

Progranulin Deficiency Induces Mitochondrial Dysfunction in Frontotemporal Lobar Degeneration with TDP-43 Inclusions

 , , ,
, , ,  ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines Culture and Treatments
2.2. Cell Lysates and Immunoblot
2.3. Quantitative Real-Time PCR
2.4. Cell Viability and Apoptosis Measurement
2.5. Measurement of Mitochondrial Membrane Potential (ΔΨm)
2.6. Reactive Oxygen Species (ROS) Measurement
2.7. Measurement of Cellular Oxygen Consumption
2.8. ATP Levels Measurement
2.9. Mitochondrial Mass
2.10. Immunofluorescence and Colocalization Analysis
2.11. Statistical Analysis
3. Results
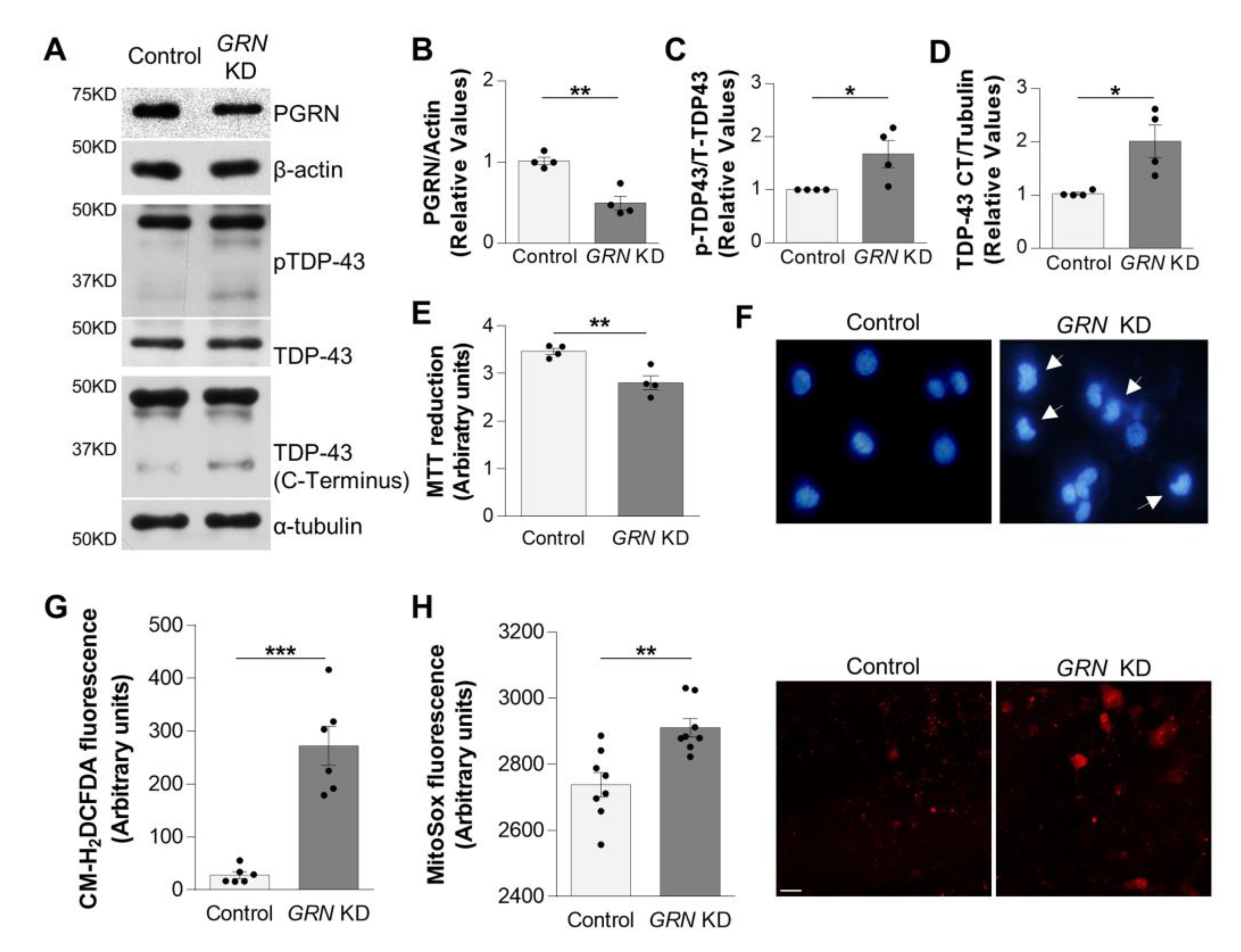
3.1. GRN KD SH-SY5Y Cells Recapitulate Pathological Characteristics of FTLD-TDP
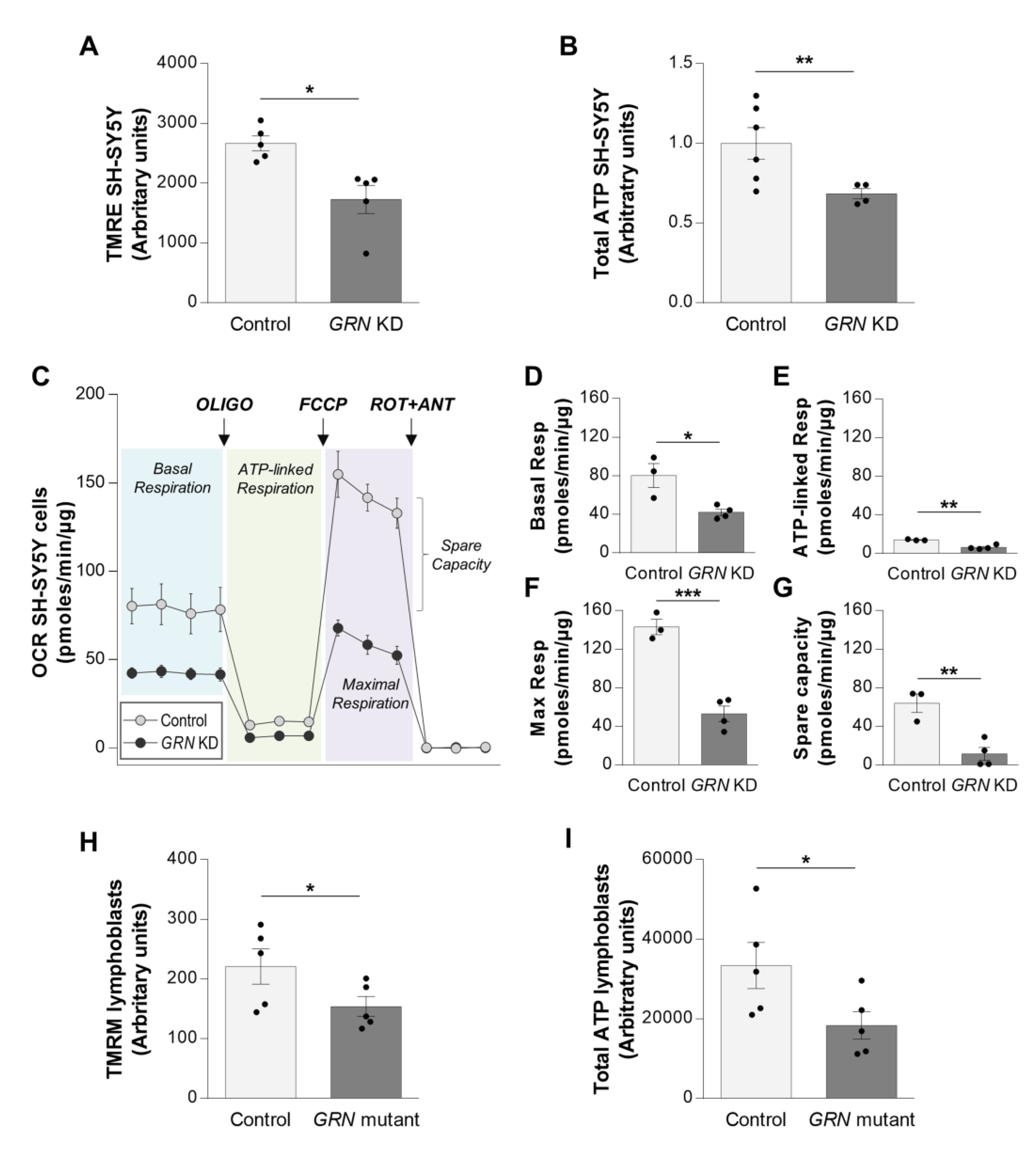
3.2. PGRN Insufficiency Impairs Mitochondrial Bioenergetics in SH-SY5Y Cells and FTLD-TDP Patient’s Lymphoblasts
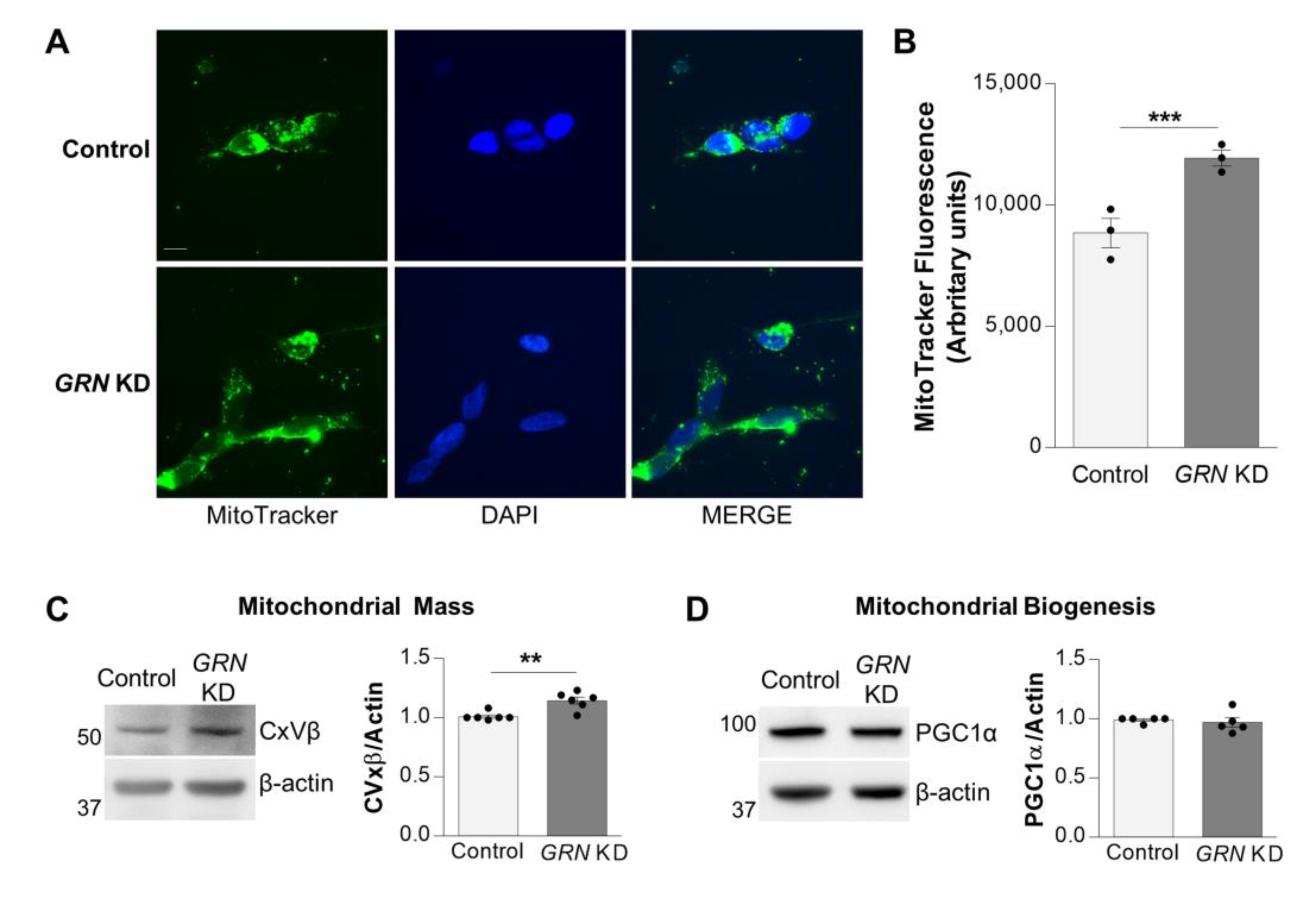
3.3. Progranulin Deficiency Increases Mitochondrial Mass
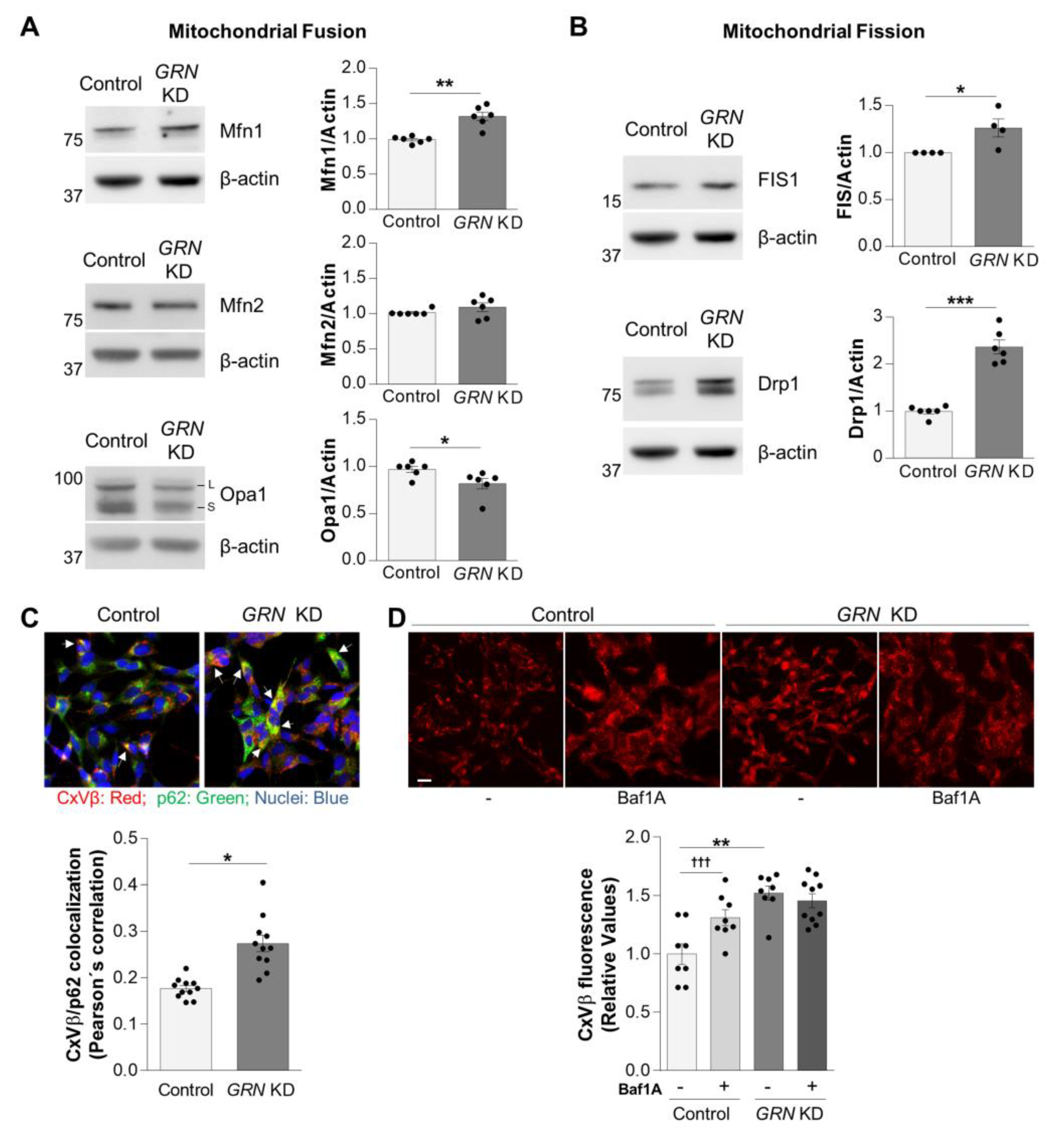
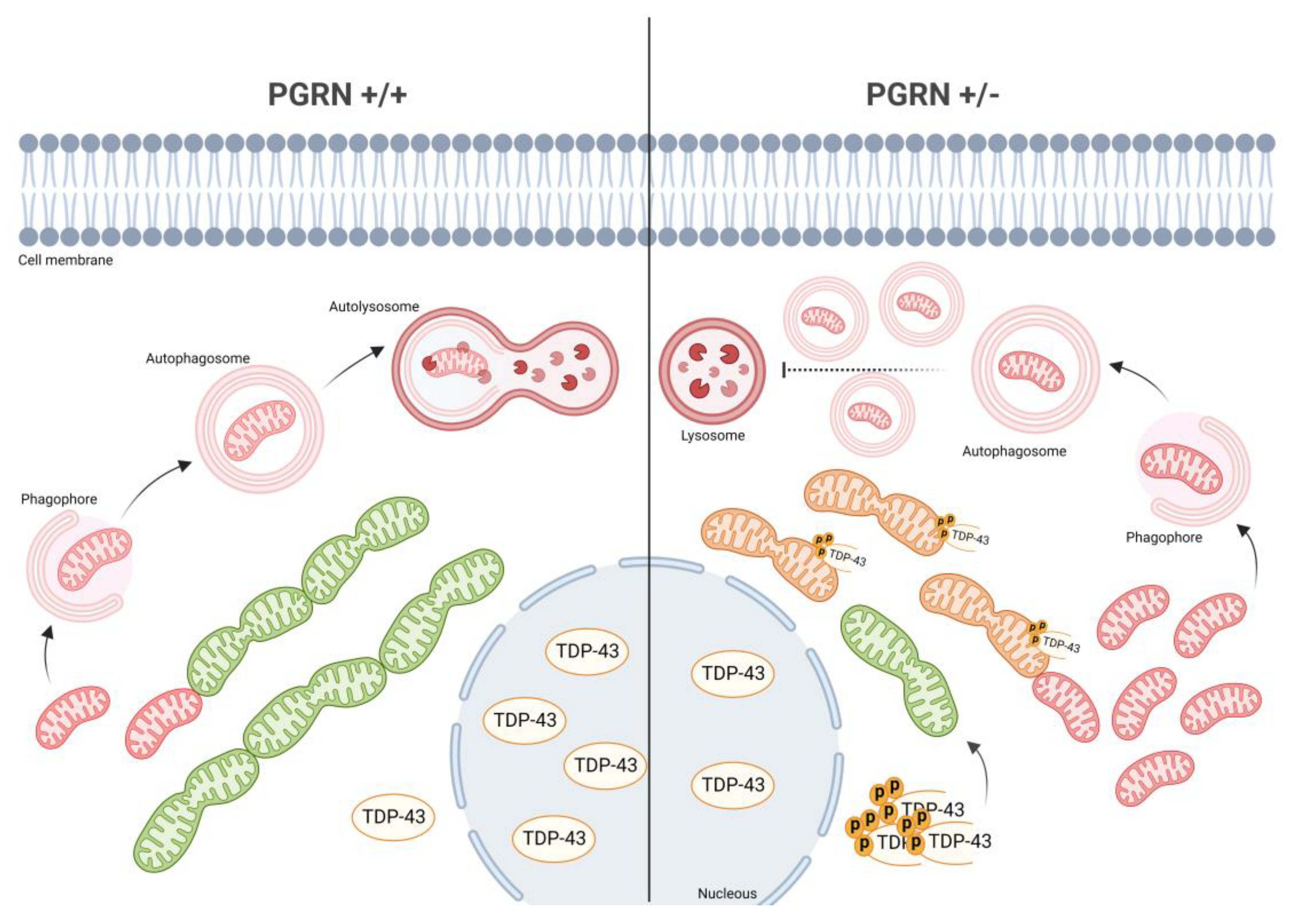
3.4. Impaired Autophagy in GRN KD Cells Blocks the Removal of Damaged Mitochondria
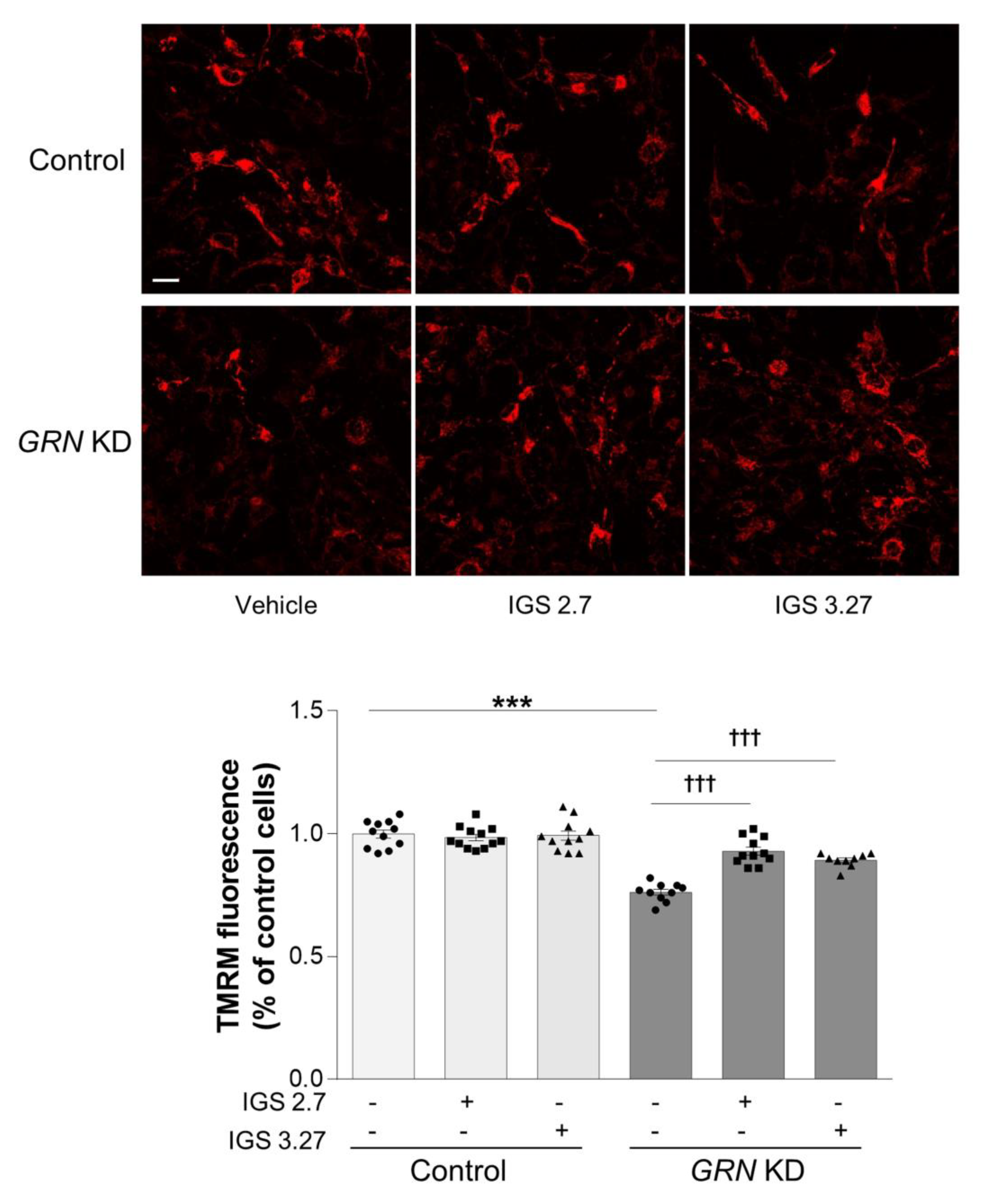
3.5. Inhibition of TDP-43 Phosphorylation Restores Mitochondrial Bioenergetics in GRN KD Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease. |
| ALS | Amyotrophic lateral sclerosis. |
| ANOVA | Analysis of variance |
| BafA1 | Bafilomycin A1 |
| BSA | Bovine serum albumin |
| BCA | Bicinchoninic acid |
| FCCP | Carbonylcyanide-p-trifluoromethoxyphenylhydrazone |
| CK-1δ | Casein kinase 1 delta |
| CxVβ | Complex V-β subunit |
| DAPI | 4,6-diamidino-2phenylindole |
| DMEM | Dulbecco’s Modified Eagle Medium |
| Drp1 | Dynamin-related protein 1 |
| EBV | Epstein Barr virus |
| FBS | Fetal bovine serum |
| FIS1 | Fission protein 1 |
| FTLD-TDP | Frontotemporal lobar degeneration with TDP-43 inclusions |
| GRN | GRN gene |
| KD | Knockdown |
| LOF | Loss-of-function |
| Opa1 | Mitochondrial dynamin-like GTPase |
| Mfn1 | Mitofusin1 |
| Mfn2 | Mitofusin2 |
| OCR | Oxygen consumption rate |
| PD | Parkinson’s disease |
| P/S. | Penicillin/streptomycin |
| PBS | Phosphate buffered saline |
| PGRN | Progranulin |
| PGC1α | Proliferator-activated receptor γ co-activator 1 α |
| ROS | Reactive Oxygen Species |
| SEM | Standard Error of the Mean |
| TDP-43 | TAR DNA-binding protein 43 |
| TMRE | Tetramethyl rhodamine ethyl ester |
