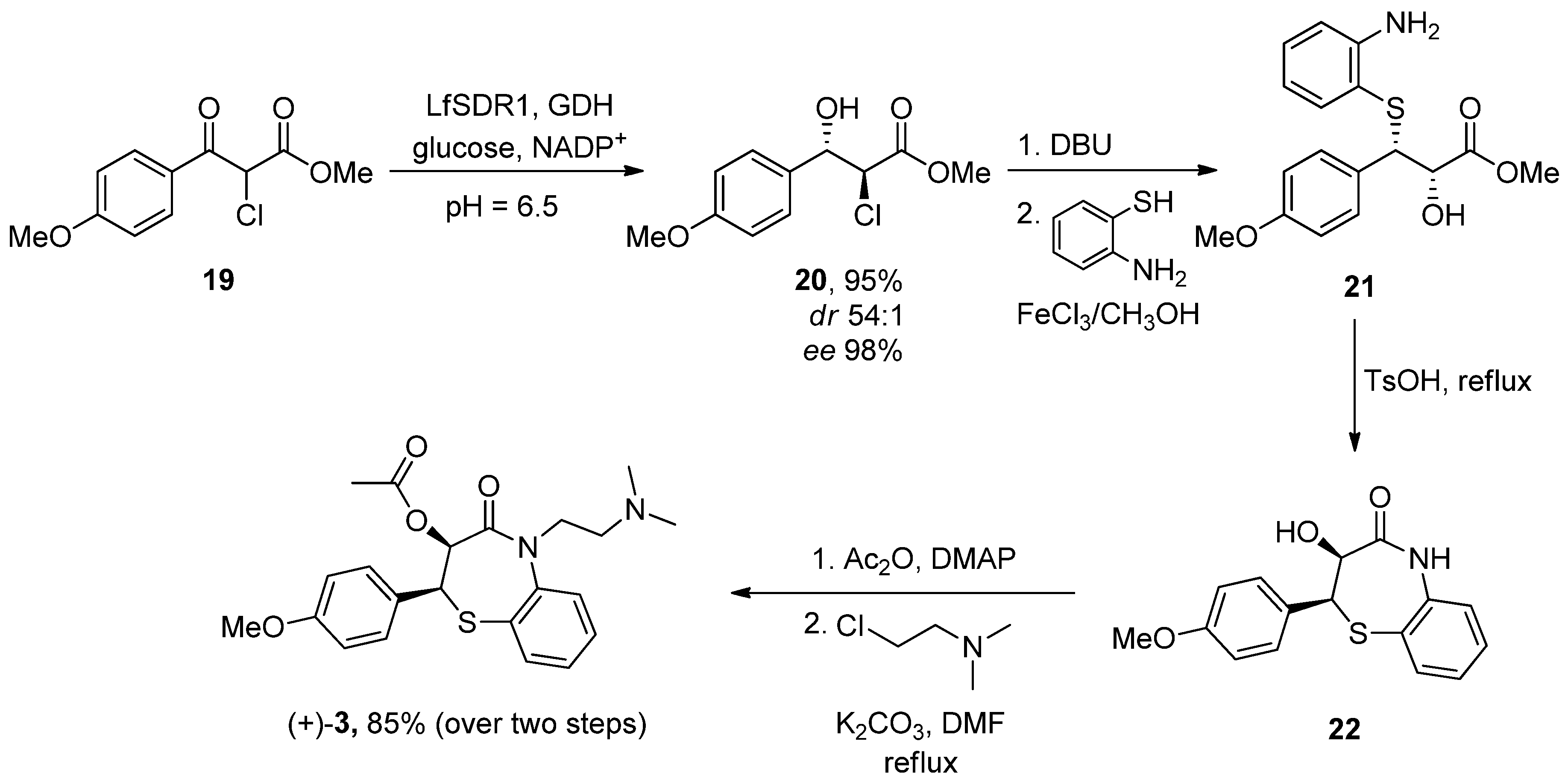
6.3. Diltiazem
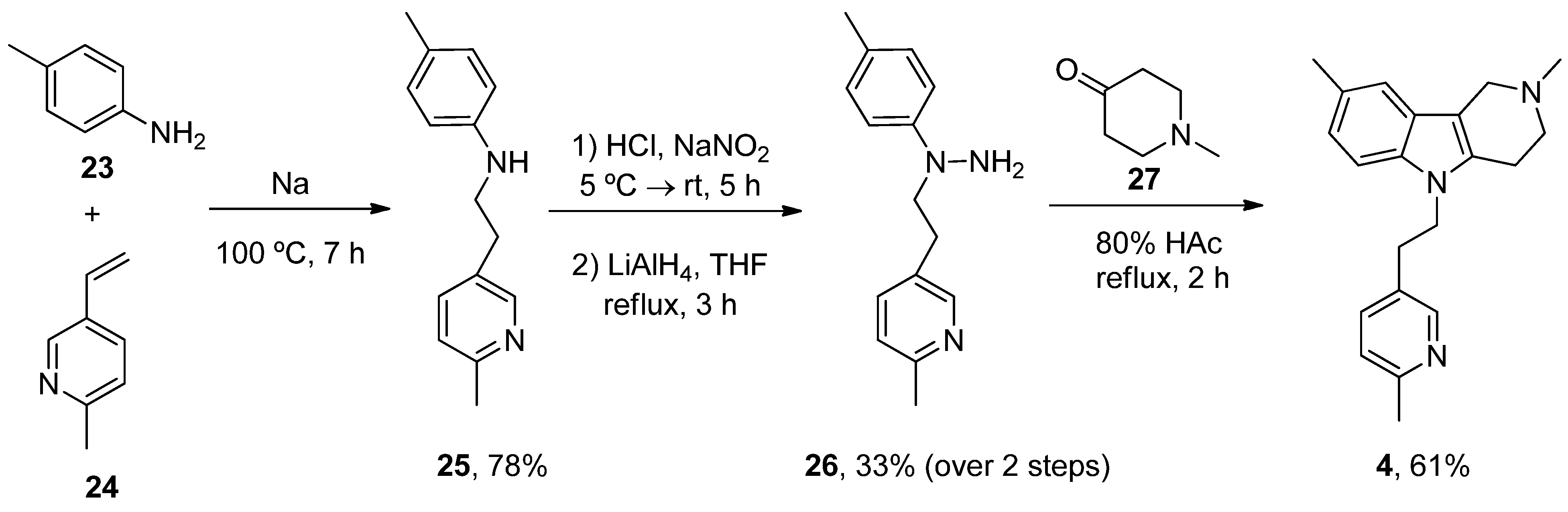
6.4. Latrepirdine
6.5. Nifedipine
6.6. Nimodipine
7. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Yoboue, E.D.; Sitia, R.; Simmen, T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018, 9, 331. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.K.; Carlson, E.A.; Yan, S.S. Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim. Biophys. Acta 2014, 1842, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Cho, S.S.; Jeong, Y.; Park, K.C.; Kang, S.J.; Kang, E.; Kim, S.E.; Lee, K.H.; Na, D.L. Glucose metabolism in early onset versus late onset Alzheimer’s disease: An SPM analysis of 120 patients. Brain 2005, 128, 1790–1801. [Google Scholar] [CrossRef] [PubMed]
- Bosetti, F.; Brizzi, F.; Barogi, S.; Mancuso, M.; Siciliano, G.; Tendi, E.A.; Murri, L.; Rapoport, S.I.; Solaini, G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol. Aging 2002, 23, 371–376. [Google Scholar] [CrossRef]
- Parker, W.D., Jr.; Filley, C.M.; Parks, J.K. Cytochrome oxidase deficiency in Alzheimer’s disease. Neurology 1990, 40, 1302–1303. [Google Scholar] [CrossRef]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Yan, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 18670–18675. [Google Scholar] [CrossRef]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar] [CrossRef]
- Cho, D.H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.I.; Lee, K.H.; Gabr, A.A.; Choi, G.E.; Kim, J.S.; Ko, S.H.; Han, H.J. Abeta-Induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis. Biochim. Biophys. Acta 2016, 1863, 2820–2834. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Hu, Y.; Wang, Z.H.; Luo, Y.; Zhang, Y.; Liu, X.P.; Feng, Q.; Wang, Q.; Ye, K.; Liu, G.P.; et al. Human wild-type full-length tau accumulation disrupts mitochondrial dynamics and the functions via increasing mitofusins. Sci. Rep. 2016, 6, 24756. [Google Scholar] [CrossRef]
- Tapias, V.; Jainuddin, S.; Ahuja, M.; Stack, C.; Elipenahli, C.; Vignisse, J.; Gerges, M.; Starkova, N.; Xu, H.; Starkov, A.A.; et al. Benfotiamine treatment activates the Nrf2/ARE pathway and is neuroprotective in a transgenic mouse model of tauopathy. Hum. Mol. Genet. 2018, 27, 2874–2892. [Google Scholar] [CrossRef] [PubMed]
- Vossel, K.A.; Xu, J.C.; Fomenko, V.; Miyamoto, T.; Suberbielle, E.; Knox, J.A.; Ho, K.; Kim, D.H.; Yu, G.Q.; Mucke, L. Tau reduction prevents Abeta-induced axonal transport deficits by blocking activation of GSK3beta. J. Cell Biol. 2015, 209, 419–433. [Google Scholar] [CrossRef]
- Bobba, A.; Amadoro, G.; Valenti, D.; Corsetti, V.; Lassandro, R.; Atlante, A. Mitochondrial respiratory chain Complexes I and IV are impaired by beta-amyloid via direct interaction and through Complex I-dependent ROS production, respectively. Mitochondrion 2013, 13, 298–311. [Google Scholar] [CrossRef]
- Lovell, M.A.; Ehmann, W.D.; Butler, S.M.; Markesbery, W.R. Elevated thiobarbituric acid-reactive substances and antioxidant enzyme activity in the brain in Alzheimer’s disease. Neurology 1995, 45, 1594–1601. [Google Scholar] [CrossRef]
- Markesbery, W.R.; Lovell, M.A. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol. Aging 1998, 19, 33–36. [Google Scholar] [CrossRef]
- Youssef, P.; Chami, B.; Lim, J.; Middleton, T.; Sutherland, G.T.; Witting, P.K. Evidence supporting oxidative stress in a moderately affected area of the brain in Alzheimer’s disease. Sci. Rep. 2018, 8, 11553. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.F.; Montine, K.S.; Moore, M.; Morrow, J.D.; Kaye, J.A.; Montine, T.J. Suppression of longitudinal increase in CSF F2-isoprostanes in Alzheimer’s disease. J. Alzheimer’s Dis. 2004, 6, 93–97. [Google Scholar] [CrossRef]
- Sultana, R.; Poon, H.F.; Cai, J.; Pierce, W.M.; Merchant, M.; Klein, J.B.; Markesbery, W.R.; Butterfield, D.A. Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiol. Dis. 2006, 22, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Reed, T.; Perluigi, M.; Sultana, R.; Pierce, W.M.; Klein, J.B.; Turner, D.M.; Coccia, R.; Markesbery, W.R.; Butterfield, D.A. Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: Insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiol. Dis. 2008, 30, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Castegna, A.; Thongboonkerd, V.; Klein, J.B.; Lynn, B.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of nitrated proteins in Alzheimer’s disease brain. J. Neurochem. 2003, 85, 1394–1401. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Weber, D.; Raupbach, J.; Dakal, T.C.; Fliessbach, K.; Ramirez, A.; Grune, T.; Wullner, U. Advanced glycation end products and protein carbonyl levels in plasma reveal sex-specific differences in Parkinson’s and Alzheimer’s disease. Redox Biol. 2020, 34, 101546. [Google Scholar] [CrossRef]
- Mecocci, P.; MacGarvey, U.; Beal, M.F. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann. Neurol. 1994, 36, 747–751. [Google Scholar] [CrossRef]
- Valero, R.A.; Senovilla, L.; Nunez, L.; Villalobos, C. The role of mitochondrial potential in control of calcium signals involved in cell proliferation. Cell Calcium. 2008, 44, 259–269. [Google Scholar] [CrossRef]
- Nunez, L.; Senovilla, L.; Sanz-Blasco, S.; Chamero, P.; Alonso, M.T.; Villalobos, C.; Garcia-Sancho, J. Bioluminescence imaging of mitochondrial Ca2+ dynamics in soma and neurites of individual adult mouse sympathetic neurons. J. Physiol. 2007, 580, 385–395. [Google Scholar] [CrossRef]
