
Protective Effects of H2S Donor Treatment in Experimental Colitis: A Focus on Antioxidants


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Preparation of Drugs
2.3. Induction of Colitis and Experimental Design
2.4. Determination of Macroscopic Damage
2.5. Measurement of SOD Activity in the Colon Tissue
2.6. Measurement of Colonic Total GSH Level
2.7. Measurement of Colonic 3-NT and Prdxs Levels by ELISA
2.8. Determination of Protein Concentrations
2.9. Statistical Analysis
3. Results
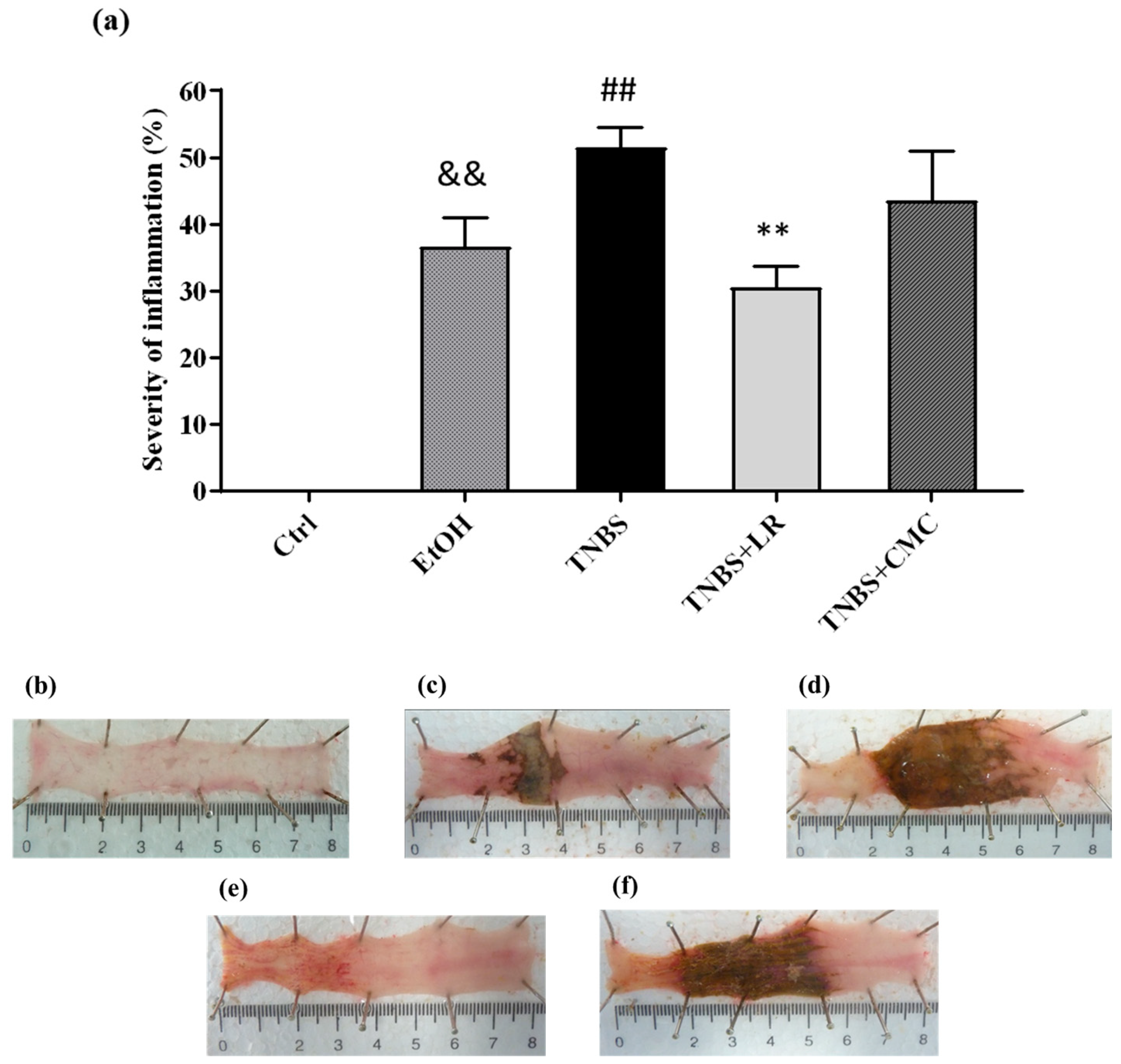
3.1. Hydrogen Sulfide Donor Ameliorates the Extent of Lesions in TNBS-Induced Rat Colitis
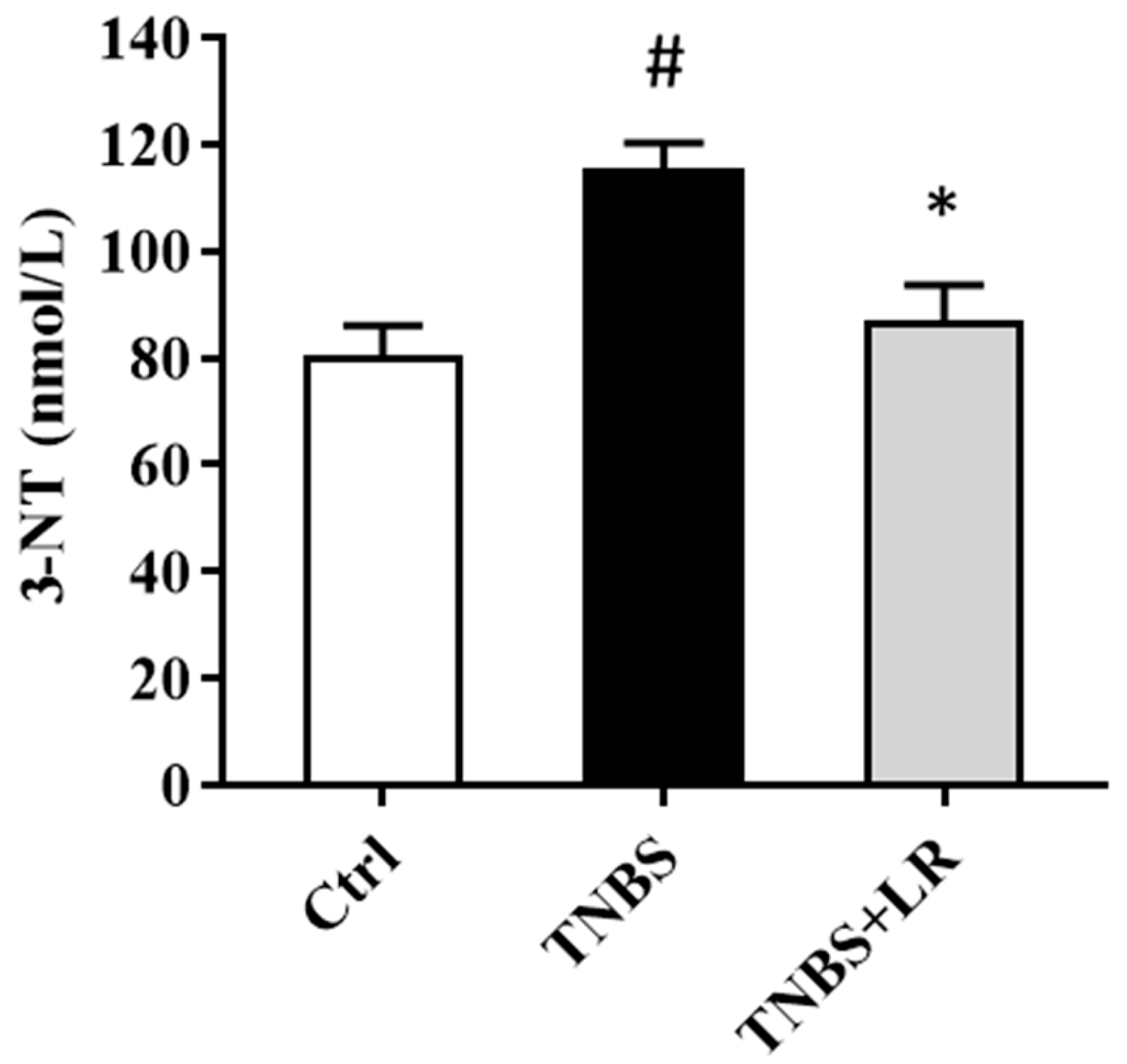
3.2. Hydrogen Sulfide Donor Attenuated the Level of Oxidative Stress Marker 3-NT
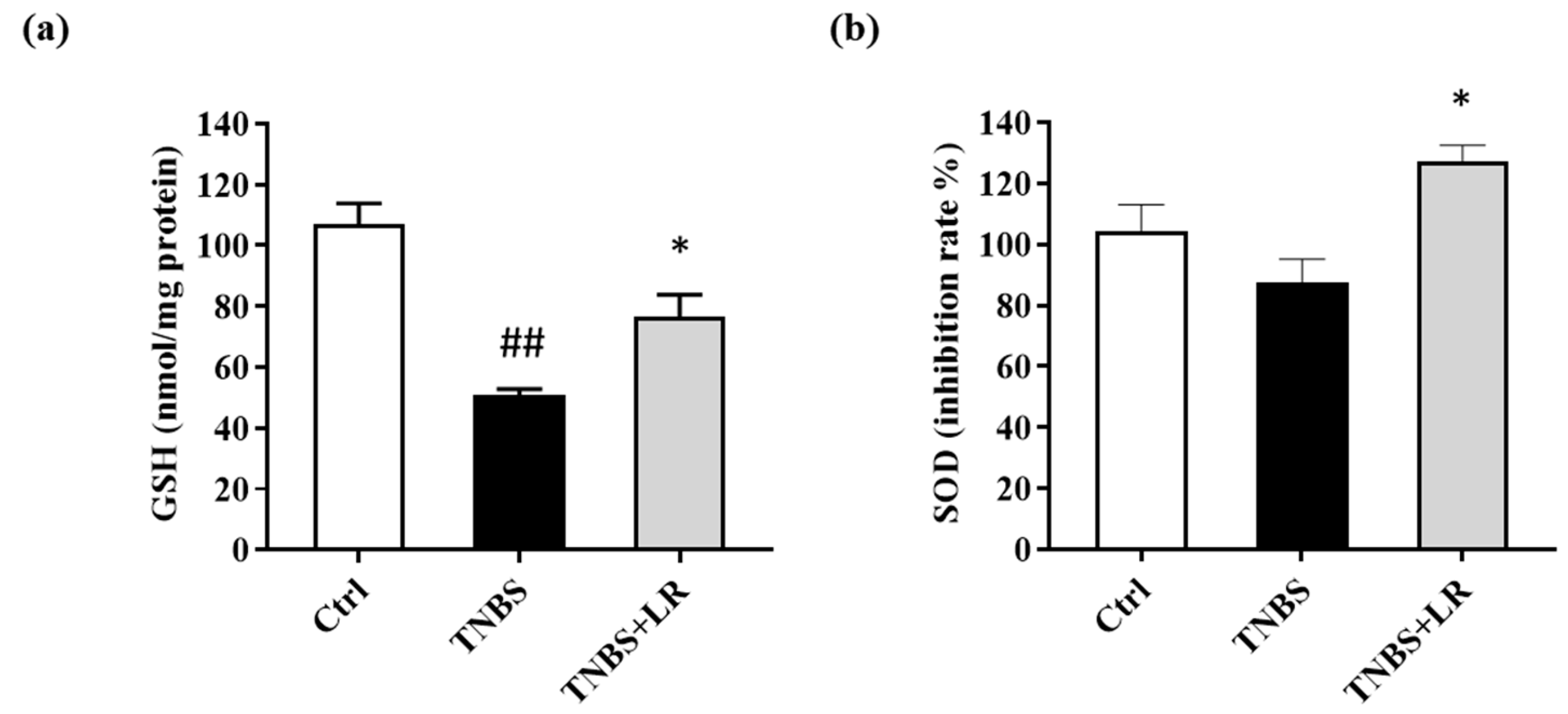
3.3. Effect of Hydrogen Sulfide Donor on the Level of Antioxidant Total GSH in TNBS Colitis
3.4. Hydrogen Sulfide Donor Treatment Elevated the SOD Activity in TNBS-Induced Colitis
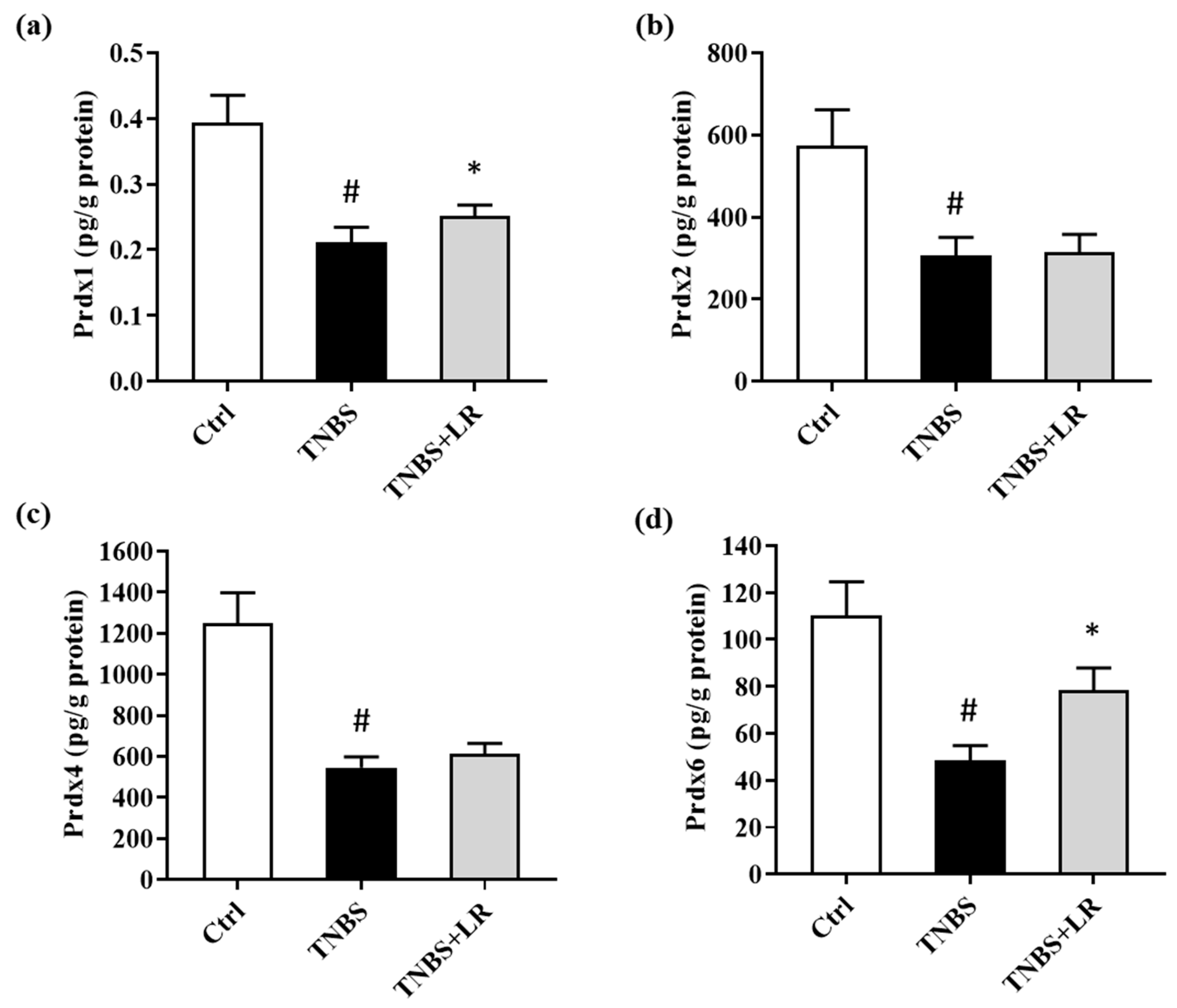
3.5. Alterations in the Levels of Prdx1, -2, -4, and -6 after Hydrogen Sulfide Donor Treatment in TNBS Colitis
4. Discussion
Limitation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Seyedian, S.S.; Nokhostin, F.; Malamir, M.D. A review of the diagnosis, prevention, and treatment methods of inflammatory bowel disease. J. Med. Life 2019, 12, 113–122. [Google Scholar] [CrossRef]
- Baumgart, D.C.; Sandborn, W.J. Crohn’s disease. Lancet 2012, 380, 1590–1605. [Google Scholar] [CrossRef] [PubMed]
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.F. Ulcerative colitis. Lancet 2017, 389, 1756–1770. [Google Scholar] [PubMed]
- Pithadia, A.B.; Jain, S. Treatment of inflammatory bowel disease (IBD). Pharmacol. Rep. 2011, 63, 629–642. [Google Scholar] [CrossRef]
