

Bacillus subtilis YtpP and Thioredoxin A Are New Players in the Coenzyme-A-Mediated Defense Mechanism against Cellular Stress

 ,
,  ,
, 
Abstract
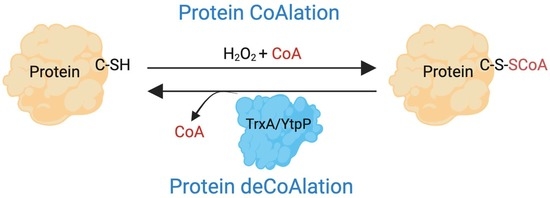
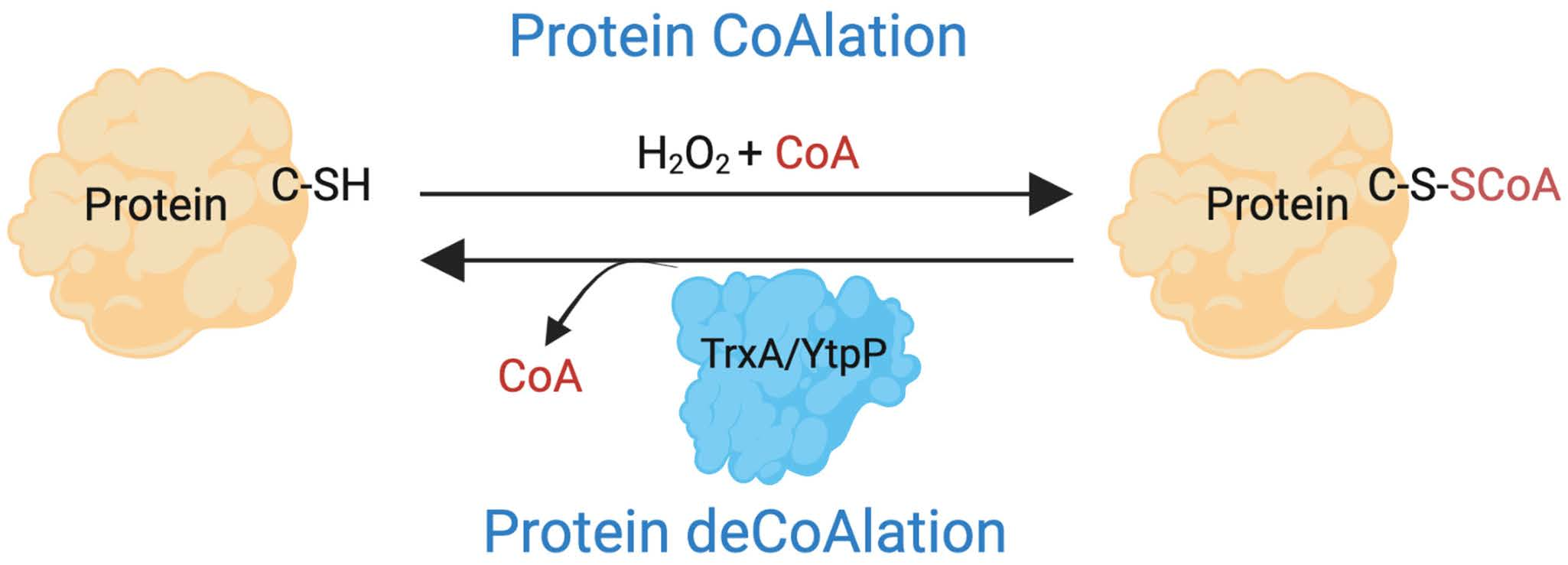{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
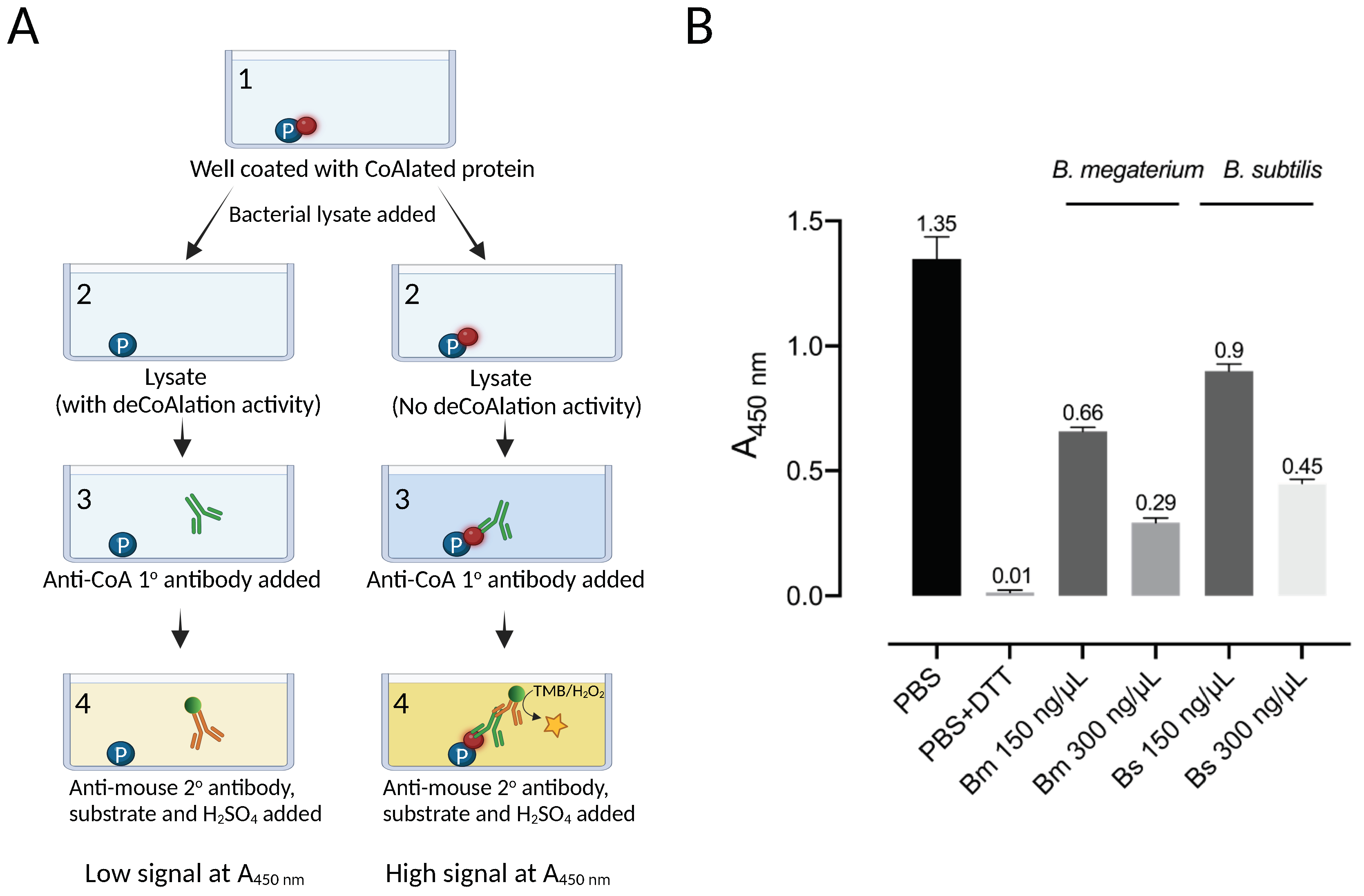
2.1. ELISA-Based deCoAlation Assay Using B. subtilis and B. megaterium Lysates
2.2. Growth, Harvesting, and Lysis of B. megaterium and B. subtilis Cells
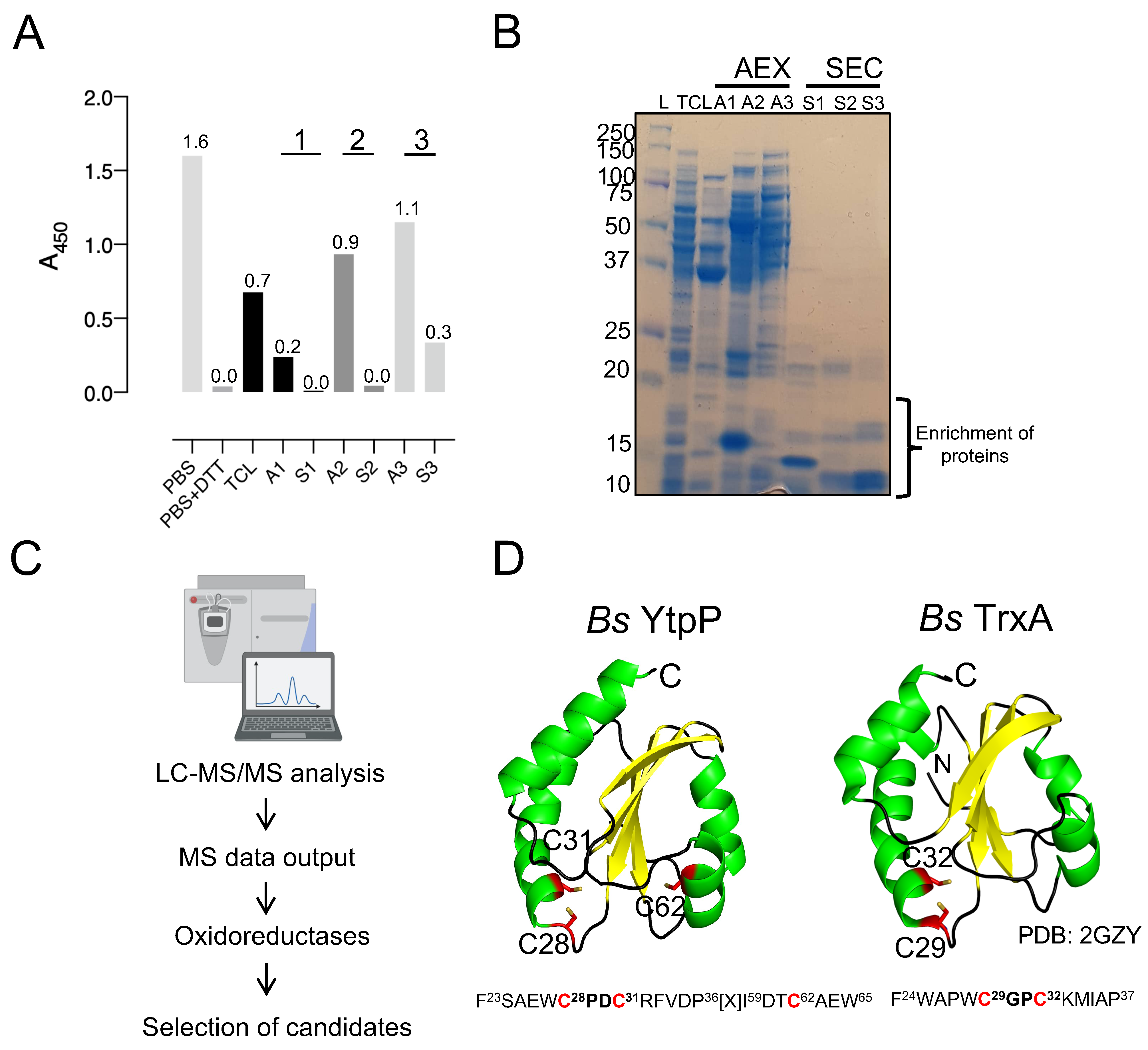
2.3. Endogenous Purification of Enzymes with deCoAlation Activity from B. megaterium Lysates
2.4. Mass-Spectrometry Analysis of Purified Fractions That Showed deCoAlation Activity
2.5. Selection of Candidate Proteins with Possible deCoAlation Activity
2.6. B. subtilis YtpP and TrxA Wild-Type Gene Design and Site-Directed Mutagenesis
2.7. Expression and Purification of WT and Cysteine Mutants of YtpP and TrxA
2.8. Expression and Purification of GAPDH, MsrA, and PRDX5
2.9. CoAlation of GAPDH, MsrA, and PRDX5
2.10. Protein deCoAlation Assay Using anti-CoA Western Blot
2.11. Detection of CoA Release following deCoAlation by Using High-Performance Liquid Chromatography
3. Results
3.1. Protein deCoAlation Activity Is Detected in B. megaterium and B. subtilis Lysates by Using an ELISA-Based deCoAlation Assay
3.2. Purification of Endogenous Enzymes with deCoAlation activity from B. megaterium Lysates
3.3. Mass Spectrometry Identification of Candidate Enzymes That deCoAlate

3.4. B. subtilis YtpP and TrxA deCoAlate CoA–PRDX5 and CoA–MsrA
3.5. Cys28 and Cys31 Are the Catalytic Cysteines of YtpP
3.6. TrxA Cys29 and Cys32 Are the Catalytic Cysteine Residues Required for Protein deCoAlation
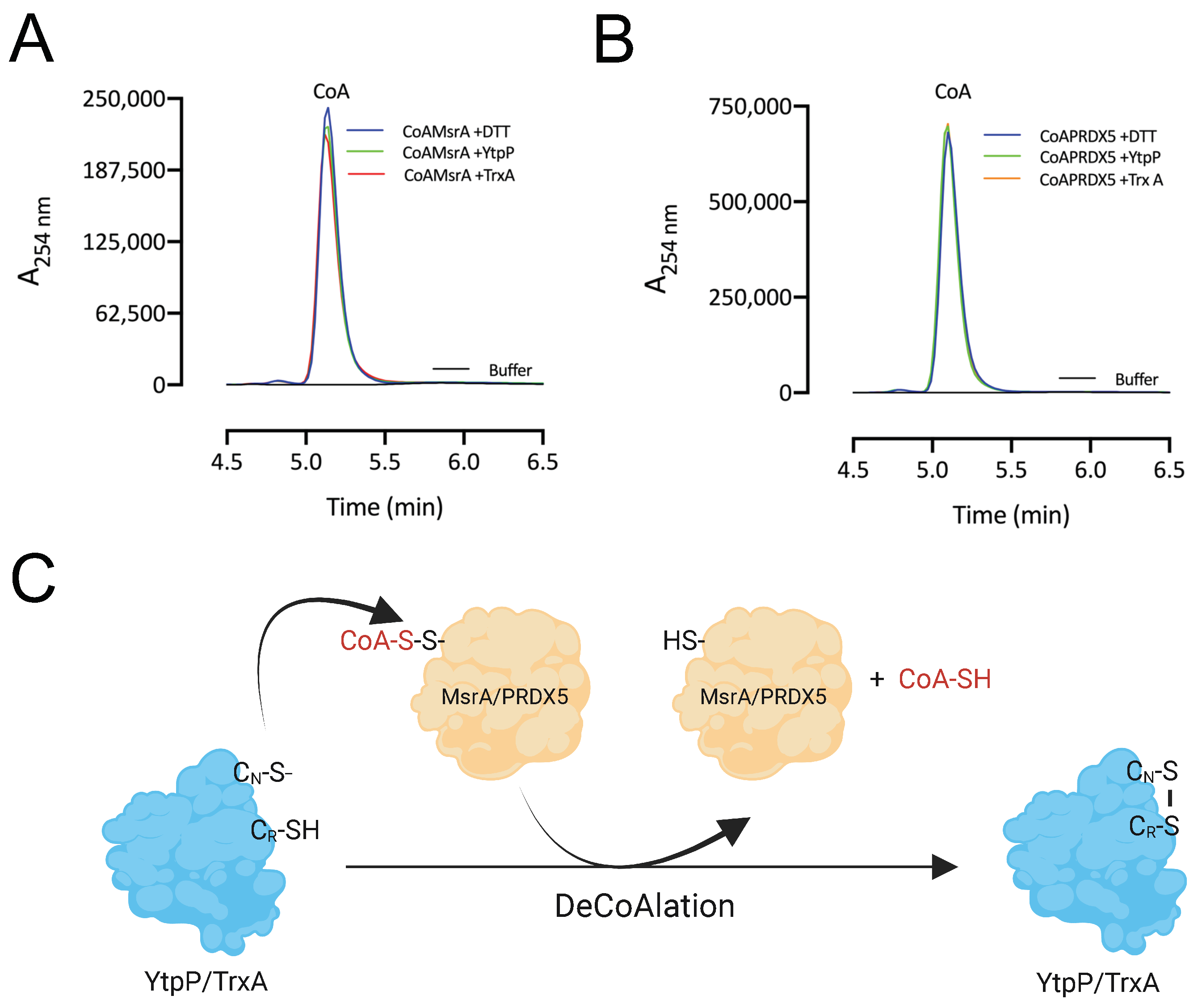
3.7. CoA Is Released following deCoAlation by YtpP and TrxA
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Van Laer, K.; Hamilton, C.J.; Messens, J. Low-molecular-weight thiols in thiol-disulfide exchange. Antioxid. Redox Signal. 2013, 18, 1642–1653. [Google Scholar] [CrossRef]
