
Endogenous and Exogenous Antioxidants in Skeletal Muscle Fatigue Development during Exercise


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Peripheral Muscle Fatigue
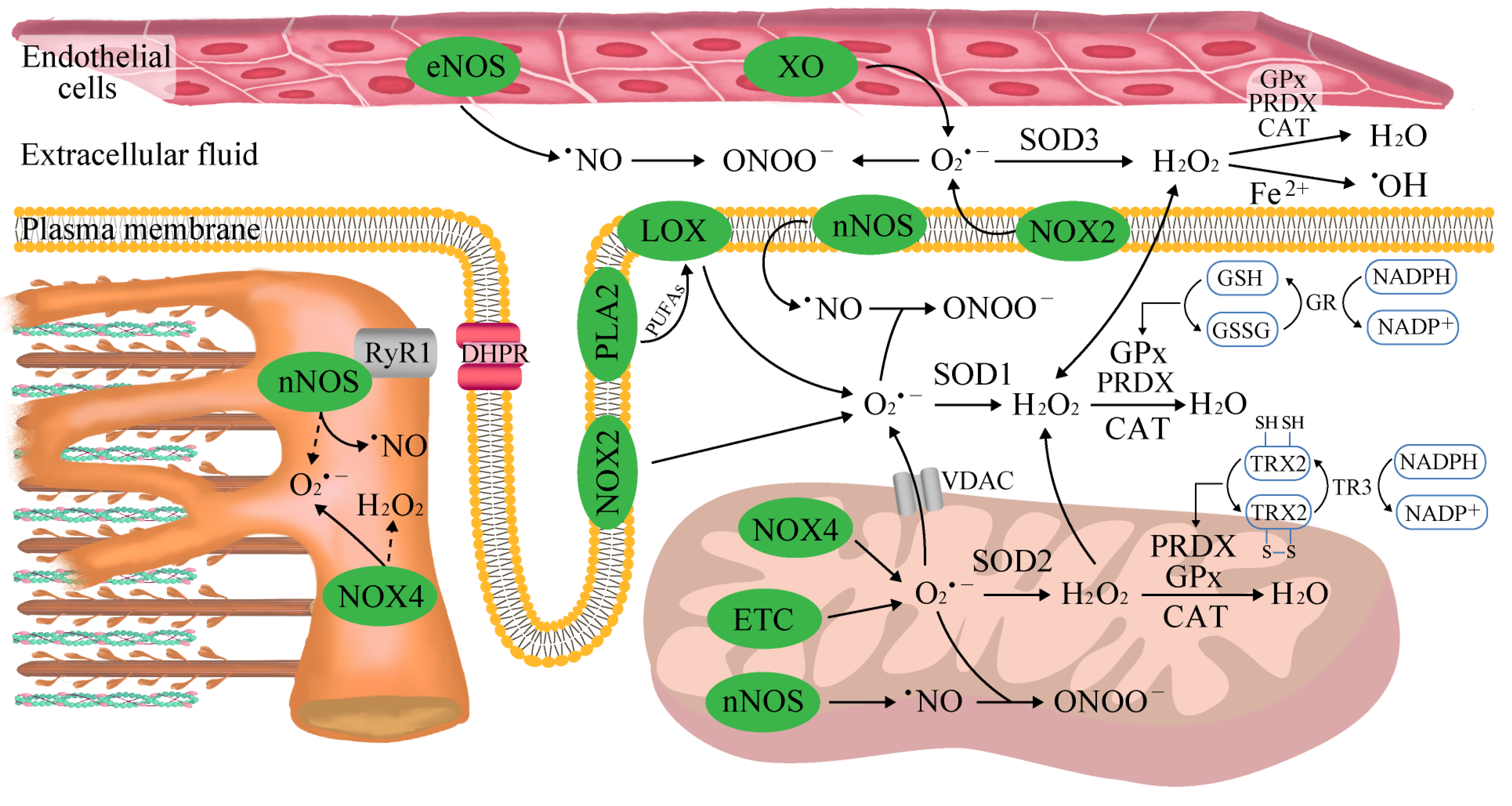
2.1. The Sources of Reactive Oxygen and Nitrogen Species (ROS/RNS) in Skeletal Muscle
2.2. Aerobic Exercise
2.3. Anaerobic Exercise
2.4. Endogenous Mechanisms of Reactive Oxygen and Nitrogen Species Detoxification
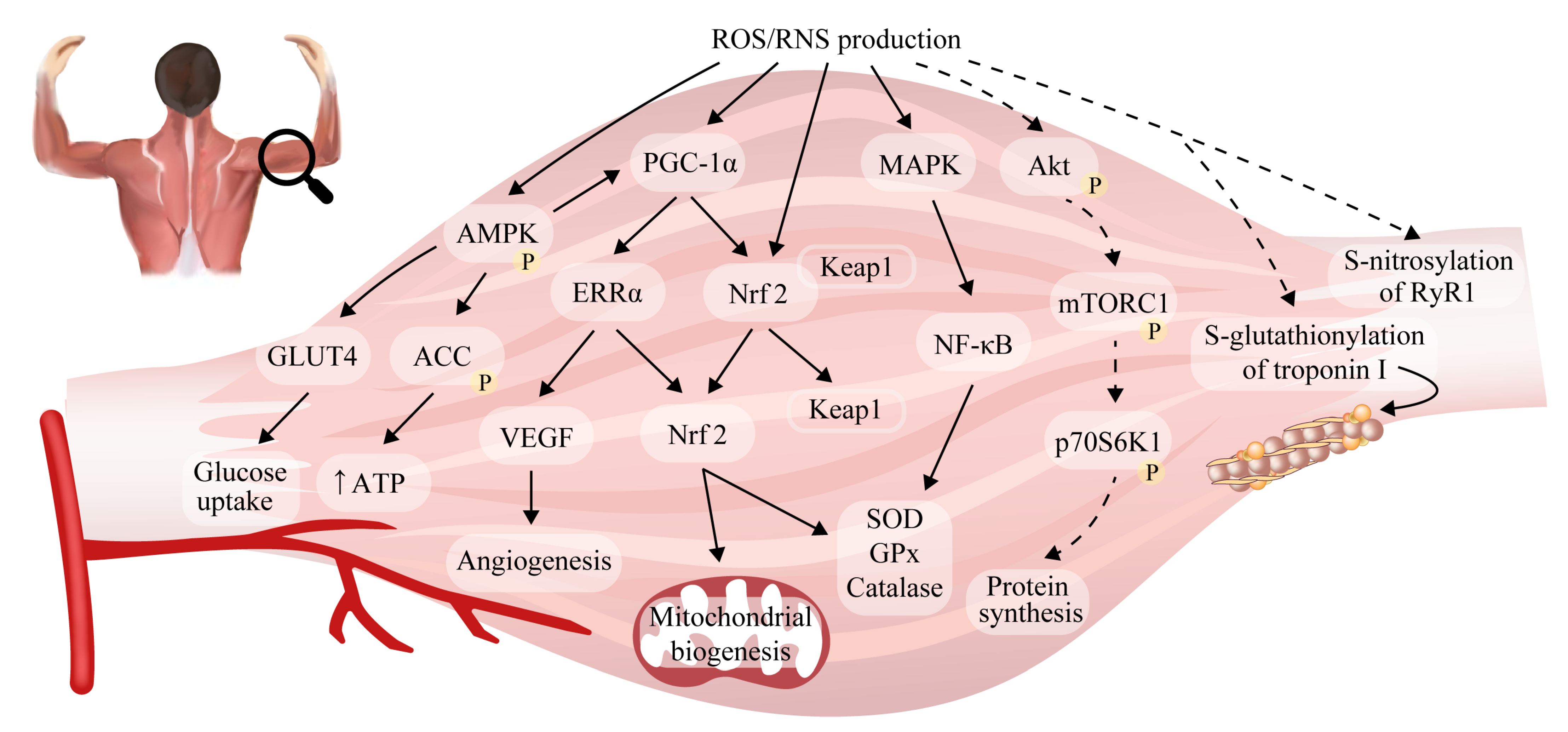
2.5. Reactive Oxygen/Nitrogen Species and Adaptations to Exercise in Skeletal Muscle
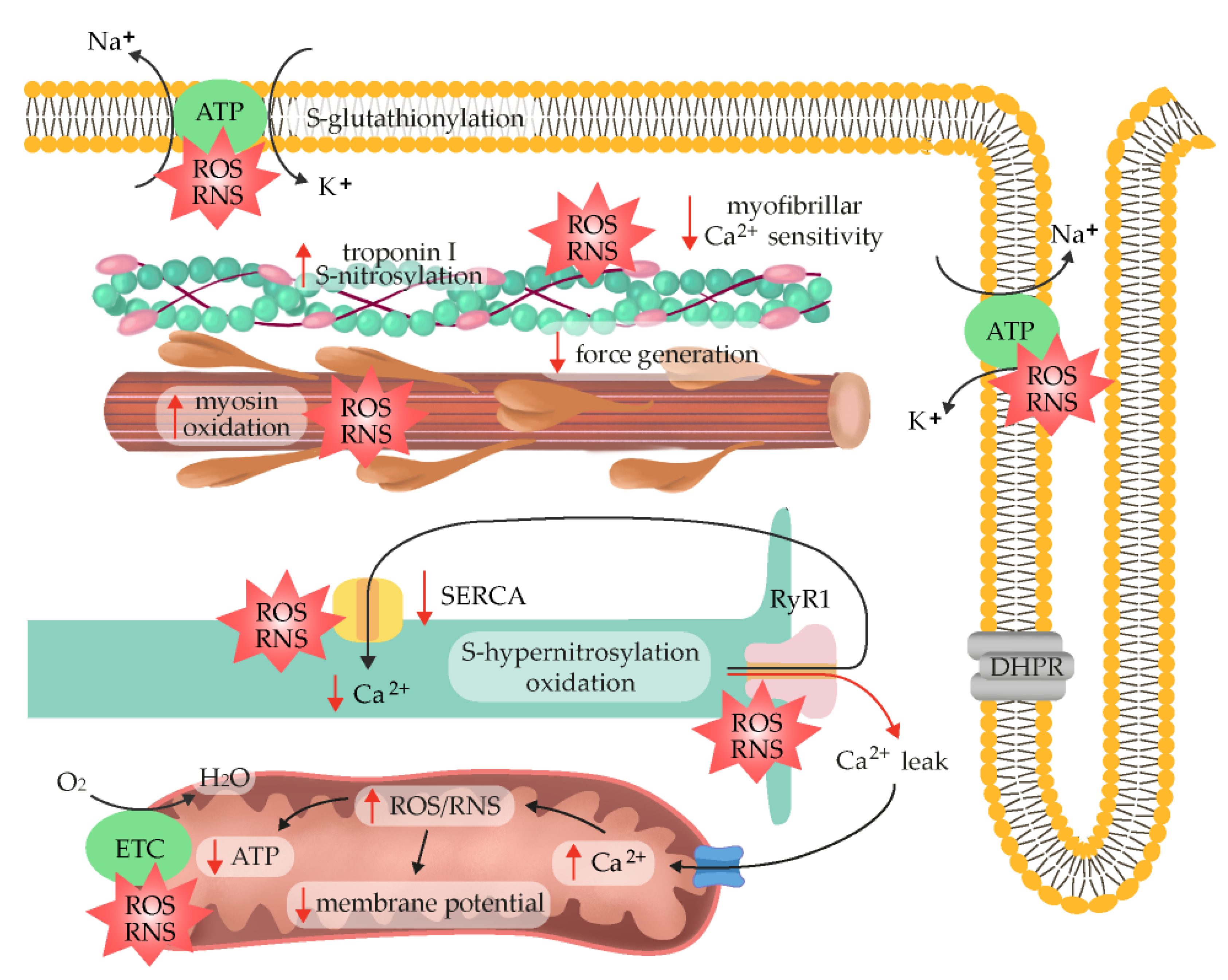
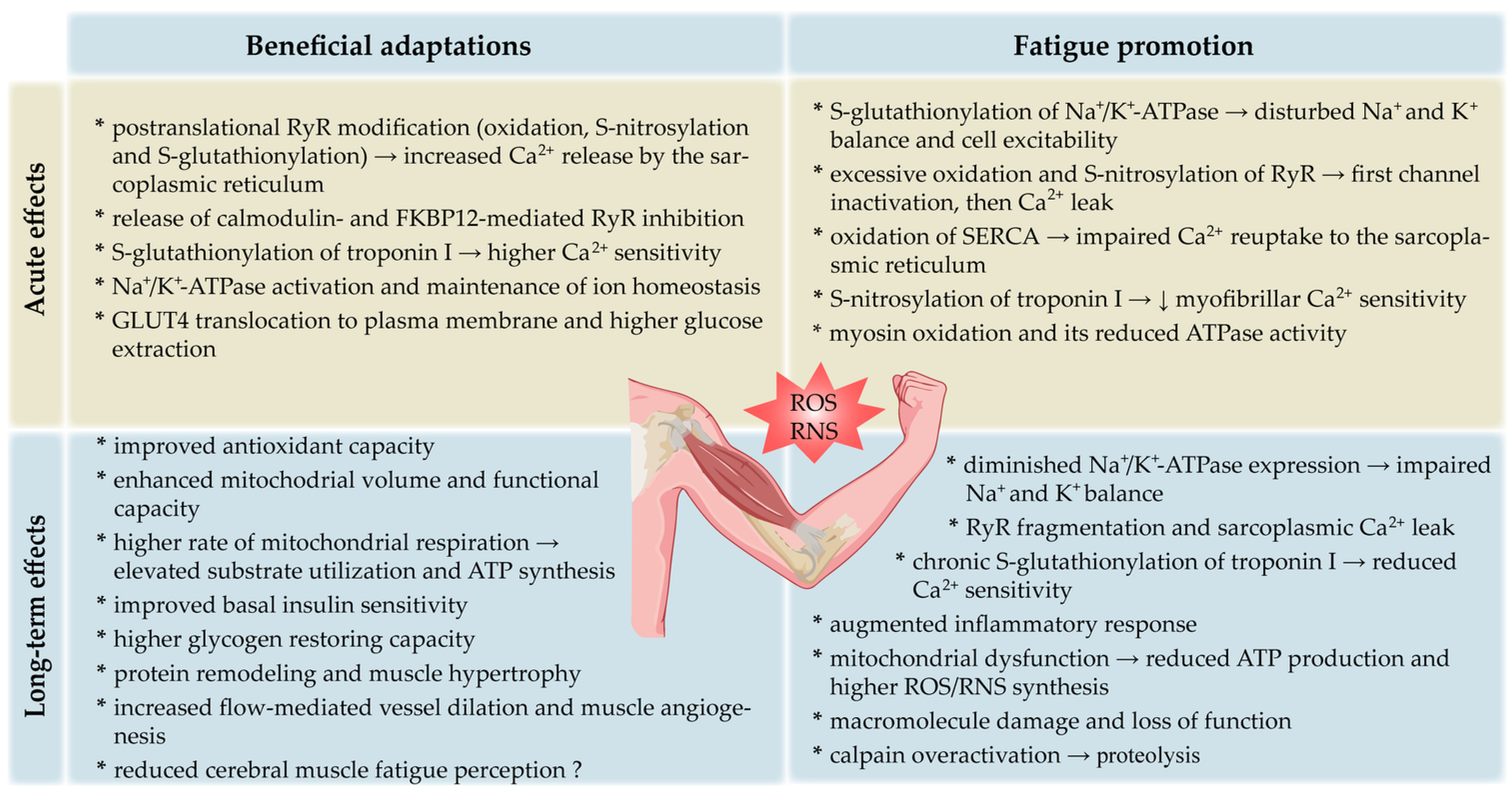
2.6. ROS/RNS as Fatigue Mediators
2.7. Exogenous Antioxidants
 [198] and GPx ↓, SOD were noticed, while exercise performance measures remained unchanged [199]. However, the use of higher doses of 100 and 300 mg/d suggested that CoQ10 can improve subjective fatigue sensation [8]. CoQ10 also exhibited therapeutic effects against fatigue in statin-treated myopathy and fibromyalgia patients [200]. A recent meta-analysis of the combined results of 13 trials revealed a statistically significant reduction with regard to fatigue symptomology after CoQ10 administration [201]. However, moderate-to-high heterogeneity was observed, especially in relation to the administered dose. A greater exercise capacity was also observed in individuals with mitochondrial disease, while healthy subjects exhibited a greater increase in anaerobic performance in a group without additional CoQ10 supplementation [202]. Therefore, there is no convincing evidence to support the anti-fatiguing properties of CoQ10.
[198] and GPx ↓, SOD were noticed, while exercise performance measures remained unchanged [199]. However, the use of higher doses of 100 and 300 mg/d suggested that CoQ10 can improve subjective fatigue sensation [8]. CoQ10 also exhibited therapeutic effects against fatigue in statin-treated myopathy and fibromyalgia patients [200]. A recent meta-analysis of the combined results of 13 trials revealed a statistically significant reduction with regard to fatigue symptomology after CoQ10 administration [201]. However, moderate-to-high heterogeneity was observed, especially in relation to the administered dose. A greater exercise capacity was also observed in individuals with mitochondrial disease, while healthy subjects exhibited a greater increase in anaerobic performance in a group without additional CoQ10 supplementation [202]. Therefore, there is no convincing evidence to support the anti-fatiguing properties of CoQ10.3. Central Fatigue
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Knez, W.L.; Coombes, J.S.; Jenkins, D.G. Ultra-Endurance Exercise and Oxidative Damage Implications for Cardiovascular Health. Sport. Med. 2006, 36, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, G.; Manning, P.; Newton, J.L. Understanding Muscle Dysfunction in Chronic Fatigue Syndrome. J. Aging Res. 2016, 2016, 2497348. [Google Scholar] [CrossRef] [PubMed]
- Lamb, G.D.; Posterino, G.S. Effects of Oxidation and Reduction on Contractile Function in Skeletal Muscle Fibres of the Rat. J. Physiol. 2003, 546, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Dutka, T.L.; Mollica, J.P.; Lamb, G.D. Differential Effects of Peroxynitrite on Contractile Protein Properties in Fast- and Slow-Twitch Skeletal Muscle Fibers of Rat. J. Appl. Physiol. 2011, 110, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Moopanar, T.R.; Allen, D.G. Reactive Oxygen Species Reduce Myofibrillar Ca2+ Sensitivity in Fatiguing Mouse Skeletal Muscle at 37 °C. J. Physiol. 2005, 564, 189–199. [Google Scholar] [CrossRef]
- Slattery, K.M.; Dascombe, B.; Wallace, L.K.; Bentley, D.J.; Coutts, A.J. Effect of N-Acetylcysteine on Cycling Performance after Intensified Training. Med. Sci. Sport. Exerc. 2014, 46, 1114–1123. [Google Scholar] [CrossRef]
- Nicol, L.M.; Rowlands, D.S.; Fazakerly, R.; Kellett, J. Curcumin Supplementation Likely Attenuates Delayed Onset Muscle Soreness (DOMS). Eur. J. Appl. Physiol. 2015, 115, 1769–1777. [Google Scholar] [CrossRef]
- Mizuno, K.; Tanaka, M.; Nozaki, S.; Mizuma, H.; Ataka, S.; Tahara, T.; Sugino, T.; Shirai, T.; Kajimoto, Y.; Kuratsune, H.; et al. Antifatigue Effects of Coenzyme Q10 during Physical Fatigue. Nutrition 2008, 24, 293–299. [Google Scholar] [CrossRef]
- Wyckelsma, V.L.; Venckunas, T.; Brazaitis, M.; Gastaldello, S.; Snieckus, A.; Eimantas, N.; Baranauskiene, N.; Subocius, A.; Skurvydas, A.; Pääsuke, M.; et al. Vitamin c and e Treatment Blunts Sprint Interval Training–Induced Changes in Inflammatory Mediator-, Calcium-, and Mitochondria-Related Signaling in Recreationally Active Elderly Humans. Antioxidants 2020, 9, 879. [Google Scholar] [CrossRef]
- Gomez-Cabrera, M.C.; Domenech, E.; Romagnoli, M.; Arduini, A.; Borras, C.; Pallardo, F.V.; Sastre, J.; Viña, J. Oral Administration of Vitamin C Decreases Muscle Mitochondrial Biogenesis and Hampers Training-Induced Adaptations in Endurance Performance. Am. J. Clin. Nutr. 2008, 87, 142–149. [Google Scholar] [CrossRef]
- Braakhuis, A.J.; Hopkins, W.G.; Lowe, T.E. Effects of Dietary Antioxidants on Training and Performance in Female Runners. Eur. J. Sport Sci. 2014, 14, 160–168. [Google Scholar] [CrossRef]
- Suzuki, K.; Tominaga, T.; Ruhee, R.T.; Ma, S. Characterization and Modulation of Systemic Inflammatory Response to Exhaustive Exercise in Relation to Oxidative Stress. Antioxidants 2020, 9, 401. [Google Scholar] [CrossRef]
- Sahlin, K.; Ren, J.M. Relationship of Contraction Capacity to Metabolic Changes during Recovery from a Fatiguing Contraction. J. Appl. Physiol. 1989, 67, 648–654. [Google Scholar] [CrossRef]
- Woodward, M.; Debold, E.P. Acidosis and Phosphate Directly Reduce Myosin’s Force-Generating Capacity through Distinct Molecular Mechanisms. Front. Physiol. 2018, 9, 862. [Google Scholar] [CrossRef]
- Andrade, F.H.; Reid, M.B.; Westerblad, H. Contractile Response to Low Peroxide Concentrations: Myofibrillar Calcium Sensitivity as a Likely Target for Redox-modulation of Skeletal Muscle Function. FASEB J. 2001, 15, 309–311. [Google Scholar] [CrossRef]
- Christiansen, D.; Eibye, K.; Hostrup, M.; Bangsbo, J. Training with Blood Flow Restriction Increases Femoral Artery Diameter and Thigh Oxygen Delivery during Knee-Extensor Exercise in Recreationally Trained Men. J. Physiol. 2020, 598, 2337–2353. [Google Scholar] [CrossRef]
- Christiansen, D. Molecular Stressors Underlying Exercise Training-Induced Improvements in K + Regulation during Exercise and Na +, K + -ATPase Adaptation in Human Skeletal Muscle. Acta Physiol. 2019, 225, e13196. [Google Scholar] [CrossRef]
- Centner, C.; Zdzieblik, D.; Dressler, P.; Fink, B.; Gollhofer, A.; König, D. Acute Effects of Blood Flow Restriction on Exercise-Induced Free Radical Production in Young and Healthy Subjects. Free Radic. Res. 2018, 52, 446–454. [Google Scholar] [CrossRef]
- Zuo, L.; Clanton, T.L. Reactive Oxygen Species Formation in the Transition to Hypoxia in Skeletal Muscle. Am. J. Physiol.-Cell Physiol. 2005, 289, 207–216. [Google Scholar] [CrossRef]
