
How to Increase Cellular Glutathione



Abstract

1. Introduction
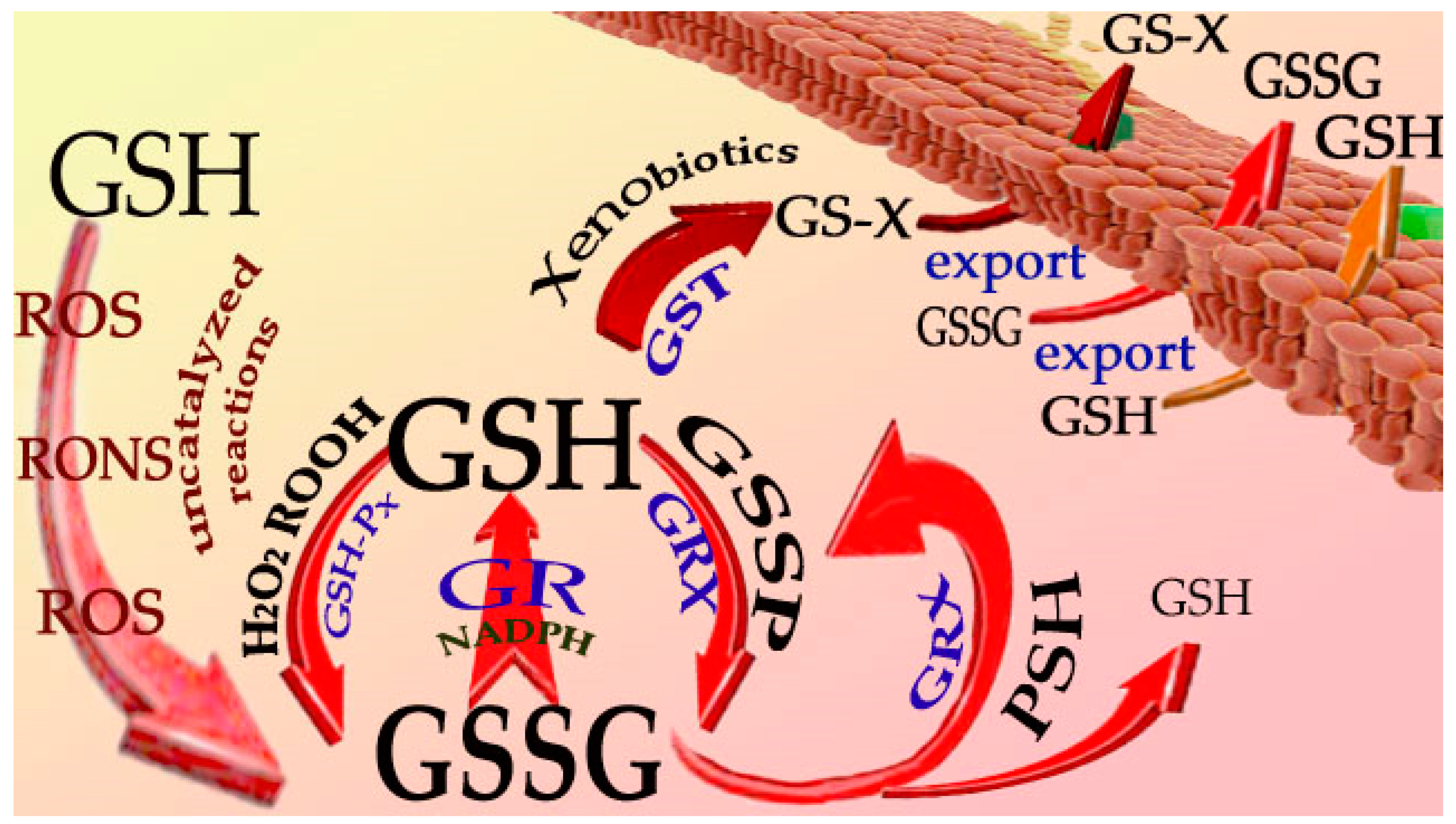
2. Reactions of GSH
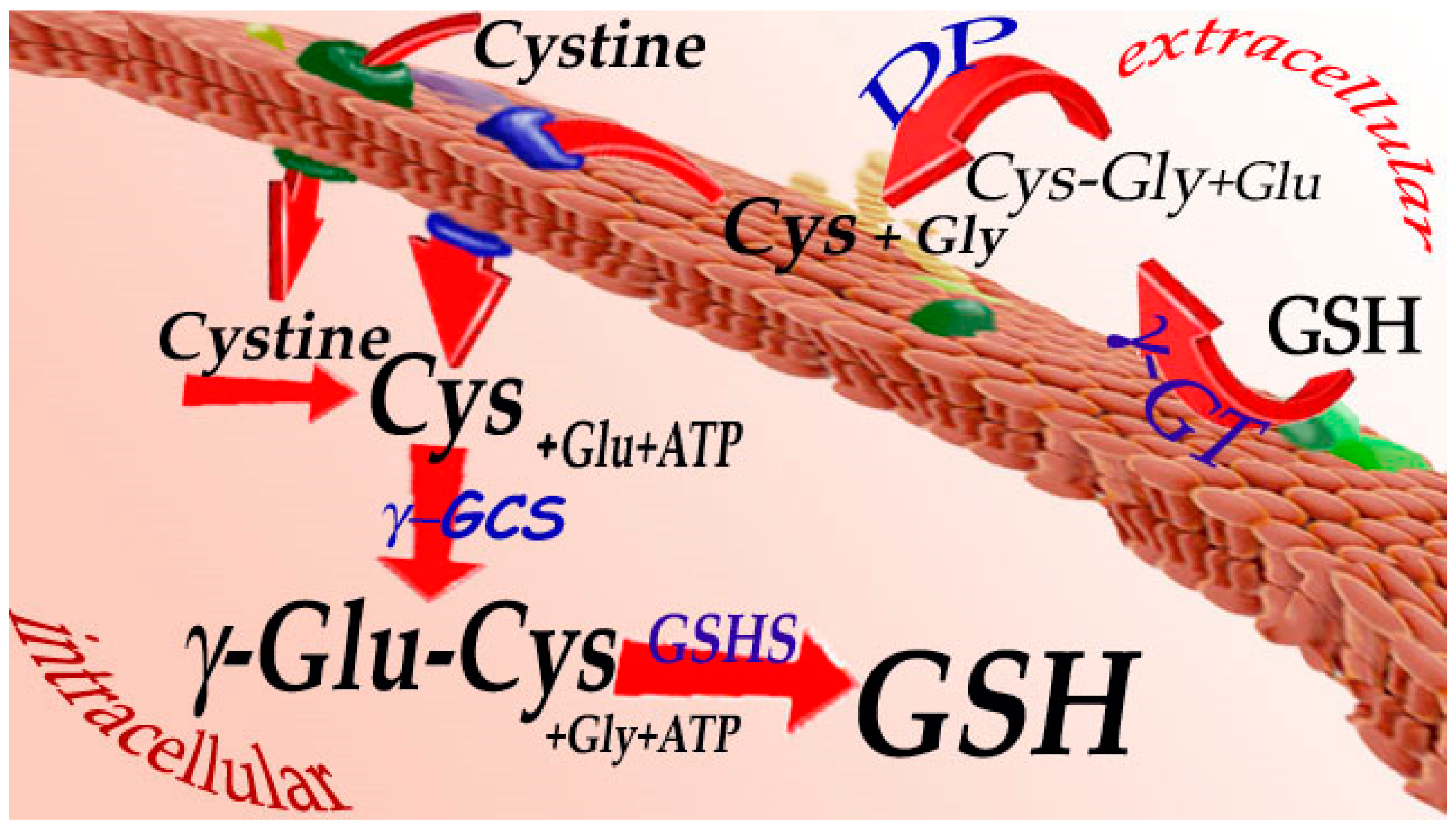
3. Synthesis and Catabolism of Glutathione
4. GSH Levels in Tissues

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | Species | Concentration | Reference |
|---|---|---|---|
| Kidney | Human | 1564 ± 106 μM | [34] |
| Muscle | Human | 2543 ± 267 μM | [35] |
| RBCs | Human | 8470 ± 1750 nmol/g Hb | [36] |
| Neutrophils | Humans | 13.2 ± 1.8 nmol/107 cells | [37] |
| Lymphocytes | Human | 5.7 ± 0.35 nmol/107 cells | [38] |
| Platelets | Human | 13.4 ± 2.64 nmol/109 platelets | [12] |
| Plasma | Human | 3.1 ± 0.26 μM | [31] |
| ELF | Human | 0.2–0.4 mM | [33] |
| Liver | Rat | 8221 ± 558 μM | [39] |
| Kidney | Rat | 2221 ± 302 μM | [39] |
| Lung | Rat | 2314 ± 182 μM | [39] |
| Heart | Rat | 1835 ± 244 μM | [39] |
| Spleen | Rat | 2648 ± 55 μM | [39] |
| Brain | Rat | 1801 ± 59 μM | [39] |
| Extracellular brain cortex | Rat | 2.10 ± 1.78 μM | [40] |
| White adipose tissue | Mouse | ~4.3 nmol/mg protein | [28] |
| Brown adipose tissue | Mouse | ~2 nmol/mg protein | [28] |
5. GSH and Disease
5.1. Inborn Alterations in GSH Metabolism
5.2. Ageing and Related Diseases
5.2.1. Age-Related Ocular Diseases
5.2.2. COPD
5.2.3. Diabetes Mellitus
5.2.4. Cardiovascular Diseases
5.2.5. Neurodegenerative Diseases
5.2.6. Other Age-Related Conditions
5.3. Cystic Fibrosis
5.4. Other Diseases
6. GSH Levels Regulation
6.1. GSH, GSH Esters, γ-Glutamylcysteine
6.2. Nrf2
6.3. Cysteine Pro-Drugs
6.3.1. N-Acetylcysteine (NAC)
6.3.2. N-Acetylcysteine Ethyl Ester (NACET)
6.3.3. N-Acetylcysteinamide (NACA)
6.3.4. Thiazolidines
6.3.5. Other Cysteine Derivatives
6.4. Taurine
6.5. Silymarin
6.6. Food and Diet
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Lushchak, V.I. Glutathione homeostasis and functions: Potential targets for medical interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef]
- Giustarini, D.; Dalle-Donne, I.; Tsikas, D.; Rossi, R. Oxidative stress and human diseases: Origin, link, measurement, mechanisms, and biomarkers. Crit. Rev. Clin. Lab. Sci. 2009, 46, 241–281. [Google Scholar] [CrossRef]
- Tajc, S.G.; Tolbert, B.S.; Basavappa, R.; Miller, B.L. Direct determination of thiol pKa by isothermal titration microcalorimetry. J. Am. Chem. Soc. 2004, 126, 10508–10509. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef]
- Makino, N.; Mochizuki, Y.; Bannai, S.; Sugita, Y. Kinetic studies on the removal of extracellular hydrogen peroxide by cultured fibroblasts. J. Biol. Chem. 1994, 269, 1020–1025. [Google Scholar] [CrossRef]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef]
- Wlodek, L. Beneficial and harmful effects of thiols. Pol. J. Pharmacol. 2002, 54, 215–223. [Google Scholar]
- Zalachoras, I.; Hollis, F.; Ramos-Fernández, E.; Trovo, L.; Sonnay, S.; Geiser, E.; Preitner, N.; Steiner, P.; Sandi, C.; Morató, L. Therapeutic potential of glutathione-enhancers in stress-related psychopathologies. Neurosci. Biobehav. Rev. 2020, 114, 134–155. [Google Scholar] [CrossRef]
- Rossi, R.; Milzani, A.; Dalle-Donne, I.; Giustarini, D.; Lusini, L.; Colombo, R.; Di Simplicio, P. Blood glutathione disulfide: In vivo factor or in vitro artifact? Clin. Chem. 2002, 48, 742–753. [Google Scholar] [CrossRef]
- Giustarini, D.; Dalle-Donne, I.; Milzani, A.; Fanti, P.; Rossi, R. Analysis of GSH and GSSG after derivatization with N-ethylmaleimide. Nat. Protoc. 2013, 8, 1660–1669. [Google Scholar] [CrossRef]
- Giustarini, D.; Colombo, G.; Garavaglia, M.L.; Astori, E.; Portinaro, N.M.; Reggiani, F.; Badalamenti, S.; Aloisi, A.M.; Santucci, A.; Rossi, R.; et al. Assessment of glutathione/glutathione disulphide ratio and S-glutathionylated proteins in human blood, solid tissues, and cultured cells. Free Radic. Biol. Med. 2017, 112, 360–375. [Google Scholar] [CrossRef]
- Giustarini, D.; Tsikas, D.; Colombo, G.; Milzani, A.; Dalle-Donne, I.; Fanti, P.; Rossi, R. Pitfalls in the analysis of the physiological antioxidant glutathione (GSH) and its disulfide (GSSG) in biological samples: An elephant in the room. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2016, 1019, 21–28. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Rossi, R.; Colombo, G.; Giustarini, D.; Milzani, A. Protein S-glutathionylation: A regulatory device from bacteria to humans. Trends Biochem. Sci. 2009, 34, 85–96. [Google Scholar] [CrossRef]