- Arner, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef] [PubMed]
- Gout, I. Coenzyme A: A protective thiol in bacterial antioxidant defence. Biochem. Soc. Trans. 2019, 47, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Gout, I. Coenzyme A, protein CoAlation and redox regulation in mammalian cells. Biochem. Soc. Trans. 2018, 46, 721–728. [Google Scholar] [CrossRef]
- Sharma, S.V.; Van Laer, K.; Messens, J.; Hamilton, C.J. Thiol redox and pKa properties of mycothiol, the predominant low-molecular-weight thiol cofactor in the actinomycetes. ChemBioChem 2016, 17, 1689–1692. [Google Scholar] [CrossRef]
- Perera, V.R.; Newton, G.L.; Pogliano, K. Bacillithiol: A key protective thiol in Staphylococcus aureus. Expert Rev. Anti-Infect. 2015, 13, 1089–1107. [Google Scholar] [CrossRef] [PubMed]
- Roos, G.; Messens, J. Protein sulfenic acid formation: From cellular damage to redox regulation. Free Radic. Biol. Med. 2011, 51, 314–326. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Carroll, K.S. Cysteine-mediated redox signaling: Chemistry, biology, and tools for discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef]
- Jung, T.; Bader, N.; Grune, T. Oxidized proteins: Intracellular distribution and recognition by the proteasome. Arch. Biochem. Biophys. 2007, 462, 231–237. [Google Scholar] [CrossRef]
- Lillig, C.H.; Berndt, C. Cellular functions of glutathione. Biochim. Biophys. Acta 2013, 1830, 3137–3138. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Glutathione and its role in cellular functions. Free Radic. Biol. Med. 1999, 27, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Giustarini, D.; Rossi, R.; Milzani, A.; Colombo, R.; Dalle-Donne, I. S-glutathionylation: From redox regulation of protein functions to human diseases. J. Cell Mol. Med. 2004, 8, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta 2013, 1830, 3217–3266. [Google Scholar] [CrossRef] [PubMed]
- Deponte, M.; Lillig, C.H. Enzymatic control of cysteinyl thiol switches in proteins. Biol. Chem. 2015, 396, 401–413. [Google Scholar] [CrossRef]
