Abstract
Carbon monoxide (CO) is a cytoprotective endogenous gas that is ubiquitously produced by the stress response enzyme heme-oxygenase. Being a gas, CO rapidly diffuses through tissues and binds to hemoglobin (Hb) increasing carboxyhemoglobin (COHb) levels. COHb can be formed in erythrocytes or in plasma from cell-free Hb. Herein, it is discussed as to whether endogenous COHb is an innocuous and inevitable metabolic waste product or not, and it is hypothesized that COHb has a biological role. In the present review, literature data are presented to support this hypothesis based on two main premises: (i) there is no direct correlation between COHb levels and CO toxicity, and (ii) COHb seems to have a direct cytoprotective and antioxidant role in erythrocytes and in hemorrhagic models in vivo. Moreover, CO is also an antioxidant by generating COHb, which protects against the pro-oxidant damaging effects of cell-free Hb. Up to now, COHb has been considered as a sink for both exogenous and endogenous CO generated during CO intoxication or heme metabolism, respectively. Hallmarking COHb as an important molecule with a biological (and eventually beneficial) role is a turning point in CO biology research, namely in CO intoxication and CO cytoprotection.
1. Hemoglobin Function
Hemoglobin (Hb) is a widely conserved protein presented in all aerobic life forms where it is responsible for the transport and storage of oxygen (O2). In red blood cells (RBC), Hb corresponds to 98% of the total amount of cellular protein [1]. Hb is a globular protein with a tetrameric structure that is formed by two α and two β polypeptide chains. Each chain contains one heme group, a protoporphyrin IX molecule bound to reduced ferrous ion (Fe2+) which can bind one molecule of O2. Thus, Hb can bind up to four molecules of O2.
The primary biological role of Hb is the transport of O2 from the lungs to all tissues in the organism. Nevertheless, Hb also presents other functions related to its ability to bind other gaseous molecules, namely carbon dioxide (CO2), and the endogenous gasotransmitters: nitric oxide (NO), hydrogen sulfide (H2S), and carbon monoxide (CO) [2,3]. For the sake of completion, a brief overview of the main roles of the first three gasotransmitters is now presented, whereas the role of CO is more extensively presented in the next section.
CO2 is a metabolic product of the tricarboxylic acid cycle (TCA cycle) that affects pH, buffers the blood and inhibits innate immune and inflammatory responses [4]. Hb facilitates CO2 elimination from the organism, thus maintaining cellular homeostasis. Likewise, Hb affinity to O2 decreases in response to milieu acidification or to CO2-Hb binding which produces an allostery change. The decrease in Hb’s affinity to O2 is particularly important for facilitating O2 delivery into tissues [5].
NO is a gasotransmitter and an important signaling molecule enzymatically produced by the activity of nitric oxide synthase (NOS) or non-enzymatic reduction from nitrite. NO regulates vasomodulation by acting on vascular tone and on the expression of endothelial adhesion proteins, plays a role in immunity and inflammation, and is also a CNS signaling molecule [6,7]. Thus, the binding of Hb to NO alters its levels, changing cell signaling. In circulation, RBCs are the main scavengers of NO due to their high amounts of Hb. In fact, NO is much more reactive than CO, reacting with Hb in a much faster and efficient manner. OxyHb-Fe2+ converts NO into nitrate forming methemoglobin (MetHb or Hb-Fe3+) (Figure 1), while NO reacts with deoxyHb-Fe2+ forming DeoxyHb-Fe+ NO iron nitrosyl Hb [8,9].
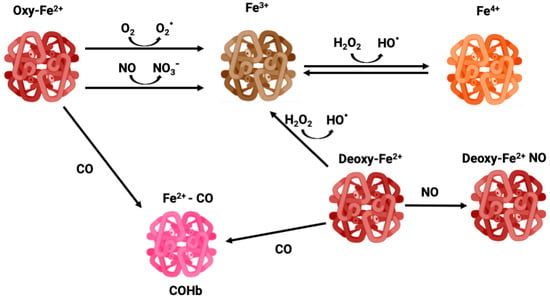
Figure 1.
Different oxidative states of iron—oxyhemoglobin, methemoglobin, carboxyhemoglobin, and ferryl-hemoglobin.
Hydrogen sulfide (H2S) is a gasotransmitter and signaling molecule constantly produced by enzymes such as 3-MST (3-mercaptopyruvate sulfurtransferase), CBS (cystathionine β-synthase) and CSE (cystathionine γ-lyase). High concentrations of H2S are cytotoxic, but at low concentrations it possesses a variety of physiological functions, such as the regulation of blood vessel constriction and dilation and neuronal activity [10]. H2S can exclusively bind to methemoglobin (Hb-Fe3+) to form a methemoglobin–sulfide compound. Nevertheless, this compound can later undergo reductive sulfhydration resulting in the reduction of methemoglobin back to hemoglobin (Hb-Fe2+), along with the formation of polysulfides and thiosulfates. Although H2S mechanisms are not fully understood, the end goal of H2S-MetHb binding seems to be H2S transport and the capacity to reduce methemoglobin back to ferrous hemoglobin (Hb-Fe2+) [3].
CO is a key endogenous signaling gasotransmitter involved in inflammation, cell death, and metabolism control, maintaining cellular homeostasis, as described in the next section. Nevertheless, not much is known about the biological role of carboxyhemoglobin (COHb) under physiological conditions and its potential protective functions under pathological conditions.
2. Carbon Monoxide (CO) and Its Biological Role
As early as 1920, carbon monoxide (CO) was found in exhaled human air, a fact attributed to pollution and smoking and a bit later (1933) to metabolism of microbiota in the gut [11]. CO was identified as a product of heme catabolism only in 1951 [12], taking about two decades more for the identification of heme-oxygenase [13]. Heme-oxygenase (HO) can be inducible (HO-1) or constitutive (HO-2), and its expression and/or activity increases in response to stress along with CO production.
In the last two decades, CO has been extensively studied as a homeostatic and protective molecule. CO’s anti-inflammatory property was the first biological role to be scrutinized. In macrophages, CO controls inflammatory response [14,15] and the ability to kill bacteria [16]. Likewise CO also modulates and limits neuroinflammation in microglia [17,18] and in vivo models of multiple sclerosis [19,20]. CO plays an anti-apoptotic role in different cell types: endothelial cells [21,22]; lung cells [23]; cardiomyocytes [24]; neurons [25,26,27]; and astrocytes [28,29,30]. Another biological process regulated by CO is cell metabolism, namely the balance between glycolysis and oxidative phosphorylation and the modulation of the pentose phosphate pathway related to ROS signaling response [31]. CO-induced reinforcement of oxidative metabolism facilitates prostate cancer treatment [32] and neuronal differentiation [33] and also prevents cell death [29] and limits neuroinflammation [34,35]. Revisions on the biological role of CO in homeostasis and cytoprotection are available [36,37].
Therefore, administration of low CO levels holds great therapeutic potential, with considerable ongoing biomedical research and clinical trials [37]. The administration of these low levels of CO in humans is made by inhalation (iCO). However, in preclinical animal studies of many animal models of disease, CO has been administered through prodrugs or CO-loaded materials. The prodrugs are generally named CO-releasing molecules (CORMs), which can be based on metal carbonyl complexes [38], organic molecules [39,40], or materials loaded with CORMs (CORMAs) [41,42]. All these developments around CO delivery methods different from simple inhalation intend to improve the selective targeting of CO to the disease tissues, which is impossible with inhalation whereby CO is transported in the blood stream without any tissue selectivity, either as dissolved gas in the plasma or as COHb. In all cases, in vivo animal studies with CORMs and/or iCO therapy have used measurements of COHb as a control of exposure and toxicity since, in humans, the latter is absent below 15% COHb with very rare, alleged exceptions. In the case of CORMs and CORMAs, efficient targeting is expected to reduce COHb to very low values just like what happens when HO-1 is activated at the site of disease. In this sense, a clear and rapid COHb rise is an unwanted event. Notwithstanding, some experimental evidence suggests that COHb is not toxic and could instead possess protective activities. Thus, the cytoprotection elicited by CO may be due not only to its direct effect on cells but may also imply COHb signaling.
3. Hemoglobin in Red Blood Cells
Erythrocytes or red blood cells (RBCs) correspond to about one quarter of human cells. They are generated and develop in bone marrow and have a rapid cellular population turnover with a lifetime of 100 to 120 days in circulation. These cells lack nuclei and most of the cellular organelles in order to maximize O2 transport by presenting great amounts of Hb (~98% of total protein). Therefore, cellular function is mainly controlled by protein–protein interactions and allosteric and post-translational modifications [43].