- Mannervik, B. Thioltransferases. In Enzymatic Basis of Detoxication; Jacoby, W.B., Ed.; Academic Press: New York, NY, USA, 1980; Volume 2, pp. 229–244. [Google Scholar]
- Axelsson, K.; Mannervik, B. An essential role of cytosolic thioltransferase in protection of pyruvate kinase from rabbit liver against oxidative inactivation. FEBS Lett. 1983, 152, 114–118. [Google Scholar] [CrossRef]
- Szczuko, M.; Ziętek, M.; Kulpa, D.; Seidler, T. Riboflavin—Properties, occurrence and its use in medicine. Pteridines 2019, 30, 33–47. [Google Scholar] [CrossRef]
- Diaz-Vivancos, P.; de Simone, A.; Kiddle, G.; Foyer, C.H. Glutathione—Linking cell proliferation to oxidative stress. Free Radic. Biol. Med. 2015, 89, 1154–1164. [Google Scholar] [CrossRef]
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef]
- Giustarini, D.; Tsikas, D.; Rossi, R. Study of the effect of thiols on the vasodilatory potency of S-nitrosothiols by using a modified aortic ring assay. Toxicol. Appl. Pharmacol. 2011, 256, 95–102. [Google Scholar] [CrossRef]
- Cai, J.; Chen, Y.; Seth, S.; Furukawa, S.; Compans, R.W.; Jones, D.P. Inhibition of influenza infection by glutathione. Free Radic. Biol. Med. 2003, 34, 928–936. [Google Scholar] [CrossRef]
- Bartolini, D.; Stabile, A.M.; Bastianelli, S.; Giustarini, D.; Pierucci, S.; Busti, C.; Vacca, C.; Gidari, A.; Francisci, D.; Castronari, R.; et al. SARS-CoV2 infection impairs the metabolism and redox function of cellular glutathione. Redox Biol. 2021, 45, 102041. [Google Scholar] [CrossRef]
- Sen, C.K. Cellular thiols and redox-regulated signal transduction. Curr. Top. Cell. Regul. 2000, 36, 1–30. [Google Scholar]
- Meister, A. Metabolism and functions of glutathione. Trends Biochem. Sci. 1981, 6, 231–234. [Google Scholar] [CrossRef]
- Meister, A. Glutathione metabolism. Methods Enzymol. 1995, 251, 3–7. [Google Scholar]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef]
- Ballatori, N.; Krance, S.M.; Marchan, R.; Hammond, C.L. Plasma membrane glutathione transporters and their roles in cell physiology and pathophysiology. Mol. Asp. Med. 2009, 30, 13–28. [Google Scholar] [CrossRef]
- Turner, M.; Mantick, N.A.; Carlson, G.P. Comparison of the depletion of glutathione in mouse liver and lung following administration of styrene and its metabolites styrene oxide and 4-vinylphenol. Toxicology 2005, 206, 383–388. [Google Scholar] [CrossRef]
- Lettieri Barbato, D.; Tatulli, G.; Cannata, S.M.; Bernardini, S.; Aquilano, K.; Ciriolo, M.R. Glutathione decrement drives thermogenic program in adipose cells. Sci. Rep. 2015, 5, 13091. [Google Scholar] [CrossRef]
- Dringen, R.; Kussmaul, L.; Gutterer, J.M.; Hirrlinger, J.; Hamprecht, B. The glutathione system of peroxide detoxification is less efficient in neurons than in astroglial cells. J. Neurochem. 1999, 72, 2523–2530. [Google Scholar] [CrossRef]
- Giustarini, D.; Galvagni, F.; Tesei, A.; Farolfi, A.; Zanoni, M.; Pignatta, S.; Milzani, A.; Marone, I.M.; Dalle-Donne, I.; Nassini, R.; et al. Glutathione, glutathione disulfide, and S-glutathionylated proteins in cell cultures. Free Radic. Biol. Med. 2015, 89, 972–981. [Google Scholar] [CrossRef]
- Grintzalis, K.; Papapostolou, I.; Zisimopoulos, D.; Stamatiou, I.; Georgiou, C.D. Multiparametric protocol for the determination of thiol redox state in living matter. Free Radic. Biol. Med. 2014, 74, 85–98. [Google Scholar] [CrossRef]
- Anderson, M.E.; Meister, A. Dynamic state of glutathione in blood plasma. J. Biol. Chem. 1980, 255, 9530–9533. [Google Scholar] [CrossRef]
- Cross, C.E.; van der Vliet, A.; O’Neill, C.A.; Louie, S.; Halliwell, B. Oxidants, antioxidants, and respiratory tract lining fluids. Environ. Health Perspect. 1994, 102, 185–191. [Google Scholar]
- Lusini, L.; Tripodi, S.A.; Rossi, R.; Giannerini, F.; Giustarini, D.; del Vecchio, M.T.; Barbanti, G.; Cintorino, M.; Tosi, P.; Di Simplicio, P. Altered glutathione anti-oxidant metabolism during tumor progression in human renal-cell carcinoma. Int. J. Cancer 2001, 91, 55–59. [Google Scholar] [CrossRef]
- Søndergård, S.D.; Cintin, I.; Kuhlman, A.B.; Morville, T.H.; Bergmann, M.L.; Kjær, L.K.; Poulsen, H.E.; Giustarini, D.; Rossi, R.; Dela, F.; et al. The effects of 3 weeks of oral glutathione supplementation on whole body insulin sensitivity in obese males with and without type 2 diabetes: A randomized trial. Appl. Physiol. Nutr. Metab. 2021, 9, 1133–1142. [Google Scholar] [CrossRef]