- Rezaie, A.; Parker, R.D.; Abdollahi, M. Oxidative stress and pathogenesis of inflammatory bowel disease: An epiphenomenon or the cause? Dig. Dis. Sci. 2007, 52, 2015–2021. [Google Scholar] [CrossRef]
- Kruidenier, L.; Verspaget, H.W. Review article: Oxidative stress as a pathogenic factor in inflammatory bowel disease--radicals or ridiculous? Aliment. Pharmacol. Ther. 2002, 16, 1997–2015. [Google Scholar] [CrossRef]
- Ahsan, H. 3-Nitrotyrosine: A biomarker of nitrogen free radical species modified proteins in systemic autoimmunogenic conditions. Hum. Immunol. 2013, 74, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Yuksel, M.; Ates, I.; Kaplan, M.; Arikan, M.F.; Ozin, Y.O.; Kilic, Z.M.Y.; Topcuoglu, C.; Kayacetin, E. Is Oxidative Stress Associated with Activation and Pathogenesis of Inflammatory Bowel Disease? J. Med. Biochem. 2017, 36, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Masella, R.; Di Benedetto, R.; Vari, R.; Filesi, C.; Giovannini, C. Novel mechanisms of natural antioxidant compounds in biological systems: Involvement of glutathione and glutathione-related enzymes. J. Nutr. Biochem. 2005, 16, 577–586. [Google Scholar]
- Diaz de Barboza, G.; Guizzardi, S.; Moine, L.; Tolosa de Talamoni, N. Oxidative stress, antioxidants and intestinal calcium absorption. World J. Gastroenterol. 2017, 23, 2841–2853. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Branicky, R.; Noe, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [PubMed]
- Galasso, M.; Gambino, S.; Romanelli, M.G.; Donadelli, M.; Scupoli, M.T. Browsing the oldest antioxidant enzyme: Catalase and its multiple regulation in cancer. Free Radic. Biol. Med. 2021, 172, 264–272. [Google Scholar]
- Rhee, S.G. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 1. [Google Scholar] [PubMed]