- Korkmaz, E.; Dönmez, G.; Uzuner, K.; Babayeva, N.; Torgutalp, Ş.Ş.; Özçakar, L. Effects of Blood Flow Restriction Training on Muscle Strength and Architecture. J. Strength Cond. Res. 2022, 36, 1396–1403. [Google Scholar] [CrossRef]
- Anderson, E.J.; Neufer, P.D. Type II Skeletal Myofibers Possess Unique Properties That Potentiate Mitochondrial H2O2 Generation. Am. J. Physiol.-Cell Physiol. 2006, 290, C844–C851. [Google Scholar] [CrossRef] [PubMed]
- Gejl, K.D.; Hvid, L.G.; Andersson, E.P.; Jensen, R.; Holmberg, H.C.; Ørtenblad, N. Contractile Properties of MHC I and II Fibers From Highly Trained Arm and Leg Muscles of Cross-Country Skiers. Front. Physiol. 2021, 12, 855. [Google Scholar] [CrossRef]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free Radicals: Properties, Sources, Targets, and Their Implication in Various Diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J.A.; Quintanilha, A.T.; Brooks, G.A.; Packer, L. Free Radicals and Tissue Damage Produced by Exercise. Biochem. Biophys. Res. Commun. 1982, 107, 1198–1205. [Google Scholar] [CrossRef]
- Dillard, C.J.; Litov, R.E.; Savin, W.M.; Dumelin, E.E.; Tappel, A.L. Effects of Exercise, Vitamin E, and Ozone on Pulmonary Function and Lipid Peroxidation. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1978, 45, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.A.; Lawrenson, L.; McEneny, J.; Young, I.S.; James, P.E.; Jackson, S.K.; Henry, R.R.; Mathieu-Costello, O.; McCord, J.M.; Richardson, R.S. Electron Paramagnetic Spectroscopic Evidence of Exercise-Induced Free Radical Accumulation in Human Skeletal Muscle. Free Radic. Res. 2007, 41, 182–190. [Google Scholar] [CrossRef]
- Powers, S.K.; Deminice, R.; Ozdemir, M.; Yoshihara, T.; Bomkamp, M.P.; Hyatt, H. Exercise-Induced Oxidative Stress: Friend or Foe? J. Sport Health Sci. 2020, 9, 415–425. [Google Scholar] [CrossRef]
- Fogarty, M.C.; Hughes, C.M.; Burke, G.; Brown, J.C.; Trinick, T.R.; Duly, E.; Bailey, D.M.; Davison, G.W. Exercise-Induced Lipid Peroxidation: Implications for Deoxyribonucleic Acid Damage and Systemic Free Radical Generation. Environ. Mol. Mutagen. 2011, 52, 35–42. [Google Scholar] [CrossRef]
- Silva, L.A.; Tromm, C.B.; Doyenart, R.; Thirupathi, A.; Silveira, P.C.L.; Pinho, R.A. Effects of Different Frequencies of Physical Training on Electron Transport Chain and Oxidative Damage in Healthy Mice. Motriz. Rev. Educ. Fis. 2018, 24, 101804. [Google Scholar] [CrossRef]
- McNeil, C.J.; Allen, M.D.; Olympico, E.; Shoemaker, J.K.; Rice, C.L. Blood Flow and Muscle Oxygenation during Low, Moderate, and Maximal Sustained Isometric Contractions. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2015, 309, R475–R481. [Google Scholar] [CrossRef]
- Christiansen, D.; Eibye, K.H.; Hostrup, M.; Bangsbo, J. Blood Flow-Restricted Training Enhances Thigh Glucose Uptake during Exercise and Muscle Antioxidant Function in Humans. Metabolism 2019, 98, 1–15. [Google Scholar] [CrossRef]
- Van Der Poel, C.; Edwards, J.N.; MacDonald, W.A.; Stephenson, D.G. Effect of Temperature-Induced Reactive Oxygen Species Production on Excitation-Contraction Coupling in Mammalian Skeletal Muscle. Clin. Exp. Pharmacol. Physiol. 2008, 35, 1482–1487. [Google Scholar] [CrossRef]
- Supruniuk, E.; Maciejczyk, M.; Zalewska, A.; Górski, J.; Chabowski, A. Blood Profile of Cytokines, Chemokines, Growth Factors, and Redox Biomarkers in Response to Different Protocols of Treadmill Running in Rats. Int. J. Mol. Sci. 2020, 21, 8071. [Google Scholar] [CrossRef]
- Bouviere, J.; Fortunato, R.S.; Dupuy, C.; Werneck-De-castro, J.P.; Carvalho, D.P.; Louzada, R.A. Exercise-Stimulated Ros Sensitive Signaling Pathways in Skeletal Muscle. Antioxidants 2021, 10, 537. [Google Scholar] [CrossRef]
- Menazza, S.; Blaauw, B.; Tiepolo, T.; Toniolo, L.; Braghetta, P.; Spolaore, B.; Reggiani, C.; di Lisa, F.; Bonaldo, P.; Canton, M. Oxidative Stress by Monoamine Oxidases Is Causally Involved in Myofiber Damage in Muscular Dystrophy. Hum. Mol. Genet. 2010, 19, 4207–4215. [Google Scholar] [CrossRef]
- Picard, M.; Hepple, R.T.; Burelle, Y. Mitochondrial Functional Specialization in Glycolytic and Oxidative Muscle Fibers: Tailoring the Organelle for Optimal Function. Am. J. Physiol.-Cell Physiol. 2012, 302, 629–641. [Google Scholar] [CrossRef]
- Goncalves, R.L.S.; Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Brand, M.D. Sites of Superoxide and Hydrogen Peroxide Production by Muscle Mitochondria Assessed Ex Vivo under Conditions Mimicking Rest and Exercise. J. Biol. Chem. 2015, 290, 209–227. [Google Scholar] [CrossRef]
- Granatiero, V.; Gherardi, G.; Vianello, M.; Salerno, E.; Zecchini, E.; Toniolo, L.; Pallafacchina, G.; Murgia, M.; Blaauw, B.; Rizzuto, R.; et al. Role of P66shc in Skeletal Muscle Function. Sci. Rep. 2017, 7, 6283. [Google Scholar] [CrossRef]
- Alves, J.O.; Pereira, L.M.; Monteiro, I.C.C.D.R.; Dos Santos, L.H.P.; Ferraz, A.S.M.; Loureiro, A.C.C.; Lima, C.C.; Leal-Cardoso, J.H.; Carvalho, D.P.; Fortunato, R.S.; et al. Strenuous Acute Exercise Induces Slow and Fast Twitch-Dependent NADPH Oxidase Expression in Rat Skeletal Muscle. Antioxidants 2020, 9, 57. [Google Scholar] [CrossRef]
- Henríquez-Olguin, C.; Knudsen, J.R.; Raun, S.H.; Li, Z.; Dalbram, E.; Treebak, J.T.; Sylow, L.; Holmdahl, R.; Richter, E.A.; Jaimovich, E.; et al. Cytosolic ROS Production by NADPH Oxidase 2 Regulates Muscle Glucose Uptake during Exercise. Nat. Commun. 2019, 10, 4623. [Google Scholar] [CrossRef]
- Sakellariou, G.K.; Vasilaki, A.; Palomero, J.; Kayani, A.; Zibrik, L.; McArdle, A.; Jackson, M.J. Studies of Mitochondrial and Nonmitochondrial Sources Implicate Nicotinamide Adenine Dinucleotide Phosphate Oxidase(s) in the Increased Skeletal Muscle Superoxide Generation That Occurs during Contractile Activity. Antioxid. Redox Signal. 2013, 18, 603–621. [Google Scholar] [CrossRef] [PubMed]
- Javeshghani, D.; Magder, S.A.; Barreiro, E.; Quinn, M.T.; Hussain, S.N.A. Molecular Characterization of a Superoxide-Generating NAD(P)H Oxidase in the Ventilatory Muscles. Am. J. Respir. Crit. Care Med. 2002, 165, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Henríquez-Olguín, C.; Renani, L.B.; Arab-Ceschia, L.; Raun, S.H.; Bhatia, A.; Li, Z.; Knudsen, J.R.; Holmdahl, R.; Jensen, T.E. Adaptations to High-Intensity Interval Training in Skeletal Muscle Require NADPH Oxidase 2. Redox Biol. 2019, 24, 101188. [Google Scholar] [CrossRef] [PubMed]
- Wojtovich, A.P.; Berry, B.J.; Galkin, A. Redox Signaling through Compartmentalization of Reactive Oxygen Species: Implications for Health and Disease. Antioxid. Redox Signal. 2019, 31, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Specht, K.S.; Kant, S.; Addington, A.K.; McMillan, R.P.; Hulver, M.W.; Learnard, H.; Campbell, M.; Donnelly, S.R.; Caliz, A.D.; Pei, Y.; et al. Nox4 Mediates Skeletal Muscle Metabolic Responses to Exercise. Mol. Metab. 2021, 45, 101160. [Google Scholar] [CrossRef]