- Núñez, L.; Villalobos, C.; García-Sancho, J. Coupling or not coupling of mitochondria to Ca2+ sources in neurones. Soma and neurites differ. Physiol. News 2008, 70, 23–24. [Google Scholar] [CrossRef]
- Sanz-Blasco, S.; Valero, R.A.; Rodriguez-Crespo, I.; Villalobos, C.; Nunez, L. Mitochondrial Ca2+ overload underlies Abeta oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS ONE 2008, 3, e2718. [Google Scholar] [CrossRef] [PubMed]
- Calvo, M.; Sanz-Blasco, S.; Caballero, E.; Villalobos, C.; Nunez, L. Susceptibility to excitotoxicity in aged hippocampal cultuRes. and neuroprotection by non-steroidal anti-inflammatory drugs: Role of mitochondrial calcium. J. Neurochem. 2015, 132, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Rodriguez, M.; Garcia-Durillo, M.; Villalobos, C.; Nunez, L. Aging Enables Ca2+ Overload and Apoptosis Induced by Amyloid-beta Oligomers in Rat Hippocampal Neurons: Neuroprotection by Non-Steroidal Anti-Inflammatory Drugs and R-Flurbiprofen in Aging Neurons. J. Alzheimer’s Dis. 2016, 54, 207–221. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Garcia-Durillo, M.; Villalobos, C.; Nunez, L. In vitro aging promotes endoplasmic reticulum (ER)-mitochondria Ca2+ cross talk and loss of store-operated Ca2+ entry (SOCE) in rat hippocampal neurons. Biochim. Biophys. Acta 2016, 1863, 2637–2649. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Hernando-Perez, E.; Nunez, L.; Villalobos, C. Amyloid beta Oligomers Increase ER-Mitochondria Ca2+ Cross Talk in Young Hippocampal Neurons and Exacerbate Aging-Induced Intracellular Ca2+ Remodeling. Front. Cell. Neurosci. 2019, 13, 22. [Google Scholar] [CrossRef] [PubMed]
- Caballero, E.; Hernando-Perez, E.; Tapias, V.; Calvo-Rodriguez, M.; Villalobos, C.; Nunez, L. Amyloid Beta Oligomers-Induced Ca2+ Entry Pathways: Role of Neuronal Networks, NMDA Receptors and Amyloid Channel Formation. Biomedicines 2022, 10, 1153. [Google Scholar] [CrossRef]
- Gonzalez-Andres, P.; Fernandez-Pena, L.; Diez-Poza, C.; Villalobos, C.; Nunez, L.; Barbero, A. Marine Heterocyclic Compounds That Modulate Intracellular Calcium Signals: Chemistry and Synthesis Approaches. Mar. Drugs 2021, 19, 78. [Google Scholar] [CrossRef]
- Edison, P.; Ahmed, I.; Fan, Z.; Hinz, R.; Gelosa, G.; Ray Chaudhuri, K.; Walker, Z.; Turkheimer, F.E.; Brooks, D.J. Microglia, amyloid, and glucose metabolism in Parkinson’s disease with and without dementia. Neuropsychopharmacology 2013, 38, 938–949. [Google Scholar] [CrossRef]
- Trimmer, P.A.; Swerdlow, R.H.; Parks, J.K.; Keeney, P.; Bennett, J.P., Jr.; Miller, S.W.; Davis, R.E.; Parker, W.D., Jr. Abnormal mitochondrial morphology in sporadic Parkinson’s and Alzheimer’s disease cybrid cell lines. Exp. Neurol. 2000, 162, 37–50. [Google Scholar] [CrossRef]
- Jimenez-Jimenez, F.J.; Molina, J.A.; Hernanz, A.; Fernandez-Vivancos, E.; de Bustos, F.; Barcenilla, B.; Gomez-Escalonilla, C.; Zurdo, M.; Berbel, A.; Villanueva, C. Cerebrospinal fluid levels of thiamine in patients with Parkinson’s disease. Neurosci. Lett. 1999, 271, 33–36. [Google Scholar] [CrossRef]
- Gibson, G.E.; Kingsbury, A.E.; Xu, H.; Lindsay, J.G.; Daniel, S.; Foster, O.J.; Lees, A.J.; Blass, J.P. Deficits in a tricarboxylic acid cycle enzyme in brains from patients with Parkinson’s disease. Neurochem. Int. 2003, 43, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Matuda, S.; Yoshino, H.; Mori, H.; Hattori, N.; Ikebe, S. An immunohistochemical study on alpha-ketoglutarate dehydrogenase complex in Parkinson’s disease. Ann. Neurol. 1994, 35, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Mallajosyula, J.K.; Chinta, S.J.; Rajagopalan, S.; Nicholls, D.G.; Andersen, J.K. Metabolic control analysis in a cellular model of elevated MAO-B: Relevance to Parkinson’s disease. Neurotox. Res. 2009, 16, 186–193. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Mann, V.M.; Cooper, J.M.; Daniel, S.E.; Srai, K.; Jenner, P.; Marsden, C.D.; Schapira, A.H. Complex I, iron, and ferritin in Parkinson’s disease substantia nigra. Ann. Neurol. 1994, 36, 876–881. [Google Scholar] [CrossRef]
- Zilocchi, M.; Finzi, G.; Lualdi, M.; Sessa, F.; Fasano, M.; Alberio, T. Mitochondrial alterations in Parkinson’s disease human samples and cellular models. Neurochem. Int. 2018, 118, 61–72. [Google Scholar] [CrossRef]
- Lynch, D.S.; Loh, S.H.Y.; Harley, J.; Noyce, A.J.; Martins, L.M.; Wood, N.W.; Houlden, H.; Plun-Favreau, H. Nonsyndromic Parkinson disease in a family with autosomal dominant optic atrophy due to OPA1 mutations. Neurol. Genet. 2017, 3, e188. [Google Scholar] [CrossRef]
- Chu, Y.; Morfini, G.A.; Langhamer, L.B.; He, Y.; Brady, S.T.; Kordower, J.H. Alterations in axonal transport motor proteins in sporadic and experimental Parkinson’s disease. Brain 2012, 135, 2058–2073. [Google Scholar] [CrossRef]
- Tapias, V.; McCoy, J.L.; Greenamyre, J.T. Phenothiazine normalizes the NADH/NAD(+) ratio, maintains mitochondrial integrity and protects the nigrostriatal dopamine system in a chronic rotenone model of Parkinson’s disease. Redox Biol. 2019, 24, 101164. [Google Scholar] [CrossRef]
- Beal, M.F.; Chiluwal, J.; Calingasan, N.Y.; Milne, G.L.; Shchepinov, M.S.; Tapias, V. Isotope-reinforced polyunsaturated fatty acids improve Parkinson’s disease-like phenotype in rats overexpressing alpha-synuclein. Acta Neuropathol. Commun. 2020, 8, 220. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultuRes. and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [PubMed]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rub, C.; Liu, Y.; Magrane, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. alpha-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef] [PubMed]