| TMRM | Tetramethyl rhodamine methyl ester |
References
- Baker, M.; Mackenzie, I.R.; Pickering-Brown, S.M.; Gass, J.; Rademakers, R.; Lindholm, C.; Snowden, J.; Adamson, J.; Sadovnick, A.D.; Rollinson, S.; et al. Mutations in Progranulin Cause Tau-Negative Frontotemporal Dementia Linked to Chromosome 17. Nature 2006, 442, 916–919. [Google Scholar] [CrossRef]
- Cruts, M.; Gijselinck, I.; van der Zee, J.; Engelborghs, S.; Wils, H.; Pirici, D.; Rademakers, R.; Vandenberghe, R.; Dermaut, B.; Martin, J.-J.; et al. Null Mutations in Progranulin Cause Ubiquitin-Positive Frontotemporal Dementia Linked to Chromosome 17q21. Nature 2006, 442, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Lee, E.B.; Mackenzie, I.R. FTLD-TDP Pathological Subtypes: Clinical and Mechanistic Significance. Adv. Exp. Med. Biol. 2021, 1281, 201–217. [Google Scholar] [CrossRef]
- Natarajan, K.; Eisfeldt, J.; Hammond, M.; Laffita-Mesa, J.M.; Patra, K.; Khoshnood, B.; Öijerstedt, L.; Graff, C. Single-Cell Multimodal Analysis in a Case with Reduced Penetrance of Progranulin-Frontotemporal Dementia. Acta Neuropathol. Commun. 2021, 9, 132. [Google Scholar] [CrossRef]
- Dominguez, J.; Ng, A.; Yu, J.; Guevarra, A.C.; Daroy, M.L.; Alfon, A.; Catindig, J.-A.; Dizon, M.; Santiago, J.; Moral, M.C.D.; et al. Autosomal Dominant Frontotemporal Lobar Degeneration in a Filipino Family with Progranulin Mutation. DEM 2020, 49, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, T.; Ishii, K.; Miura, T.; Mezaki, N.; Kasuga, K.; Ikeuchi, T.; Tamaoka, A. A Novel Frameshift GRN Mutation Results in Frontotemporal Lobar Degeneration with a Distinct Clinical Phenotype in Two Siblings: Case Report and Literature Review. BMC Neurol. 2017, 17, 182. [Google Scholar] [CrossRef] [PubMed]
- Cabezas-Opazo, F.A.; Vergara-Pulgar, K.; Pérez, M.J.; Jara, C.; Osorio-Fuentealba, C.; Quintanilla, R.A. Mitochondrial Dysfunction Contributes to the Pathogenesis of Alzheimer’s Disease. Oxid. Med. Cell Longev. 2015, 2015, 509654. [Google Scholar] [CrossRef] [PubMed]
- Johri, A.; Beal, M.F. Mitochondrial Dysfunction in Neurodegenerative Diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Stichel, C.C.; Zhu, X.-R.; Bader, V.; Linnartz, B.; Schmidt, S.; Lübbert, H. Mono- and Double-Mutant Mouse Models of Parkinson’s Disease Display Severe Mitochondrial Damage. Hum. Mol. Genet. 2007, 16, 2377–2393. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, L.; Lin, W.-L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS Disease-Associated Mutant TDP-43 Impairs Mitochondrial Dynamics and Function in Motor Neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, X.; Huo, Z.; Chen, Y.; Liu, J.; Zhao, Z.; Meng, F.; Su, Q.; Bao, W.; Zhang, L.; et al. The Impact of Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis. Cells 2022, 11, 2049. [Google Scholar] [CrossRef] [PubMed]