- Leonardi, R.; Zhang, Y.M.; Rock, C.O.; Jackowski, S. Coenzyme A: Back in action. Prog. Lipid Res. 2005, 44, 125–153. [Google Scholar] [CrossRef]
- Aloum, L.; Brimson, C.A.; Zhyvoloup, A.; Baines, R.; Bakovic, J.; Filonenko, V.; Thompson, C.R.L.; Gout, I. Coenzyme A and protein CoAlation levels are regulated in response to oxidative stress and during morphogenesis in Dictyostelium discoideum. Biochem. Biophys. Res. Commun. 2019, 511, 294–299. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Zhyvoloup, A.; Bakovic, J.; Thomas, N.; Yu, B.Y.K.; Das, S.; Orengo, C.; Newell, C.; Ward, J.; Saladino, G.; et al. Protein CoAlation and antioxidant function of coenzyme A in prokaryotic cells. Biochem. J. 2018, 475, 1909–1937. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Peak-Chew, S.Y.; Newell, C.; Miller-Aidoo, S.; Mangal, S.; Zhyvoloup, A.; Bakovic, J.; Malanchuk, O.; Pereira, G.C.; Kotiadis, V.; et al. Protein CoAlation: A redox-regulated protein modification by coenzyme A in mammalian cells. Biochem. J. 2017, 474, 2489–2508. [Google Scholar] [CrossRef]
- Tossounian, M.A.; Baczynska, M.; Dalton, W.; Newell, C.; Ma, Y.; Das, S.; Semelak, J.A.; Estrin, D.A.; Filonenko, V.; Trujillo, M.; et al. Profiling the site of protein CoAlation and coenzyme A stabilization interactions. Antioxidants 2022, 11, 1362. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.Y.K.; Tossounian, M.A.; Hristov, S.D.; Lawrence, R.; Arora, P.; Tsuchiya, Y.; Peak-Chew, S.Y.; Filonenko, V.; Oxenford, S.; Angell, R.; et al. Regulation of metastasis suppressor NME1 by a key metabolic cofactor coenzyme A. Redox Biol. 2021, 44, 101978. [Google Scholar] [CrossRef]
- Bakovic, J.; Yu, B.Y.K.; Silva, D.; Baczynska, M.; Peak-Chew, S.Y.; Switzer, A.; Burchell, L.; Wigneshweraraj, S.; Vandanashree, M.; Gopal, B.; et al. Redox regulation of the quorum-sensing transcription factor AgrA by coenzyme A. Antioxidants 2021, 10, 841. [Google Scholar] [CrossRef] [PubMed]