- Tapias, V.; Hu, X.; Luk, K.C.; Sanders, L.H.; Lee, V.M.; Greenamyre, J.T. Synthetic alpha-synuclein fibrils cause mitochondrial impairment and selective dopamine neurodegeneration in part via iNOS-mediated nitric oxide production. Cell. Mol. Life Sci. 2017, 74, 2851–2874. [Google Scholar] [CrossRef]
- Pozo Devoto, V.M.; Dimopoulos, N.; Alloatti, M.; Pardi, M.B.; Saez, T.M.; Otero, M.G.; Cromberg, L.E.; Marin-Burgin, A.; Scassa, M.E.; Stokin, G.B.; et al. alphaSynuclein control of mitochondrial homeostasis in human-derived neurons is disrupted by mutations associated with Parkinson’s disease. Sci. Rep. 2017, 7, 5042. [Google Scholar] [CrossRef] [PubMed]
- Ulusoy, A.; Rusconi, R.; Perez-Revuelta, B.I.; Musgrove, R.E.; Helwig, M.; Winzen-Reichert, B.; Di Monte, D.A. Caudo-rostral brain spreading of alpha-synuclein through vagal connections. EMBO Mol. Med. 2013, 5, 1119–1127. [Google Scholar] [CrossRef]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. alpha-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra78. [Google Scholar] [CrossRef]
- Chan, C.S.; Guzman, J.N.; Ilijic, E.; Mercer, J.N.; Rick, C.; Tkatch, T.; Meredith, G.E.; Surmeier, D.J. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 2007, 447, 1081–1086. [Google Scholar] [CrossRef]
- Parkinson Study Group STEADY-PD III Investigators. Isradipine Versus Placebo in Early Parkinson Disease: A Randomized Trial. Ann. Intern. Med. 2020, 172, 591–598. [Google Scholar] [CrossRef]
- Abe, K.; Kimura, H. Amyloid beta toxicity consists of a Ca2+-independent early phase and a Ca2+-dependent late phase. J. Neurochem. 1996, 67, 2074–2078. [Google Scholar] [CrossRef]
- Verma, M.; Callio, J.; Otero, P.A.; Sekler, I.; Wills, Z.P.; Chu, C.T. Mitochondrial Calcium Dysregulation Contributes to Dendrite Degeneration Mediated by PD/LBD-Associated LRRK2 Mutants. J. Neurosci. 2017, 37, 11151–11165. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Williams, C.; Etcheto, A.; Goodsaid, F.; Parmentier, F.; Sallantin, J.; Kaufmann, W.E.; Missling, C.U.; Afshar, M. A precision medicine framework using artificial intelligence for the identification and confirmation of genomic biomarkers of response to an Alzheimer’s disease therapy: Analysis of the blarcamesine (ANAVEX2-73) Phase 2a clinical study. Alzheimer’s Dement. 2020, 6, e12013. [Google Scholar] [CrossRef] [PubMed]
- Villard, V.; Espallergues, J.; Keller, E.; Vamvakides, A.; Maurice, T. Anti-amnesic and neuroprotective potentials of the mixed muscarinic receptor/sigma 1 (sigma1) ligand ANAVEX2-73, a novel aminotetrahydrofuran derivative. J. Psychopharmacol. 2011, 25, 1101–1117. [Google Scholar] [CrossRef] [PubMed]
- Lahmy, V.; Meunier, J.; Malmstrom, S.; Naert, G.; Givalois, L.; Kim, S.H.; Villard, V.; Vamvakides, A.; Maurice, T. Blockade of Tau hyperphosphorylation and Aβ1-42 generation by the aminotetrahydrofuran derivative ANAVEX2-73, a mixed muscarinic and sigma(1) receptor agonist, in a nontransgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 2013, 38, 1706–1723. [Google Scholar] [CrossRef] [PubMed]
- Lahmy, V.; Long, R.; Morin, D.; Villard, V.; Maurice, T. Mitochondrial protection by the mixed muscarinic/sigma1 ligand ANAVEX2-73, a tetrahydrofuran derivative, in Abeta25-35 peptide-injected mice, a nontransgenic Alzheimer’s disease model. Front. Cell. Neurosci. 2015, 8, 463. [Google Scholar] [CrossRef]
- Goguadze, N.; Zhuravliova, E.; Morin, D.; Mikeladze, D.; Maurice, T. Sigma-1 Receptor Agonists Induce Oxidative Stress in Mitochondria and Enhance Complex I Activity in Physiological Condition but Protect Against Pathological Oxidative Stress. Neurotox. Res. 2019, 35, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Maurice, T. Protection by sigma-1 receptor agonists is synergic with donepezil, but not with memantine, in a mouse model of amyloid-induced memory impairments. Behav. Brain Res. 2016, 296, 270–278. [Google Scholar] [CrossRef]
- Christ, M.G.; Huesmann, H.; Nagel, H.; Kern, A.; Behl, C. Sigma-1 Receptor Activation Induces Autophagy and Increases Proteostasis Capacity In Vitro and In Vivo. Cells 2019, 8, 211. [Google Scholar] [CrossRef]
- Foscolos, G.B.; Kolocouris, N.; Fytas, G.; Marakos, P.; Pouli, N.; Vamvakides, A. Synthesis and pharmacological study of some new beta-(dialkylaminomethyl)- gamma-butyrolactones and their tetrahydrofuran analogues. Farmaco 1996, 51, 19–26. [Google Scholar] [CrossRef]
- Han, M.E.; Kim, H.J.; Lee, Y.S.; Kim, D.H.; Choi, J.T.; Pan, C.S.; Yoon, S.; Baek, S.Y.; Kim, B.S.; Kim, J.B.; et al. Regulation of cerebrospinal fluid production by caffeine consumption. BMC Neurosci. 2009, 10, 110. [Google Scholar] [CrossRef]
- Zeitlin, R.; Patel, S.; Burgess, S.; Arendash, G.W.; Echeverria, V. Caffeine induces beneficial changes in PKA signaling and JNK and ERK activities in the striatum and cortex of Alzheimer’s transgenic mice. Brain Res. 2011, 1417, 127–136. [Google Scholar] [CrossRef]
- Han, K.; Jia, N.; Li, J.; Yang, L.; Min, L.Q. Chronic caffeine treatment reverses memory impairment and the expression of brain BNDF and TrkB in the PS1/APP double transgenic mouse model of Alzheimer’s disease. Mol. Med. Rep. 2013, 8, 737–740. [Google Scholar] [CrossRef]
- Larsson, S.C.; Woolf, B.; Gill, D. Plasma Caffeine Levels and Risk of Alzheimer’s Disease and Parkinson’s Disease: Mendelian Randomization Study. Nutrients 2022, 14, 1697. [Google Scholar] [CrossRef]