- Gong, M.C.; Arbogast, S.; Guo, Z.; Mathenia, J.; Su, W.; Reid, M.B. Calcium-Independent Phospholipase A2 Modulates Cytosolic Oxidant Activity and Contractile Function in Murine Skeletal Muscle Cells. J. Appl. Physiol. 2006, 100, 399–405. [Google Scholar] [CrossRef]
- Nethery, D.; Stofan, D.; Callahan, L.; DiMarco, A.; Supinski, G. Formation of Reactive Oxygen Species by the Contracting Diaphragm Is PLA2 Dependent. J. Appl. Physiol. 1999, 87, 792–800. [Google Scholar] [CrossRef]
- Zhao, X.; Bey, E.A.; Wientjes, F.B.; Cathcart, M.K. Cytosolic Phospholipase A2 (CPLA2) Regulation of Human Monocyte NADPH Oxidase Activity: CPLA2 Affects Translocation but Not Phosphorylation of P67phox and P47phox. J. Biol. Chem. 2002, 277, 25385–25392. [Google Scholar] [CrossRef]
- Zuo, L.; Christofi, F.L.; Wright, V.P.; Bao, S.; Clanton, T.L. Lipoxygenase-Dependent Superoxide Release in Skeletal Muscle. J. Appl. Physiol. 2004, 97, 661–668. [Google Scholar] [CrossRef]
- Gomez-Cabrera, M.C.; Borrás, C.; Pallardo, F.V.; Sastre, J.; Ji, L.L.; Viña, J. Decreasing Xanthine Oxidase-Mediated Oxidative Stress Prevents Useful Cellular Adaptations to Exercise in Rats. J. Physiol. 2005, 567, 113–120. [Google Scholar] [CrossRef]
- Sutkowy, P.; Wróblewska, J.; Wróblewski, M.; Nuszkiewicz, J.; Modrzejewska, M.; Woźniak, A. The Impact of Exercise on Redox Equilibrium in Cardiovascular Diseases. J. Clin. Med. 2022, 11, 4833. [Google Scholar] [CrossRef]
- Tidball, J.G.; Wehling-Henricks, M. Nitric Oxide Synthase Deficiency and the Pathophysiology of Muscular Dystrophy. J. Physiol. 2014, 592, 4627–4638. [Google Scholar] [CrossRef]
- Joyner, M.J.; Coyle, E.F. Endurance Exercise Performance: The Physiology of Champions. J. Physiol. 2008, 586, 35–44. [Google Scholar] [CrossRef]
- Cheng, A.J.; Yamada, T.; Rassier, D.E.; Andersson, D.C.; Westerblad, H.; Lanner, J.T. Reactive Oxygen/Nitrogen Species and Contractile Function in Skeletal Muscle during Fatigue and Recovery. J. Physiol. 2016, 594, 5149–5160. [Google Scholar] [CrossRef]
- Kawamura, T.; Muraoka, I. Exercise-Induced Oxidative Stress and the Effects of Antioxidant Intake from a Physiological Viewpoint. Antioxidants 2018, 7, 119. [Google Scholar] [CrossRef]
- Beckman, J.S.; Koppenol, W.H. Nitric Oxide, Superoxide, and Peroxynitrite: The Good, the Bad, and the Ugly. Am. J. Physiol.-Cell Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef]
- Powers, S.K.; Jackson, M.J. Exercise-Induced Oxidative Stress: Cellular Mechanisms and Impact on Muscle Force Production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef]
- Wiecek, M.; Maciejczyk, M.; Szymura, J.; Kantorowicz, M.; Szygula, Z. Impact of Single Anaerobic Exercise on Delayed Activation of Endothelial Xanthine Oxidase in Men and Women. Redox Rep. 2017, 22, 367–376. [Google Scholar] [CrossRef]
- Alessio, H.M.; Hagerman, A.E.; Fulkerson, B.K.; Ambrose, J.; Rice, R.E.; Wiley, R.L. Generation of Reactive Oxygen Species after Exhaustive Aerobic and Isometric Exercise. Med. Sci. Sport. Exerc. 2000, 32, 1576–1581. [Google Scholar] [CrossRef]
- Groussard, C.; Rannou-Bekono, F.; Machefer, G.; Chevanne, M.; Vincent, S.; Sergent, O.; Cillard, J.; Gratas-Delamarche, A. Changes in Blood Lipid Peroxidation Markers and Antioxidants after a Single Sprint Anaerobic Exercise. Eur. J. Appl. Physiol. 2003, 89, 14–20. [Google Scholar] [CrossRef]
- He, F.; Li, J.; Liu, Z.; Chuang, C.C.; Yang, W.; Zuo, L. Redox Mechanism of Reactive Oxygen Species in Exercise. Front. Physiol. 2016, 7, e0185993. [Google Scholar] [CrossRef] [PubMed]
- Morales-Alamo, D.; Calbet, J.A.L. Free Radicals and Sprint Exercise in Humans. Free Radic. Res. 2014, 48, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Asano, K.; Inoue, M.; Kizaki, T.; Oh-Ishi, S.; Suzuki, K.; Taniguchi, N.; Ohno, H. Superoxide Dismutase Derivative Reduces Oxidative Damage in Skeletal Muscle of Rats during Exhaustive Exercise. J. Appl. Physiol. 1995, 79, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Sloboda, D.D.; Brooks, S.V. Reactive Oxygen Species Generation Is Not Different during Isometric and Lengthening Contractions of Mouse Muscle. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2013, 305, R832. [Google Scholar] [CrossRef] [PubMed]
- Pye, D.; Palomero, J.; Kabayo, T.; Jackson, M.J. Real-Time Measurement of Nitric Oxide in Single Mature Mouse Skeletal Muscle Fibres during Contractions. J. Physiol. 2007, 581, 309–318. [Google Scholar] [CrossRef]
- Steinbacher, P.; Eckl, P. Impact of Oxidative Stress on Exercising Skeletal Muscle. Biomolecules 2015, 5, 356–377. [Google Scholar] [CrossRef]
- Vincent, H.K.; Powers, S.K.; Stewart, D.J.; Demirel, H.A.; Shanely, R.A.; Naito, H. Short-Term Exercise Training Improves Diaphragm Antioxidant Capacity and Endurance. Eur. J. Appl. Physiol. Occup. Physiol. 2000, 81, 67–74. [Google Scholar] [CrossRef]
- Vincent, H.K.; Powers, S.K.; Demirel, H.A.; Coombes, J.S.; Naito, H. Exercise Training Protects against Contraction-Induced Lipid Peroxidation in the Diaphragm. Eur. J. Appl. Physiol. Occup. Physiol. 1999, 79, 268–273. [Google Scholar] [CrossRef]
- García, J.J.; Berzosa, C.; Cebrián, I.; Fuentes-Broto, L.; Gómez-Trullén, E.; Piedrafita, E.; Martínez-Ballarín, E.; López-Pingarrn, L.; Reiter, R.J. Acute Exercise Increases Plasma Total Antioxidant Status and Antioxidant Enzyme Activities in Untrained Men. J. Biomed. Biotechnol. 2011, 2011, 540458. [Google Scholar] [CrossRef]
- Ferraro, E.; Giammarioli, A.M.; Chiandotto, S.; Spoletini, I.; Rosano, G. Exercise-Induced Skeletal Muscle Remodeling and Metabolic Adaptation: Redox Signaling and Role of Autophagy. Antioxid. Redox Signal. 2014, 21, 154–176. [Google Scholar] [CrossRef]
- Larkin, L.M.; Davis, C.S.; Sims-Robinson, C.; Kostrominova, T.Y.; van Remmen, H.; Richardson, A.; Feldman, E.L.; Brooks, S.V. Skeletal Muscle Weakness Due to Deficiency of CuZn-Superoxide Dismutase Is Associated with Loss of Functional Innervation. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2011, 301, R1400–R1407. [Google Scholar] [CrossRef]
- Sakellariou, G.K.; McDonagh, B.; Porter, H.; Giakoumaki, I.I.; Earl, K.E.; Nye, G.A.; Vasilaki, A.; Brooks, S.V.; Richardson, A.; Van Remmen, H.; et al. Comparison of Whole Body SOD1 Knockout with Muscle-Specific SOD1 Knockout Mice Reveals a Role for Nerve Redox Signaling in Regulation of Degenerative Pathways in Skeletal Muscle. Antioxid. Redox Signal. 2018, 28, 275–295. [Google Scholar] [CrossRef]
- Kuwahara, H.; Horie, T.; Ishikawa, S.; Tsuda, C.; Kawakami, S.; Noda, Y.; Kaneko, T.; Tahara, S.; Tachibana, T.; Okabe, M.; et al. Oxidative Stress in Skeletal Muscle Causes Severe Disturbance of Exercise Activity without Muscle Atrophy. Free Radic. Biol. Med. 2010, 48, 1252–1262. [Google Scholar] [CrossRef]
- Brooks, S.V.; Vasilaki, A.; Larkin, L.M.; McArdle, A.; Jackson, M.J. Repeated Bouts of Aerobic Exercise Lead to Reductions in Skeletal Muscle Free Radical Generation and Nuclear Factor ΚB Activation. J. Physiol. 2008, 586, 3979–3990. [Google Scholar] [CrossRef]