Because of the high O2 tension and abundant iron content, RBCs are prone to generate reactive oxygen species (ROS), which trigger oxidative stress and cell damage. In fact, Hb can suffer slow autooxidation, reducing O2 to the superoxide anion (O2∙−) and oxidating Fe2+ to Fe3+, thus leading to the formation of MetHb, which is unable to bind O2 (Figure 1). The auto-oxidation of Hb is a slow process, but can be accelerated under hypoxia [44]. Likewise, dismutation of O2∙− produces hydrogen peroxide (H2O2) which via the Fenton reaction oxidizes Fe2+ to Fe3+ along with hydroxyl radical (HO∙) production, functioning as an amplification loop. In addition, Hb and MetHb can also function as pseudo peroxidases converting H2O2 into H2O under uncontrolled pathological conditions [45]. In particular, Fe3+ in MetHb is a dangerous molecule that can react with H2O2 leading to Fe4+ ferrylHb formation, which is a powerful oxidant [46] (Figure 1). Endothelial vessel cells and macrophages produce NO which can cross membranes reaching RBC. Once there, NO can oxidize Hb to MetHb, generating nitrate, as well as react with a superoxide, forming peroxinitrite, which is a highly reactive and tissue damaging molecule [44].
4. RBC Anti-Oxidant Machinery
In order to avoid the oxidative processes mentioned above, the RBC environment must be very reducing with strong antioxidant systems able to maintain Hb at the functional reduced Fe2+ state. Accordingly, the RBC antioxidant machinery is constituted by enzymes such as catalase, superoxide dismutase, glutathione peroxidase, and peroxiredoxin-2 enzymes [44]. Catalase and glutathione peroxidase are the main antioxidant enzymes involved in the maintenance of a reducing environment in RBCs [47]. Moreover, high levels of intracellular reduced GSH and ascorbic acid are key elements for maintaining iron at its reduced state [48]. In RBCs, ascorbic acid (AA) can reach concentrations as high as 2 mM. In response to an oxidant, AA is oxidized to dehydroascorbic acid (DHA), which must be recycled by reacting with reduced glutathione (GSH). Likewise, AA is also a critical reducing molecule in plasma, where its oxidation product, DHA, is transported into RBCs via GLUT-type D-glucose transporters for reduction into AA by GSH [46]. RBCs do not contain mitochondria, with glycolysis being their main source of ATP. Glycolysis is linked to the pentose phosphate pathway (PPP), which is the key metabolic pathway for the generation of the reducing factor NADPH needed for GSSG reduction into GSH [49] (Figure 2). Alternatively, Ogasawara and colleagues also demonstrated that in response to tert-butylhydroperoxide treatment (oxidative stress) the reduction of MetHb to Hb was not supported by increased levels of PPP but by glycolysis and NADH generation. In fact, there is an increase in glucose metabolism via glycolysis, generating more pyruvate and NADH via glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [50]. In conclusion, glucose metabolism can be antioxidant in RBCs through different metabolic pathways (Figure 2).
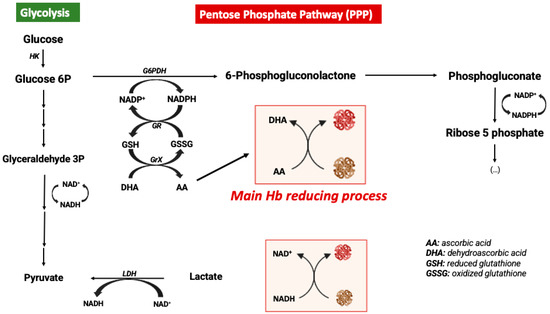
Figure 2.
Metabolic modulation of hemoglobin redox state.
Finally, the Hb protein itself also presents antioxidant functions. Whenever its β93 cysteine (β93Cys) residue is changed for alanine, there is an increase in ROS generation in response to stressful stimuli such as inflammation in mice [51].
5. Cell-Free Hemoglobin
The presence of cell-free Hb in the plasma occurs following hemolysis with the leakage of Hb from RBCs. Pathological hemolysis is a consequence of hemolytic diseases, namely hemorrhage, sickle cell disease, autoimmune induced anemia, infection-induced anemia, thalassemia, massive blood transfusion, among others [52,53]. Under pathological hemolysis, the toxicity of cell free-Hb and heme is not limited to blood vessels but also affects organ parenchyma. In contrast, under physiological conditions, vascular hemolysis also occurs as a consequence of normal RBC turnover. It is well established that the concentration of intracellular Hb (inside erythrocytes) is 10 mM, while the extracellular Hb concentration is around 2 μM in healthy humans [54]. Nevertheless, a study performed in 1959 calculated that about 10% of total RBC breakdown occurred in blood vessels [55]. Despite these controversial data, one can speculate that the systems for scavenging cell-free Hb are quite efficient (please see Section 6).
Inside RBCs, Hb is an α2β2tetramer protein with reduced Fe2+ and an efficient cellular machinery for the maintenance of a reducing environment. In contrast, in an extracellular environment, tetramer Hb is in balance with αβdimer Hb, with a predominant dimer state depending on the cell-free Hb concentration [56]. αβdimer Hb is smaller, which facilitates its extravasation into perivascular tissue, increasing oxidative stress in vulnerable organ parenchyma (the kidneys and liver) [56]. In addition, there is NO-induced oxidative stress by oxidizing oxyHb-Fe2+ to Hb-Fe3+, which easily releases the reactive ferric protoporphyrin-IX group (heme group). In fact, heme is a strong prooxidant and pro-inflammatory molecule, exacerbating tissue damage [56]. Cell-free Hb-Fe2+ also scavenges endogenous NO produced by endothelial cells. Thus, NO does not reach smooth muscle cells, and vasodilation is inhibited [57,58]. In fact, some hemolytic diseases are associated with hypertension since the lack of NO promotes vasoconstriction [59] (Figure 3). Interestingly, cell-free methemoglobin can suffer auto-reduction, generating cell-free Hb-Fe2+, which in turn scavenges NO and promotes vasoconstriction [60].
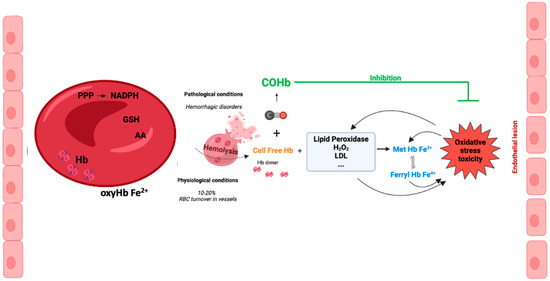
Figure 3.
Cell-free hemoglobin and cell-free carboxyhemoglobin—metabolism and reactions.
In vascular endothelial cells, cell-free Hb induces oxidative stress, upregulation of heme-oxygenase and ferritin, and cell injury, with the effect being much more pronounced whenever MetHb is present [61,62]. Cell-free ferrylHb (Hb-Fe4+) triggers inflammation and permeability of vascular endothelial cells, changing actin cytoskeleton organization and increasing the expression of pro-inflammatory genes [63]. Likewise, interaction between cell-free Hb and atheroma lipids promotes an amplification loop of vessel lesions, involving oxidative stress and inflammation. In fact, oxidized LDL (low density lipoprotein) and lipids derived from atherosclerosis patients facilitates oxidation of MetHb to ferrylHb [64]. Likewise, treatment with ascorbic acid can eventually decrease the cell-free-Hb-induced permeability and lesions in the endothelium [65]. Finally, being a pro-inflammatory molecule, ferrylHb also activates macrophages, promoting a proinflammatory environment in tissues [66].
Great amounts of plasma are filtered by the kidneys daily; therefore, cell-free Hb and its metabolites are highly nephrotoxic, inducing oxidative stress, apoptosis, and inflammation [67,68], which can lead to acute and eventually chronic kidney disease [69,70].
Cerebral microvascular endothelial cells along with astrocytes and pericytes form the blood-brain barrier (BBB), which is a key structure with higher selectivity and limited permeability for the maintenance of brain homeostasis as a sanctuary organ [71]. Intracerebral or subarachnoid hemorrhage are the main causes of cell-free Hb affecting the brain, although vascular hemolysis with systemic circulating cell-free Hb also disturbs the BBB and cerebral parenchyma [72]. Systemic exposure to Hb alters the organization of tight junction proteins and promotes lipid peroxidation and iron deposition in the BBB, leading to its permeabilization [73]. Moreover, rat intracerebral injection of Hb solution promotes BBB leakage by inducing endothelial and inducible NO synthase activity and NO production, which in turn alter tight junction proteins’ organization [74]. Similarly, in hemorrhagic stroke models based on cerebral Hb injection, BBB permeability occurs by peroxinitrite formation following Hb’s reaction with NO [75] or by activation of matrix metalloproteinase-9 (MMP-9) inducing apoptosis in the EC of the BBB [76].
Hb dimer and free heme are small molecules that can easily cross endothelial barriers and infiltrate into tissues; thus, their deleterious effect can also reach other compartment than just the blood or endothelium [56,77]. In particular, in the CNS, cell-free Hb is neurotoxic when reaching the brain parenchyma by triggering neuronal cell death [78] and by inducing neuroinflammation with microglial activation and the release of many proinflammatory cytokines [79,80]. Likewise, Hb breakdown products actively participate in brain injury following intracerebral hemorrhage [81]. The presence of cell-free MetHb in cerebrospinal fluid (CSF) following preterm cerebral intraventricular hemorrhage is associated with pro-neuroinflammatory response with higher levels of TNF-α, IL-1β, and TRL-4 [82]. In astrocytic cell culture, MetHb treatment also increases TNF-α expression, while oxyHb does not [82].