- Kim, J.W.; Byun, M.S.; Yi, D.; Lee, J.H.; Jeon, S.Y.; Jung, G.; Lee, H.N.; Sohn, B.K.; Lee, J.Y.; Kim, Y.K.; et al. Coffee intake and decreased amyloid pathology in human brain. Transl. Psychiatry 2019, 9, 270. [Google Scholar] [CrossRef]
- Eskelinen, M.H.; Kivipelto, M. Caffeine as a protective factor in dementia and Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20 (Suppl. 1), S167–S174. [Google Scholar] [CrossRef]
- Arendash, G.W.; Schleif, W.; Rezai-Zadeh, K.; Jackson, E.K.; Zacharia, L.C.; Cracchiolo, J.R.; Shippy, D.; Tan, J. Caffeine protects Alzheimer’s mice against cognitive impairment and reduces brain beta-amyloid production. Neuroscience 2006, 142, 941–952. [Google Scholar] [CrossRef]
- Cheung, K.H.; Shineman, D.; Muller, M.; Cardenas, C.; Mei, L.; Yang, J.; Tomita, T.; Iwatsubo, T.; Lee, V.M.; Foskett, J.K. Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron 2008, 58, 871–883. [Google Scholar] [CrossRef]
- Cao, C.; Cirrito, J.R.; Lin, X.; Wang, L.; Verges, D.K.; Dickson, A.; Mamcarz, M.; Zhang, C.; Mori, T.; Arendash, G.W.; et al. Caffeine suppresses amyloid-beta levels in plasma and brain of Alzheimer’s disease transgenic mice. J. Alzheimer’s Dis. 2009, 17, 681–697. [Google Scholar] [CrossRef] [PubMed]
- Stazi, M.; Lehmann, S.; Sakib, M.S.; Pena-Centeno, T.; Buschgens, L.; Fischer, A.; Weggen, S.; Wirths, O. Long-term caffeine treatment of Alzheimer mouse models ameliorates behavioural deficits and neuron loss and promotes cellular and molecular markers of neurogenesis. Cell. Mol. Life Sci. 2021, 79, 55. [Google Scholar] [CrossRef] [PubMed]
- Laurent, C.; Eddarkaoui, S.; Derisbourg, M.; Leboucher, A.; Demeyer, D.; Carrier, S.; Schneider, M.; Hamdane, M.; Muller, C.E.; Buee, L.; et al. Beneficial effects of caffeine in a transgenic model of Alzheimer’s disease-like tau pathology. Neurobiol. Aging 2014, 35, 2079–2090. [Google Scholar] [CrossRef]
- Ross, G.W.; Abbott, R.D.; Petrovitch, H.; Morens, D.M.; Grandinetti, A.; Tung, K.H.; Tanner, C.M.; Masaki, K.H.; Blanchette, P.L.; Curb, J.D.; et al. Association of coffee and caffeine intake with the risk of Parkinson disease. JAMA 2000, 283, 2674–2679. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Zhang, S.M.; Hernan, M.A.; Kawachi, I.; Colditz, G.A.; Speizer, F.E.; Willett, W.C. Prospective study of caffeine consumption and risk of Parkinson’s disease in men and women. Ann. Neurol. 2001, 50, 56–63. [Google Scholar] [CrossRef]
- Altman, R.D.; Lang, A.E.; Postuma, R.B. Caffeine in Parkinson’s disease: A pilot open-label, dose-escalation study. Mov. Disord. 2011, 26, 2427–2431. [Google Scholar] [CrossRef]
- Postuma, R.B.; Lang, A.E.; Munhoz, R.P.; Charland, K.; Pelletier, A.; Moscovich, M.; Filla, L.; Zanatta, D.; Rios Romenets, S.; Altman, R.; et al. Caffeine for treatment of Parkinson disease: A randomized controlled trial. Neurology 2012, 79, 651–658. [Google Scholar] [CrossRef]
- Saaksjarvi, K.; Knekt, P.; Rissanen, H.; Laaksonen, M.A.; Reunanen, A.; Mannisto, S. Prospective study of coffee consumption and risk of Parkinson’s disease. Eur. J. Clin. Nutr. 2008, 62, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Li, S. Dose-response meta-analysis on coffee, tea and caffeine consumption with risk of Parkinson’s disease. Geriatr Gerontol. Int. 2014, 14, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Chen, H.; Schwarzschild, M.A.; Zhang, S.M.; Colditz, G.A.; Speizer, F.E. Caffeine, postmenopausal estrogen, and risk of Parkinson’s disease. Neurology 2003, 60, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Powers, K.M.; Kay, D.M.; Factor, S.A.; Zabetian, C.P.; Higgins, D.S.; Samii, A.; Nutt, J.G.; Griffith, A.; Leis, B.; Roberts, J.W.; et al. Combined effects of smoking, coffee, and NSAIDs on Parkinson’s disease risk. Mov. Disord. 2008, 23, 88–95. [Google Scholar] [CrossRef]
- Khadrawy, Y.A.; Salem, A.M.; El-Shamy, K.A.; Ahmed, E.K.; Fadl, N.N.; Hosny, E.N. Neuroprotective and Therapeutic Effect of Caffeine on the Rat Model of Parkinson’s Disease Induced by Rotenone. J. Diet. Suppl. 2017, 14, 553–572. [Google Scholar] [CrossRef]
- Singh, S.; Singh, K.; Patel, S.; Patel, D.K.; Singh, C.; Nath, C.; Singh, M.P. Nicotine and caffeine-mediated modulation in the expression of toxicant responsive genes and vesicular monoamine transporter-2 in 1-methyl 4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease phenotype in mouse. Brain Res. 2008, 1207, 193–206. [Google Scholar] [CrossRef]
- Bagga, P.; Chugani, A.N.; Patel, A.B. Neuroprotective effects of caffeine in MPTP model of Parkinson’s disease: A (13)C NMR study. Neurochem. Int. 2016, 92, 25–34. [Google Scholar] [CrossRef]
- Aguiar, L.M.; Nobre, H.V., Jr.; Macedo, D.S.; Oliveira, A.A.; Freitas, R.M.; Vasconcelos, S.M.; Cunha, G.M.; Sousa, F.C.; Viana, G.S. Neuroprotective effects of caffeine in the model of 6-hydroxydopamine lesion in rats. Pharmacol. Biochem. Behav. 2006, 84, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.; Ren, X.; Zheng, W.; Zeng, Z.; Guo, Y.; Hou, Z.; Guo, W.; Chen, X.; Li, F.; Chen, J.F. Chronic Caffeine Treatment Protects Against alpha-Synucleinopathy by Reestablishing Autophagy Activity in the Mouse Striatum. Front. Neurosci. 2018, 12, 301. [Google Scholar] [CrossRef] [PubMed]
- Zajac, M.A.; Zakrzewski, A.G.; Kowal, M.G.; Narayan, S. A Novel Method of Caffeine Synthesis from Uracil. Synth. Commun. 2003, 33, 3291–3297. [Google Scholar] [CrossRef]