- Giustarini, D.; Dalle-Donne, I.; Milzani, A.; Rossi, R. Detection of glutathione in whole blood after stabilization with N-ethylmaleimide. Anal. Biochem. 2011, 415, 81–83. [Google Scholar] [CrossRef]
- Voetman, A.A.; Loos, J.A.; Roos, D. Changes in the levels of glutathione in phagocytosing human neutrophils. Blood 1980, 55, 741–747. [Google Scholar] [CrossRef]
- Hamilos, D.L.; Zelarney, P.; Mascali, J.J. Lymphocyte proliferation in glutathione-depleted lymphocytes: Direct relationship between glutathione availability and the proliferative response. Immunopharmacology 1989, 18, 223–235. [Google Scholar] [CrossRef]
- Giustarini, D.; Dalle-Donne, I.; Milzani, A.; Rossi, R. Low molecular mass thiols, disulfides and protein mixed disulfides in rat tissues: Influence of sample manipulation, oxidative stress and ageing. Mech. Ageing Dev. 2011, 132, 141–148. [Google Scholar] [CrossRef]
- Yang, C.S.; Chou, S.T.; Lin, N.N.; Liu, L.; Tsai, P.J.; Kuo, J.S.; Lai, J.S. Determination of extracellular glutathione in rat brain by microdialysis and high-performance liquid chromatography with fluorescence detection. J. Chromatogr. B Biomed. Appl. 1994, 661, 231–235. [Google Scholar] [CrossRef]
- Ristoff, E.; Larsson, A. Inborn errors in the metabolism of glutathione. Orphanet J. Rare Dis. 2007, 2, 16. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Bhat, K.S.; John, A.; Reddy, P.R.; Reddy, P.S.; Reddy, V.N. Effect of pigmentation on glutathione redox cycle antioxidant defense in whole as well as different regions of human cataractous lens. Exp. Eye Res. 1991, 52, 715–721. [Google Scholar] [CrossRef]
- Tanito, M.; Nishiyama, A.; Tanaka, T.; Masutani, H.; Nakamura, H.; Yodoi, J.; Ohira, A. Change of redox status and modulation by thiol replenishment in retinal photooxidative damage. Investig. Ophthalmol Vis. Sci. 2002, 43, 2392–2400. [Google Scholar]
- Rahman, I.; Li, X.Y.; Donaldson, K.; Harrison, D.J.; MacNee, W. Glutathione homeostasis in alveolar epithelial cells in vitro and lung in vivo under oxidative stress. Am. J. Physiol. 1995, 269, L285–L292. [Google Scholar] [CrossRef]
- Rangasamy, T.; Cho, C.Y.; Thimmulappa, R.K.; Zhen, L.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Investig. 2004, 114, 1248–1259. [Google Scholar] [CrossRef]
- Malhotra, D.; Thimmulappa, R.; Navas-Acien, A.; Sandford, A.; Elliott, M.; Singh, A.; Chen, L.; Zhuang, X.; Hogg, J.; Pare, P.; et al. Decline in NRF2-regulated antioxidant in chronic obstructive pulmonary disease lungs due to loss of its positive regulator DJ-1. Am. J. Respir. Crit. Care Med. 2008, 178, 592–604. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Shin, A.H.; Oh, C.J.; Park, J.W. Glycation-induced inactivation of antioxidant enzymes and modulation of cellular redox status in lens cells. Arch. Pharm. Res. 2006, 29, 577–581. [Google Scholar] [CrossRef]
- Sekhar, R.V.; McKay, S.V.; Patel, S.G.; Guthikonda, A.P.; Reddy, V.T.; Balasubramanyam, A.; Jahoor, F. Glutathione synthesis is diminished in patients with uncontrolled diabetes and restored by dietary supplementation with cysteine and glycine. Diabetes Care 2011, 34, 162–167. [Google Scholar] [CrossRef]
- Memisogullari, R.; Taysi, S.; Bakan, E.; Capoglu, I. Antioxidant status and lipid peroxidation in type II diabetes mellitus. Cell Biochem. Funct. 2003, 21, 291–296. [Google Scholar] [CrossRef]
- Giustarini, D.; Dalle-Donne, I.; Colombo, R.; Petralia, S.; Giampaoletti, S.; Milzani, A.; Rossi, R. Protein glutathionylation in erythrocytes. Clin. Chem. 2003, 49, 327–330. [Google Scholar] [CrossRef]
- Shimizu, H.; Kiyohara, Y.; Kato, I.; Kitazono, T.; Tanizaki, Y.; Kubo, M.; Ueno, H.; Ibayashi, S.; Fujishima, M.; Iida, M. Relationship between plasma glutathione levels and cardiovascular disease in a defined population: The Hisayama study. Stroke 2004, 35, 2072–2077. [Google Scholar] [CrossRef]
- Damy, T.; Kirsch, M.; Khouzami, L.; Caramelle, P.; Le Corvoisier, P.; Roudot-Thoraval, F.; Dubois-Randé, J.L.; Hittinger, L.; Pavoine, C.; Pecker, F. Glutathione deficiency in cardiac patients is related to the functional status and structural cardiac abnormalities. PLoS ONE 2009, 4, e4871. [Google Scholar] [CrossRef]
- Julius, M.; Lang, C.A.; Gleiberman, L.; Harburg, E.; Difranceisco, W.; Schork, A. Glutathione and. morbidity in a community-based sample of elderly. J. Clin. Epidemiol. 1994, 47, 1021–1026. [Google Scholar] [CrossRef]