In summary, cell-free Hb is a highly reactive molecule, with it becoming a dimer protein with oxidized iron, that in turn is extremely toxic, with the endothelium being the first target.
6. Haptoglobin and Hemopexin
During evolution, nature has developed systems to protect organisms from cell-free Hb and free heme: haptoglobin and hemopexin, respectively. Haptoglobin (Hp) is an α2- sialoglycoprotein and is the primary Hb-binding protein in the plasma [83]. Hp binds to dimers of Hb, which is rapidly formed following Hb’s release from RBCs [84]. Under physiological conditions, circulating Hp scavenges free Hb originating from normal RBC turnover, thus inhibiting Hb-mediated toxicity. Then, the complex Hb-Hp interacts with CD163 receptors of monocytes/macrophages for its internalization and further clearance in the lysosomes, with upregulation of heme-oxygenase (HO) for heme degradation [85]. Whenever excessive and pathological hemolysis occurs, Hp is depleted in the plasma, and cell-free Hb leads to oxidative stress and endothelium and tissue lesions [83]. Interestingly, there are many studies targeting the levels and the genotype of circulating Hp as a potential biomarker for disease severity, in particular in hemorrhagic diseases [86,87].
In contrast, free heme is first bound to albumin and then is transferred to hemopexin (Hpx). The complex heme-Hpx is primarily eliminated in hepatocytes after its internalization via LDL-receptor-related protein 1 (LRP1) [83]. In fact, the presence of hemopexin reduces endothelial oxidative stress and brain lesions following atherosclerosis [88] and intracerebral hemorrhage [89]. Interestingly, α1-microglobulin is a heme-binding protein also able to scavenge free heme following hemorrhage in atherosclerosis lesions. In fact, the pro-inflammatory ferrylHb and free heme promote expression of α1-microglobulin [90].
7. Is Carboxyhemoglobin (COHb) Toxic?
Since the ancient era, coal fumes are known to be toxic. In the 18th century, the concept of a gas being produced by combustion was established, and a more systematic characterization of gas intoxication in humans was carried out. Still in 1796, this gas was described to have more affinity to “animal fibers” than oxygen, but only in 1800 was CO’s structure found [11]. Claude Bernard discovered that CO displaces oxygen leading to body hypoxia in 1846, and in the 20th century, Haldane and colleagues carried out a quantitative study on CO’s affinity to hemoglobin. For an interesting and complete historical review about CO, please see [11]. With a long role in human history, carbon monoxide (CO) is widely known to the public as a silent killer due to its high affinity to Hb, forming carboxyhemoglobin (COHb). CO presents 240-fold greater affinity for Hb than O2, limiting O2 delivery into tissues, which can lead to systemic hypoxia [11,91]. Interestingly, CO partially binding to Hb increases the affinity of the remaining O2 molecules to Hb, limiting their release into the tissues [92].
With the high affinity of CO to Hb, it is expectable that the storage of endogenously produced CO occurs under the form of COHb. Under physiological conditions, CO is continuously produced by the catabolism of free heme corresponding to more than 90% of endogenous CO production, with the remaining CO coming from lipid and xenobiotic metabolism [93]. Thus, an adult generates 20–50 mL of exhaled CO gas per day under physiological conditions. On the other side, 10–15% of normal erythrocytes’ turnover occurs by vascular hemolysis, leaking hemoglobin (Hb) and free heme into the plasma [55,56]. Thus, endogenous CO is scavenged by Hb, leading to basal COHb production. Indeed, the human basal levels of circulating COHb in a non-smoker subject are 0.1 to 2%, depending on the environmental conditions [11,94]. Moreover, under pathological conditions (in response to inflammation, oxidative stress, hypoxia, etc.), the levels of HO-1 expression increase along with CO generation. Therefore, elevation in COHb circulating levels also occurs.
Since in biology nothing is produced without purpose, it is hypothesized that COHb is not an inert or waste by-product of CO metabolism. Rather, COHb might present an active biological role and thus, COHb could be considered as an integral part of the cytoprotective HO/CO axis previously described. In the next sections, literature data will be presented to support this hypothesis.
8. COHb Is Not a Measure of CO Toxicity
In biological systems, CO binds to heme proteins, changing their function. In fact, CO-induced intoxication is usually associated with deficient systemic oxygen delivery as previously mentioned, while cytotoxicity is mostly associated with CO binding to cytochrome c oxidase which in turn inhibits mitochondrial respiration leading to ATP deprivation and oxidative stress [95].
For decades, the levels of circulating COHb have been considered a measure of CO intoxication. Nevertheless, a great variety of COHb concentrations are associated with fatal CO intoxication, ranging from 3% to 98% [96]. It is important to note that with such a wide range of values, its statistical significance is almost meaningless. Additionally, the highest reported value of COHb levels in an individual who survived is 73% [97], reinforcing the point that COHb does not accurately correlate with CO poisoning. Thus, COHb should only be used as a confirming biomarker of recent CO exposure and not as a measure of CO poisoning severity [98].
Moreover, in 1975, an important in vivo study on systemic CO toxicity showed no correlation between COHb levels and CO toxicity [99]. When a group of dogs was exposed to 13% of CO gas by inhalation, COHb levels reached 54% to 90%, and all animals died within 1 h. In the second group, the dogs received a transfusion of CO-loaded blood, leading to a final value of 80% COHb. In this case, all animals survived [99]. These results indicate that COHb levels cannot be taken as a measure or predictor of systemic CO toxicity. If anything, they show that exogenously administered COHb is not toxic and that, in the case of CO inhalation, toxicity is most likely due to free CO. Accordingly, in 2021, Mao and colleagues developed an accurate and sensitive colorimetric assay for CO quantification in tissues and blood following CO inhalation [100]. This assay is based on the synthetic supramolecule hemoCD1, synthesized by Kitagishi and colleagues, composed of an iron(II)porphyrin, meso-tetrakis(4-sulfonatophenyl) porphinatoiron-(II), and a per-O-methylated β-cyclodextrin dimer, which binds to CO with an affinity higher than that of Hb [101]. Following CO gas inhalation at 400 ppm, the blood COHb concentration linearly increases over time, while CO levels in different tissues (the liver, lungs, cerebrum, cerebellum, and heart) rapidly reach a plateau below the tissue’s saturation capacity. These data suggest that COHb is formed to prevent toxic accumulation of free CO in tissues, ultimately resulting in protection [100].
In summary, COHb in circulation is not toxic and circulating COHb concentration must not be considered a measure of systemic CO toxicity. Likewise, CO-induced toxicity may be mostly associated with CO gas which binds to cellular heme proteins, in particular cytochrome c oxidase. Nevertheless, one can also envisage a scenario where COHb would deliver CO to other heme proteins depending on its affinity, but this needs further investigation.
9. How Can COHb Be (cyto)Protective?
Several hypotheses can be considered for endogenous COHb to have a biological role that can be systemically protective and/or cytoprotective.
9.1. COHb Formation Is a Protection Mechanism against Cell-Free Hb Oxidation and Toxicity
Upon hemolysis, cell-free Hb is easily oxidized from Hb-Fe2+ into Hb-Fe3+ (methemoglobin, MetHb) or Hb-Fe4+ (ferrylHb), which in turn promote oxidative stress, lipid peroxidation, inflammation, and BBB disruption [56,72]. Cell-free COHb is more stable and less toxic than cell-free Hb [102]. For example, in vitro studies mimicking plasma conditions demonstrated that CO reduces Hb-Fe3+ into carboxyHb-Fe2+ in the presence of peroxide as an electron donor [102]. Likewise, in a model of hemolytic malaria (P. berghei-infected mice), CO gas exposure decreased the levels of circulating MetHb and free heme, which limits oxidative stress [103]. Furthermore, the Kirklareli mutation (H58L) in human Hb promotes mild anemia because the mutated Hb is susceptible to high auto-oxidation [104]. Interestingly, smokers carrying the mutation are protected against anemia when compared to non-smokers. This is due to the fact that the CO derived from cigarette smoke tightly binds to the mutated Hb and prevents its oxidation into MetHb [104].
Administered as a mouse intraperitoneal injection, hemoCD binds first to free endogenously produced CO and then captures CO from circulating COHb. Thus, hemoCD generates a depletion of endogenous CO, which in turn upregulates liver HO-1 expression to compensate for the lack of CO. In fact, after losing CO, plasma oxyHb is rapidly oxidized into MetHb, releasing free heme which triggers HO-1 expression [101].
Cell-free or RBC COHb levels may also represent novel potential biomarkers of disease. In fact, high levels of cell-free Hb in human plasma have been found to be associated with worse prognosis in sepsis [105]. Likewise, in intensive care units, COHb is a biomarker for hemolysis. In fact, hemolysis releases cell-free Hb, which is eliminated by macrophagic phagocytosis, increasing the expression of HO-1 for heme degradation which increases CO, thus forming COHb [106]. Thus, COHb holds potential as a diagnostic or prognostic circulating biomarker.