- Scimmi, C.; Cardinali, M.; Abenante, L.; Amatista, M.; Nacca, F.G.; Lenardao, E.J.; Sancineto, L.; Santi, C. Q-Tube®-Assisted Alkylation and Arylation of Xanthines and Other N-H-Containing Heterocycles in Water. Chemistry 2021, 3, 1126–1137. [Google Scholar] [CrossRef]
- Templ, J.; Gjata, E.; Getzner, F.; Schnürch, M. Monoselective N-Methylation of Amides, Indoles, and Related StructuRes. Using Quaternary Ammonium Salts as Solid Methylating Agents. Org. Lett. 2022, 24, 7315–7319. [Google Scholar] [CrossRef]
- Sberna, G.; Saez-Valero, J.; Beyreuther, K.; Masters, C.L.; Small, D.H. The amyloid beta-protein of Alzheimer’s disease increases acetylcholinesterase expression by increasing intracellular calcium in embryonal carcinoma P19 cells. J. Neurochem. 1997, 69, 1177–1184. [Google Scholar] [CrossRef]
- Paris, D.; Bachmeier, C.; Patel, N.; Quadros, A.; Volmar, C.H.; Laporte, V.; Ganey, J.; Beaulieu-Abdelahad, D.; Ait-Ghezala, G.; Crawford, F.; et al. Selective antihypertensive dihydropyridines lower Abeta accumulation by targeting both the production and the clearance of Abeta across the blood-brain barrier. Mol. Med. 2011, 17, 149–162. [Google Scholar] [CrossRef]
- Mok, S.S.; Clippingdale, A.B.; Beyreuther, K.; Masters, C.L.; Barrow, C.J.; Small, D.H. A beta peptides and calcium influence secretion of the amyloid protein precursor from chick sympathetic neurons in culture. J. Neurosci. Res. 2000, 61, 449–457. [Google Scholar] [CrossRef]
- Bouras, C.; Giannakopoulos, P.; Good, P.F.; Hsu, A.; Hof, P.R.; Perl, D.P. A laser microprobe mass analysis of brain aluminum and iron in dementia pugilistica: Comparison with Alzheimer’s disease. Eur. Neurol. 1997, 38, 53–58. [Google Scholar] [CrossRef]
- Rani, A.; Neha; Sodhi, R.K.; Kaur, A. Protective effect of a calcium channel blocker “diltiazem” on aluminum chloride-induced dementia in mice. Naunyn Schmiedebergs Arch. Pharm. 2015, 388, 1151–1161. [Google Scholar] [CrossRef]
- Ritz, B.; Rhodes, S.L.; Qian, L.; Schernhammer, E.; Olsen, J.H.; Friis, S. L-type calcium channel blockers and Parkinson disease in Denmark. Ann. Neurol. 2010, 67, 600–606. [Google Scholar] [CrossRef]
- Guzman, J.N.; Sanchez-Padilla, J.; Chan, C.S.; Surmeier, D.J. Robust pacemaking in substantia nigra dopaminergic neurons. J. Neurosci. 2009, 29, 11011–11019. [Google Scholar] [CrossRef]
- Mosharov, E.V.; Larsen, K.E.; Kanter, E.; Phillips, K.A.; Wilson, K.; Schmitz, Y.; Krantz, D.E.; Kobayashi, K.; Edwards, R.H.; Sulzer, D. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron 2009, 62, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Anjaneyulu, M.; Chopra, K. Diltiazem attenuates oxidative stress in diabetic rats. Ren Fail 2005, 27, 335–344. [Google Scholar] [CrossRef][Green Version]
- Koller, P.T.; Bergmann, S.R. Reduction of lipid peroxidation in reperfused isolated rabbit hearts by diltiazem. Circ. Res. 1989, 65, 838–846. [Google Scholar] [CrossRef]
- Gizur, T.; Harsànyi, K. Some Applications of Isopropenyl Acetate To O-, N-and C-Acetylation. Synth. Commun. 1990, 20, 2365–2371. [Google Scholar] [CrossRef]
- Miyata, O.; Shinada, T.; Ninomiya, I.; Naito, T. Asymmetric induction at two contiguous stereogenic centers by diastereoface differentiating nucleophilic addition reaction. Tetrahedron Lett. 1991, 32, 3519–3522. [Google Scholar] [CrossRef]
- Schwartz, A.; Madan, P.B.; Mohacsi, E.; O’Brien, J.P.; Todaro, L.J.; Coffen, D.L. Enantioselective synthesis of calcium channel blockers of the diltiazem group. J. Org. Chem. 1992, 57, 851–856. [Google Scholar] [CrossRef]
- Jacobsen, E.N.; Deng, L.; Furukawa, Y.; Martínez, L.E. Enantioselective catalytic epoxidation of cinnamate esters. Tetrahedron 1994, 50, 4323–4334. [Google Scholar] [CrossRef]
- Yue, X.; Li, Y.; Liu, M.; Sang, D.; Huang, Z.; Chen, F. Biocatalytic dynamic reductive kinetic resolution of aryl α-chloro β-keto esters: Divergent, stereocontrolled synthesis of diltiazem, clentiazem, and siratiazem. Chem. Commun. 2022, 58, 9010–9013. [Google Scholar] [CrossRef] [PubMed]
- Bachurin, S.; Bukatina, E.; Lermontova, N.; Tkachenko, S.; Afanasiev, A.; Grigoriev, V.; Grigorieva, I.; Ivanov, Y.; Sablin, S.; Zefirov, N. Antihistamine agent Dimebon as a novel neuroprotector and a cognition enhancer. Ann. N. Y. Acad. Sci. 2001, 939, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Gavrilova, S.I.; Sano, M.; Thomas, R.G.; Aisen, P.S.; Bachurin, S.O.; Seely, L.; Hung, D.; dimebon, i. Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer’s disease: A randomised, double-blind, placebo-controlled study. Lancet 2008, 372, 207–215. [Google Scholar] [CrossRef]
- Cano-Cuenca, N.; Solis-Garcia del Pozo, J.E.; Jordan, J. Evidence for the efficacy of latrepirdine (Dimebon) treatment for improvement of cognitive function: A meta-analysis. J. Alzheimer’s Dis. 2014, 38, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Lermontova, N.N.; Lukoyanov, N.V.; Serkova, T.P.; Lukoyanova, E.A.; Bachurin, S.O. Dimebon improves learning in animals with experimental Alzheimer’s disease. Bull. Exp. Biol. Med. 2000, 129, 544–546. [Google Scholar] [CrossRef] [PubMed]
- Webster, S.J.; Wilson, C.A.; Lee, C.H.; Mohler, E.G.; Terry, A.V., Jr.; Buccafusco, J.J. The acute effects of dimebolin, a potential Alzheimer’s disease treatment, on working memory in rhesus monkeys. Br. J. Pharmacol. 2011, 164, 970–978. [Google Scholar] [CrossRef]
- Peters, O.M.; Shelkovnikova, T.; Tarasova, T.; Springe, S.; Kukharsky, M.S.; Smith, G.A.; Brooks, S.; Kozin, S.A.; Kotelevtsev, Y.; Bachurin, S.O.; et al. Chronic administration of Dimebon does not ameliorate amyloid-beta pathology in 5xFAD transgenic mice. J. Alzheimer’s Dis. 2013, 36, 589–596. [Google Scholar] [CrossRef]