- Dedert, C.; Mishra, V.; Aggarwal, G.; Nguyen, A.D.; Xu, F. Progranulin Preserves Autophagy Flux and Mitochondrial Function in Rat Cortical Neurons Under High Glucose Stress. Front. Cell. Neurosci. 2022, 16, 874258. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Zhou, M.; Wang, Z.; Fu, Y.; Jia, M.; Wang, X.; Liu, M.; Zhang, Y.; Sun, Y.; Lu, Y.; et al. PGRN Acts as a Novel Regulator of Mitochondrial Homeostasis by Facilitating Mitophagy and Mitochondrial Biogenesis to Prevent Podocyte Injury in Diabetic Nephropathy. Cell Death Dis. 2019, 10, 524. [Google Scholar] [CrossRef]
- de la Encarnación, A.; Alquézar, C.; Esteras, N.; Martín-Requero, Á. Progranulin Deficiency Reduces CDK4/6/PRb Activation and Survival of Human Neuroblastoma SH-SY5Y Cells. Mol. Neurobiol. 2015, 52, 1714–1725. [Google Scholar] [CrossRef] [PubMed]
- de la Encarnación, A.; Alquézar, C.; Martín-Requero, Á. Increased Wnt Signaling and Reduced Viability in a Neuronal Model of Progranulin-Deficient Frontotemporal Lobar Degeneration. Mol. Neurobiol. 2016, 53, 7107–7118. [Google Scholar] [CrossRef]
- Alquezar, C.; Esteras, N.; Alzualde, A.; Moreno, F.; Ayuso, M.S.; de Munain, A.L.; Martín-Requero, Á. Inactivation of CDK/PRb Pathway Normalizes Survival Pattern of Lymphoblasts Expressing the FTLD-Progranulin Mutation c.709-1G>A. PLoS ONE 2012, 7, e37057. [Google Scholar] [CrossRef]
- Gao, X.; Joselin, A.P.; Wang, L.; Kar, A.; Ray, P.; Bateman, A.; Goate, A.M.; Wu, J.Y. Progranulin Promotes Neurite Outgrowth and Neuronal Differentiation by Regulating GSK-3β. Protein Cell 2010, 1, 552–562. [Google Scholar] [CrossRef]
- Alquezar, C.; Salado, I.G.; De La Encarnación, A.; Pérez, D.I.; Moreno, F.; Gil, C.; De Munain, A.L.; Martínez, A.; Martín-Requero, Á. Targeting TDP-43 Phosphorylation by Casein Kinase-1δ Inhibitors: A Novel Strategy for the Treatment of Frontotemporal Dementia. Mol. Neurodegener. 2016, 11, 36. [Google Scholar] [CrossRef]
- Salado, I.G.; Redondo, M.; Bello, M.L.; Perez, C.; Liachko, N.F.; Kraemer, B.C.; Miguel, L.; Lecourtois, M.; Gil, C.; Martinez, A.; et al. Protein Kinase CK-1 Inhibitors As New Potential Drugs for Amyotrophic Lateral Sclerosis. J. Med. Chem. 2014, 57, 2755–2772. [Google Scholar] [CrossRef]