- Sofic, E.; Lange, K.; Jellinger, K.; Riederer, P. Reduced and oxidized glutathione in the substantia nigra of patients ith Parkinson’s disease. Neurosci. Lett. 1992, 142, 128–130. [Google Scholar] [CrossRef]
- Bonneh-Barkay, D.; Reaney, S.H.; Langston, W.J.; Di Monte, D.A. Redox cycling of the herbicide paraquat in microglial cultures. Brain Res. Mol. Brain Res. 2005, 134, 52–56. [Google Scholar] [CrossRef]
- Konnova, E.A.; Swanberg, M. Animal Models of Parkinson’s Disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018; Chapter 5. [Google Scholar]
- Aaseth, J.; Dusek, P.; Roos, P.M. Prevention of progression in Parkinson’s disease. Biometals 2018, 31, 737–747. [Google Scholar] [CrossRef]
- Levine, M.E.; Suarez, J.A.; Brandhorst, S.; Balasubramanian, P.; Cheng, C.W.; Madia, F.; Fontana, L.; Mirisola, M.G.; Guevara-Aguirre, J.; Wan, J.; et al. Low protein intake is associated with a major reduction in IGF-1, cancer, and overall mortality in the 65 and younger but not older population. Cell Metab. 2014, 19, 407–417. [Google Scholar] [CrossRef]
- Dickerhof, N.; Pearson, J.F.; Hoskin, T.S.; Berry, L.J.; Turner, R.; Sly, P.D.; Kettle, A.J.; Arest, C.F. Oxidative stress in early cystic fibrosis lung disease is exacerbated by airway glutathione deficiency. Free Radic. Biol. Med. 2017, 113, 236–243. [Google Scholar] [CrossRef]
- Roum, H.; Buhl, R.; McElvaney, N.G.; Borok, Z.R.; Crystal, G. Systemic deficiency of glutathione in cystic fibrosis. J. Appl. Physiol. 1993, 75, 2419–2424. [Google Scholar] [CrossRef]
- Tirouvanziam, R.; Conrad, C.K.; Bottiglieri, T.; Herzenberg, L.A.; Moss, R.B. High dose oral N-acetylcysteine, a glutathione prodrug, modulates inflammation in cystic fibrosis. Proc. Natl. Acad. Sci. USA 2006, 103, 4628–4633. [Google Scholar] [CrossRef]
- Gao, L.; Broughman, J.R.; Iwamoto, T.; Tomich, J.M.; Venglarik, C.J.; Forman, H.J. Synthetic chloride channel restores glutathione secretion in cystic fibrosis airway epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L24–L30. [Google Scholar] [CrossRef]
- Teskey, G.; Abrahem, R.; Cao, R.; Gyurjian, K.; Islamoglu, H.; Lucero, M.; Martinez, A.; Paredes, E.; Salaiz, O.; Robinson, B.; et al. Glutathione as a marker for human disease. In Advances in Clinical Chemistry; Makowski, G.S., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 87, pp. 141–159. [Google Scholar]
- Sinha, R.; Sinha, I.; Calcagnotto, A.; Trushin, N.; Haley, J.S.; Schell, T.D.; Richie, J.P., Jr. Oral supplementation with liposomal glutathione elevates body stores of glutathione and markers of immune function. Eur. J. Clin. Nutr. 2018, 72, 105–111. [Google Scholar] [CrossRef]
- Mischley, L.K.; Lau, R.C.; Shankland, E.G.; Wilbur, T.K.; Padowski, J.M. Phase IIb study of intranasal glutathione in Parkinson’s disease. J. Park. Dis. 2017, 7, 289–299. [Google Scholar] [CrossRef]
- Zarka, M.H.; Bridge, W.J. Oral administration of γ-glutamylcysteine increases intracellular glutathione levels above homeostasis in a randomised human trial pilot study. Redox Biol. 2017, 11, 631–636. [Google Scholar] [CrossRef]
- De Rosa, S.C.; Zaretsky, M.D.; Dubs, J.G.; Roederer, M.; Anderson, M.; Green, A.; Mitra, D.; Watanabe, N.; Nakamura, H.; Tjioe, I.; et al. N-acetylcysteine replenishes glutathione in HIV infection. Eur. J. Clin. Investig. 2000, 30, 915–929. [Google Scholar] [CrossRef]
- Moberly, J.B.; Logan, J.; Borum, P.R.; Story, K.O.; Webb, L.E.; Jassal, S.V.; Oreopoulos, D.G. Elevation of whole-blood glutathione in peritoneal dialysis patients by L-2-oxothiazolidine-4-carboxylate, a cysteine prodrug (procysteine). J. Am. Soc. Nephrol. 1998, 9, 1093–1099. [Google Scholar] [CrossRef]
- Basu, A.; Betts, N.M.; Mulugeta, A.; Tong, C.; Newman, E.; Lyons, T.J. Green tea supplementation increases glutathione and plasma antioxidant capacity in adults with the metabolic syndrome. Nutr. Res. 2013, 33, 180–187. [Google Scholar] [CrossRef]
- Pirouzeh, R.; Heidarzadeh-Esfahani, N.; Morvaridzadeh, M.; Izadi, A.; Yosaee, S.; Potter, E.; Heshmati, J.; Pizarro, A.B.; Omidi, A.; Heshmati, S. Effect of DASH diet on oxidative stress parameters: A systematic review and meta-analysis of randomized clinical trials. Diabetes Metab. Syndr. 2020, 14, 2131–2138. [Google Scholar] [CrossRef]