- Liberali, R.; Wilhelm Filho, D.; Petroski, E.L. Aerobic and Anaerobic Training Sessions Promote Antioxidant Changes in Young Male Soccer Players. Med. Express 2016, 3. [Google Scholar] [CrossRef]
- Xu, H.; Ranjit, R.; Richardson, A.; Van Remmen, H. Muscle Mitochondrial Catalase Expression Prevents Neuromuscular Junction Disruption, Atrophy, and Weakness in a Mouse Model of Accelerated Sarcopenia. J. Cachexia. Sarcopenia Muscle 2021, 12, 1582–1596. [Google Scholar] [CrossRef]
- McClung, J.M.; DeRuisseau, K.C.; Whidden, M.A.; Van Remmen, H.; Richardson, A.; Song, W.; Vrabas, I.S.; Powers, S.K. Overexpression of Antioxidant Enzymes in Diaphragm Muscle Does Not Alter Contraction-Induced Fatigue or Recovery. Exp. Physiol. 2010, 95, 222–231. [Google Scholar] [CrossRef]
- Jacob, C. A Scent of Therapy: Pharmacological Implications of Natural Products Containing Redox-Active Sulfur Atoms. Nat. Prod. Rep. 2006, 23, 851–863. [Google Scholar] [CrossRef]
- Elokda, A.S.; Nielsen, D.H. Effects of Exercise Training on the Glutathione Antioxidant System. Eur. J. Prev. Cardiol. 2007, 14, 630–637. [Google Scholar] [CrossRef]
- Seifi-Skishahr, F.; Damirchi, A.; Farjaminezhad, M.; Babaei, P. Physical Training Status Determines Oxidative Stress and Redox Changes in Response to an Acute Aerobic Exercise. Biochem. Res. Int. 2016, 2016, 3757623. [Google Scholar] [CrossRef]
- Margonis, K.; Fatouros, I.G.; Jamurtas, A.Z.; Nikolaidis, M.G.; Douroudos, I.; Chatzinikolaou, A.; Mitrakou, A.; Mastorakos, G.; Papassotiriou, I.; Taxildaris, K.; et al. Oxidative Stress Biomarkers Responses to Physical Overtraining: Implications for Diagnosis. Free Radic. Biol. Med. 2007, 43, 901–910. [Google Scholar] [CrossRef]
- Steinberg, J.G.; Delliaux, S.; Jammes, Y. Reliability of Different Blood Indices to Explore the Oxidative Stress in Response to Maximal Cycling and Static Exercises. Clin. Physiol. Funct. Imaging 2006, 26, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Kerksick, C.; Willoughby, D. The Antioxidant Role of Glutathione and N-Acetyl-Cysteine Supplements and Exercise-Induced Oxidative Stress. J. Int. Soc. Sport. Nutr. 2005, 2, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Layec, G.; Blain, G.M.; Rossman, M.J.; Park, S.Y.; Hart, C.R.; Trinity, J.D.; Gifford, J.R.; Sidhu, S.K.; Weavil, J.C.; Hureau, T.J.; et al. Acute High-Intensity Exercise Impairs Skeletal Muscle Respiratory Capacity. Med. Sci. Sport. Exerc. 2018, 50, 2409–2417. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Criswell, D.; Lawler, J.; Li Li, J.; Martin, D.; Herb, R.A.; Dudley, G. Influence of Exercise and Fiber Type on Antioxidant Enzyme Activity in Rat Skeletal Muscle. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 1994, 266, R375–R380. [Google Scholar] [CrossRef]
- Mollica, J.P.; Dutka, T.L.; Merry, T.L.; Lamboley, C.R.; Mcconell, G.K.; Mckenna, M.J.; Murphy, R.M.; Lamb, G.D. S-Glutathionylation of Troponin I (Fast) Increases Contractile Apparatus Ca2+ Sensitivity in Fast-Twitch Muscle Fibres of Rats and Humans. J. Physiol. 2012, 590, 1443–1463. [Google Scholar] [CrossRef]
- Lamboley, C.R.; Rouffet, D.M.; Dutka, T.L.; McKenna, M.J.; Lamb, G.D. Effects of High-Intensity Intermittent Exercise on the Contractile Properties of Human Type I and Type II Skeletal Muscle Fibers. J. Appl. Physiol. 2020, 128, 1207–1216. [Google Scholar] [CrossRef]
- Dutka, T.L.; Mollica, J.P.; Lamboley, C.R.; Weerakkody, V.C.; Greening, D.W.; Posterino, G.S.; Murphy, R.M.; Lamb, G.D. S-Nitrosylation and S-Glutathionylation of Cys134 on Troponin I Have Opposing Competitive Actions on Ca2+ Sensitivity in Rat Fast-Twitch Muscle Fibers. Am. J. Physiol.-Cell Physiol. 2017, 312, C316–C327. [Google Scholar] [CrossRef]
- Christiansen, D.; Eibye, K.H.; Rasmussen, V.; Voldbye, H.M.; Thomassen, M.; Nyberg, M.; Gunnarsson, T.G.P.; Skovgaard, C.; Lindskrog, M.S.; Bishop, D.J.; et al. Cycling with Blood Flow Restriction Improves Performance and Muscle K+ Regulation and Alters the Effect of Anti-Oxidant Infusion in Humans. J. Physiol. 2019, 597, 2421–2444. [Google Scholar] [CrossRef]
- Pearson, T.; Kabayo, T.; Ng, R.; Chamberlain, J.; McArdle, A.; Jackson, M.J. Skeletal Muscle Contractions Induce Acute Changes in Cytosolic Superoxide, but Slower Responses in Mitochondrial Superoxide and Cellular Hydrogen Peroxide. PLoS ONE 2014, 9, e96378. [Google Scholar] [CrossRef]
- Balsera, M.; Buchanan, B.B. Evolution of the Thioredoxin System as a Step Enabling Adaptation to Oxidative Stress. Free Radic. Biol. Med. 2019, 140, 28–35. [Google Scholar] [CrossRef]
- Radak, Z.; Zhao, Z.; Koltai, E.; Ohno, H.; Atalay, M. Oxygen Consumption and Usage during Physical Exercise: The Balance between Oxidative Stress and ROS-Dependent Adaptive Signaling. Antioxid. Redox Signal. 2013, 18, 1208–1246. [Google Scholar] [CrossRef]
- Dimauro, I.; Pearson, T.; Caporossi, D.; Jackson, M.J. In Vitro Susceptibility of Thioredoxins and Glutathione to Redox Modification and Aging-Related Changes in Skeletal Muscle. Free Radic. Biol. Med. 2012, 53, 2017–2027. [Google Scholar] [CrossRef]
- Chaves, A.B.; Miranda, E.R.; Mey, J.T.; Blackburn, B.K.; Fuller, K.N.Z.; Stearns, B.; Ludlow, A.; Williamson, D.L.; Houmard, J.A.; Haus, J.M. Exercise Reduces the Protein Abundance of TXNIP and Its Interacting Partner REDD1 in Skeletal Muscle: Potential Role for a PKA-Mediated Mechanism. J. Appl. Physiol. 2022, 132, 357–366. [Google Scholar] [CrossRef]
- Trewin, A.J.; Parker, L.; Shaw, C.S.; Hiam, D.S.; Garnham, A.; Levinger, I.; McConell, G.K.; Stepto, N.K. Acute HIIE Elicits Similar Changes in Human Skeletal Muscle Mitochondrial H2O2 Release, Respiration, and Cell Signaling as Endurance Exercise Even with Less Work. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2018, 315, R1003–R1016. [Google Scholar] [CrossRef]
- Manabe, Y.; Takagi, M.; Nakamura-Yamada, M.; Goto-Inoue, N.; Taoka, M.; Isobe, T.; Fujii, N.L. Redox Proteins Are Constitutively Secreted by Skeletal Muscle. J. Physiol. Sci. 2014, 64, 401–409. [Google Scholar] [CrossRef]
- Wadley, A.J.; Chen, Y.W.; Bennett, S.J.; Lip, G.Y.H.; Turner, J.E.; Fisher, J.P.; Aldred, S. Monitoring Changes in Thioredoxin and Over-Oxidised Peroxiredoxin in Response to Exercise in Humans. Free Radic. Res. 2015, 49, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Wadley, A.J.; Aldred, S.; Coles, S.J. An Unexplored Role for Peroxiredoxin in Exercise-Induced Redox Signalling? Redox Biol. 2016, 8, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.P.; Shin, Y.J.; Cho, S.C.; Lee, S.M.; Bahn, Y.J.; Kim, J.Y.; Kwon, E.S.; Jeong, D.Y.; Park, S.C.; Rhee, S.G.; et al. Peroxiredoxin 3 Has a Crucial Role in the Contractile Function of Skeletal Muscle by Regulating Mitochondrial Homeostasis. Free Radic. Biol. Med. 2014, 77, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.; Ranjit, R.; Kneis, P.; Xu, H.; Piekarz, K.M.; Freeman, W.M.; Kinter, M.; Richardson, A.; Ran, Q.; Brooks, S.V.; et al. Scavenging Mitochondrial Hydrogen Peroxide by Peroxiredoxin 3 Overexpression Attenuates Contractile Dysfunction and Muscle Atrophy in a Murine Model of Accelerated Sarcopenia. Aging Cell 2022, 21, e13569. [Google Scholar] [CrossRef] [PubMed]