- Alquezar, C.; Esteras, N.; Bartolomé, F.; Merino, J.J.; Alzualde, A.; de Munain, A.L.; Martín-Requero, Á. Alteration in Cell Cycle-Related Proteins in Lymphoblasts from Carriers of the c.709-1G>A PGRN Mutation Associated with FTLD-TDP Dementia. Neurobiol. Aging 2012, 33, 429.e7–429.e20. [Google Scholar] [CrossRef]
- Alquezar, C.; Esteras, N.; de la Encarnación, A.; Moreno, F.; de Munain, A.L.; Martín-Requero, Á. Increasing Progranulin Levels and Blockade of the ERK1/2 Pathway: Upstream and Downstream Strategies for the Treatment of Progranulin Deficient Frontotemporal Dementia. Eur. Neuropsychopharmacol. 2015, 25, 386–403. [Google Scholar] [CrossRef] [PubMed]
- Bartolome, F.; Wu, H.-C.; Burchell, V.S.; Preza, E.; Wray, S.; Mahoney, C.J.; Fox, N.C.; Calvo, A.; Canosa, A.; Moglia, C.; et al. Pathogenic VCP Mutations Induce Mitochondrial Uncoupling and Reduced ATP Levels. Neuron 2013, 78, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Oleinik, N.; Kim, J.; Roth, B.M.; Selvam, S.P.; Gooz, M.; Johnson, R.H.; Lemasters, J.J.; Ogretmen, B. Mitochondrial Protein Import is Regulated by P17/PERMIT to Mediate Lipid Metabolism and Cellular Stress. Sci. Adv. 2019, 5, eaax1978. [Google Scholar] [CrossRef] [PubMed]
- Imamura, H.; Huynh Nhat, K.P.; Togawa, H.; Saito, K.; Iino, R.; Kato-Yamada, Y.; Nagai, T.; Noji, H. Visualization of ATP Levels Inside Single Living Cells with Fluorescence Resonance Energy Transfer-Based Genetically Encoded indicators. Proc. Natl. Acad. Sci. USA 2009, 106, 15651–15656. [Google Scholar] [CrossRef]
- de la Cueva, M.; Antequera, D.; Ordoñez-Gutierrez, L.; Wandosell, F.; Camins, A.; Carro, E.; Bartolome, F. Amyloid-β Impairs Mitochondrial Dynamics and Autophagy in Alzheimer’s Disease Experimental Models. Sci. Rep. 2022, 12, 10092. [Google Scholar] [CrossRef]
- Bartolome, F.; de la Cueva, M.; Pascual, C.; Antequera, D.; Fernandez, T.; Gil, C.; Martinez, A.; Carro, E. Amyloid β-Induced Impairments on Mitochondrial Dynamics, Hippocampal Neurogenesis, and Memory are Restored by Phosphodiesterase 7 Inhibition. Alzheimers Res. Ther. 2018, 10, 24. [Google Scholar] [CrossRef]
- Bartolome, F.; Antequera, D.; Tavares, E.; Pascual, C.; Maldonado, R.; Camins, A.; Carro, E. Obesity and Neuroinflammatory Phenotype in Mice Lacking Endothelial Megalin. J. Neuroinflamm. 2017, 14, 26. [Google Scholar] [CrossRef]