- Anderson, M.E.; Powrie, F.; Puri, R.N.; Meister, A. Glutathione monoethyl ester: Preparation, uptake by tissues, and conversion to glutathione. Arch. Biochem. Biophys. 1985, 239, 538–548. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Z.; Li, B.; Yao, H.; Zarka, M.; Welch, J.; Sachdev, P.; Bridge, W.; Braidy, N. Supplementation with γ-glutamylcysteine (γ-GC) lessens oxidative stress, brain inflammation and amyloid pathology and improves spatial memory in a murine model of AD. Neurochem. Int. 2021, 144, 104931. [Google Scholar] [CrossRef]
- Tosi, G.M.; Giustarini, D.; Franci, L.; Minetti, A.; Imperatore, F.; Caldi, E.; Fiorenzani, P.; Aloisi, A.M.; Sparatore, A.; Rossi, R.; et al. Superior properties of N-acetylcysteine ethyl ester over N-acetyl cysteine to prevent retinal pigment epithelial cells oxidative damage. Int. J. Mol. Sci. 2021, 22, 600. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.M.; Yin, M.C.; Hsu, C.C.; Liu, T.C. Antioxidative and anti-inflammatory effects of four cysteine-containing agents in striatum of MPTP-treated mice. Nutrition 2007, 23, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Shi, H.; Zhang, C.; Ren, M.; Han, M.; Wei, X.; Zhang, X.; Lou, H. Dimethyl fumarate attenuates 6-OHDA-induced neurotoxicity in SH-SY5Y cells and in animal model of Parkinson’s disease by enhancing Nrf2 activity. Neuroscience 2015, 286, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Tu, D.G.; Chang, Y.L.; Chou, C.H.; Lin, Y.L.; Chiang, C.C.; Chang, Y.Y.; Chen, Y.C. Preventive effects of taurine against d-galactose-induced cognitive dysfunction and brain damage. Food Funct. 2018, 9, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Chtourou, Y.; Garoui, E.; Boudawara, T.; Zeghal, N. Therapeutic efficacy of silymarin from milk thistle in reducing manganese-induced hepatic damage and apoptosis in rats. Hum. Exp. Toxicol. 2013, 32, 70–81. [Google Scholar] [CrossRef]
- Giustarini, D.; Milzani, A.; Dalle-Donne, I.; Tsikas, D.; Rossi, R. N-Acetylcysteine ethyl ester (NACET): A novel lipophilic cell-permeable cysteine derivative with an unusual pharmacokinetic feature and remarkable antioxidant potential. Biochem. Pharmacol. 2012, 84, 1522–1533. [Google Scholar] [CrossRef]
- Giustarini, D.; Fanti, P.; Sparatore, A.; Matteucci, E.; Rossi, R. Anethole dithiolethione lowers the homocysteine and raises the glutathione levels in solid tissues and plasma of rats: A novel non-vitamin homocysteine-lowering agent. Biochem. Pharmacol. 2014, 89, 246–254. [Google Scholar] [CrossRef]
- Reisman, S.A.; Chertow, G.M.; Hebbar, S.; Vaziri, N.D.; Ward, K.W.; Meyer, C.J. Bardoxolone methyl decreases megalin and activates nrf2 in the kidney. J. Am. Soc. Nephrol. 2012, 23, 1663–1673. [Google Scholar] [CrossRef]
- Anderson, M.E.; Underwood, M.; Bridges, R.J.; Meister, A. Glutathione metabolism at the blood-cerebrospinal fluid barrier. FASEB J. 1989, 3, 2527–2531. [Google Scholar] [CrossRef]
- Giustarini, D.; Galvagni, F.; Dalle Donne, I.; Milzani, A.; Severi, F.M.; Santucci, A.; Rossi, R. N-acetylcysteine ethyl ester as GSH enhancer in human primary endothelial cells: A comparative study with other drugs. Free Radic. Biol. Med. 2018, 126, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.J.; Anderson, M.E.; Meister, A. Transport of glutathione diethyl ester into human cells. Proc. Natl. Acad. Sci. USA 1993, 90, 9171–9175. [Google Scholar] [CrossRef]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef] [PubMed]
- Nioi, P.; McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD(P)H:quinone oxidoreductase 1 gene: Reassessment of the ARE consensus sequence. Biochem. J. 2003, 374, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Sekhar, K.R.; Rachakonda, G.; Freeman, M.L. Cysteine-based regulation of the CUL3 adaptor protein Keap1. Toxicol. Appl. Pharmacol. 2010, 244, 21–26. [Google Scholar] [CrossRef]
- Perez, H.L.; Junnotula, V.; Knecht, D.; Nie, H.; Sanchez, Y.; Boehm, J.C.; Booth-Genthe, C.; Yan, H.; Davis, R.; Callahan, J.F. Analytical approaches for quantification of a Nrf2 pathway activator: Overcoming bioanalytical challenges to support a toxicity study. Analyst 2014, 139, 1902–1912. [Google Scholar] [CrossRef]
- Kode, A.; Rajendrasozhan, S.; Caito, S.; Yang, S.R.; Megson, I.L.; Rahman, I. Resveratrol induces glutathione synthesis by activation of Nrf2 and protects against cigarette smoke-mediated oxidative stress in human lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L478–L488. [Google Scholar] [CrossRef] [PubMed]
- Shay, K.P.; Michels, A.J.; Li, W.; Kong, A.T.; Hagen, T.M. Cap-independent Nrf2 translation is part of a lipoic acid-stimulated detoxification stress response. Biochim. Biophys. Acta 2012, 1823, 1101–1109. [Google Scholar] [CrossRef]