Altogether, these data point to another CO mechanism of action: the generation of COHb protecting against cell-free Hb’s deleterious effects (Figure 3).
9.2. COHb Is an Antioxidant
In vitro, the exposure of red blood cells (RBCs) to CO promotes a great increase in the levels of intracellular reduced GSH, from about 2 mM to 3 mM [107]. CO does not promote glycolysis but partially increases the pentose phosphate pathway. Nevertheless, the main origin of this great increase in GSH levels is Hb de-glutathionylation, meaning the release of glutathione from cysteine residues, in particular Cys93 and Cys112 [107]. COHb formation alters Hb conformation and releases GSH, which in turn plays a key antioxidant role. Therefore, COHb formation in RBCs due to the presence of CO reinforces their antioxidant defenses (Figure 4).
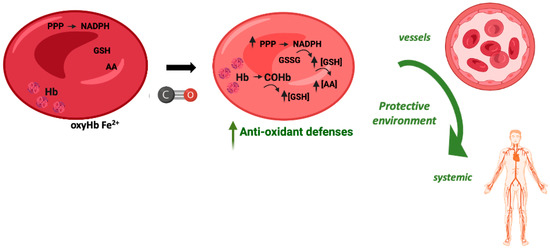
Figure 4.
Potential cytoprotective and antioxidant effects of carboxyhemoglobin.
Using an in vivo rat model of hemorrhagic shock, transfusion of RBCs or vesicles containing Hb exposed to CO gas, that is containing COHb, showed better systemic parameters (arterial blood pressure, lactate, or PO2 and PCO2) than whenever a transfusion was performed with RBCs or vesicles containing Hb [108]. In addition, COHb vesicles or CO-RBCs decreased oxidative damage in the lungs and liver when compared to Hb vesicles or RBC transfusion. Moreover, the circulating COHb levels decreased from 26–39% immediately after transfusion to 3% after 6 h [108]. RBC transfusion is the gold standard treatment for massive hemorrhage. Despite this, this therapy is associated with great hepatic oxidative stress and a decrease in cytochrome P450 activity. Whenever RBCs exposed to CO (CO-RBCs) were used for transfusion, there was a reversion of the deleterious effects of RBCs, with a decrease in nitric oxide and free heme levels, an increase in cytochrome P450 activity, and lower oxidative stress biomarkers [109,110]. Likewise, at a late stage after resuscitation, CO-RBCa also limited the increased levels of the plasma pro-inflammatory markers IL-6 and TNF-α [111]. Finally, in a rat model of acute kidney injury triggered by traumatic rhabdomyolysis and hemorrhage, transfusion of CO-RBCs was revealed to be renoprotective, avoiding the oxidative stress induced by free heme [112]. In summary, transfusion of CO-RBCs confers systemic protection against oxidative stress in the context of hemorrhagic disorders. It should also be tested in other models, including organ transplant or blood transfusion after surgery.
9.3. COHb May Present Other Protective Functions
The most frequently used concentration of CO gas inhalation in animal models is 250 ppm, which promotes protection in several different tissues and pathologies [93]. Depending on the exposure time and the animal model, 250 to 500 ppm leads to circulating COHb levels between 5 and 30% [113]. Thus, the protective effect may be due to free CO reaching tissues or due to circulating COHb or even both.
In addition, ALF186 is a CO-releasing molecule (CORM) that readily delivers its CO load to RBC, reproducibly raising COHb within minutes after mouse injection, with most of the released CO binding to COHb [114]. Interestingly, in vivo i.v. administration of ALF186 protects retinal ganglion cells against ischemia and reperfusion [115]. Because practically all CO molecules released from ALF186 bind to Hb, one can speculate on a direct effect of COHb in tissues, in this case a protective effect in the retina [115]. Nevertheless, this experiment should be repeated with COHb administration instead of CO to confirm the role of COHb.
Finally, another clue indicating that COHb might have a protective role is disclosed by smokers or the phenomenon called the smoking paradox. In fact, smokers present on average higher circulating COHb levels than non-smokers, going from 2% to 10% [116,117]. Despite being extremely injurious and harmful, epidemiological data show that smokers present lower levels of incidence of Parkinson’s disease [118], pre-eclampsia [119], and some skin cancers [120], among other diseases [11]. These protective effects have been attributed to free CO; however, it can be speculated that the higher levels of COHb may also play a role. Of course, molecules other than CO may also be involved in the smoking paradox, since tobacco smoke has a complex composition.
10. Impact and Future Perspectives
The research about COHb as a protective molecule opens a new paradigm in the CO biology field. For 20 years, scientists have searched for the underlying CO mechanisms of cytoprotection, without paying attention to COHb. Moreover, the development of CORMs has always been guided by the idea of avoiding extensive COHb formation to keep the CORM structure intact until it reaches the tissues in need to deliver CO locally. It is urgent now to expand scientific knowledge on the physiological role of COHb, which may also open new avenues for therapy development.
In fact, COHb presents advantages as a therapeutic agent since it is an endogenous molecule that exists permanently in organisms, meaning that our body is fully adapted to it. The advantages of using COHb instead of CO gas or any type of CORMs are obvious. In the former case, the opposition to the use of CO gas due to the widespread, deeply rooted perception of its potential toxicity would be removed. In the latter case, beyond CO, the added risks posed by xenobiotic molecules from scaffolds, carriers, or metabolites would be absent. Many blood substitute products based on Hb have been developed to replace blood and deliver oxygen into tissue. Following this paradigm, PEGylated COHb emerges as a potential therapeutic agent against hemorrhagic and ischemic stroke since it delivers CO and O2 into tissues [121].
Funding
The funding agency that supported the work “Fundação para a Ciência e Tecnologia” (FCT) with 2 projects: Applied Molecular Biosciences Unit-UCIBIO (UID/Multi/04378/2019) and the Associate Laboratory Institute for Health and Bioeconomy (LA/P/0140/2020).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Goodman, S.R.; Kurdia, A.; Ammann, L.; Kakhniashvili, D.; Daescu, O. The Human Red Blood Cell Proteome and Interactome. Exp. Biol. Med. 2007, 232, 1391–1408. [Google Scholar] [CrossRef]
- Quaye, I.K. Extracellular Hemoglobin: The Case of a Friend Turned Foe. Front. Physiol. 2015, 6, 96. [Google Scholar] [CrossRef] [PubMed]
- Jensen, B.; Fago, A. A Novel Possible Role for Met Hemoglobin as Carrier of Hydrogen Sulfide in the Blood. Antioxid. Redox Signal. 2020, 32, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Cummins, E.P.; Selfridge, A.C.; Sporn, P.H.; Sznajder, J.I.; Taylor, C.T. Carbon Dioxide-Sensing in Organisms and Its Implications for Human Disease. Cell. Mol. Life Sci. 2014, 71, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Mairbäurl, H.; Weber, R.E. Oxygen Transport by Hemoglobin. Compr. Physiol. 2011, 2, 1463–1489. [Google Scholar]
- Bloch, K.D.; Ichinose, F.; Roberts, J.D.; Zapol, W.M. Inhaled NO as a Therapeutic Agent. Cardiovasc. Res. 2007, 75, 339–348. [Google Scholar] [CrossRef]