- Olthoff, J.T.; Lindsay, A.; Abo-Zahrah, R.; Baltgalvis, K.A.; Patrinostro, X.; Belanto, J.J.; Yu, D.Y.; Perrin, B.J.; Garry, D.J.; Rodney, G.G.; et al. Loss of Peroxiredoxin-2 Exacerbates Eccentric Contraction-Induced Force Loss in Dystrophin-Deficient Muscle. Nat. Commun. 2018, 9, 5104. [Google Scholar] [CrossRef]
- Pengam, M.; Amérand, A.; Simon, B.; Guernec, A.; Inizan, M.; Moisan, C. How Do Exercise Training Variables Stimulate Processes Related to Mitochondrial Biogenesis in Slow and Fast Trout Muscle Fibres? Exp. Physiol. 2021, 106, 938–957. [Google Scholar] [CrossRef]
- MacInnis, M.J.; Gibala, M.J. Physiological Adaptations to Interval Training and the Role of Exercise Intensity. J. Physiol. 2017, 595, 2915. [Google Scholar] [CrossRef]
- Bishop, D.J.; Botella, J.; Granata, C. CrossTalk Opposing View: Exercise Training Volume Is More Important than Training Intensity to Promote Increases in Mitochondrial Content. J. Physiol. 2019, 597, 4115–4118. [Google Scholar] [CrossRef]
- Egan, B.; Zierath, J.R. Exercise Metabolism and the Molecular Regulation of Skeletal Muscle Adaptation. Cell Metab. 2013, 17, 162–184. [Google Scholar] [CrossRef]
- Yavari, A.; Javadi, M.; Mirmiran, P.; Bahadoran, Z. Exercise-Induced Oxidative Stress and Dietary Antioxidants. Asian J. Sport. Med. 2015, 6, 24898. [Google Scholar] [CrossRef]
- Ruas, J.L.; White, J.P.; Rao, R.R.; Kleiner, S.; Brannan, K.T.; Harrison, B.C.; Greene, N.P.; Wu, J.; Estall, J.L.; Irving, B.A.; et al. A PGC-1α Isoform Induced by Resistance Training Regulates Skeletal Muscle Hypertrophy. Cell 2012, 151, 1319–1331. [Google Scholar] [CrossRef]
- Irrcher, I.; Ljubicic, V.; Hood, D.A. Interactions between ROS and AMP Kinase Activity in the Regulation of PGC-1α Transcription in Skeletal Muscle Cells. Am. J. Physiol.-Cell Physiol. 2009, 296, C116–C123. [Google Scholar] [CrossRef]
- Christiansen, D.; Murphy, R.M.; Bangsbo, J.; Stathis, C.G.; Bishop, D.J. Increased FXYD1 and PGC-1α MRNA after Blood Flow-Restricted Running Is Related to Fibre Type-Specific AMPK Signalling and Oxidative Stress in Human Muscle. Acta Physiol. 2018, 223, e13045. [Google Scholar] [CrossRef]
- Christiansen, D.; Bishop, D.J. Aerobic-Interval Exercise with Blood Flow Restriction Potentiates Early Markers of Metabolic Health in Man. Acta Physiol. 2022, 234, e13769. [Google Scholar] [CrossRef]
- Thompson, H.S.; Maynard, E.B.; Morales, E.R.; Scordilis, S.P. Exercise-Induced HSP27, HSP70 and MAPK Responses in Human Skeletal Muscle. Acta Physiol. Scand. 2003, 178, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Supruniuk, E.; Mikłosz, A.; Chabowski, A. Pyrroloquinoline Quinone Modifies Lipid Profile, but Not Insulin Sensitivity, of Palmitic Acid-Treated L6 Myotubes. Int. J. Mol. Sci. 2020, 21, 8382. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed]
- Tauffenberger, A.; Magistretti, P.J. Reactive Oxygen Species: Beyond Their Reactive Behavior. Neurochem. Res. 2021, 46, 77–87. [Google Scholar] [CrossRef]
- Gouspillou, G.; Sgarioto, N.; Norris, B.; Barbat-Artigas, S.; Aubertin-Leheudre, M.; Morais, J.A.; Burelle, Y.; Taivassalo, T.; Hepple, R.T. The Relationship between Muscle Fiber Type-Specific PGC-1α Content and Mitochondrial Content Varies between Rodent Models and Humans. PLoS ONE 2014, 9, e103044. [Google Scholar] [CrossRef]
- Grgic, J.; Homolak, J.; Mikulic, P.; Botella, J.; Schoenfeld, B.J. Inducing Hypertrophic Effects of Type I Skeletal Muscle Fibers: A Hypothetical Role of Time under Load in Resistance Training Aimed at Muscular Hypertrophy. Med. Hypotheses 2018, 112, 40–42. [Google Scholar] [CrossRef]
- Yoon, M.S. MTOR as a Key Regulator in Maintaining Skeletal Muscle Mass. Front. Physiol. 2017, 8, 788. [Google Scholar] [CrossRef]
- Gallego-Selles, A.; Martin-Rincon, M.; Martinez-Canton, M.; Perez-Valera, M.; Martín-Rodríguez, S.; Gelabert-Rebato, M.; Santana, A.; Morales-Alamo, D.; Dorado, C.; Calbet, J.A.L. Regulation of Nrf2/Keap1 Signalling in Human Skeletal Muscle during Exercise to Exhaustion in Normoxia, Severe Acute Hypoxia and Post-Exercise Ischaemia: Influence of Metabolite Accumulation and Oxygenation. Redox Biol. 2020, 36, 101627. [Google Scholar] [CrossRef]
- Loh, K.; Deng, H.; Fukushima, A.; Cai, X.; Boivin, B.; Galic, S.; Bruce, C.; Shields, B.J.; Skiba, B.; Ooms, L.M.; et al. Reactive Oxygen Species Enhance Insulin Sensitivity. Cell Metab. 2009, 10, 260–272. [Google Scholar] [CrossRef]
- Uruno, A.; Yagishita, Y.; Katsuoka, F.; Kitajima, Y.; Nunomiya, A.; Nagatomi, R.; Pi, J.; Biswal, S.S.; Yamamoto, M. Nrf2-Mediated Regulation of Skeletal Muscle Glycogen Metabolism. Mol. Cell. Biol. 2016, 36, 1655–1672. [Google Scholar] [CrossRef]
- Sylow, L.; Jensen, T.E.; Kleinert, M.; Mouatt, J.R.; Maarbjerg, S.J.; Jeppesen, J.; Prats, C.; Chiu, T.T.; Boguslavsky, S.; Klip, A.; et al. Rac1 Is a Novel Regulator of Contraction-Stimulated Glucose Uptake in Skeletal Muscle. Diabetes 2013, 62, 1139–1151. [Google Scholar] [CrossRef]
- Xirouchaki, C.E.; Jia, Y.; McGrath, M.J.; Greatorex, S.; Tran, M.; Merry, T.L.; Hong, D.; Eramo, M.J.; Broome, S.C.; Woodhead, J.S.T.; et al. Skeletal Muscle NOX4 Is Required for Adaptive Responses That Prevent Insulin Resistance. Sci. Adv. 2021, 7, 4988. [Google Scholar] [CrossRef]
- Scheele, C.; Nielsen, S.; Pedersen, B.K. ROS and Myokines Promote Muscle Adaptation to Exercise. Trends Endocrinol. Metab. 2009, 20, 95–99. [Google Scholar] [CrossRef]
- Nielsen, A.R.; Mounier, R.; Plomgaard, P.; Mortensen, O.H.; Penkowa, M.; Speerschneider, T.; Pilegaard, H.; Pedersen, B.K. Expression of Interleukin-15 in Human Skeletal Muscle-Effect of Exercise and Muscle Fibre Type Composition. J. Physiol. 2007, 584, 305–312. [Google Scholar] [CrossRef]
- Li, F.; Li, Y.; Tang, Y.; Lin, B.; Kong, X.; Oladele, O.A.; Yin, Y. Protective Effect of Myokine IL-15 against H2O2-Mediated Oxidative Stress in Skeletal Muscle Cells. Mol. Biol. Rep. 2014, 41, 7715–7722. [Google Scholar] [CrossRef]
- Nisoli, E.; Falcone, S.; Tonello, C.; Cozzi, V.; Palomba, L.; Fiorani, M.; Pisconti, A.; Brunelli, S.; Cardile, A.; Francolini, M.; et al. Mitochondrial Biogenesis by NO Yields Functionally Active Mitochondria in Mammals. Proc. Natl. Acad. Sci. USA 2004, 101, 16507–16512. [Google Scholar] [CrossRef]
- Tadashi, S.; Suga, K.; Taisuke, O.; Yokota, T.; Takada, S.; Mochamad, A.S.; Takahashi, M.; Fukushima, A.; Homma, T.; Kadoguchi, T.; et al. Nitric Oxide Is Involved the Beneficial Effects of Exercise Training via Activating Skeletal Muscle Mitochondrial Biogenesis in Mice|Circulation. Available online: https://www.ahajournals.org/doi/10.1161/circ.124.suppl_21.A12803 (accessed on 4 December 2022).
- Hart, J.D.E.; Dulhunty, A.F. Nitric Oxide Activates or Inhibits Skeletal Muscle Ryanodine Receptors Depending on Its Concentration, Membrane Potential and Ligand Binding. J. Membr. Biol. 2000, 173, 227–236. [Google Scholar] [CrossRef]