- Guo, A.; Tapia, L.; Bamji, S.X.; Cynader, M.S.; Jia, W. Progranulin Deficiency Leads to Enhanced Cell Vulnerability and TDP-43 Translocation in Primary Neuronal Cultures. Brain Res. 2010, 1366, 1–8. [Google Scholar] [CrossRef]
- Valdez, C.; Wong, Y.C.; Schwake, M.; Bu, G.; Wszolek, Z.K.; Krainc, D. Progranulin-Mediated Deficiency of Cathepsin D Results in FTD and NCL-Like Phenotypes in Neurons Derived from FTD Patients. Hum. Mol. Genet. 2017, 26, 4861–4872. [Google Scholar] [CrossRef]
- Li, Q.; Yokoshi, M.; Okada, H.; Kawahara, Y. The Cleavage Pattern of TDP-43 Determines Its Rate of Clearance and Cytotoxicity. Nat. Commun. 2015, 6, 6183. [Google Scholar] [CrossRef]
- Phan, K.; He, Y.; Pickford, R.; Bhatia, S.; Katzeff, J.S.; Hodges, J.R.; Piguet, O.; Halliday, G.M.; Kim, W.S. Uncovering Pathophysiological Changes in Frontotemporal Dementia Using Serum Lipids. Sci. Rep. 2020, 10, 3640. [Google Scholar] [CrossRef] [PubMed]
- Yousef, A.; Robinson, J.L.; Irwin, D.J.; Byrne, M.D.; Kwong, L.K.; Lee, E.B.; Xu, Y.; Xie, S.X.; Rennert, L.; Suh, E.; et al. Neuron Loss and Degeneration in the Progression of TDP-43 in Frontotemporal Lobar Degeneration. Acta. Neuropathol. Commun. 2017, 5, 68. [Google Scholar] [CrossRef] [PubMed]
- López de Munain, A.; Alzualde, A.; Gorostidi, A.; Otaegui, D.; Ruiz-Martínez, J.; Indakoetxea, B.; Ferrer, I.; Pérez-Tur, J.; Sáenz, A.; Bergareche, A.; et al. Mutations in Progranulin Gene: Clinical, Pathological, and Ribonucleic Acid Expression Findings. Biol. Psychiatry 2008, 63, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-Mediated Mitophagy is Dependent on VDAC1 and P62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Suzuki, G.; Matsuwaki, T.; Hosokawa, M.; Serrano, G.; Beach, T.G.; Yamanouchi, K.; Hasegawa, M.; Nishihara, M. Progranulin Regulates Lysosomal Function and Biogenesis through Acidification of Lysosomes. Hum. Mol. Genet. 2017, 26, 969–988. [Google Scholar] [CrossRef]
- Chang, M.C.; Srinivasan, K.; Friedman, B.A.; Suto, E.; Modrusan, Z.; Lee, W.P.; Kaminker, J.S.; Hansen, D.V.; Sheng, M. Progranulin Deficiency Causes Impairment of Autophagy and TDP-43 Accumulation. J. Exp. Med. 2017, 214, 2611–2628. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Kametani, F.; Nonaka, T.; Suzuki, T.; Arai, T.; Dohmae, N.; Akiyama, H.; Hasegawa, M. Identification of Casein Kinase-1 Phosphorylation Sites on TDP-43. Biochem. Biophys. Res. Commun. 2009, 382, 405–409. [Google Scholar] [CrossRef]