- Moncada, S. Nitric Oxide: Discovery and Impact on Clinical Medicine. J. R. Soc. Med. 1999, 92, 164–169. [Google Scholar] [CrossRef]
- Premont, R.T.; Reynolds, J.D.; Zhang, R.; Stamler, J.S. Role of Nitric Oxide Carried by Hemoglobin in Cardiovascular Physiology. Circ. Res. 2020, 126, 129–158. [Google Scholar] [CrossRef]
- Umbreit, J. Methemoglobin—It’s Not Just Blue: A Concise Review. Am. J. Hematol. 2007, 82, 134–144. [Google Scholar] [CrossRef]
- Pietri, R.; Román-Morales, E.; López-Garriga, J. Hydrogen Sulfide and Hemeproteins: Knowledge and Mysteries. Antioxid. Redox Signal. 2011, 15, 393–404. [Google Scholar] [CrossRef]
- Hopper, C.P.; Zambrana, P.N.; Goebel, U.; Wollborn, J. A Brief History of Carbon Monoxide and Its Therapeutic Origins. Nitric Oxide 2021, 111–112, 45–63. [Google Scholar] [CrossRef] [PubMed]
- Sjöstrand, T. Formation of Carbon Monoxide in Connexion with Hæmoglobin Catabolism. Nature 1951, 168, 1118–1119. [Google Scholar] [CrossRef] [PubMed]
- Tenhunen, R.; Marver, H.S.; Schmid, R. The Enzymatic Conversion of Heme to Bilirubin by Microsomal Heme Oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755. [Google Scholar] [CrossRef]
- Bilban, M.; Bach, F.H.; Otterbein, S.L.; Ifedigbo, E.; de Costa d’Avila, J.; Esterbauer, H.; Chin, B.Y.; Usheva, A.; Robson, S.C.; Wagner, O.; et al. Carbon Monoxide Orchestrates a Protective Response through PPARγ. Immunity 2006, 24, 601–610. [Google Scholar] [CrossRef]
- Jung, S.S.; Moon, J.S.; Xu, J.F.; Ifedigbo, E.; Ryter, S.W.; Choi, A.M.K.; Nakahira, K. Carbon Monoxide Negatively Regulates NLRP3 Inflammasome Activation in Macrophages. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2015, 308, L1058–L1067. [Google Scholar] [CrossRef]
- Wegiel, B.; Larsen, R.; Gallo, D.; Chin, B.Y.; Harris, C.; Mannam, P.; Kaczmarek, E.; Lee, P.J.; Zuckerbraun, B.S.; Flavell, R.; et al. Macrophages Sense and Kill Bacteria through Carbon Monoxide–Dependent Inflammasome Activation. J. Clin. Investig. 2014, 124, 4926–4940. [Google Scholar] [CrossRef]
- Bani-Hani, M.G.; Greenstein, D.; Mann, B.E.; Green, C.J.; Motterlini, R. Modulation of Thrombin-Induced Neuroinflammation in BV-2 Microglia by Carbon Monoxide-Releasing Molecule 3. J. Pharmacol. Exp. Ther. 2006, 318, 1315–1322. [Google Scholar] [CrossRef]
- Soares, N.L.; Paiva, I.; Bravo, J.; Queiroga, C.S.F.; Melo, B.F.; Conde, S.V.; Romão, C.C.; Summavielle, T.; Vieira, H.L.A. Carbon Monoxide Modulation of Microglia-Neuron Communication: Anti-Neuroinflammatory and Neurotrophic Role. Mol. Neurobiol. 2022, 59, 872–889. [Google Scholar] [CrossRef]
- Chora, Â.A.; Fontoura, P.; Cunha, A.; Pais, T.F.; Cardoso, S.; Ho, P.P.; Lee, L.Y.; Sobel, R.A.; Steinman, L.; Soares, M.P. Heme Oxygenase—1 and Carbon Monoxide Suppress Autoimmune Neuroinflammation. J. Clin. Investig. 2007, 117, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Fagone, P.; Mangano, K.; Quattrocchi, C.; Motterlini, R.; Di Marco, R.; Magro, G.; Penacho, N.; Romao, C.C.; Nicoletti, F. Prevention of Clinical and Histological Signs of Proteolipid Protein (PLP)-Induced Experimental Allergic Encephalomyelitis (EAE) in Mice by the Water-Soluble Carbon Monoxide-Releasing Molecule (CORM)-A1. Clin. Exp. Immunol. 2011, 163, 368–374. [Google Scholar] [CrossRef]
- Brouard, S.; Otterbein, L.E.; Anrather, J.; Tobiasch, E.; Bach, F.H.; Choi, A.M.; Soares, M.P. Carbon Monoxide Generated by Heme Oxygenase 1 Suppresses Endothelial Cell Apoptosis. J. Exp. Med. 2000, 192, 1015–1026. [Google Scholar] [CrossRef]
- Basuroy, S.; Tcheranova, D.; Bhattacharya, S.; Leffler, C.W.; Parfenova, H. Nox4 NADPH Oxidase-Derived Reactive Oxygen Species, via Endogenous Carbon Monoxide, Promote Survival of Brain Endothelial Cells during TNF-α-Induced Apoptosis. Am. J. Physiol. Physiol. 2011, 300, C256–C265. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shan, P.; Alam, J.; Davis, R.J.; Flavell, R.A.; Lee, P.J. Carbon Monoxide Modulates Fas/Fas Ligand, Caspases, and Bcl-2 Family Proteins via the P38?? Mitogen-Activated Protein Kinase Pathway during Ischemia-Reperfusion Lung Injury. J. Biol. Chem. 2003, 278, 22061–22070. [Google Scholar] [CrossRef]
- Suliman, H.B.; Carraway, M.S.; Ali, A.S.; Reynolds, C.M.; Welty-wolf, K.E.; Piantadosi, C.A. The CO/HO System Reverses Inhibition of Mitochondrial Biogenesis and Prevents Murine Doxorubicin Cardiomyopathy. J. Clin. Investig. 2007, 117, 3730–3741. [Google Scholar] [CrossRef]
- Vieira, H.L.A.; Queiroga, C.S.F.; Alves, P.M. Pre-Conditioning Induced by Carbon Monoxide Provides Neuronal Protection against Apoptosis. J. Neurochem. 2008, 107, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Schallner, N.; Romão, C.C.; Biermann, J.; Lagrèze, W.A.; Otterbein, L.E.; Buerkle, H.; Loop, T.; Goebel, U. Carbon Monoxide Abrogates Ischemic Insult to Neuronal Cells via the Soluble Guanylate Cyclase-CGMP Pathway. PLoS ONE 2013, 8, e60672. [Google Scholar] [CrossRef]
- Almeida, A.S.; Soares, N.L.; Vieira, M.; Gramsbergen, J.B.; Vieira, H.L.A. Carbon Monoxide Releasing Molecule-A1 (CORM-A1) Improves Neurogenesis: Increase of Neuronal Differentiation Yield by Preventing Cell Death. PLoS ONE 2016, 11, e0154781. [Google Scholar] [CrossRef]
- Queiroga, C.S.F.; Almeida, A.S.; Martel, C.; Brenner, C.; Alves, P.M.; Vieira, H.L.A. Glutathionylation of Adenine Nucleotide Translocase Induced by Carbon Monoxide Prevents Mitochondrial Membrane Permeabilization and Apoptosis. J. Biol. Chem. 2010, 285, 17077–17088. [Google Scholar] [CrossRef]
- Almeida, A.S.; Queiroga, C.S.F.; Sousa, M.F.Q.; Alves, P.M.; Vieira, H.L.A. Carbon Monoxide Modulates Apoptosis by Reinforcing Oxidative Metabolism in Astrocytes: Role of Bcl-2. J. Biol. Chem. 2012, 287, 10761–10770. [Google Scholar] [CrossRef]
- Oliveira, S.R.; Figueiredo-Pereira, C.; Duarte, C.B.; Vieira, H.L.A. P2X7 Receptors Mediate CO-Induced Alterations in Gene Expression in Cultured Cortical Astrocytes—Transcriptomic Study. Mol. Neurobiol. 2019, 56, 3159–3174. [Google Scholar] [CrossRef]
- Figueiredo-Pereira, C.; Dias-Pedroso, D.; Soares, N.L.; Vieira, H.L.A. CO-Mediated Cytoprotection Is Dependent on Cell Metabolism Modulation. Redox Biol. 2020, 32, 101470. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, B.; Gallo, D.; Csizmadia, E.; Harris, C.; Belcher, J.; Vercellotti, G.M.; Penacho, N.; Seth, P.; Sukhatme, V.; Ahmed, A.; et al. Carbon Monoxide Expedites Metabolic Exhaustion to Inhibit Tumor Growth. Cancer Res. 2013, 73, 7009–7021. [Google Scholar] [CrossRef]
- Almeida, A.S.; Sonnewald, U.; Alves, P.M.; Vieira, H.L.A. Carbon Monoxide Improves Neuronal Differentiation and Yield by Increasing the Functioning and Number of Mitochondria. J. Neurochem. 2016, 138, 423–435. [Google Scholar] [CrossRef]
- Dias-Pedroso, D.; Ramalho, J.S.; Sardão, V.A.; Jones, J.G.; Romão, C.C.; Oliveira, P.J.; Vieira, H.L.A. Carbon Monoxide-Neuroglobin Axis Targeting Metabolism Against Inflammation in BV-2 Microglial Cells. Mol. Neurobiol. 2022, 59, 916–931. [Google Scholar] [CrossRef]