- Horie, K.; Mikami, T.; Yoshida, T.; Sato, Y.; Okayasu, I. Peroxiredoxin 1 expression in active ulcerative colitis mucosa identified by proteome analysis and involvement of thioredoxin based on immunohistochemistry. Oncol. Lett. 2018, 15, 2364–2372. [Google Scholar] [CrossRef] [PubMed]
- Senhaji, N.; Zaid, Y.; El Khalfi, B.; Fahimi, M.; Martin, J.; Badre, W.; Nadifi, S.; Soukri, A. Peroxiredoxin-2 up-regulation in inflammatory bowel disease: Friend or foe? J. Gastroenterol. Hepatol. 2017, 32, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Takagi, T.; Homma, T.; Fujii, J.; Shirasawa, N.; Yoriki, H.; Hotta, Y.; Higashimura, Y.; Mizushima, K.; Hirai, Y.; Katada, K.; et al. Elevated ER stress exacerbates dextran sulfate sodium-induced colitis in PRDX4-knockout mice. Free Radic. Biol. Med. 2019, 134, 153–164. [Google Scholar] [CrossRef]
- Melhem, H.; Spalinger, M.R.; Cosin-Roger, J.; Atrott, K.; Lang, S.; Wojtal, K.A.; Vavricka, S.R.; Rogler, G.; Frey-Wagner, I. Prdx6 Deficiency Ameliorates DSS Colitis: Relevance of Compensatory Antioxidant Mechanisms. J. Crohns Colitis 2017, 11, 871–884. [Google Scholar] [CrossRef]
- Zhu, H.; Li, Y.R. Oxidative stress and redox signaling mechanisms of inflammatory bowel disease: Updated experimental and clinical evidence. Exp. Biol. Med. 2012, 237, 474–480. [Google Scholar] [CrossRef]
- Wang, R. Two’s company, three’s a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef]
- Nagy, P. Mechanistic chemical perspective of hydrogen sulfide signaling. Methods Enzymol. 2015, 554, 3–29. [Google Scholar] [PubMed]