- Bakovic, J.; Yu, B.Y.K.; Silva, D.; Chew, S.P.; Kim, S.; Ahn, S.H.; Palmer, L.; Aloum, L.; Stanzani, G.; Malanchuk, O.; et al. A key metabolic integrator, coenzyme A, modulates the activity of peroxiredoxin 5 via covalent modification. Mol. Cell Biochem. 2019, 461, 91–102. [Google Scholar] [CrossRef]
- Tossounian, M.A.; Zhang, B.; Gout, I. The writers, readers, and erasers in redox regulation of GAPDH. Antioxidants 2020, 9, 1288. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Byrne, D.P.; Burgess, S.G.; Bormann, J.; Bakovic, J.; Huang, Y.; Zhyvoloup, A.; Yu, B.Y.K.; Peak-Chew, S.; Tran, T.; et al. Covalent Aurora A regulation by the metabolic integrator coenzyme A. Redox Biol. 2020, 28, 101318. [Google Scholar] [CrossRef]
- Tsuji, K.; Yoon, K.S.; Ogo, S. Glyceraldehyde-3-phosphate dehydrogenase from Citrobacter sp. S-77 is post-translationally modified by CoA (protein CoAlation) under oxidative stress. FEBS Open Bio. 2019, 9, 53–73. [Google Scholar] [CrossRef] [PubMed]
- Lashley, T.; Tossounian, M.A.; Costello Heaven, N.; Wallworth, S.; Peak-Chew, S.; Bradshaw, A.; Cooper, J.M.; de Silva, R.; Srai, S.K.; Malanchuk, O.; et al. Extensive anti-CoA immunostaining in Alzheimer’s disease and covalent modification of Tau by a key cellular metabolite coenzyme A. Front. Cell Neurosci. 2021, 15, 739425. [Google Scholar] [CrossRef]
- Zhyvoloup, A.; Yu, B.Y.K.; Bakovic, J.; Davis-Lunn, M.; Tossounian, M.A.; Thomas, N.; Tsuchiya, Y.; Peak-Chew, S.Y.; Wigneshweraraj, S.; Filonenko, V.; et al. Analysis of disulphide bond linkage between CoA and protein cysteine thiols during sporulation and in spores of Bacillus species. FEMS Microbiol. Lett. 2020, 367, fnaa174. [Google Scholar] [CrossRef]
- Chi, B.K.; Gronau, K.; Mader, U.; Hessling, B.; Becher, D.; Antelmann, H. S-bacillithiolation protects against hypochlorite stress in Bacillus subtilis as revealed by transcriptomics and redox proteomics. Mol. Cell Proteom. 2011, 10, M111.009506. [Google Scholar] [CrossRef]