- Hidalgo, C.; Sánchez, G.; Barrientos, G.; Aracena-Parks, P. A Transverse Tubule NADPH Oxidase Activity Stimulates Calcium Release from Isolated Triads via Ryanodine Receptor Type 1 S-Glutathionylation. J. Biol. Chem. 2006, 281, 26473–26482. [Google Scholar] [CrossRef]
- Witherspoon, J.W.; Meilleur, K.G. Review of RyR1 Pathway and Associated Pathomechanisms. Acta Neuropathol. Commun. 2016, 4, 121. [Google Scholar] [CrossRef]
- Percival, J.M.; Anderson, K.N.E.; Huang, P.; Adams, M.E.; Froehner, S.C. Golgi and Sarcolemmal Neuronal NOS Differentially Regulate Contraction-Induced Fatigue and Vasoconstriction in Exercising Mouse Skeletal Muscle. J. Clin. Investig. 2010, 120, 816–826. [Google Scholar] [CrossRef]
- Sonveaux, P.; Copetti, T.; de Saedeleer, C.J.; Végran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frérart, F.; et al. Targeting the Lactate Transporter MCT1 in Endothelial Cells Inhibits Lactate-Induced HIF-1 Activation and Tumor Angiogenesis. PLoS ONE 2012, 7, e33418. [Google Scholar] [CrossRef] [PubMed]
- Gliemann, L.; Vestergaard Hansen, C.; Rytter, N.; Hellsten, Y. Regulation of Skeletal Muscle Blood Flow during Exercise. Curr. Opin. Physiol. 2019, 10, 146–155. [Google Scholar] [CrossRef]
- Gliemann, L.; Olesen, J.; Biensø, R.S.; Schmidt, J.F.; Akerstrom, T.; Nyberg, M.; Lindqvist, A.; Bangsbo, J.; Hellsten, Y. Resveratrol Modulates the Angiogenic Response to Exercise Training in Skeletal Muscles of Aged Men. Am. J. Physiol.-Heart Circ. Physiol. 2014, 307, H1111–H1119. [Google Scholar] [CrossRef] [PubMed]
- Novelli, G.P.; Bracciotti, G.; Falsini, S. Spin-Trappers and Vitamin E Prolong Endurance to Muscle Fatigue in Mice. Free Radic. Biol. Med. 1990, 8, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, L.; Figueiredo-Freitas, C.; Casimiro-Lopes, G.; Magdesian, M.H.; Assreuy, J.; Sorenson, M.M. Myosin Is Reversibly Inhibited by S-Nitrosylation. Biochem. J. 2009, 424, 221–231. [Google Scholar] [CrossRef]
- Ji, L.L.; Fu, R.; Mitchell, E.W. Glutathione and Antioxidant Enzymes in Skeletal Muscle: Effects of Fiber Type and Exercise Intensity. J. Appl. Physiol. 1992, 73, 1854–1859. [Google Scholar] [CrossRef]
- Fauler, M.; Jurkat-Rott, K.; Lehmann-Horn, F. Membrane Excitability and Excitation-Contraction Uncoupling in Muscle Fatigue. Neuromuscul. Disord. 2012, 22, S162–S167. [Google Scholar] [CrossRef]
- Watanabe, D.; Wada, M.; Hogan, M.; Hepple, R.; Watanabe, D.; Wada, M. Fatigue-Induced Change in T-System Excitability and Its Major Cause in Rat Fast-Twitch Skeletal Muscle In Vivo. J. Physiol. 2020, 598, 5195–5211. [Google Scholar] [CrossRef]
- Moon, Y.; Balke, J.E.; Madorma, D.; Siegel, M.P.; Knowels, G.; Brouckaert, P.; Buys, E.S.; Marcinek, D.J.; Percival, J.M. Nitric Oxide Regulates Skeletal Muscle Fatigue, Fiber Type, Microtubule Organization, and Mitochondrial ATP Synthesis Efficiency Through CGMP-Dependent Mechanisms. Antioxid. Redox Signal. 2017, 26, 966–985. [Google Scholar] [CrossRef]
- McKenna, M.J.; Medved, I.; Goodman, C.A.; Brown, M.J.; Bjorksten, A.R.; Murphy, K.T.; Petersen, A.C.; Sostaric, S.; Gong, X. N-Acetylcysteine Attenuates the Decline in Muscle Na+, K+-Pump Activity and Delays Fatigue during Prolonged Exercise in Humans. J. Physiol. 2006, 576, 279–288. [Google Scholar] [CrossRef]
- Place, N.; Yamada, T.; Zhang, S.J.; Westerblad, H.; Bruton, J.D. High Temperature Does Not Alter Fatigability in Intact Mouse Skeletal Muscle Fibres. J. Physiol. 2009, 587, 4717–4724. [Google Scholar] [CrossRef]
- Xia, R.; Webb, J.A.; Gnall, L.L.M.; Cutler, K.; Abramson, J.J. Skeletal Muscle Sarcoplasmic Reticulum Contains a NADH-Dependent Oxidase That Generates Superoxide. Am. J. Physiol.-Cell Physiol. 2003, 285, C215–C221. [Google Scholar] [CrossRef]
- Marengo, J.J.; Hidalgo, C.; Bull, R. Sulfhydryl Oxidation Modifies the Calcium Dependence of Ryanodine- Sensitive Calcium Channels of Excitable Cells. Biophys. J. 1998, 74, 1263–1277. [Google Scholar] [CrossRef]
- Aghdasi, B.; Reid, M.B.; Hamilton, S.L. Nitric Oxide Protects the Skeletal Muscle Ca2+ Release Channel from Oxidation Induced Activation. J. Biol. Chem. 1997, 272, 25462–25467. [Google Scholar] [CrossRef]
- Favero, T.G.; Zable, A.C.; Abramson, J.J. Hydrogen Peroxide Stimulates the Ca2+ Release Channel from Skeletal Muscle Sarcoplasmic Reticulum. J. Biol. Chem. 1995, 270, 25557–25563. [Google Scholar] [CrossRef]
- Moon, Y.; Cao, Y.; Zhu, J.; Xu, Y.; Balkan, W.; Buys, E.S.; Diaz, F.; Kerrick, W.G.; Hare, J.M.; Percival, J.M. GSNOR Deficiency Enhances In Situ Skeletal Muscle Strength, Fatigue Resistance, and RyR1 S-Nitrosylation Without Impacting Mitochondrial Content and Activity. Antioxid. Redox Signal. 2017, 26, 165–181. [Google Scholar] [CrossRef]
- Van Der Poel, C.; Stephenson, D.G. Effects of Elevated Physiological Temperatures on Sarcoplasmic Reticulum Function in Mechanically Skinned Muscle Fibers of the Rat. Am. J. Physiol.-Cell Physiol. 2007, 293, C133–C141. [Google Scholar] [CrossRef]
- Kobayashi, T.; Kurebayashi, N.; Murayama, T. The Ryanodine Receptor as a Sensor for Intracellular Environments in Muscles. Int. J. Mol. Sci. 2021, 22, 10795. [Google Scholar] [CrossRef]
- Radak, Z.; Ishihara, K.; Tekus, E.; Varga, C.; Posa, A.; Balogh, L.; Boldogh, I.; Koltai, E. Exercise, Oxidants, and Antioxidants Change the Shape of the Bell-Shaped Hormesis Curve. Redox Biol. 2017, 12, 285–290. [Google Scholar] [CrossRef]
- Stammers, A.N.; Susser, S.E.; Hamm, N.C.; Hlynsky, M.W.; Kimber, D.E.; Kehler, D.S.; Duhamel, T.A. The Regulation of Sarco(Endo)Plasmic Reticulum Calcium-ATPases (SERCA)1. Can. J. Physiol. Pharmacol. 2015, 93, 843–854. [Google Scholar] [CrossRef]
- Andrade, F.H.; Reid, M.B.; Allen, D.G.; Westerblad, H. Effect of Hydrogen Peroxide and Dithiothreitol on Contractile Function of Single Skeletal Muscle Fibres from the Mouse. J. Physiol. 1998, 509, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Reardon, T.F.; Allen, D.G. Time to Fatigue Is Increased in Mouse Muscle at 37 °C; the Role of Iron and Reactive Oxygen Species. J. Physiol. 2009, 587, 4705–4716. [Google Scholar] [CrossRef] [PubMed]
- Darnley, G.M.; Duke, A.M.; Steele, D.S.; MacFarlane, N.G. Effects of Reactive Oxygen Species on Aspects of Excitation-Contraction Coupling in Chemically Skinned Rabbit Diaphragm Muscle Fibres. Exp. Physiol. 2001, 86, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Reardon, T.F.; Allen, D.G. Iron Injections in Mice Increase Skeletal Muscle Iron Content, Induce Oxidative Stress and Reduce Exercise Performance. Exp. Physiol. 2009, 94, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.M.; Dutka, T.L.; Lamb, G.D. Hydroxyl Radical and Glutathione Interactions Alter Calcium Sensitivity and Maximum Force of the Contractile Apparatus in Rat Skeletal Muscle Fibres. J. Physiol. 2008, 586, 2203–2216. [Google Scholar] [CrossRef]
- Spencer, T.; Posterino, G.S. Sequential Effects of GSNO and H2O2 on the Ca 2+ Sensitivity of the Contractile Apparatus of Fast- and Slow-Twitch Skeletal Muscle Fibers from the Rat. Am. J. Physiol.-Cell Physiol. 2009, 296, 1015–1023. [Google Scholar] [CrossRef]