- Zaichko, N.V.; Melnik, A.V.; Yoltukhivskyy, M.M.; Olhovskiy, A.S.; Palamarchuk, I.V. Hydrogen sulfide: Metabolism, biological and medical role. Ukr. Biochem. J. 2014, 86, 5–25. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.Z.; Liu, Y.; Bian, J.S. Hydrogen Sulfide and Cellular Redox Homeostasis. Oxid. Med. Cell. Longev. 2016, 2016, 6043038. [Google Scholar] [CrossRef]
- Morris, G.P.; Beck, P.L.; Herridge, M.S.; Depew, W.T.; Szewczuk, M.R.; Wallace, J.L. Hapten-induced model of chronic inflammation and ulceration in the rat colon. Gastroenterology 1989, 96, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Kupai, K.; Almasi, N.; Kosa, M.; Nemcsok, J.; Murlasits, Z.; Torok, S.; Al-Awar, A.; Barath, Z.; Posa, A.; Varga, C. H(2)S confers colonoprotection against TNBS-induced colitis by HO-1 upregulation in rats. Inflammopharmacology 2018, 26, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Mancardi, D.; Penna, C.; Merlino, A.; Del Soldato, P.; Wink, D.A.; Pagliaro, P. Physiological and pharmacological features of the novel gasotransmitter: Hydrogen sulfide. Biochim. Biophys. Acta 2009, 1787, 864–872. [Google Scholar] [CrossRef]
- Gemici, B.; Wallace, J.L. Anti-inflammatory and cytoprotective properties of hydrogen sulfide. Methods Enzymol. 2015, 555, 169–193. [Google Scholar]
- Sparatore, A.; Santus, G.; Giustarini, D.; Rossi, R.; Del Soldato, P. Therapeutic potential of new hydrogen sulfide-releasing hybrids. Expert. Rev. Clin. Pharmacol. 2011, 4, 109–121. [Google Scholar] [CrossRef]
- Marutani, E.; Ichinose, F. Emerging pharmacological tools to control hydrogen sulfide signaling in critical illness. Intensive Care Med. Exp. 2020, 8, 5. [Google Scholar] [CrossRef]
- Antoniou, E.; Margonis, G.A.; Angelou, A.; Pikouli, A.; Argiri, P.; Karavokyros, I.; Papalois, A.; Pikoulis, E. The TNBS-induced colitis animal model: An overview. Ann. Med. Surg. 2016, 11, 9–15. [Google Scholar] [CrossRef]