- Gao, J.; Wang, L.; Yan, T.; Perry, G.; Wang, X. TDP-43 Proteinopathy and Mitochondrial Abnormalities in Neurodegeneration. Mol. Cell. Neurosci. 2019, 100, 103396. [Google Scholar] [CrossRef]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; da Rocha, E.L.; et al. The Inhibition of TDP-43 Mitochondrial Localization Blocks Its Neuronal Toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef]
- Wang, P.; Deng, J.; Dong, J.; Liu, J.; Bigio, E.H.; Mesulam, M.; Wang, T.; Sun, L.; Wang, L.; Lee, A.Y.-L.; et al. TDP-43 Induces Mitochondrial Damage and Activates the Mitochondrial Unfolded Protein Response. PLoS Genet. 2019, 15, e1007947. [Google Scholar] [CrossRef]
- Rabinovici, G.D.; Miller, B.L. Frontotemporal Lobar Degeneration: Epidemiology, Pathophysiology, Diagnosis and Management. CNS Drugs 2010, 24, 375. [Google Scholar] [CrossRef] [PubMed]
- Bartolome, F.; Esteras, N.; Martin-Requero, A.; Boutoleau-Bretonniere, C.; Vercelletto, M.; Gabelle, A.; Le Ber, I.; Honda, T.; Dinkova-Kostova, A.T.; Hardy, J.; et al. Pathogenic P62/SQSTM1 Mutations Impair Energy Metabolism through Limitation of Mitochondrial Substrates. Sci. Rep. 2017, 7, 1666. [Google Scholar] [CrossRef] [PubMed]
- Bobba, A.; Amadoro, G.; Valenti, D.; Corsetti, V.; Lassandro, R.; Atlante, A. Mitochondrial Respiratory Chain Complexes I and IV are Impaired by β-Amyloid via Direct Interaction and through Complex I-Dependent ROS Production, respectively. Mitochondrion 2013, 13, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Downward, J.; Latchman, D.S.; Tabrizi, S.J.; Wood, N.W.; et al. PINK1-Associated Parkinson’s Disease Is Caused by Neuronal Vulnerability to Calcium-Induced Cell Death. Mol. Cell 2009, 33, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Su, J.H.; Nichol, K.E.; Sitch, T.; Sheu, P.; Chubb, C.; Miller, B.L.; Tomaselli, K.J.; Kim, R.C.; Cotman, C.W. DNA Damage and Activated Caspase-3 Expression in Neurons and Astrocytes: Evidence for Apoptosis in Frontotemporal Dementia. Exp. Neurol. 2000, 163, 9–19. [Google Scholar] [CrossRef]
- Morán, M.; Rivera, H.; Sánchez-Aragó, M.; Blázquez, A.; Merinero, B.; Ugalde, C.; Arenas, J.; Cuezva, J.M.; Martín, M.A. Mitochondrial Bioenergetics and Dynamics Interplay in Complex I-Deficient Fibroblasts. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2010, 1802, 443–453. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Angelova, P.R. Cellular Mechanisms of Complex I-Associated Pathology. Biochem. Soc. Trans. 2019, 47, 1963–1969. [Google Scholar] [CrossRef]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 1963–1969. [Google Scholar] [CrossRef]