- Malanchuk, O.M.; Panasyuk, G.G.; Serbin, N.M.; Gout, I.T.; Filonenko, V.V. Generation and characterization of monoclonal antibodies specific to coenzyme A. Biopolym. Cell 2015, 31, 187–192. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Bandeira, N.; Perez-Riverol, Y.; Sharma, V.; Carver, J.J.; Mendoza, L.; Kundu, D.J.; Wang, S.; Bandla, C.; Kamatchinathan, S.; et al. The ProteomeXchange consortium at 10 years: 2023 update. Nucleic Acids Res. 2023, 51, D1539–D1548. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Xu, Q.W.; Wang, R.; Uszkoreit, J.; Griss, J.; Sanchez, A.; Reisinger, F.; Csordas, A.; Ternent, T.; Del-Toro, N.; et al. PRIDE Inspector Toolsuite: Moving Toward a Universal Visualization Tool for Proteomics Data Standard Formats and Quality Assessment of ProteomeXchange Datasets. Mol. Cell Proteom. 2016, 15, 305–317. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Pham, U.; Hu, W.; Ohnuma, S.; Gout, I. Changes in acetyl CoA levels during the early embryonic development of Xenopus laevis. PLoS ONE 2014, 9, e97693. [Google Scholar] [CrossRef] [PubMed]
- Nakano, S.; Kuster-Schock, E.; Grossman, A.D.; Zuber, P. Spx-dependent global transcriptional control is induced by thiol-specific oxidative stress in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 2003, 100, 13603–13608. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold protein structure database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Brandes, N.; Schmitt, S.; Jakob, U. Thiol-based redox switches in eukaryotic proteins. Antioxid. Redox Signal. 2009, 11, 997–1014. [Google Scholar] [CrossRef]
- Jothivasan, V.K.; Hamilton, C.J. Mycothiol: Synthesis, biosynthesis and biological functions of the major low molecular weight thiol in actinomycetes. Nat. Prod. Rep. 2008, 25, 1091–1117. [Google Scholar] [CrossRef]
- Newton, G.L.; Buchmeier, N.; Fahey, R.C. Biosynthesis and functions of mycothiol, the unique protective thiol of actinobacteria. Microbiol. Mol. Biol. Rev. 2008, 72, 471–494. [Google Scholar] [CrossRef]