- Matsunami, M.; Kirishi, S.; Okui, T.; Kawabata, A. Hydrogen sulfide-induced colonic mucosal cytoprotection involves T-type calcium channel-dependent neuronal excitation in rats. J. Physiol. Pharmacol. 2012, 63, 61–68. [Google Scholar] [PubMed]
- Chen, X.; Liu, X.S. Hydrogen sulfide from a NaHS source attenuates dextran sulfate sodium (DSS)-induced inflammation via inhibiting nuclear factor-kappaB. J. Zhejiang Univ. Sci. B 2016, 17, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell Longev. 2016, 2016, 1245049. [Google Scholar]
- Almasi, N.; Torok, S.; Valkusz, Z.; Tajti, M.; Csonka, A.; Murlasits, Z.; Posa, A.; Varga, C.; Kupai, K. Sigma-1 Receptor Engages an Anti-Inflammatory and Antioxidant Feedback Loop Mediated by Peroxiredoxin in Experimental Colitis. Antioxidants 2020, 9, 1081. [Google Scholar] [CrossRef] [PubMed]
- Gochman, E.; Mahajna, J.; Shenzer, P.; Dahan, A.; Blatt, A.; Elyakim, R.; Reznick, A.Z. The expression of iNOS and nitrotyrosine in colitis and colon cancer in humans. Acta Histochem. 2012, 114, 827–835. [Google Scholar] [CrossRef]
- Mishra, P.K.; Tyagi, N.; Sen, U.; Givvimani, S.; Tyagi, S.C. H2S ameliorates oxidative and proteolytic stresses and protects the heart against adverse remodeling in chronic heart failure. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H451–H456. [Google Scholar] [CrossRef]
- Scammahorn, J.J.; Nguyen, I.T.N.; Bos, E.M.; Van Goor, H.; Joles, J.A. Fighting Oxidative Stress with Sulfur: Hydrogen Sulfide in the Renal and Cardiovascular Systems. Antioxidants 2021, 10, 373. [Google Scholar] [CrossRef]
- Dziabowska-Grabias, K.; Sztanke, M.; Zajac, P.; Celejewski, M.; Kurek, K.; Szkutnicki, S.; Korga, P.; Bulikowski, W.; Sztanke, K. Antioxidant Therapy in Inflammatory Bowel Diseases. Antioxidants 2021, 10, 412. [Google Scholar] [CrossRef]