- Bruton, J.D.; Place, N.; Yamada, T.; Silva, J.P.; Andrade, F.H.; Dahlstedt, A.J.; Zhang, S.J.; Katz, A.; Larsson, N.G.; Westerblad, H. Reactive Oxygen Species and Fatigue-Induced Prolonged Low-Frequency Force Depression in Skeletal Muscle Fibres of Rats, Mice and SOD2 Overexpressing Mice. J. Physiol. 2008, 586, 175–184. [Google Scholar] [CrossRef]
- Van der Poel, C.; Stephenson, D.G. Reversible Changes in Ca2+—Activation Properties of Rat Skeletal Muscle Exposed to Elevated Physiological Temperatures. J. Physiol. 2002, 544, 765–776. [Google Scholar] [CrossRef]
- Lee, J.A.; Westerblad, H.; Allen, D.G. Changes in Tetanic and Resting [Ca2+]i during Fatigue and Recovery of Single Muscle Fibres from Xenopus Laevis. J. Physiol. 1991, 433, 307–326. [Google Scholar] [CrossRef]
- Wilson, G.J.; dos Remedios, C.G.; Stephenson, D.G.; Williams, D.A. Effects of Sulphydryl Modification on Skinned Rat Skeletal Muscle Fibres Using 5,5′-dithiobis(2-nitrobenzoic Acid). J. Physiol. 1991, 437, 409–430. [Google Scholar] [CrossRef]
- Prochniewicz, E.; Lowe, D.A.; Spakowicz, D.J.; Higgins, L.A.; O’Conor, K.; Thompson, L.D.V.; Ferrington, D.A.; Thomas, D.D. Functional, Structural, and Chemical Changes in Myosin Associated with Hydrogen Peroxide Treatment of Skeletal Muscle Fibers. Am. J. Physiol.-Cell Physiol. 2008, 294, C613–C626. [Google Scholar] [CrossRef]
- Moen, R.J.; Cornea, S.; Oseid, D.E.; Binder, B.P.; Klein, J.C.; Thomas, D.D. Redox-Sensitive Residue in the Actin-Binding Interface of Myosin. Biochem. Biophys. Res. Commun. 2014, 453, 345–349. [Google Scholar] [CrossRef]
- Yamada, T.; Mishima, T.; Sakamoto, M.; Sugiyama, M.; Matsunaga, S.; Wada, M. Oxidation of Myosin Heavy Chain and Reduction in Force Production in Hyperthyroid Rat Soleus. J. Appl. Physiol. 2006, 100, 1520–1526. [Google Scholar] [CrossRef]
- Li LI, J.; Gomezcabrera, M.; Steinhafel, N.; Vina, J. Acute Exercise Activates Nuclear Factor (NF)-ΚB Signaling Pathway in Rat Skeletal Muscle. FASEB J. 2004, 18, 1499–1506. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.G. Crosstalk of Reactive Oxygen Species and NF-ΚB Signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef]
- VanderVeen, B.N.; Fix, D.K.; Montalvo, R.N.; Counts, B.R.; Smuder, A.J.; Murphy, E.A.; Koh, H.J.; Carson, J.A. The Regulation of Skeletal Muscle Fatigability and Mitochondrial Function by Chronically Elevated Interleukin-6. Exp. Physiol. 2019, 104, 385–397. [Google Scholar] [CrossRef]
- Janssen Duijghuijsen, L.M.; Keijer, J.; Mensink, M.; Lenaerts, K.; Ridder, L.; Nierkens, S.; Kartaram, S.W.; Verschuren, M.C.M.; Pieters, R.H.H.; Bas, R.; et al. Adaptation of Exercise-Induced Stress in Well-Trained Healthy Young Men. Exp. Physiol. 2017, 102, 86–99. [Google Scholar] [CrossRef]
- Jin, C.-H.; Paik, I.-Y.; Kwak, Y.-S.; Jee, Y.-S.; Kim, J.-Y. Exhaustive Submaximal Endurance and Resistance Exercises Induce Temporary Immunosuppression via Physical and Oxidative Stress. J. Exerc. Rehabil. 2015, 11, 198–203. [Google Scholar] [CrossRef]
- Prieto-Hinojosa, A.; Knight, A.; Compton, C.; Gleeson, M.; Travers, P.J. Reduced Thymic Output in Elite Athletes. Brain. Behav. Immun. 2014, 39, 75–79. [Google Scholar] [CrossRef]
- Wang, F.; Wang, X.; Liu, Y.; Zhang, Z. Effects of Exercise-Induced ROS on the Pathophysiological Functions of Skeletal Muscle. Oxid. Med. Cell. Longev. 2021, 2021, 3846122. [Google Scholar] [CrossRef]
- Place, N.; Ivarsson, N.; Venckunas, T.; Neyroud, D.; Brazaitis, M.; Cheng, A.J.; Ochala, J.; Kamandulis, S.; Girard, S.; Volungevičius, G.; et al. Ryanodine Receptor Fragmentation and Sarcoplasmic Reticulum Ca2+ Leak after One Session of High-Intensity Interval Exercise. Proc. Natl. Acad. Sci. USA 2015, 112, 15492–15497. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.M.; Snow, R.J.; Lamb, G.D. μ-Calpain and Calpain-3 Are Not Autolyzed with Exhaustive Exercise in Humans. Am. J. Physiol.-Cell Physiol. 2006, 290, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, H.W.; Powers, S.K. The Role of Calpains in Skeletal Muscle Remodeling with Exercise and Inactivity-Induced Atrophy. Int. J. Sport. Med. 2020, 41, 994–1008. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Zhao, A.; Yan, X.; Gao, H.; Zhang, C.; Liu, X.; Luo, Q.; Xie, F.; Liu, S.; Shi, D. Hepatic AMPK Signaling Activation in Response to Dynamic REDOX Balance Is a Biomarker of Exercise to Improve Blood Glucose Control. eLife 2022, 11, e79939. [Google Scholar] [CrossRef]
- Gomez-Cabrera, M.C.; Domenech, E.; Viña, J. Moderate Exercise Is an Antioxidant: Upregulation of Antioxidant Genes by Training. Free Radic. Biol. Med. 2008, 44, 126–131. [Google Scholar] [CrossRef]
- Trapp, D.; Knez, W.; Sinclair, W. Could a Vegetarian Diet Reduce Exercise-Induced Oxidative Stress? A Review of the Literature. J. Sport. Sci. 2010, 28, 1261–1268. [Google Scholar] [CrossRef]
- Peternelj, T.T.; Coombes, J.S. Antioxidant Supplementation during Exercise Training: Beneficial or Detrimental? Sport. Med. 2011, 41, 1043–1069. [Google Scholar] [CrossRef]
- Nieman, D.C.; Henson, D.A.; McAnulty, S.R.; McAnulty, L.S.; Morrow, J.D.; Ahmed, A.; Heward, C.B. Vitamin E and Immunity after the Kona Triathlon World Championship. Med. Sci. Sport. Exerc. 2004, 36, 1328–1335. [Google Scholar] [CrossRef]
- Sacheck, J.M.; Milbury, P.E.; Cannon, J.G.; Roubenoff, R.; Blumberg, J.B. Effect of Vitamin E and Eccentric Exercise on Selected Biomarkers of Oxidative Stress in Young and Elderly Men. Free Radic. Biol. Med. 2003, 34, 1575–1588. [Google Scholar] [CrossRef]
- Close, G.L.; Ashton, T.; Cable, T.; Doran, D.; Holloway, C.; McArdle, F.; MacLaren, D.P.M. Ascorbic Acid Supplementation Does Not Attenuate Post-Exercise Muscle Soreness Following Muscle-Damaging Exercise but May Delay the Recovery Process. Br. J. Nutr. 2006, 95, 976–981. [Google Scholar] [CrossRef]
- Ristow, M.; Zarse, K.; Oberbach, A.; Klöting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Blüher, M. Antioxidants Prevent Health-Promoting Effects of Physical Exercise in Humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef]
- Paulsen, G.; Cumming, K.T.; Holden, G.; Hallén, J.; Rønnestad, B.R.; Sveen, O.; Skaug, A.; Paur, I.; Bastani, N.E.; Østgaard, H.N.; et al. Vitamin C and E Supplementation Hampers Cellular Adaptation to Endurance Training in Humans: A Double-Blind, Randomised, Controlled Trial. J. Physiol. 2014, 592, 1887–1901. [Google Scholar] [CrossRef]
- Makanae, Y.; Kawada, S.; Sasaki, K.; Nakazato, K.; Ishii, N. Vitamin C Administration Attenuates Overload-Induced Skeletal Muscle Hypertrophy in Rats. Acta Physiol. 2013, 208, 57–65. [Google Scholar] [CrossRef]
- Theodorou, A.A.; Nikolaidis, M.G.; Paschalis, V.; Koutsias, S.; Panayiotou, G.; Fatouros, I.G.; Koutedakis, Y.; Jamurtas, A.Z. No Effect of Antioxidant Supplementation on Muscle Performance and Blood Redox Status Adaptations to Eccentric Training. Am. J. Clin. Nutr. 2011, 93, 1373–1383. [Google Scholar] [CrossRef]
- Richardson, R.S.; Donato, A.J.; Uberoi, A.; Wray, D.W.; Lawrenson, L.; Nishiyama, S.; Bailey, D.M. Exercise-Induced Brachial Artery Vasodilation: Role of Free Radicals. Am. J. Physiol.-Heart Circ. Physiol. 2007, 292, H1516–H1522. [Google Scholar] [CrossRef]