- Newton, G.L.; Rawat, M.; La Clair, J.J.; Jothivasan, V.K.; Budiarto, T.; Hamilton, C.J.; Claiborne, A.; Helmann, J.D.; Fahey, R.C. Bacillithiol is an antioxidant thiol produced in Bacilli. Nat. Chem. Biol. 2009, 5, 625–627. [Google Scholar] [CrossRef]
- Peralta, D.; Bronowska, A.K.; Morgan, B.; Doka, E.; Van Laer, K.; Nagy, P.; Grater, F.; Dick, T.P. A proton relay enhances H2O2 sensitivity of GAPDH to facilitate metabolic adaptation. Nat. Chem. Biol. 2015, 11, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Hillion, M.; Imber, M.; Pedre, B.; Bernhardt, J.; Saleh, M.; Loi, V.V.; Maass, S.; Becher, D.; Astolfi Rosado, L.; Adrian, L.; et al. The glyceraldehyde-3-phosphate dehydrogenase GapDH of Corynebacterium diphtheriae is redox-controlled by protein S-mycothiolation under oxidative stress. Sci. Rep. 2017, 7, 5020. [Google Scholar] [CrossRef] [PubMed]
- Imber, M.; Huyen, N.T.T.; Pietrzyk-Brzezinska, A.J.; Loi, V.V.; Hillion, M.; Bernhardt, J.; Tharichen, L.; Kolsek, K.; Saleh, M.; Hamilton, C.J.; et al. Protein S-bacillithiolation functions in thiol protection and redox regulation of the glyceraldehyde-3-phosphate dehydrogenase Gap in Staphylococcus aureus under hypochlorite stress. Antioxid. Redox Signal. 2018, 28, 410–430. [Google Scholar] [CrossRef]
- Antelmann, H.; Helmann, J.D. Thiol-based redox switches and gene regulation. Antioxid. Redox Signal. 2011, 14, 1049–1063. [Google Scholar] [CrossRef]
- Zuber, P.; Chauhan, S.; Pilaka, P.; Nakano, M.M.; Gurumoorthy, S.; Lin, A.A.; Barendt, S.M.; Chi, B.K.; Antelmann, H.; Mader, U. Phenotype enhancement screen of a regulatory spx mutant unveils a role for the ytpQ gene in the control of iron homeostasis. PLoS ONE 2011, 6, e25066. [Google Scholar] [CrossRef] [PubMed]
- Kouwen, T.R.; Dubois, J.Y.; Freudl, R.; Quax, W.J.; van Dijl, J.M. Modulation of thiol-disulfide oxidoreductases for increased production of disulfide-bond-containing proteins in Bacillus subtilis. Appl. Env. Microbiol. 2008, 74, 7536–7545. [Google Scholar] [CrossRef] [PubMed]