- Shinkai, Y.; Sumi, D.; Fukami, I.; Ishii, T.; Kumagai, Y. Sulforaphane, an activator of Nrf2, suppresses cellular accumulation of arsenic and its cytotoxicity in primary mouse hepatocytes. FEBS Lett. 2006, 580, 1771–1774. [Google Scholar] [CrossRef]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, X.; Fan, H.; Liu, Y. Curcumin upregulates transcription factor Nrf2, HO-1 expression and protects rat brains against focal ischemia. Brain Res. 2009, 1282, 133–141. [Google Scholar] [CrossRef] [PubMed]
- de Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ct2/show/NCT02036970?term=bardoxolone (accessed on 6 February 2023).
- Sheremata, W.; Brown, A.D.; Rammohan, K.W. Dimethyl fumarate for treating relapsing multiple sclerosis. Expert Opin. Drug Saf. 2015, 14, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Gonsette, R.E. Neurodegeneration in multiple sclerosis: The role of oxidative stress and excitotoxicity. J. Neurol. Sci. 2008, 274, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhang, F.; Sun, F.; Gu, K.; Dong, S.; He, D. Dimethyl fumarate for multiple sclerosis. Cochrane Database Syst. Rev. 2015, 22, CD011076. [Google Scholar] [CrossRef] [PubMed]
- Raftos, J.E.; Whillier, S.; Chapman, B.E.; Kuchel, P.W. Kinetics of uptake and deacetylation of N-acetylcysteine by human erythrocytes. Int. J. Biochem. Cell Biol. 2007, 39, 1698–1706. [Google Scholar] [CrossRef]
- Aitio, M.L. N-acetylcysteine—Passe-partout or much ado about nothing? Br. J. Clin. Pharmacol. 2005, 61, 5–15. [Google Scholar] [CrossRef]
- Olsson, B.; Johansson, M.; Gabrielsson, J.; Bolme, P. Pharmacokinetics and bioavailability of reduced and oxidized N-acetylcysteine. Eur. J. Clin. Pharmacol. 1988, 34, 77–82. [Google Scholar] [CrossRef]
- Phelps, D.T.; Deneke, S.M.; Daley, D.L.; Fanburg, B.L. Elevation of glutathione levels in bovine pulmonary artery endothelial cells by N-acetylcysteine. Am. J. Respir. Cell Mol. Biol. 1992, 7, 293–299. [Google Scholar] [CrossRef]
- Issels, R.D.; Nagele, A.; Eckert, K.G.; Wilmanns, W. Promotion of cysteine uptake and its utilization for glutathione biosynthesis induced by cysteamine and N-acetylcysteine. Biochem. Pharmacol. 1988, 37, 881–888. [Google Scholar] [CrossRef]
- de Quay, B.; Malinverni, R.; Lauterburg, B.H. Glutathione depletion in HIV-infected patients: Role of cysteine deficiency and effect of oral N-acetylcysteine. Aids 1992, 6, 815–819. [Google Scholar] [CrossRef]
- Meyer, A.; Buhl, R.; Kampf, S.; Magnussen, H. Intravenous N-acetylcysteine and lung glutathione of patients with pulmonary fibrosis and normal. Am. J. Respir. Crit. Care Med. 1995, 152, 1055–1060. [Google Scholar] [CrossRef]
- Lasram, M.M.; Dhouib, I.B.; Annabi, A.; El Fazaa, S.; Gharbi, N. A review on the possible molecular mechanism of action of N-acetylcysteine against insulin resistance and type-2 diabetes development. Clin. Biochem. 2015, 48, 1200–1208. [Google Scholar] [CrossRef] [PubMed]
- Holmay, M.J.; Terpstra, M.; Coles, L.D.; Mishra, U.; Ahlskog, M.; Öz, G.; Cloyd, J.C.; Tuite, P.J. N-Acetylcysteine boosts brain and blood glutathione in Gaucher and Parkinson diseases. Clin. Neuropharmacol. 2013, 36, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, R.V.; Patel, S.G.; Guthikonda, A.P.; Reid, M.; Balasubramanyam, A.; Taffet, G.E.; Jahoor, F. Deficient synthesis of glutathione underlies oxidative stress in aging and can be corrected by dietary cysteine and glycine supplementation. Am. J. Clin. Nutr. 2011, 94, 847–853. [Google Scholar] [CrossRef]
- Kumar, P.; Liu, C.; Hsu, J.W.; Chacko, S.; Minard, C.G.; Jahoor, F.; Sekhar, R.V. Glycine and N-acetylcysteine (GlyNAC) supplementation in older adults improves glutathione deficiency, oxidative stress, mitochondrial dysfunction, inflammation, insulin resistance, endothelial dysfunction, genotoxicity, muscle strength, and cognition: Results of a pilot clinical trial. Clin. Transl. Med. 2021, 11, e372. [Google Scholar]
- Sekhar, R.V. GlyNAC Supplementation improves glutathione deficiency, oxidative stress, mitochondrial dysfunction, inflammation, aging hallmarks, metabolic defects, muscle strength, cognitive decline, and body composition: Implications for healthy aging. J. Nutr. 2021, 151, 3606–3616. [Google Scholar] [CrossRef]