- Sido, B.; Hack, V.; Hochlehnert, A.; Lipps, H.; Herfarth, C.; Droge, W. Impairment of intestinal glutathione synthesis in patients with inflammatory bowel disease. Gut 1998, 42, 485–492. [Google Scholar] [CrossRef]
- Guo, C.; Liang, F.; Shah Masood, W.; Yan, X. Hydrogen sulfide protected gastric epithelial cell from ischemia/reperfusion injury by Keap1 s-sulfhydration, MAPK dependent anti-apoptosis and NF-kappaB dependent anti-inflammation pathway. Eur. J. Pharmacol. 2014, 725, 70–78. [Google Scholar] [CrossRef]
- Cui, J.; Liu, L.; Zou, J.; Qiao, W.; Liu, H.; Qi, Y.; Yan, C. Protective effect of endogenous hydrogen sulfide against oxidative stress in gastric ischemia-reperfusion injury. Exp. Ther. Med. 2013, 5, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Segui, J.; Gironella, M.; Sans, M.; Granell, S.; Gil, F.; Gimeno, M.; Coronel, P.; Pique, J.M.; Panes, J. Superoxide dismutase ameliorates TNBS-induced colitis by reducing oxidative stress, adhesion molecule expression, and leukocyte recruitment into the inflamed intestine. J. Leukoc. Biol. 2004, 76, 537–544. [Google Scholar] [CrossRef]
- Knoops, B.; Argyropoulou, V.; Becker, S.; Ferte, L.; Kuznetsova, O. Multiple Roles of Peroxiredoxins in Inflammation. Mol. Cells 2016, 39, 60–64. [Google Scholar] [PubMed]
- Hsieh, S.Y.; Shih, T.C.; Yeh, C.Y.; Lin, C.J.; Chou, Y.Y.; Lee, Y.S. Comparative proteomic studies on the pathogenesis of human ulcerative colitis. Proteomics 2006, 6, 5322–5331. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Du, X.; Peng, Z.; Zhang, Z.; Cui, L.; Li, D.; Wang, R.; Ma, M. Silencing of peroxiredoxin 1 expression ameliorates ulcerative colitis in a rat model. J. Int. Med. Res. 2021, 49, 300060520986313. [Google Scholar] [CrossRef]