- Scharf, C.; Riethdorf, S.; Ernst, H.; Engelmann, S.; Volker, U.; Hecker, M. Thioredoxin is an essential protein induced by multiple stresses in Bacillus subtilis. J. Bacteriol. 1998, 180, 1869–1877. [Google Scholar] [CrossRef] [PubMed]
- Smits, W.K.; Dubois, J.Y.; Bron, S.; van Dijl, J.M.; Kuipers, O.P. Tricksy business: Transcriptome analysis reveals the involvement of thioredoxin A in redox homeostasis, oxidative stress, sulfur metabolism, and cellular differentiation in Bacillus subtilis. J. Bacteriol. 2005, 187, 3921–3930. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A. Thioredoxin structure and mechanism: Conformational changes on oxidation of the active-site sulfhydryls to a disulfide. Structure 1995, 3, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Kouwen, T.R.; Andrell, J.; Schrijver, R.; Dubois, J.Y.; Maher, M.J.; Iwata, S.; Carpenter, E.P.; van Dijl, J.M. Thioredoxin A active-site mutants form mixed disulfide dimers that resemble enzyme-substrate reaction intermediates. J. Mol. Biol. 2008, 379, 520–534. [Google Scholar] [CrossRef]
- Tossounian, M.A.; Khanh Truong, A.C.; Buts, L.; Wahni, K.; Mourenza, A.; Leermakers, M.; Vertommen, D.; Mateos, L.M.; Volkov, A.N.; Messens, J. Methionine sulfoxide reductase B from Corynebacterium diphtheriae catalyzes sulfoxide reduction via an intramolecular disulfide cascade. J. Biol. Chem. 2020, 295, 3664–3677. [Google Scholar] [CrossRef] [PubMed]
- Tossounian, M.A.; Pedre, B.; Wahni, K.; Erdogan, H.; Vertommen, D.; Van Molle, I.; Messens, J. Corynebacterium diphtheriae methionine sulfoxide reductase a exploits a unique mycothiol redox relay mechanism. J. Biol. Chem. 2015, 290, 11365–11375. [Google Scholar] [CrossRef] [PubMed]
- Pedre, B.; Van Molle, I.; Villadangos, A.F.; Wahni, K.; Vertommen, D.; Turell, L.; Erdogan, H.; Mateos, L.M.; Messens, J. The Corynebacterium glutamicum mycothiol peroxidase is a reactive oxygen species-scavenging enzyme that shows promiscuity in thiol redox control. Mol. Microbiol. 2015, 96, 1176–1191. [Google Scholar] [CrossRef]