- Bachurin, S.O.; Shelkovnikova, T.A.; Ustyugov, A.A.; Peters, O.; Khritankova, I.; Afanasieva, M.A.; Tarasova, T.V.; Alentov, I.I.; Buchman, V.L.; Ninkina, N.N. Dimebon slows progression of proteinopathy in gamma-synuclein transgenic mice. Neurotox. Res. 2012, 22, 33–42. [Google Scholar] [CrossRef][Green Version]
- Wang, J.; Ferruzzi, M.G.; Varghese, M.; Qian, X.; Cheng, A.; Xie, M.; Zhao, W.; Ho, L.; Pasinetti, G.M. Preclinical study of dimebon on beta-amyloid-mediated neuropathology in Alzheimer’s disease. Mol. Neurodegener. 2011, 6, 7. [Google Scholar] [CrossRef]
- Day, M.; Chandran, P.; Luo, F.; Rustay, N.R.; Markosyan, S.; LeBlond, D.; Fox, G.B. Latrepirdine increases cerebral glucose utilization in aged mice as measured by [18F]-fluorodeoxyglucose positron emission tomography. Neuroscience 2011, 189, 299–304. [Google Scholar] [CrossRef]
- Zhang, S.; Hedskog, L.; Petersen, C.A.; Winblad, B.; Ankarcrona, M. Dimebon (latrepirdine) enhances mitochondrial function and protects neuronal cells from death. J. Alzheimer’s Dis. 2010, 21, 389–402. [Google Scholar] [CrossRef]
- Bachurin, S.O.; Shevtsova, E.P.; Kireeva, E.G.; Oxenkrug, G.F.; Sablin, S.O. Mitochondria as a target for neurotoxins and neuroprotective agents. Ann. N. Y. Acad. Sci. 2003, 993, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Shevtzova, E.F.; Kireeva, E.G.; Bachurin, S.O. Effect of beta-amyloid peptide fragment 25-35 on nonselective permeability of mitochondria. Bull. Exp. Biol. Med. 2001, 132, 1173–1176. [Google Scholar] [CrossRef] [PubMed]
- Steele, J.W.; Gandy, S. Latrepirdine (Dimebon(R)), a potential Alzheimer therapeutic, regulates autophagy and neuropathology in an Alzheimer mouse model. Autophagy 2013, 9, 617–618. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Wang, M.; Hutchins, G.D.; Zheng, Q.-H. [11C]Dimebon, radiosynthesis and lipophilicity of a new potential PET agent for imaging of Alzheimer’s disease and Huntington’s disease. Bioorg. Med. Chem. Lett. 2010, 20, 2529–2532. [Google Scholar] [CrossRef] [PubMed]
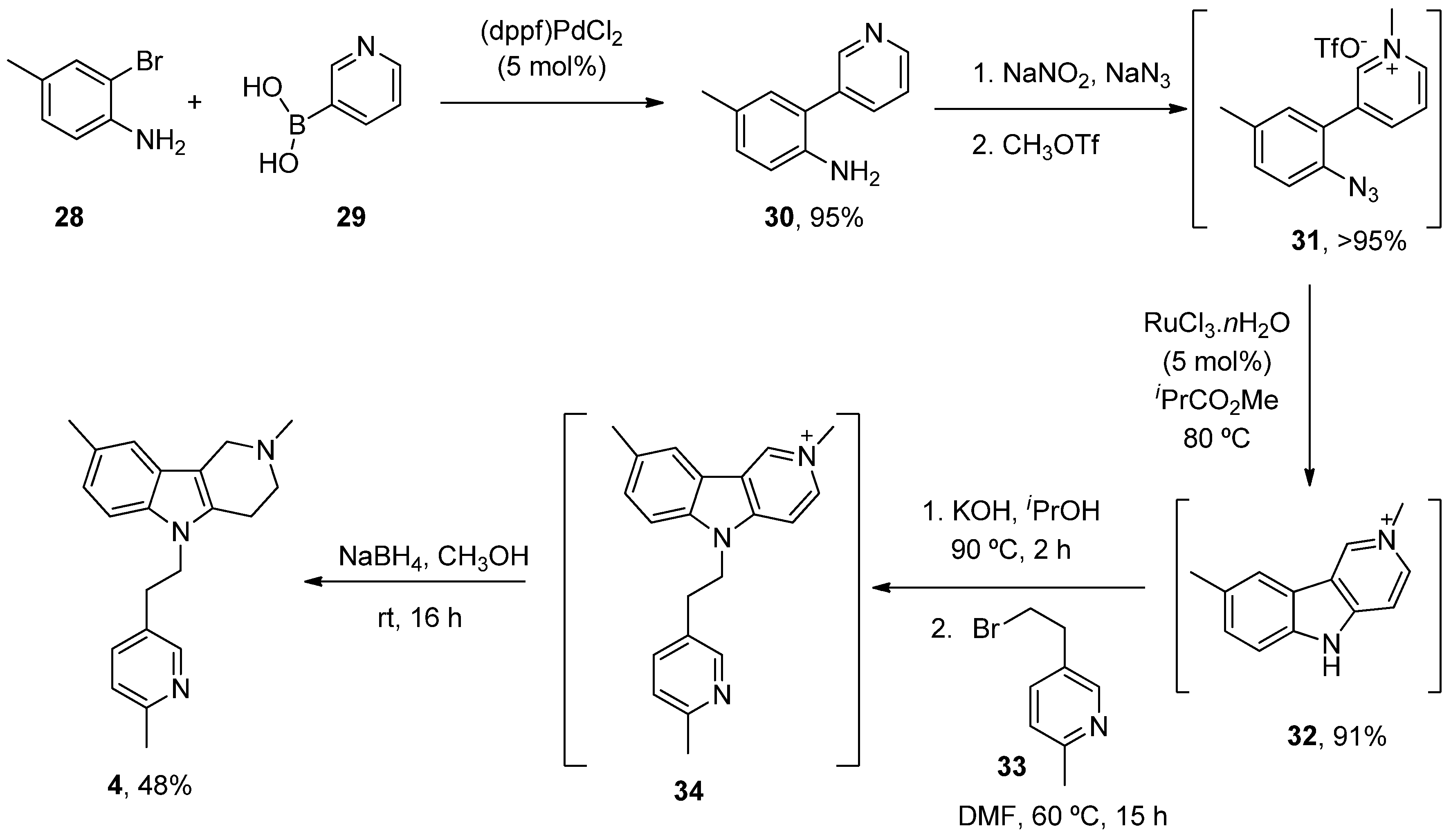
- Dong, H.; Latka, R.T.; Driver, T.G. Ruthenium-Catalyzed γ-Carbolinium Ion Formation from Aryl Azides; Synthesis of Dimebolin. Org. Lett. 2011, 13, 2726–2729. [Google Scholar] [CrossRef]
- Liu, S.J.; Gasperini, R.; Foa, L.; Small, D.H. Amyloid-beta decreases cell-surface AMPA receptors by increasing intracellular calcium and phosphorylation of GluR2. J. Alzheimer’s Dis. 2010, 21, 655–666. [Google Scholar] [CrossRef]
- Lovell, M.A.; Abner, E.; Kryscio, R.; Xu, L.; Fister, S.X.; Lynn, B.C. Calcium Channel Blockers, Progression to Dementia, and Effects on Amyloid Beta Peptide Production. Oxid. Med. Cell Longev. 2015, 2015, 787805. [Google Scholar] [CrossRef]
- Nakajima, M.; Miura, M.; Aosaki, T.; Shirasawa, T. Deficiency of presenilin-1 increases calcium-dependent vulnerability of neurons to oxidative stress in vitro. J. Neurochem. 2001, 78, 807–814. [Google Scholar] [CrossRef][Green Version]
- Hefter, D.; Kaiser, M.; Weyer, S.W.; Papageorgiou, I.E.; Both, M.; Kann, O.; Muller, U.C.; Draguhn, A. Amyloid Precursor Protein Protects Neuronal Network Function after Hypoxia via Control of Voltage-Gated Calcium Channels. J. Neurosci. 2016, 36, 8356–8371. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.R.; Lyckman, A.; Oddo, S.; Laferla, F.M.; Querfurth, H.W.; Shtifman, A. Increased intraneuronal resting [Ca2+] in adult Alzheimer’s disease mice. J. Neurochem. 2008, 105, 262–271. [Google Scholar] [CrossRef]
- Wang, Y.; Mattson, M.P. L-type Ca2+ currents at CA1 synapses, but not CA3 or dentate granule neuron synapses, are increased in 3xTgAD mice in an age-dependent manner. Neurobiol. Aging 2014, 35, 88–95. [Google Scholar] [CrossRef]
- Furukawa, K.; Wang, Y.; Yao, P.J.; Fu, W.; Mattson, M.P.; Itoyama, Y.; Onodera, H.; D’Souza, I.; Poorkaj, P.H.; Bird, T.D.; et al. Alteration in calcium channel properties is responsible for the neurotoxic action of a familial frontotemporal dementia tau mutation. J. Neurochem. 2003, 87, 427–436. [Google Scholar] [CrossRef]
- Pasternak, B.; Svanstrom, H.; Nielsen, N.M.; Fugger, L.; Melbye, M.; Hviid, A. Use of calcium channel blockers and Parkinson’s disease. Am. J. Epidemiol. 2012, 175, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Beurrier, C.; Congar, P.; Bioulac, B.; Hammond, C. Subthalamic nucleus neurons switch from single-spike activity to burst-firing mode. J. Neurosci. 1999, 19, 599–609. [Google Scholar] [CrossRef]
- Garcia, L.; Audin, J.; D’Alessandro, G.; Bioulac, B.; Hammond, C. Dual effect of high-frequency stimulation on subthalamic neuron activity. J. Neurosci. 2003, 23, 8743–8751. [Google Scholar] [CrossRef]
- Eaton, M.E.; Macias, W.; Youngs, R.M.; Rajadhyaksha, A.; Dudman, J.T.; Konradi, C. L-type Ca2+ channel blockers promote Ca2+ accumulation when dopamine receptors are activated in striatal neurons. Brain Res. Mol. Brain Res. 2004, 131, 65–72. [Google Scholar] [CrossRef][Green Version]
- Wang, R.; Ma, Z.; Wang, J.; Xie, J. L-type Cav1.2 calcium channel is involved in 6-hydroxydopamine-induced neurotoxicity in rats. Neurotox. Res. 2012, 21, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Yabuki, Y.; Ohizumi, Y.; Yokosuka, A.; Mimaki, Y.; Fukunaga, K. Nobiletin treatment improves motor and cognitive deficits seen in MPTP-induced Parkinson model mice. Neuroscience 2014, 259, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, S.; Flatman, J.A.; Engberg, I. Nifedipine- and omega-conotoxin-sensitive Ca2+ conductances in guinea-pig substantia nigra pars compacta neurones. J. Physiol. 1993, 466, 727–747. [Google Scholar]