- Wilson, J.L.J.L.; Bouillaud, F.; Almeida, A.S.A.S.; Vieira, H.L.; Ouidja, M.O.M.O.; Dubois-Randé, J.L.J.-L.; Foresti, R.; Motterlini, R. Carbon Monoxide Reverses the Metabolic Adaptation of Microglia Cells to an Inflammatory Stimulus. Free Radic. Biol. Med. 2017, 104, 311–323. [Google Scholar] [CrossRef]
- Queiroga, C.S.F.; Vercelli, A.; Vieira, H.L.A. Carbon Monoxide and the CNS: Challenges and Achievements. Br. J. Pharmacol. 2015, 172, 1533–1545. [Google Scholar] [CrossRef] [PubMed]
- Motterlini, R.; Foresti, R. Biological Signaling by Carbon Monoxide and Carbon Monoxide-Releasing Molecules. Am. J. Physiol. Physiol. 2017, 312, C302–C313. [Google Scholar] [CrossRef] [PubMed]
- Ling, K.; Men, F.; Wang, W.C.; Zhou, Y.Q.; Zhang, H.W.; Ye, D.W. Carbon Monoxide and Its Controlled Release: Therapeutic Application, Detection, and Development of Carbon Monoxide Releasing Molecules (CORMs). J. Med. Chem. 2018, 61, 2611–2635. [Google Scholar] [CrossRef]
- Ji, X.; Wang, B. Strategies toward Organic Carbon Monoxide Prodrugs. Acc. Chem. Res. 2018, 51, 1377–1385. [Google Scholar] [CrossRef]
- Lazarus, L.S.; Dederich, C.T.; Anderson, S.N.; Benninghoff, A.D.; Berreau, L.M. Flavonol-Based Carbon Monoxide Delivery Molecule with Endoplasmic Reticulum, Mitochondria, And Lysosome Localization. ACS Med. Chem. Lett. 2022, 13, 236–242. [Google Scholar] [CrossRef]
- Inaba, H.; Fujita, K.; Ueno, T. Design of Biomaterials for Intracellular Delivery of Carbon Monoxide. Biomater. Sci. 2015, 3, 1423–1438. [Google Scholar] [CrossRef]
- Yan, H.; Du, J.; Zhu, S.; Nie, G.; Zhang, H.; Gu, Z.; Zhao, Y. Emerging Delivery Strategies of Carbon Monoxide for Therapeutic Applications: From CO Gas to CO Releasing Nanomaterials. Small 2019, 15, 1904382. [Google Scholar] [CrossRef]
- Sae-Lee, W.; McCafferty, C.L.; Verbeke, E.J.; Havugimana, P.C.; Papoulas, O.; McWhite, C.D.; Houser, J.R.; Vanuytsel, K.; Murphy, G.J.; Drew, K.; et al. The Protein Organization of a Red Blood Cell. Cell Rep. 2022, 40, 111103. [Google Scholar] [CrossRef]
- Rifkind, J.M.; Nagababu, E. Hemoglobin Redox Reactions and Red Blood Cell Aging. Antioxid. Redox Signal. 2013, 18, 2274–2283. [Google Scholar] [CrossRef]
- Reeder, B.J. Redox and Peroxidase Activities of the Hemoglobin Superfamily: Relevance to Health and Disease. Antioxid. Redox Signal. 2017, 26, 763–776. [Google Scholar] [CrossRef]
- Buehler, P.W.; Alayash, A.I. Redox Biology of Blood Revisited: The Role of Red Blood Cells in Maintaining Circulatory Reductive Capacity. Antioxid. Redox Signal. 2005, 7, 1755–1760. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.M.; Ho, Y.-S.; Yu, D.-Y.; Kuypers, F.A.; Ravindranath, Y.; Goyette, G.W. The Effects of Disruption of Genes for Peroxiredoxin-2, Glutathione Peroxidase-1, and Catalase on Erythrocyte Oxidative Metabolism. Free Radic. Biol. Med. 2010, 48, 519–525. [Google Scholar] [CrossRef]
- Çimen, M.Y.B. Free Radical Metabolism in Human Erythrocytes. Clin. Chim. Acta 2008, 390, 1–11. [Google Scholar] [CrossRef]
- May, J.M.; Qu, Z.; Morrow, J.D. Mechanisms of Ascorbic Acid Recycling in Human Erythrocytes. Biochim. Biophys. Acta-Gen. Subj. 2001, 1528, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, Y.; Funakoshi, M.; Ishii, K. Glucose Metabolism Is Accelerated by Exposure to T-Butylhydroperoxide during NADH Consumption in Human Erythrocytes. Blood Cells Mol. Dis. 2008, 41, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Vitturi, D.A.; Sun, C.-W.; Harper, V.M.; Thrash-Williams, B.; Cantu-Medellin, N.; Chacko, B.K.; Peng, N.; Dai, Y.; Wyss, J.M.; Townes, T.; et al. Antioxidant Functions for the Hemoglobin Β93 Cysteine Residue in Erythrocytes and in the Vascular Compartment in Vivo. Free Radic. Biol. Med. 2013, 55, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Rother, R.P.; Bell, L.; Hillmen, P. Of Intravascular Hemolysis A Novel Mechanism of Human Disease. J. Am. Med. Assoc. 2005, 293, 1653–1662. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.J.; Moestrup, S.K. Receptor Targeting of Hemoglobin Mediated by the Haptoglobins: Roles beyond Heme Scavenging. Blood 2009, 114, 764–771. [Google Scholar] [CrossRef]
- Yeo, T.W.; Lampah, D.A.; Tjitra, E.; Gitawati, R.; Kenangalem, E.; Piera, K.; Granger, D.L.; Lopansri, B.K.; Weinberg, J.B.; Price, R.N.; et al. Relationship of Cell-Free Hemoglobin to Impaired Endothelial Nitric Oxide Bioavailability and Perfusion in Severe Falciparum Malaria. J. Infect. Dis. 2009, 200, 1522–1529. [Google Scholar] [CrossRef]
- Garby, L.; Noyes, W.D. Studies of Hemoglobin Metabolism. I. The Kinetic Properties of the Plasma Hemoglobin Pool in Normal Man. J. Clin. Investig. 1959, 38, 1479–1483. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.J.; Buehler, P.W.; Alayash, A.I.; Belcher, J.D.; Vercellotti, G.M. Hemolysis and Free Hemoglobin Revisited: Exploring Hemoglobin and Hemin Scavengers as a Novel Class of Therapeutic Proteins. Blood 2013, 121, 1276–1284. [Google Scholar] [CrossRef]
- Gladwin, M.T.; Crawford, J.H.; Patel, R.P. The Biochemistry of Nitric Oxide, Nitrite, and Hemoglobin: Role in Blood Flow Regulation. Free Radic. Biol. Med. 2004, 36, 707–717. [Google Scholar] [CrossRef]
- Doherty, D.H.; Doyle, M.P.; Curry, S.R.; Vali, R.J.; Fattor, T.J.; Olson, J.S.; Lemon, D.D. Rate of Reaction with Nitric Oxide Determines the Hypertensive Effect of Cell-Free Hemoglobin. Nat. Biotechnol. 1998, 16, 672–676. [Google Scholar] [CrossRef]
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O.; Schechter, A.N.; Gladwin, M.T. Cell-Free Hemoglobin Limits Nitric Oxide Bioavailability in Sickle-Cell Disease. Nat. Med. 2002, 8, 1383–1389. [Google Scholar] [CrossRef]
- Wang, D.; Piknova, B.; Solomon, S.B.; Cortes-Puch, I.; Kern, S.J.; Sun, J.; Kanias, T.; Gladwin, M.T.; Helms, C.; Kim-Shapiro, D.B.; et al. In Vivo Reduction of Cell-Free Methemoglobin to Oxyhemoglobin Results in Vasoconstriction in Canines. Transfusion 2013, 53, 3149–3163. [Google Scholar] [CrossRef]
- Balla, J.; Jacob, H.S.; Balla, G.; Nath, K.; Eaton, J.W.; Vercellotti, G.M. Endothelial-Cell Heme Uptake from Heme Proteins: Induction of Sensitization and Desensitization to Oxidant Damage. Proc. Natl. Acad. Sci. USA 1993, 90, 9285–9289. [Google Scholar] [CrossRef]
- Motterlini, R.; Foresti, R.; Vandegriff, K.; Intaglietta, M.; Winslow, R.M. Oxidative-Stress Response in Vascular Endothelial Cells Exposed to Acellular Hemoglobin Solutions. Am. J. Physiol. Circ. Physiol. 1995, 269, H648–H655. [Google Scholar] [CrossRef]
- Silva, G.; Jeney, V.; Chora, Â.; Larsen, R.; Balla, J.; Soares, M.P. Oxidized Hemoglobin Is an Endogenous Proinflammatory Agonist That Targets Vascular Endothelial Cells. J. Biol. Chem. 2009, 284, 29582–29595. [Google Scholar] [CrossRef]
- Potor, L.; Bányai, E.; Becs, G.; Soares, M.P.; Balla, G.; Balla, J.; Jeney, V. Atherogenesis May Involve the Prooxidant and Proinflammatory Effects of Ferryl Hemoglobin. Oxid. Med. Cell. Longev. 2013, 2013, 676425. [Google Scholar] [CrossRef] [PubMed]