- He, R.; Zheng, W.; Ginman, T.; Ottosson, H.; Norgren, S.; Zhao, Y.; Hassan, M. Pharmacokinetic profile of N-acetylcysteine amide and its main metabolite in mice using new analytical method. Eur. J. Pharm. Sci. 2020, 143, 105158. [Google Scholar] [CrossRef]
- Grinberg, L.; Fibach, E.; Amer, J.; Atlas, D. N-acetylcysteine amide, a novel cell-permeating thiol, restores cellular glutathione and protects human red blood cells from oxidative stress. Free Radic. Biol. Med. 2005, 38, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Banks, M.F.; Stipanuk, M.H. The utilization of N-acetylcysteine and 2-oxothiazolidine-4-carboxylate by rat hepatocytes is limited by their rate of uptake and conversion to cysteine. J. Nutr. 1994, 124, 378–387. [Google Scholar] [CrossRef]
- Bernard, G.R.; Wheeler, A.P.; Arons, M.M.; Morris, P.E.; Paz, H.L.; Russell, J.A.; Wright, P.E. A trial of antioxidants N-acetylcysteine and procysteine in ARDS. The Antioxidant in ARDS Study Group. Chest 1997, 112, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.C.; Huang, C.N.; Hung, Y.C.; Yin, M.C. Five cysteine-containing compounds have antioxidative activity in Balb/cA mice. J. Nutr. 2004, 134, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Goc, Z.; Kapusta, E.; Formicki, G.; Martiniaková, M.; Omelka, R. Effect of taurine on ethanol-induced oxidative stress in mouse liver and kidney. Chin. J. Physiol. 2019, 62, 148–156. [Google Scholar] [CrossRef]
- Jafri, A.J.A.; Agarwal, R.; Iezhitsa, I.; Agarwal, P.; Ismail, N.M. Taurine protects against NMDA-induced retinal damage by reducing retinal oxidative stress. Amino Acids 2019, 51, 641–646. [Google Scholar] [CrossRef]
- Takke, A.; Shende, P. Nanotherapeutic silibinin: An insight of phytomedicine in healthcare reformation. Nanomedicine 2019, 21, 102057. [Google Scholar] [CrossRef]
- Surai, P.F. Silymarin as a natural antioxidant: An overview of the current evidence and perspectives. Antioxidants 2015, 4, 204–247. [Google Scholar] [CrossRef]
- Shaker, E.; Mahmoud, H.; Mnaa, S. Silymarin, the antioxidant component and Silybum marianum extracts prevent liver damage. Food Chem. Toxicol. 2010, 48, 803–806. [Google Scholar] [CrossRef]
- Mansour, H.H.; Hafez, H.F.; Fahmy, N.M. Silymarin modulates Cisplatin-induced oxidative stress and hepatotoxicity in rats. J. Biochem. Mol. Biol. 2006, 39, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Mehrab-Mohseni, M.; Sendi, H.; Steuerwald, N.; Ghosh, S.; Schrum, L.W.; Bonkovsky, H.L. Legalon-SIL downregulates HCV core and NS5A in human hepatocytes expressing full-length HCV. World J. Gastroenterol. 2011, 17, 1694–1700. [Google Scholar] [CrossRef]
- Kwon, D.Y.; Jung, Y.S.; Kim, S.J.; Kim, Y.S.; Choi, D.W.; Kim, Y.C. Alterations in sulfur amino acid metabolism in mice treated with silymarin: A novel mechanism of its action involved in enhancement of the antioxidant defense in liver. Planta Med. 2013, 79, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Morr, C.V.; Ha, E.Y.W. Whey protein concentrates and isolates: Processing and functional properties. Crit. Rev. Food Sci. Nutr. 1993, 33, 431–476. [Google Scholar] [CrossRef] [PubMed]
- Zavorsky, G.S.; Kubow, S.; Grey, V.; Riverin, V.; Lands, L.C. An open-label dose-response study of lymphocyte glutathione levels in healthy men and women receiving pressurized whey protein isolate supplements. Int. J. Food Sci. Nutr. 2007, 58, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Chitapanarux, T.; Tienboon, P.; Pojchamarnwiputh, S.; Leelarungrayub, D. Open-labeled pilot study of cysteine-rich whey protein isolate supplementation for nonalcoholic steatohepatitis patients. J. Gastroenterol. Hepatol. 2009, 24, 1045–1050. [Google Scholar] [CrossRef]
- Bumrungpert, A.; Pavadhgul, P.; Nunthanawanich, P.; Sirikanchanarod, A.; Adulbhan, A. Whey protein supplementation improves nutritional status, glutathione levels, and immune function in cancer patients: A randomized, double-blind controlled trial. J. Med. Food 2018, 21, 612–616. [Google Scholar] [CrossRef]
- Tosukhowong, P.; Boonla, C.; Dissayabutra, T.; Kaewwilai, L.; Muensri, S.; Chotipanich, C.; Joutsa, J.; Rinne, J.; Bhidayasiri, R. Biochemical and clinical effects of Whey protein supplementation in Parkinson’s disease: A pilot study. J. Neurol. Sci. 2016, 367, 162–170. [Google Scholar] [CrossRef]
- Ross, E.K.; Gray, J.J.; Winter, A.N.; Linseman, D.A. Immunocal® and preservation of glutathione as a novel neuroprotective strategy for degenerative disorders of the nervous system. Recent Pat. CNS Drug Discov. 2012, 7, 230–235. [Google Scholar] [CrossRef]