- Liu, M.H.; Lin, X.L.; Yuan, C.; He, J.; Tan, T.P.; Wu, S.J.; Yu, S.; Chen, L.; Liu, J.; Tian, W.; et al. Hydrogen sulfide attenuates doxorubicin-induced cardiotoxicity by inhibiting the expression of peroxiredoxin III in H9c2 cells. Mol. Med. Rep. 2016, 13, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhao, X.; Cai, H.; Sun, H.; Hu, Y.; Huang, X.; Kong, W.; Kong, W. The role of sodium hydrosulfide in attenuating the aging process via PI3K/AKT and CaMKKbeta/AMPK pathways. Redox Biol. 2017, 12, 987–1003. [Google Scholar] [CrossRef]
- Unuma, K.; Yoshikawa, A.; Aki, T.; Uemura, K. Increased circulating peroxiredoxin-4 in sepsis model rats involves secretion from hepatocytes and is mitigated by GYY4137. J. Toxicol. Pathol. 2019, 32, 305–310. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Török, S.; Almási, N.; Veszelka, M.; Börzsei, D.; Szabó, R.; Varga, C. Protective Effects of H2S Donor Treatment in Experimental Colitis: A Focus on Antioxidants. Antioxidants 2023, 12, 1025. https://doi.org/10.3390/antiox12051025
Török S, Almási N, Veszelka M, Börzsei D, Szabó R, Varga C. Protective Effects of H2S Donor Treatment in Experimental Colitis: A Focus on Antioxidants. Antioxidants. 2023; 12(5):1025. https://doi.org/10.3390/antiox12051025
Chicago/Turabian StyleTörök, Szilvia, Nikoletta Almási, Médea Veszelka, Denise Börzsei, Renáta Szabó, and Csaba Varga. 2023. "Protective Effects of H2S Donor Treatment in Experimental Colitis: A Focus on Antioxidants" Antioxidants 12, no. 5: 1025. https://doi.org/10.3390/antiox12051025
APA StyleTörök, S., Almási, N., Veszelka, M., Börzsei, D., Szabó, R., & Varga, C. (2023). Protective Effects of H2S Donor Treatment in Experimental Colitis: A Focus on Antioxidants. Antioxidants, 12(5), 1025. https://doi.org/10.3390/antiox12051025