- Kuck, J.L.; Bastarache, J.A.; Shaver, C.M.; Fessel, J.P.; Dikalov, S.I.; May, J.M.; Ware, L.B. Ascorbic Acid Attenuates Endothelial Permeability Triggered by Cell-Free Hemoglobin. Biochem. Biophys. Res. Commun. 2018, 495, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Nyakundi, B.B.; Tóth, A.; Balogh, E.; Nagy, B.; Erdei, J.; Ryffel, B.; Paragh, G.; Cordero, M.D.; Jeney, V. Oxidized Hemoglobin Forms Contribute to NLRP3 Inflammasome-Driven IL-1β Production upon Intravascular Hemolysis. Biochim. Biophys. Acta-Mol. Basis Dis. 2019, 1865, 464–475. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Navarro, A.; Vázquez-Carballo, C.; Guerrero-Hue, M.; García-Caballero, C.; Herencia, C.; Gutiérrez, E.; Yuste, C.; Sevillano, Á.; Praga, M.; Egea, J.; et al. Nrf2 Plays a Protective Role Against Intravascular Hemolysis-Mediated Acute Kidney Injury. Front. Pharmacol. 2019, 10, 740. [Google Scholar] [CrossRef]
- Gonzalez-Michaca, L.; Farrugia, G.; Croatt, A.J.; Alam, J.; Nath, K.A. Heme: A Determinant of Life and Death in Renal Tubular Epithelial Cells. Am. J. Physiol. Physiol. 2004, 286, F370–F377. [Google Scholar] [CrossRef]
- Moreno, J.A.; Martín-Cleary, C.; Gutiérrez, E.; Toldos, O.; Blanco-Colio, L.M.; Praga, M.; Ortiz, A.; Egido, J. AKI Associated with Macroscopic Glomerular Hematuria. Clin. J. Am. Soc. Nephrol. 2012, 7, 175–184. [Google Scholar] [CrossRef]
- Naik, R.P.; Derebail, V.K.; Grams, M.E.; Franceschini, N.; Auer, P.L.; Peloso, G.M.; Young, B.A.; Lettre, G.; Peralta, C.A.; Katz, R.; et al. Association of Sickle Cell Trait With Chronic Kidney Disease and Albuminuria in African Americans. JAMA 2014, 312, 2115. [Google Scholar] [CrossRef]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Keep, R.F.; Andjelkovic, A.V.; Xiang, J.; Stamatovic, S.M.; Antonetti, D.A.; Hua, Y.; Xi, G. Brain Endothelial Cell Junctions after Cerebral Hemorrhage: Changes, Mechanisms and Therapeutic Targets. J. Cereb. Blood Flow Metab. 2018, 38, 1255–1275. [Google Scholar] [CrossRef]
- Butt, O.I.; Buehler, P.W.; D’Agnillo, F. Blood-Brain Barrier Disruption and Oxidative Stress in Guinea Pig after Systemic Exposure to Modified Cell-Free Hemoglobin. Am. J. Pathol. 2011, 178, 1316–1328. [Google Scholar] [CrossRef]
- Yang, S.; Chen, Y.; Deng, X.; Jiang, W.; Li, B.; Fu, Z.; Du, M.; Ding, R. Hemoglobin-Induced Nitric Oxide Synthase Overexpression and Nitric Oxide Production Contribute to Blood–Brain Barrier Disruption in the Rat. J. Mol. Neurosci. 2013, 51, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.; Feng, L.; He, L.; Chen, Y.; Wen, P.; Fu, Z.; Lin, C.; Yang, S.; Deng, X.; Zeng, J.; et al. Peroxynitrite Decomposition Catalyst Prevents Matrix Metalloproteinase-9 Activation and Neurovascular Injury after Hemoglobin Injection into the Caudate Nucleus of Rats. Neuroscience 2015, 297, 182–193. [Google Scholar] [CrossRef]
- Katsu, M.; Niizuma, K.; Yoshioka, H.; Okami, N.; Sakata, H.; Chan, P.H. Hemoglobin-Induced Oxidative Stress Contributes to Matrix Metalloproteinase Activation and Blood-Brain Barrier Dysfunction in Vivo. J. Cereb. Blood Flow Metab. 2010, 30, 1939–1950. [Google Scholar] [CrossRef] [PubMed]
- Gáll, T.; Pethő, D.; Nagy, A.; Balla, G.; Balla, J. Therapeutic Potential of Carbon Monoxide (CO) and Hydrogen Sulfide (H2S) in Hemolytic and Hemorrhagic Vascular Disorders—Interaction between the Heme Oxygenase and H2S-Producing Systems. Int. J. Mol. Sci. 2020, 22, 47. [Google Scholar] [CrossRef]
- Kaiser, S.; Frase, S.; Selzner, L.; Lieberum, J.-L.; Wollborn, J.; Niesen, W.-D.; Foit, N.A.; Heiland, D.H.; Schallner, N. Neuroprotection after Hemorrhagic Stroke Depends on Cerebral Heme Oxygenase-1. Antioxidants 2019, 8, 496. [Google Scholar] [CrossRef]
- Taylor, R.A.; Chang, C.-F.; Goods, B.A.; Hammond, M.D.; Grory, B.M.; Ai, Y.; Steinschneider, A.F.; Renfroe, S.C.; Askenase, M.H.; McCullough, L.D.; et al. TGF-Β1 Modulates Microglial Phenotype and Promotes Recovery after Intracerebral Hemorrhage. J. Clin. Investig. 2017, 127, 280–292. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, Y.; Wang, J.; Anne Stetler, R.; Yang, Q.-W. Inflammation in Intracerebral Hemorrhage: From Mechanisms to Clinical Translation. Prog. Neurobiol. 2014, 115, 25–44. [Google Scholar] [CrossRef]
- Xi, G.; Keep, R.F.; Hoff, J.T. Mechanisms of Brain Injury after Intracerebral Haemorrhage. Lancet Neurol. 2006, 5, 53–63. [Google Scholar] [CrossRef]
- Gram, M.; Sveinsdottir, S.; Ruscher, K.; Hansson, S.R.; Cinthio, M.; Åkerström, B.; Ley, D. Hemoglobin Induces Inflammation after Preterm Intraventricular Hemorrhage by Methemoglobin Formation. J. Neuroinflamm. 2013, 10, 867. [Google Scholar] [CrossRef]
- Schaer, D.J.; Vinchi, F.; Ingoglia, G.; Tolosano, E.; Buehler, P.W. Haptoglobin, Hemopexin, and Related Defense Pathways—basic Science, Clinical Perspectives, and Drug Development. Front. Physiol. 2014, 5, 415. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Polticelli, F.; Coletta, M. Oxygen Dissociation from Ferrous Oxygenated Human Hemoglobin:Haptoglobin Complexes Confirms That in the R-State α and β Chains Are Functionally Heterogeneous. Sci. Rep. 2019, 9, 6780. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, J.H.; Etzerodt, A.; Svendsen, P.; Moestrup, S.K. The Haptoglobin-CD163-Heme Oxygenase-1 Pathway for Hemoglobin Scavenging. Oxid. Med. Cell. Longev. 2013, 2013, 523652. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, H.; Iihara, K.; Kaku, Y.; Yamauchi, K.; Fukuda, K.; Nishimura, K.; Nakai, M.; Satow, T.; Nakajima, N.; Ikegawa, M. Haptoglobin Phenotype Predicts Cerebral Vasospasm and Clinical Deterioration after Aneurysmal Subarachnoid Hemorrhage. J. Stroke Cerebrovasc. Dis. 2013, 22, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Kantor, E.; Bayır, H.; Ren, D.; Provencio, J.J.; Watkins, L.; Crago, E.; Horowitz, M.B.; Ferrell, R.E.; Conley, Y.P.; Alexander, S.A. Haptoglobin Genotype and Functional Outcome after Aneurysmal Subarachnoid Hemorrhage. J. Neurosurg. 2014, 120, 386–390. [Google Scholar] [CrossRef]
- Nagy, E.; Eaton, J.W.; Jeney, V.; Soares, M.P.; Varga, Z.; Galajda, Z.; Szentmiklósi, J.; Méhes, G.; Csonka, T.; Smith, A.; et al. Red Cells, Hemoglobin, Heme, Iron, and Atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1347–1353. [Google Scholar] [CrossRef]
- Ma, B.; Day, J.P.; Phillips, H.; Slootsky, B.; Tolosano, E.; Doré, S. Deletion of the Hemopexin or Heme Oxygenase-2 Gene Aggravates Brain Injury Following Stroma-Free Hemoglobin-Induced Intracerebral Hemorrhage. J. Neuroinflamm. 2016, 13, 26. [Google Scholar] [CrossRef]
- Pethő, D.; Gáll, T.; Hendrik, Z.; Nagy, A.; Beke, L.; Gergely, A.P.; Méhes, G.; Tóth, C.; Gram, M.; Åkerström, B.; et al. Ferryl Hemoglobin and Heme Induce A1-Microglobulin in Hemorrhaged Atherosclerotic Lesions with Inhibitory Function against Hemoglobin and Lipid Oxidation. Int. J. Mol. Sci. 2021, 22, 6668. [Google Scholar] [CrossRef]
- Prockop, L.D.; Chichkova, R.I. Carbon Monoxide Intoxication: An Updated Review. J. Neurol. Sci. 2007, 262, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Choi, A.M.K. Carbon Monoxide: Present and Future Indications for a Medical Gas. Korean J. Intern. Med. 2013, 28, 123–140. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A.M.K. Heme Oxygenase-1/Carbon Monoxide: From Basic Science to Therapeutic Applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef]