- Sai, Y.; Chen, J.; Ye, F.; Zhao, Y.; Zou, Z.; Cao, J.; Dong, Z. Dopamine Release Suppression Dependent on an Increase of Intracellular Ca2+ Contributed to Rotenone-induced Neurotoxicity in PC12 Cells. J. Toxicol. Pathol. 2013, 26, 149–157. [Google Scholar] [CrossRef]
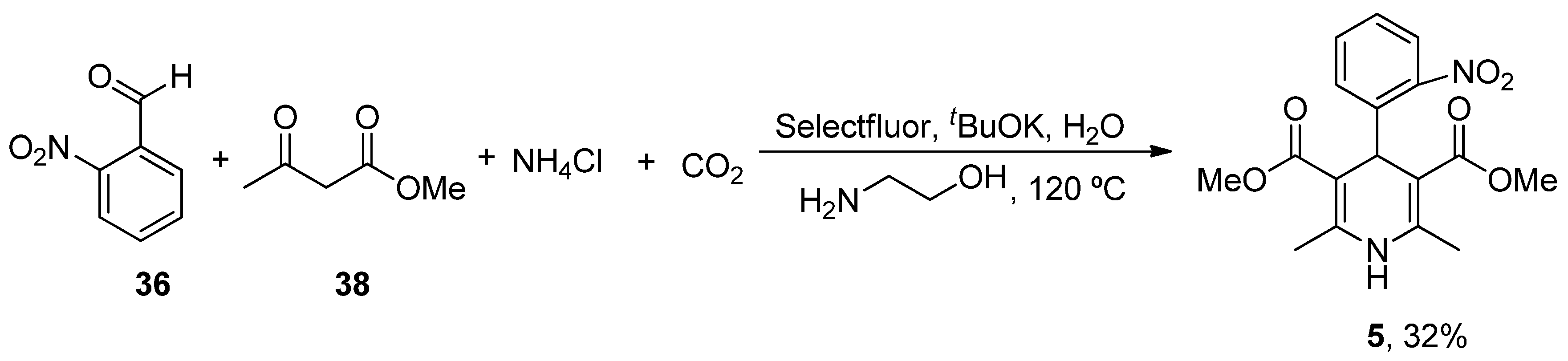
- Singh, H.; Singh, K. Carbon transfer reactions with heterocycles – V. A facile synthesis of nifedipine and analogues. Tetrahedron 1989, 45, 3967–3974. [Google Scholar] [CrossRef]
- Paraskar, A.; Sudalai, A. Cu(OTf)2 Catalyzed High Yield Synthesis of Hantzsch 1,4-Dihydropyridines. Cheminform 2007, 38, 23130. [Google Scholar] [CrossRef]
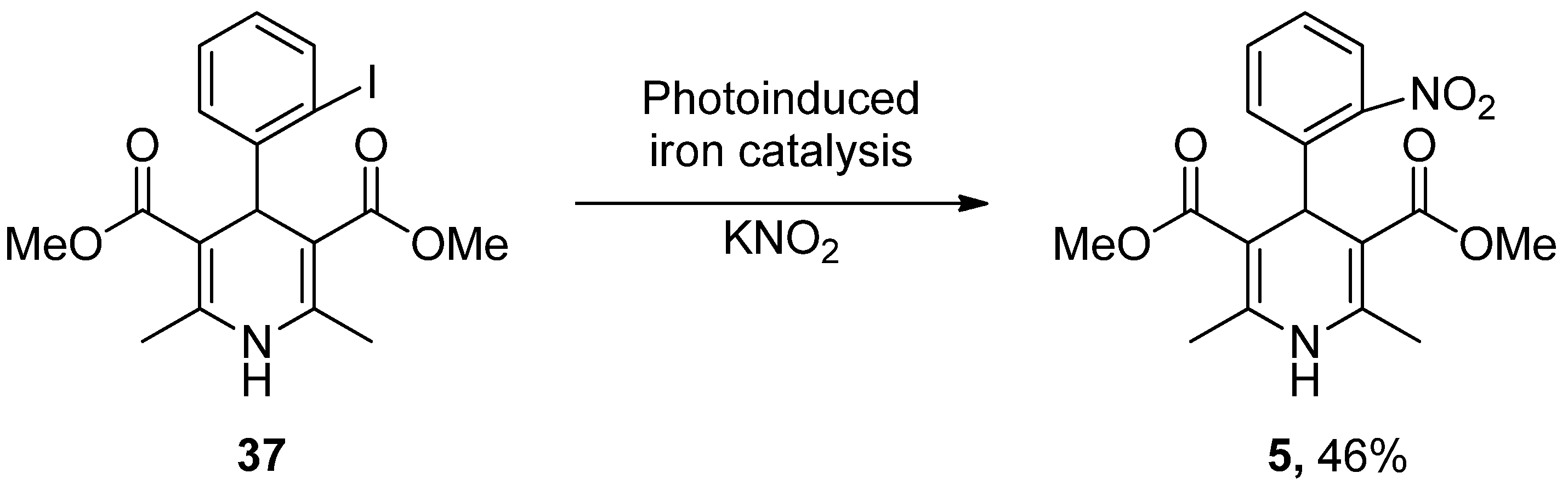
- Wu, C.; Bian, Q.; Ding, T.; Tang, M.; Zhang, W.; Xu, Y.; Liu, B.; Xu, H.; Li, H.-B.; Fu, H. Photoinduced Iron-Catalyzed ipso-Nitration of Aryl Halides via Single-Electron Transfer. ACS Catal. 2021, 11, 9561–9568. [Google Scholar] [CrossRef]
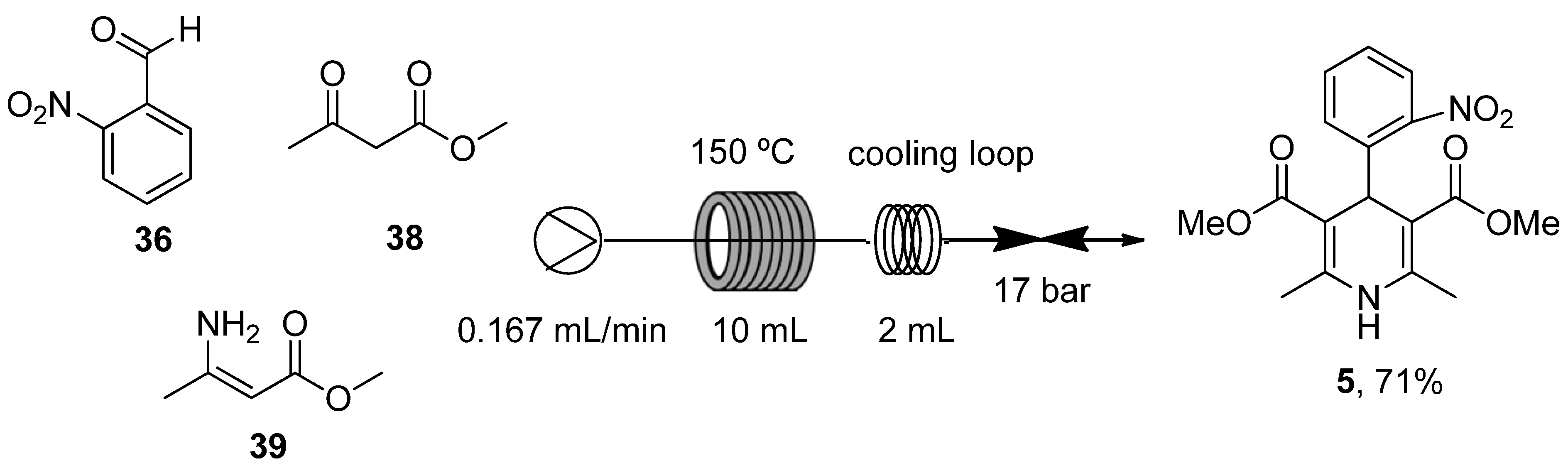
- Guidi, M.; Moon, S.; Anghileri, L.; Cambié, D.; Seeberger, P.H.; Gilmore, K. Combining radial and continuous flow synthesis to optimize and scale-up the production of medicines. React. Chem. Eng. 2021, 6, 220–224. [Google Scholar] [CrossRef]
- Xiang, S.; Fan, W.; Zhang, W.; Li, Y.; Guo, S.; Huang, D. Aqueous CO2 fixation: Construction of pyridine skeletons in cooperation with ammonium cations. Green Chem. 2021, 23, 7950–7955. [Google Scholar] [CrossRef]
- Ban, T.A.; Morey, L.; Aguglia, E.; Azzarelli, O.; Balsano, F.; Marigliano, V.; Caglieris, N.; Sterlicchio, M.; Capurso, A.; Tomasi, N.A.; et al. Nimodipine in the treatment of old age dementias. Prog. Neuropsychopharmacol. Biol. Psychiatry 1990, 14, 525–551. [Google Scholar] [CrossRef]
- Lopez-Arrieta, J.M.; Birks, J. Nimodipine for primary degenerative, mixed and vascular dementia. Cochrane Database Syst. Rev. 2002, 3, CD000147. [Google Scholar] [CrossRef]
- Batuecas, A.; Pereira, R.; Centeno, C.; Pulido, J.A.; Hernandez, M.; Bollati, A.; Bogonez, E.; Satrustegui, J. Effects of chronic nimodipine on working memory of old rats in relation to defects in synaptosomal calcium homeostasis. Eur. J. Pharmacol. 1998, 350, 141–150. [Google Scholar] [CrossRef]
- Gholami Pourbadie, H.; Naderi, N.; Janahmadi, M.; Mehranfard, N.; Motamedi, F. Calcium channel blockade attenuates abnormal synaptic transmission in the dentate gyrus elicited by entorhinal amyloidopathy. Synapse 2016, 70, 408–417. [Google Scholar] [CrossRef]
- Veng, L.M.; Mesches, M.H.; Browning, M.D. Age-related working memory impairment is correlated with increases in the L-type calcium channel protein alpha1D (Cav1.3) in area CA1 of the hippocampus and both are ameliorated by chronic nimodipine treatment. Brain Res. Mol. Brain Res. 2003, 110, 193–202. [Google Scholar] [CrossRef]
- Topcu, A.; Saral, S.; Ozturk, A.; Saral, O.; Kaya, A.K. The effect of the calcium channel blocker nimodipine on hippocampal BDNF/Ach levels in rats with experimental cognitive impairment. Neurol. Res. 2023, 45, 544–553. [Google Scholar] [CrossRef]
- Sandin, M.; Jasmin, S.; Levere, T.E. Aging and cognition: Facilitation of recent memory in aged nonhuman primates by nimodipine. Neurobiol. Aging 1990, 11, 573–575. [Google Scholar] [CrossRef]
- Pierrot, N.; Ghisdal, P.; Caumont, A.S.; Octave, J.N. Intraneuronal amyloid-beta1-42 production triggered by sustained increase of cytosolic calcium concentration induces neuronal death. J. Neurochem. 2004, 88, 1140–1150. [Google Scholar] [CrossRef]
- Ekinci, F.J.; Ortiz, D.; Shea, T.B. Okadaic acid mediates tau phosphorylation via sustained activation of the L-voltage-sensitive calcium channel. Brain Res. Mol. Brain Res. 2003, 117, 145–151. [Google Scholar] [CrossRef]
- Higham, J.P.; Hidalgo, S.; Buhl, E.; Hodge, J.J.L. Restoration of Olfactory Memory in Drosophila Overexpressing Human Alzheimer’s Disease Associated Tau by Manipulation of L-Type Ca2+ Channels. Front. Cell. Neurosci. 2019, 13, 409. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Wu, K.; Ruan, Q.; Li, D.; Liu, W.; Wang, M.; Li, Y.; Xia, J.; Yang, D.; Guo, J. Suppression of Selective Voltage-Gated Calcium Channels Alleviates Neuronal Degeneration and Dysfunction through Glutathione S-Transferase-Mediated Oxidative Stress Resistance in a Caenorhabditis elegans Model of Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2022, 2022, 8287633. [Google Scholar] [CrossRef] [PubMed]
- Hopp, S.C.; Royer, S.E.; D’Angelo, H.M.; Kaercher, R.M.; Fisher, D.A.; Wenk, G.L. Differential neuroprotective and anti-inflammatory effects of L-type voltage dependent calcium channel and ryanodine receptor antagonists in the substantia nigra and locus coeruleus. J. Neuroimmune Pharmacol. 2015, 10, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Putzier, I.; Kullmann, P.H.; Horn, J.P.; Levitan, E.S. Cav1.3 channel voltage dependence, not Ca2+ selectivity, drives pacemaker activity and amplifies bursts in nigral dopamine neurons. J. Neurosci. 2009, 29, 15414–15419. [Google Scholar] [CrossRef]
- Singh, A.; Verma, P.; Balaji, G.; Samantaray, S.; Mohanakumar, K.P. Nimodipine, an L-type calcium channel blocker attenuates mitochondrial dysfunctions to protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonism in mice. Neurochem. Int. 2016, 99, 221–232. [Google Scholar] [CrossRef]