- Elmore, S.P.; Qian, T.; Grissom, S.F.; Lemasters, J.J. The Mitochondrial Permeability Transition Initiates Autophagy in Rat Hepatocytes. FASEB J. 2001, 15, 2286–2287. [Google Scholar] [CrossRef]
- Priault, M.; Salin, B.; Schaeffer, J.; Vallette, F.M.; di Rago, J.-P.; Martinou, J.-C. Impairing the Bioenergetic Status and the Biogenesis of Mitochondria Triggers Mitophagy in Yeast. Cell Death Differ. 2005, 12, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and Selective Fusion Govern Mitochondrial Segregation and Elimination by Autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-F.; Gendron, T.F.; Zhang, Y.-J.; Lin, W.-L.; D’Alton, S.; Sheng, H.; Casey, M.C.; Tong, J.; Knight, J.; Yu, X.; et al. Wild-Type Human TDP-43 Expression Causes TDP-43 Phosphorylation, Mitochondrial Aggregation, Motor Deficits, and Early Mortality in Transgenic Mice. J. Neurosci. 2010, 30, 10851–10859. [Google Scholar] [CrossRef] [PubMed]
- Alquézar, C.; Esteras, N.; de la Encarnación, A.; Alzualde, A.; Moreno, F.; López de Munain, A.; Martín-Requero, Á. PGRN Haploinsufficiency Increased Wnt5a Signaling in Peripheral Cells from Frontotemporal Lobar Degeneration-Progranulin Mutation Carriers. Neurobiol. Aging 2014, 35, 886–898. [Google Scholar] [CrossRef] [PubMed]
- Alquézar, C.; de la Encarnación, A.; Moreno, F.; López de Munain, A.; Martín-Requero, Á. Progranulin Deficiency Induces Overactivation of WNT5A Expression via TNF-α/NF-ΚB Pathway in Peripheral Cells from Frontotemporal Dementia-Linked Granulin Mutation Carriers. J. Psychiatry Neurosci. 2016, 41, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Ansari, M.; Angelidis, I.; Theis, F.; Schiller, H.; Königshoff, M.; Lehmann, M. Chronic WNT/ß-Catenin Signaling Impairs Mitochondrial Function in Lung Epithelial Cells. Eur. Respir. J. 2021, 58, OA2699. [Google Scholar] [CrossRef]
- Silva-Alvarez, C.; Arrazola, M.; Godoy, J.; Ordenes, D.; Inestrosa, N. Canonical Wnt Signaling Protects Hippocampal Neurons from Aβ Oligomers: Role of Non-Canonical Wnt-5a/Ca2+ in Mitochondrial Dynamics. Front. Cell. Neurosci. 2013, 7, 97. [Google Scholar] [CrossRef]
- Yoon, J.C.; Ng, A.; Kim, B.H.; Bianco, A.; Xavier, R.J.; Elledge, S.J. Wnt Signaling Regulates Mitochondrial Physiology and Insulin Sensitivity. Genes. Dev. 2010, 24, 1507–1518. [Google Scholar] [CrossRef]