- Rudra, C.B.; Williams, M.A.; Sheppard, L.; Koenig, J.Q.; Schiff, M.A.; Frederick, I.O.; Dills, R. Relation of Whole Blood Carboxyhemoglobin Concentration to Ambient Carbon Monoxide Exposure Estimated Using Regression. Am. J. Epidemiol. 2010, 171, 942–951. [Google Scholar] [CrossRef][Green Version]
- D’Amico, G.; Lam, F.; Hagen, T.; Moncada, S. Inhibition of Cellular Respiration by Endogenously Produced Carbon Monoxide. J. Cell Sci. 2006, 119, 2291–2298. [Google Scholar] [CrossRef] [PubMed]
- Weaver, L.K.; Howe, S.; Hopkins, R.; Chan, K.J. Carboxyhemoglobin Half-Life in Carbon Monoxide-Poisoned Patients Treated With 100% Oxygen at Atmospheric Pressure. Chest 2000, 117, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.E.; Stewart, R.D. Absorption and Elimination of Carbon Monoxide by Inactive Young Men. Arch. Environ. Health An Int. J. 1970, 21, 165–171. [Google Scholar] [CrossRef]
- Hampson, N.B. Carboxyhemoglobin: A Primer for Clinicians. Undersea Hyperb. Med. 2018, 45, 165–171. [Google Scholar] [CrossRef]
- Goldbaum, L.R.; Ramirez, R.G.; Absalon, K.B. What Is the Mechanism of Carbon Monoxide Toxicity? Aviat. Space Environ. Med. 1975, 46, 1289–1291. [Google Scholar]
- Mao, Q.; Kawaguchi, A.T.; Mizobata, S.; Motterlini, R.; Foresti, R.; Kitagishi, H. Sensitive Quantification of Carbon Monoxide in Vivo Reveals a Protective Role of Circulating Hemoglobin in CO Intoxication. Commun. Biol. 2021, 4, 425. [Google Scholar] [CrossRef]
- Kitagishi, H.; Minegishi, S.; Yumura, A.; Negi, S.; Taketani, S.; Amagase, Y.; Mizukawa, Y.; Urushidani, T.; Sugiura, Y.; Kano, K. Feedback Response to Selective Depletion of Endogenous Carbon Monoxide in the Blood. J. Am. Chem. Soc. 2016, 138, 5417–5425. [Google Scholar] [CrossRef] [PubMed]
- Sher, E.A.; Shaklai, M.; Shaklai, N. Carbon Monoxide Promotes Respiratory Hemoproteins Iron Reduction Using Peroxides as Electron Donors. PLoS ONE 2012, 7, e33039. [Google Scholar] [CrossRef] [PubMed]
- Pamplona, A.; Ferreira, A.; Balla, J.; Jeney, V.; Balla, G.; Epiphanio, S.; Chora, Â.; Rodrigues, C.D.; Gregoire, I.P.; Cunha-Rodrigues, M.; et al. Heme Oxygenase-1 and Carbon Monoxide Suppress the Pathogenesis of Experimental Cerebral Malaria. Nat. Med. 2007, 13, 703–710. [Google Scholar] [CrossRef]
- Bissé, E.; Schaeffer-Reiss, C.; Van Dorsselaer, A.; Alayi, T.D.; Epting, T.; Winkler, K.; Benitez Cardenas, A.S.; Soman, J.; Birukou, I.; Samuel, P.P.; et al. Hemoglobin Kirklareli (α H58L), a New Variant Associated with Iron Deficiency and Increased CO Binding. J. Biol. Chem. 2017, 292, 2542–2555. [Google Scholar] [CrossRef]
- Janz, D.R.; Bastarache, J.A.; Peterson, J.F.; Sills, G.; Wickersham, N.; May, A.K.; Roberts, L.J.; Ware, L.B. Association Between Cell-Free Hemoglobin, Acetaminophen, and Mortality in Patients With Sepsis. Crit. Care Med. 2013, 41, 784–790. [Google Scholar] [CrossRef]
- Hariri, G.; Hodjat Panah, K.; Beneteau-Burnat, B.; Chaquin, M.; Mekinian, A.; Ait-Oufella, H. Carboxyhemoglobin, a Reliable Diagnosis Biomarker for Hemolysis in Intensive Care Unit: A Retrospective Study. Crit. Care 2021, 25, 7. [Google Scholar] [CrossRef] [PubMed]
- Metere, A.; Iorio, E.; Scorza, G.; Camerini, S.; Casella, M.; Crescenzi, M.; Minetti, M.; Pietraforte, D. Carbon Monoxide Signaling in Human Red Blood Cells: Evidence for Pentose Phosphate Pathway Activation and Protein Deglutathionylation. Antioxid. Redox Signal. 2014, 20, 403–416. [Google Scholar] [CrossRef]
- Sakai, H.; Horinouchi, H.; Tsuchida, E.; Kobayashi, K. Hemoglobin Vesicles and Red Blood Cells as Carriers of Carbon Monoxide Prior to Oxygen for Resuscitation after Hemorrhagic Shock in a Rat Model. Shock 2009, 31, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Ogaki, S.; Taguchi, K.; Watanabe, H.; Otagiri, M.; Maruyama, T. Carbon Monoxide–Bound Red Blood Cells Protect Red Blood Cell Transfusion-Induced Hepatic Cytochrome P450 Impairment in Hemorrhagic-Shock Rats. Drug Metab. Dispos. 2013, 41, 141–148. [Google Scholar] [CrossRef]
- Ogaki, S.; Taguchi, K.; Watanabe, H.; Ishima, Y.; Otagiri, M.; Maruyama, T. Carbon Monoxide-Bound Red Blood Cell Resuscitation Ameliorates Hepatic Injury Induced by Massive Hemorrhage and Red Blood Cell Resuscitation via Hepatic Cytochrome P450 Protection in Hemorrhagic Shock Rats. J. Pharm. Sci. 2014, 103, 2199–2206. [Google Scholar] [CrossRef]
- Ogaki, S.; Taguchi, K.; Maeda, H.; Watanabe, H.; Ishima, Y.; Otagiri, M.; Maruyama, T. Kupffer Cell Inactivation by Carbon Monoxide Bound to Red Blood Cells Preserves Hepatic Cytochrome P450 via Anti-Oxidant and Anti-Inflammatory Effects Exerted through the HMGB1/TLR-4 Pathway during Resuscitation from Hemorrhagic Shock. Biochem. Pharmacol. 2015, 97, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Ogaki, S.; Nagasaki, T.; Yanagisawa, H.; Nishida, K.; Maeda, H.; Enoki, Y.; Matsumoto, K.; Sekijima, H.; Ooi, K.; et al. Carbon Monoxide Rescues the Developmental Lethality of Experimental Rat Models of Rhabdomyolysis-Induced Acute Kidney Injury. J. Pharmacol. Exp. Ther. 2020, 372, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Choi, A.M.K. Targeting Heme Oxygenase-1 and Carbon Monoxide for Therapeutic Modulation of Inflammation. Transl. Res. 2016, 167, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Seixas, J.D.; Mukhopadhyay, A.; Santos-Silva, T.; Otterbein, L.E.; Gallo, D.J.; Rodrigues, S.S.; Guerreiro, B.H.; Gonçalves, A.M.L.; Penacho, N.; Marques, A.R.; et al. Characterization of a Versatile Organometallic Pro-Drug (CORM) for Experimental CO Based Therapeutics. Dalt. Trans. 2013, 42, 5985–5998. [Google Scholar] [CrossRef]
- Ulbrich, F.; Kaufmann, K.B.; Meske, A.; Lagrèze, W.A.; Augustynik, M.; Buerkle, H.; Ramao, C.C.; Biermann, J.; Goebel, U. The CORM ALF-186 Mediates Anti-Apoptotic Signaling via an Activation of the P38 MAPK after Ischemia and Reperfusion Injury in Retinal Ganglion Cells. PLoS ONE 2016, 11, e0165182. [Google Scholar] [CrossRef] [PubMed]
- Castleden, C.M.; Cole, P.V. Carboxyhaemoglobin Levels of Smokers and Non-Smokers Working in the City of London. Occup. Environ. Med. 1975, 32, 115–118. [Google Scholar] [CrossRef][Green Version]
- Turner, J.A.; McNicol, M.W.; Sillett, R.W. Distribution of Carboxyhaemoglobin Concentrations in Smokers and Non-Smokers. Thorax 1986, 41, 25–27. [Google Scholar] [CrossRef]
- Chen, H.; Huang, X.; Guo, X.; Mailman, R.B.; Park, Y.; Kamel, F.; Umbach, D.M.; Xu, Q.; Hollenbeck, A.; Schatzkin, A.; et al. Smoking Duration, Intensity, and Risk of Parkinson Disease. Neurology 2010, 74, 878–884. [Google Scholar] [CrossRef]
- England, L. Smoking and Risk of Preeclampsia: A Systematic Review. Front. Biosci. 2007, 12, 2471. [Google Scholar] [CrossRef]
- Arafa, A.; Mostafa, A.; Navarini, A.A.; Dong, J.-Y. The Association between Smoking and Risk of Skin Cancer: A Meta-Analysis of Cohort Studies. Cancer Causes Control 2020, 31, 787–794. [Google Scholar] [CrossRef]
- Abuchowski, A. SANGUINATE (PEGylated Carboxyhemoglobin Bovine): Mechanism of Action and Clinical Update. Artif. Organs 2017, 41, 346–350. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).