- Li, Y.; Hu, Y.; Zhang, X.; Xu, H.; Lescop, E.; Xia, B.; Jin, C. Conformational fluctuations coupled to the thiol-disulfide transfer between thioredoxin and arsenate reductase in Bacillus subtilis. J. Biol. Chem. 2007, 282, 11078–11083. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Clore, G.M.; Kennedy, W.P.; Kuszewski, J.; Gronenborn, A.M. The solution structure of human thioredoxin complexed with its target from Ref-1 reveals peptide chain reversal. Structure 1996, 4, 613–620. [Google Scholar] [CrossRef][Green Version]
- Fahey, R.C. Novel thiols of prokaryotes. Annu. Rev. Microbiol. 2001, 55, 333–356. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tossounian, M.-A.; Baczynska, M.; Dalton, W.; Peak-Chew, S.Y.; Undzenas, K.; Korza, G.; Filonenko, V.; Skehel, M.; Setlow, P.; Gout, I. Bacillus subtilis YtpP and Thioredoxin A Are New Players in the Coenzyme-A-Mediated Defense Mechanism against Cellular Stress. Antioxidants 2023, 12, 938. https://doi.org/10.3390/antiox12040938
Tossounian M-A, Baczynska M, Dalton W, Peak-Chew SY, Undzenas K, Korza G, Filonenko V, Skehel M, Setlow P, Gout I. Bacillus subtilis YtpP and Thioredoxin A Are New Players in the Coenzyme-A-Mediated Defense Mechanism against Cellular Stress. Antioxidants. 2023; 12(4):938. https://doi.org/10.3390/antiox12040938
Chicago/Turabian StyleTossounian, Maria-Armineh, Maria Baczynska, William Dalton, Sew Yeu Peak-Chew, Kipras Undzenas, George Korza, Valeriy Filonenko, Mark Skehel, Peter Setlow, and Ivan Gout. 2023. "Bacillus subtilis YtpP and Thioredoxin A Are New Players in the Coenzyme-A-Mediated Defense Mechanism against Cellular Stress" Antioxidants 12, no. 4: 938. https://doi.org/10.3390/antiox12040938
APA StyleTossounian, M.-A., Baczynska, M., Dalton, W., Peak-Chew, S. Y., Undzenas, K., Korza, G., Filonenko, V., Skehel, M., Setlow, P., & Gout, I. (2023). Bacillus subtilis YtpP and Thioredoxin A Are New Players in the Coenzyme-A-Mediated Defense Mechanism against Cellular Stress. Antioxidants, 12(4), 938. https://doi.org/10.3390/antiox12040938