- Kupsch, A.; Gerlach, M.; Pupeter, S.C.; Sautter, J.; Dirr, A.; Arnold, G.; Opitz, W.; Przuntek, H.; Riederer, P.; Oertel, W.H. Pretreatment with nimodipine prevents MPTP-induced neurotoxicity at the nigral, but not at the striatal level in mice. Neuroreport 1995, 6, 621–625. [Google Scholar] [CrossRef]
- Kupsch, A.; Sautter, J.; Schwarz, J.; Riederer, P.; Gerlach, M.; Oertel, W.H. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity in non-human primates is antagonized by pretreatment with nimodipine at the nigral, but not at the striatal level. Brain Res. 1996, 741, 185–196. [Google Scholar] [CrossRef]
- Soderstrom, K.E.; O’Malley, J.A.; Levine, N.D.; Sortwell, C.E.; Collier, T.J.; Steece-Collier, K. Impact of dendritic spine preservation in medium spiny neurons on dopamine graft efficacy and the expression of dyskinesias in parkinsonian rats. Eur. J. Neurosci. 2010, 31, 478–490. [Google Scholar] [CrossRef]
- Schuster, S.; Doudnikoff, E.; Rylander, D.; Berthet, A.; Aubert, I.; Ittrich, C.; Bloch, B.; Cenci, M.A.; Surmeier, D.J.; Hengerer, B.; et al. Antagonizing L-type Ca2+ channel reduces development of abnormal involuntary movement in the rat model of L-3,4-dihydroxyphenylalanine-induced dyskinesia. Biol. Psychiatry 2009, 65, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hu, X.; Liu, Y.; Bao, Y.; An, L. Nimodipine protects dopaminergic neurons against inflammation-mediated degeneration through inhibition of microglial activation. Neuropharmacology 2009, 56, 580–589. [Google Scholar] [CrossRef]
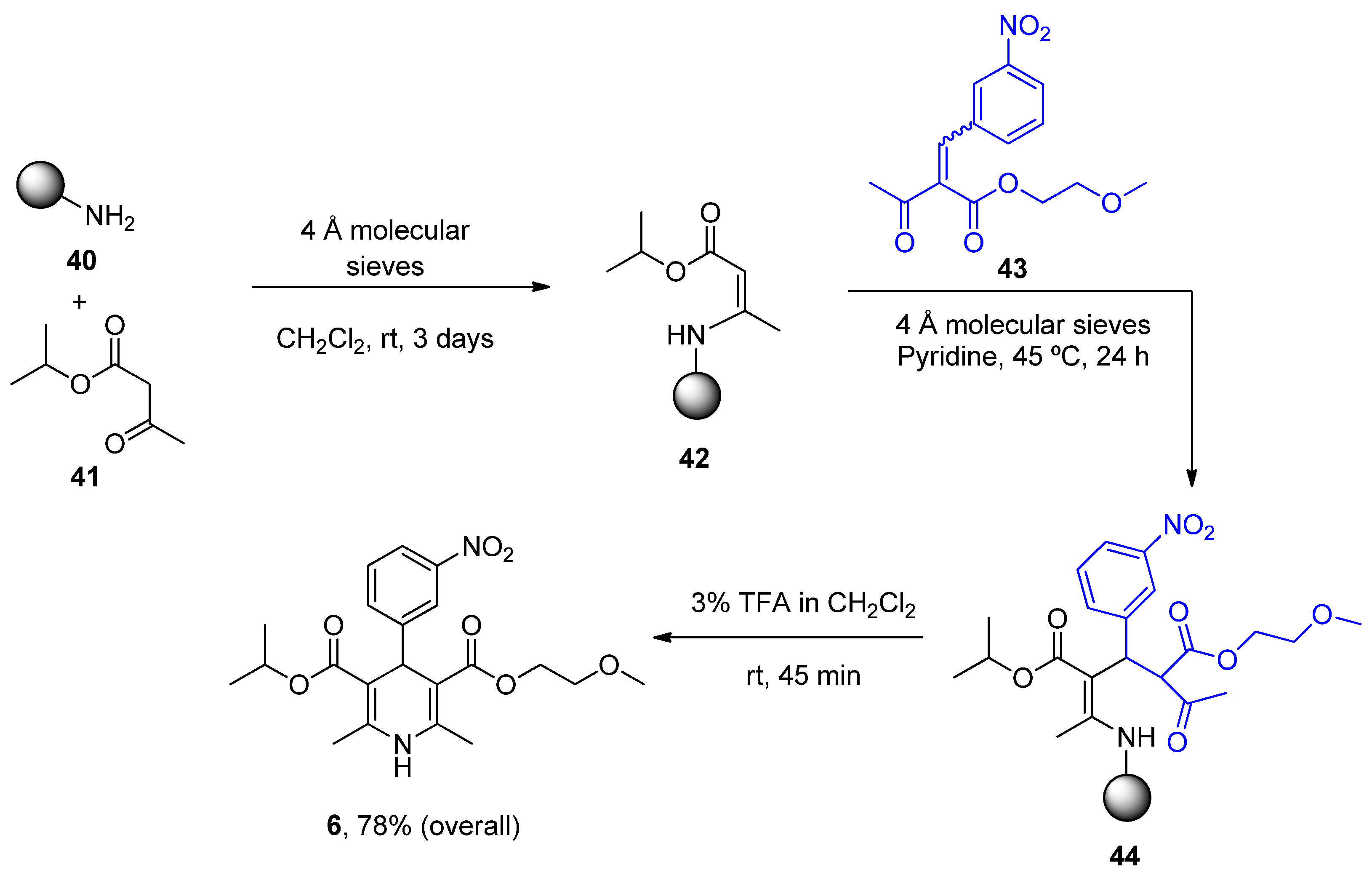
- Gordeev, M.F.; Patel, D.V.; Gordon, E.M. Approaches to Combinatorial Synthesis of Heterocycles: A Solid-Phase Synthesis of 1,4-Dihydropyridines. J. Org. Chem. 1996, 61, 924–928. [Google Scholar] [CrossRef]
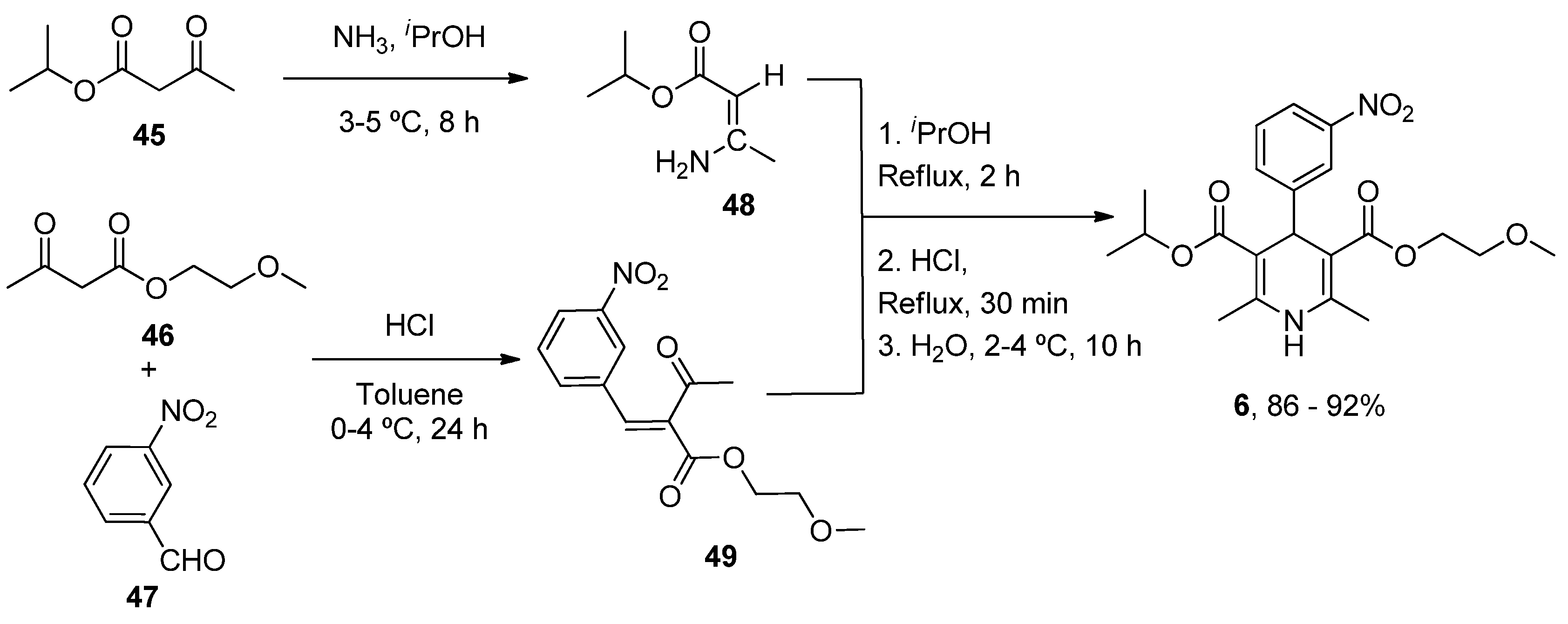
- Balaev, A.N.; Osipov, V.N.; Fedorov, V.E. Development of nimodipine production technology. Pharm. Chem. J. 2012, 46, 285–287. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
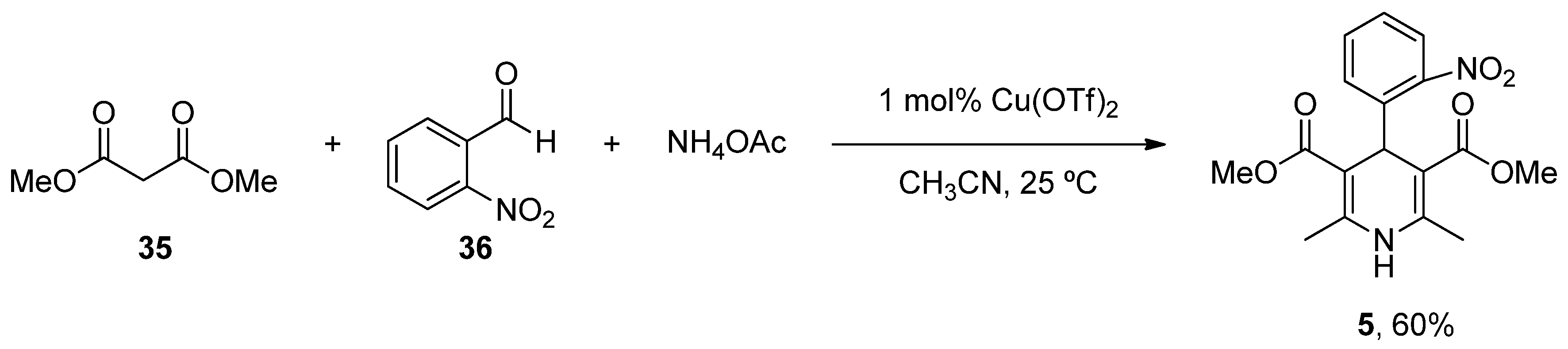{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Heterocyclic Compound | Chemical Name | Target | Proposed Mechanism | Effect on Neurodeg. Dis. Clinical Trial Status | References (Clinical Trials) |
|---|---|---|---|---|---|
| ANAVEX2-73 (Blarcamesine) | Tetrahydro-N,N-dimethyl-2,2-diphenyl-3-furanmethanamine hydrochloride | mAChRs S1R | ↓ Ca2+ release ↓ Mitochondrial stress Activated antioxidant response pathways Limited apoptosis | ↓ Risk of developing AD and PD AD (phase IIb/3) AD (phase III) PDD (phase II) PDD (phase III) | [62] ANAVEX2-73-AD-004 NCT03790709 NCT04575259 NCT03774459 |
| Caffeine (mateine) | 1,3,7-Trimethyl-3,7-dihydro-1H-purine-2,6-dione | A2AR RyR (+) | ↓ Oxidative damage ↓ Aβ levels ↓ α-Syn aggregates Restored AChE and Na+/K+ ATPase activity | ↓ Risk of developing AD and PD Epidemiological studies: motor benefits in PD | [73,74,75] [81,82,83,84,85,86,87,88] |
| Diltiazen (Cardizem) | (2S,3S)-5-(2-(Dimethylamino)ethyl)-2-(4-methoxyphenyl)-4-oxo-2,3,4,5-tetrahydrobenzo[b][1,4]thiazepin-3-yl acetate | Non-dihydro-pyridine VOCC | ↓ Ca2+ entry ↓ Oxidative damage ↓ Inflammation | ↓ Risk of developing PD Epidemiological and randomization studies | [102] |
| Latrepirdine (Dimebon) | 3,6-Dimethyl-9-(2-methyl-pyridyl-5)-ethyl-1,2,3,4-tetrahydro-γ-carboline dihydrochloride | H1R Other Ca2+ Channels | ↓ Ca2+ release and entry ↓ Aβ levels ↓ Oxidative damage to lipids Inhibited AChE ↓ α-Syn | Cognitive and psychiatric benefits in AD Phase III AD: discontinued | [112,113,114] NCT00838110 |
| Nifedipine (Procardia) | 3,5-Dimethyl 2,6-dimethyl-4-(2-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate | L-Type VOCC | ↓ Ca2+ entry ↓ Aβ production ↓ Oxidative damage | Does not ↓ risk of developing PD Epidemiological and randomization studies | [134] |
| Nimodipine (Nimotop) | 3-Isopropyl 5-(2-methoxyethyl) 2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate | L-Type VOCC | ↓ Ca2+ entry Neuroprotection ↓ Aβ toxicity ↓ Oxidative damage Abolished pacemaking activity in DA neurons | ↓ Risk of developing AD Epidemiological and randomization studies | [147,148] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tapias, V.; González-Andrés, P.; Peña, L.F.; Barbero, A.; Núñez, L.; Villalobos, C. Therapeutic Potential of Heterocyclic Compounds Targeting Mitochondrial Calcium Homeostasis and Signaling in Alzheimer’s Disease and Parkinson’s Disease. Antioxidants 2023, 12, 1282. https://doi.org/10.3390/antiox12061282
Tapias V, González-Andrés P, Peña LF, Barbero A, Núñez L, Villalobos C. Therapeutic Potential of Heterocyclic Compounds Targeting Mitochondrial Calcium Homeostasis and Signaling in Alzheimer’s Disease and Parkinson’s Disease. Antioxidants. 2023; 12(6):1282. https://doi.org/10.3390/antiox12061282
Chicago/Turabian StyleTapias, Victor, Paula González-Andrés, Laura F. Peña, Asunción Barbero, Lucía Núñez, and Carlos Villalobos. 2023. "Therapeutic Potential of Heterocyclic Compounds Targeting Mitochondrial Calcium Homeostasis and Signaling in Alzheimer’s Disease and Parkinson’s Disease" Antioxidants 12, no. 6: 1282. https://doi.org/10.3390/antiox12061282
APA StyleTapias, V., González-Andrés, P., Peña, L. F., Barbero, A., Núñez, L., & Villalobos, C. (2023). Therapeutic Potential of Heterocyclic Compounds Targeting Mitochondrial Calcium Homeostasis and Signaling in Alzheimer’s Disease and Parkinson’s Disease. Antioxidants, 12(6), 1282. https://doi.org/10.3390/antiox12061282