- Minich, D.M.; Brown, B.I. A Review of dietary (phyto)nutrients for glutathione support. Nutrients 2019, 11, 2073. [Google Scholar] [CrossRef]
- Johnston, C.S.; Meyer, C.G.; Srilakshmi, J.C. Vitamin C elevates red blood cell glutathione in healthy adults. Am. J. Clin. Nutr. 1993, 58, 103–105. [Google Scholar] [CrossRef]
- Lenton, K.J.; Sané, A.T.; Therriault, H.; Cantin, A.M.; Payette, H.; Wagner, J.R. Vitamin C augments lymphocyte glutathione in subjects with ascorbate deficiency. Am. J. Clin. Nutr. 2003, 77, 189–195. [Google Scholar] [CrossRef]
- May, J.M.; Cobb, C.E.; Mendiratta, S.; Hill, K.E.; Burk, R.F. Reduction of the ascorbyl free radical to ascorbate by thioredoxin reductase. J. Biol. Chem. 1998, 273, 23039–23045. [Google Scholar] [CrossRef] [PubMed]
- Demirkol, O.; Adams, C.; Ercal, N. Biologically important thiols in various vegetables and fruits. J. Agric. Food Chem. 2004, 52, 8151–8154. [Google Scholar] [CrossRef]
- Wierzbicka, G.T.; Hagen, T.M.; Tones, D.P. Glutathione in food. J. Food Compost. Anal. 1989, 2, 327–337. [Google Scholar] [CrossRef]
- Ansher, S.S.; Dolan, P.; Bueding, E. Chemoprotective effects of two dithiolthiones and of butylhydroxyanisole against carbon tetrachloride and acetaminophen toxicity. Hepathology 1983, 3, 932–935. [Google Scholar] [CrossRef]
- Giustarini, D.; Galvagni, F.; Dalle-Donne, I.; Milzani, A.; Lucattelli, M.; De Cunto, G.; Bartolini, D.; Galli, F.; Santucci, A.; Rossi, R. Anethole dithiolethione increases glutathione in kidney by inhibiting γ-glutamyltranspeptidase: Biochemical interpretation and pharmacological consequences. Oxid. Med. Cell. Longev. 2020, 2020, 3562972. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Munday, R. Dithiolethiones for cancer chemoprevention: Where do we stand? Mol. Cancer Ther. 2008, 7, 3470–3479. [Google Scholar] [CrossRef]
- Gupta, E.; Olopade, O.I.; Ratain, M.J.; Mick, R.; Baker, T.M.; Berezin, F.K.; Benson, A.B.; Dolan, M.E. Pharmacokinetics and pharmacodynamics of oltipraz as a chemopreventive agent. Clin. Cancer Res. 1995, 1, 1133–1138. [Google Scholar] [PubMed]
- Appel, L.J.; Brands, M.W.; Daniels, S.R.; Karanja, N.; Elmer, P.J.; Sacks, F.M. Dietary approaches to prevent and treat hypertension: A scientific statement from the American Heart Association. Hypertension 2006, 47, 296–308. [Google Scholar] [CrossRef] [PubMed]



| Molecule | Treatment | Species | Mechanism | Reference |
|---|---|---|---|---|
| GSH | Liposomal | Human | Direct | [66] |
| GSH | Intranasal | Human | Direct | [67] |
| GSH | Endovenous | Human | Direct | [67] |
| GSH | Oral | Human | Direct | [67] |
| γ-Glu-Cys | Oral | Human | Enzymatic synthesis | [68] |
| NAC | Oral | Human | Cys delivery | [69] |
| OTC | Oral | Human | Cys indirect formation | [70] |
| Green tea | Oral | Human | GSH sparing? Up-regulation antioxidant enzymes? | [71] |
| DASH diet | Oral | Human | Unknown | [72] |
| GSH mono ester | Oral | Mouse | Enzymatic release | [73] |
| γ-Glu-Cys | Diet | Mouse | Enzymatic synthesis | [74] |
| NACET | Oral | Mouse | NAC and Cys delivery | [75] |
| S-ethyl cysteine | Oral | Mouse | Cys delivery | [76] |
| S-methyl cysteine | Oral | Mouse | Cys delivery | [76] |
| S-propyl cysteine | Oral | Mouse | Cys delivery | [76] |
| Fumaric acid | Oral | Mouse | Nrf2 inducer | [77] |
| Taurine | Oral | Mouse | Cys sparing | [78] |
| Silymarin | Oral | Rat | Antioxidant/Nrf2 inducer/cysteine sparing | [79] |
| NACET | Oral | Rat | NAC and Cys delivery | [80] |
| ADT | Oral | Rat | γ-GT inhibition? Nrf2 inducer? | [81] |
| Bardoxolone | Oral | Monkey | Nrf2 inducer | [82] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giustarini, D.; Milzani, A.; Dalle-Donne, I.; Rossi, R. How to Increase Cellular Glutathione. Antioxidants 2023, 12, 1094. https://doi.org/10.3390/antiox12051094
Giustarini D, Milzani A, Dalle-Donne I, Rossi R. How to Increase Cellular Glutathione. Antioxidants. 2023; 12(5):1094. https://doi.org/10.3390/antiox12051094
Chicago/Turabian StyleGiustarini, Daniela, Aldo Milzani, Isabella Dalle-Donne, and Ranieri Rossi. 2023. "How to Increase Cellular Glutathione" Antioxidants 12, no. 5: 1094. https://doi.org/10.3390/antiox12051094
APA StyleGiustarini, D., Milzani, A., Dalle-Donne, I., & Rossi, R. (2023). How to Increase Cellular Glutathione. Antioxidants, 12(5), 1094. https://doi.org/10.3390/antiox12051094