- Costa, R.; Peruzzo, R.; Bachmann, M.; Montà, G.D.; Vicario, M.; Santinon, G.; Mattarei, A.; Moro, E.; Quintana-Cabrera, R.; Scorrano, L.; et al. Impaired Mitochondrial ATP Production Downregulates Wnt Signaling via ER Stress Induction. Cell Rep. 2019, 28, 1949–1960.e6. [Google Scholar] [CrossRef]
- Delgado-Deida, Y.; Alula, K.M.; Theiss, A.L. The Influence of Mitochondrial-Directed Regulation of Wnt Signaling on Tumorigenesis. Gastroenterol. Rep. 2020, 8, 215–223. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject ID | Sex | Age at Blood Collection | Age Onset | Clinical Presentation | c.709-1G > A Mutation Carrier |
|---|---|---|---|---|---|
| C8 | M | 46 | - | Healthy | No |
| C13 | M | 70 | - | Healthy | No |
| C18 | F | 58 | - | Healthy | No |
| C20 | M | 69 | - | Healthy | No |
| C22 | F | 64 | - | Healthy | No |
| P5 | F | 70 | 64 | FTD-bv; CBS | Yes |
| P10 | F | 70 | 65 | PNFA; CBS | Yes |
| P25 | F | 65 | 63 | FTD-bv; CBS | Yes |
| P30 | F | 54 | 54 | FTD-bv | Yes |
| P31 | F | 70 | 63 | FTD-bv; CBS | Yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Periñán, G.; de la Encarnación, A.; Moreno, F.; López de Munain, A.; Martínez, A.; Martín-Requero, Á.; Alquézar, C.; Bartolomé, F. Progranulin Deficiency Induces Mitochondrial Dysfunction in Frontotemporal Lobar Degeneration with TDP-43 Inclusions. Antioxidants 2023, 12, 581. https://doi.org/10.3390/antiox12030581
Rodríguez-Periñán G, de la Encarnación A, Moreno F, López de Munain A, Martínez A, Martín-Requero Á, Alquézar C, Bartolomé F. Progranulin Deficiency Induces Mitochondrial Dysfunction in Frontotemporal Lobar Degeneration with TDP-43 Inclusions. Antioxidants. 2023; 12(3):581. https://doi.org/10.3390/antiox12030581
Chicago/Turabian StyleRodríguez-Periñán, Guiomar, Ana de la Encarnación, Fermín Moreno, Adolfo López de Munain, Ana Martínez, Ángeles Martín-Requero, Carolina Alquézar, and Fernando Bartolomé. 2023. "Progranulin Deficiency Induces Mitochondrial Dysfunction in Frontotemporal Lobar Degeneration with TDP-43 Inclusions" Antioxidants 12, no. 3: 581. https://doi.org/10.3390/antiox12030581
APA StyleRodríguez-Periñán, G., de la Encarnación, A., Moreno, F., López de Munain, A., Martínez, A., Martín-Requero, Á., Alquézar, C., & Bartolomé, F. (2023). Progranulin Deficiency Induces Mitochondrial Dysfunction in Frontotemporal Lobar Degeneration with TDP-43 Inclusions. Antioxidants, 12(3), 581. https://doi.org/10.3390/antiox12030581