- Wray, D.W.; Uberoi, A.; Lawrenson, L.; Bailey, D.M.; Richardson, R.S. Oral Antioxidants and Cardiovascular Health in the Exercise-Trained and Untrained Elderly: A Radically Different Outcome. Clin. Sci. 2009, 116, 433–441. [Google Scholar] [CrossRef]
- Sindler, A.L.; Delp, M.D.; Reyes, R.; Wu, G.; Muller-Delp, J.M. Effects of Ageing and Exercise Training on ENOS Uncoupling in Skeletal Muscle Resistance Arterioles. J. Physiol. 2009, 587, 3885–3897. [Google Scholar] [CrossRef]
- Rochette, L.; Ghibu, S.; Richard, C.; Zeller, M.; Cottin, Y.; Vergely, C. Direct and Indirect Antioxidant Properties of α-Lipoic Acid and Therapeutic Potential. Mol. Nutr. Food Res. 2013, 57, 114–125. [Google Scholar] [CrossRef]
- Henriksen, E.J. Exercise Training and the Antioxidant α-Lipoic Acid in the Treatment of Insulin Resistance and Type 2 Diabetes. Free Radic. Biol. Med. 2006, 40, 3–12. [Google Scholar] [CrossRef]
- Maciejczyk, M.; Żebrowska, E.; Nesterowicz, M.; Supruniuk, E.; Choromańska, B.; Chabowski, A.; Żendzian-Piotrowska, M.; Zalewska, A. α-Lipoic Acid Reduces Ceramide Synthesis and Neuroinflammation in the Hypothalamus of Insulin-Resistant Rats, While in the Cerebral Cortex Diminishes the β-Amyloid Accumulation. J. Inflamm. Res. 2022, 15, 2295–2312. [Google Scholar] [CrossRef]
- Pingitore, A.; Lima, G.P.P.; Mastorci, F.; Quinones, A.; Iervasi, G.; Vassalle, C. Exercise and Oxidative Stress: Potential Effects of Antioxidant Dietary Strategies in Sports. Nutrition 2015, 31, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Isenmann, E.; Trittel, L.; Diel, P. The Effects of Alpha Lipoic Acid on Muscle Strength Recovery after a Single and a Short-Term Chronic Supplementation—A Study in Healthy Well-Trained Individuals after Intensive Resistance and Endurance Training. J. Int. Soc. Sport. Nutr. 2020, 17, 1–13. [Google Scholar] [CrossRef]
- Coombes, J.S.; Powers, S.K.; Rowell, B.; Hamilton, K.L.; Dodd, S.L.; Shanely, R.A.; Sen, C.K.; Packer, L. Effects of Vitamin E and α-Lipoic Acid on Skeletal Muscle Contractile Properties. J. Appl. Physiol. 2001, 90, 1424–1430. [Google Scholar] [CrossRef] [PubMed]
- Sangsefidi, Z.S.; Yaghoubi, F.; Hajiahmadi, S.; Hosseinzadeh, M. The Effect of Coenzyme Q10 Supplementation on Oxidative Stress: A Systematic Review and Meta-Analysis of Randomized Controlled Clinical Trials. Food Sci. Nutr. 2020, 8, 1766–1776. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Feng, Y.; Chen, G.C.; Qin, L.Q.; Fu, C.L.; Chen, L.H. Effects of Coenzyme Q10 Supplementation on Inflammatory Markers: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Pharmacol. Res. 2017, 119, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Camacho, J.D.; Bernier, M.; López-Lluch, G.; Navas, P. Coenzyme Q10 Supplementation in Aging and Disease. Front. Physiol. 2018, 9, 44. [Google Scholar] [CrossRef]
- Díaz-Castro, J.; Guisado, R.; Kajarabille, N.; García, C.; Guisado, I.M.; De Teresa, C.; Ochoa, J.J. Coenzyme Q10 Supplementation Ameliorates Inflammatory Signaling and Oxidative Stress Associated with Strenuous Exercise. Eur. J. Nutr. 2012, 51, 791–799. [Google Scholar] [CrossRef]
- Gokbel, H.; Turk, S.; Okudan, N.; Atalay, H.; Belviranli, M.; Gaipov, A.; Solak, Y. Effects of Coenzyme Q10 Supplementation on Exercise Performance and Markers of Oxidative Stress in Hemodialysis Patients: A Double-Blind Placebo-Controlled Crossover Trial. Am. J. Ther. 2016, 23, e1736–e1743. [Google Scholar] [CrossRef]
- Mehrabani, S.; Askari, G.; Miraghajani, M.; Tavakoly, R.; Arab, A. Effect of Coenzyme Q10 Supplementation on Fatigue: A Systematic Review of Interventional Studies. Complement. Ther. Med. 2019, 43, 181–187. [Google Scholar] [CrossRef]
- Tsai, I.C.; Hsu, C.W.; Chang, C.H.; Tseng, P.T.; Chang, K.V. Effectiveness of Coenzyme Q10 Supplementation for Reducing Fatigue: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Front. Pharmacol. 2022, 13, 24. [Google Scholar] [CrossRef]
- Glover, E.I.; Martin, J.; Maher, A.; Thornhill, R.E.; Moran, G.R.; Tarnopolsky, M.A. A Randomized Trial of Coenzyme Q10 in Mitochondrial Disorders. Muscle Nerve 2010, 42, 739–748. [Google Scholar] [CrossRef]
- Matuszczak, Y.; Farid, M.; Jones, J.; Lansdowne, S.; Smith, M.A.; Taylor, A.A.; Reid, M.B. Effects of N-Acetylcysteine on Glutathione Oxidation and Fatigue during Handgrip Exercise. Muscle Nerve 2005, 32, 633–638. [Google Scholar] [CrossRef]
- Terrill, J.R.; Radley-Crabb, H.G.; Grounds, M.D.; Arthur, P.G. N-Acetylcysteine Treatment of Dystrophic Mdx Mice Results in Protein Thiol Modifications and Inhibition of Exercise Induced Myofibre Necrosis. Neuromuscul. Disord. 2012, 22, 427–434. [Google Scholar] [CrossRef]
- Supinski, G.S.; Stofan, D.; Ciufo, R.; DiMarco, A. N-Acetylcysteine Administration Alters the Response to Inspiratory Loading in Oxygen-Supplemented Rats. J. Appl. Physiol. 1997, 82, 1119–1125. [Google Scholar] [CrossRef]
- Reid, M.B.; Stokić, D.S.; Koch, S.M.; Khawli, F.A.; Leis, A.A. N-Acetylcysteine Inhibits Muscle Fatigue in Humans. J. Clin. Investig. 1994, 94, 2468. [Google Scholar] [CrossRef]
- Medved, I.; Brown, M.J.; Bjorksten, A.R.; Murphy, K.T.; Petersen, A.C.; Sostaric, S.; Gong, X.; McKenna, M.J. N-Acetylcysteine Enhances Muscle Cysteine and Glutathione Availability and Attenuates Fatigue during Prolonged Exercise in Endurance-Trained Individuals. J. Appl. Physiol. 2004, 97, 1477–1485. [Google Scholar] [CrossRef]
- Medved, I.; Brown, M.J.; Bjorksten, A.R.; McKenna, M.J. Effects of Intravenous N-Acetylcysteine Infusion on Time to Fatigue and Potassium Regulation during Prolonged Cycling Exercise. J. Appl. Physiol. 2004, 96, 211–217. [Google Scholar] [CrossRef]
- Corn, S.D.; Barstow, T.J. Effects of Oral N-Acetylcysteine on Fatigue, Critical Power, and W’ in Exercising Humans. Respir. Physiol. Neurobiol. 2011, 178, 261–268. [Google Scholar] [CrossRef]
- Braakhuis, A.J.; Hopkins, W.G. Impact of Dietary Antioxidants on Sport Performance: A Review. Sport. Med. 2015, 45, 939–955. [Google Scholar] [CrossRef]
- Ryan, M.J.; Jackson, J.R.; Hao, Y.; Leonard, S.S.; Alway, S.E. Inhibition of Xanthine Oxidase Reduces Oxidative Stress and Improves Skeletal Muscle Function in Response to Electrically Stimulated Isometric Contractions in Aged Mice. Free Radic. Biol. Med. 2011, 51, 38–52. [Google Scholar] [CrossRef]
- Gomez-Cabrera, M.C.; Close, G.L.; Kayani, A.; McArdle, A.; Viña, J.; Jackson, M.J. Effect of Xanthine Oxidase-Generated Extracellular Superoxide on Skeletal Muscle Force Generation. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2010, 298, R2–R8. [Google Scholar] [CrossRef] [PubMed]
- Akimoto, T.; Pohnert, S.C.; Li, P.; Zhang, M.; Gumbs, C.; Rosenberg, P.B.; Williams, R.S.; Yan, Z. Exercise Stimulates Pgc-1α Transcription in Skeletal Muscle through Activation of the P38 MAPK Pathway. J. Biol. Chem. 2005, 280, 19587–19593. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; O’Moore, K.M.; Dickman, J.R.; Ji, L.L. Exercise Activation of Muscle Peroxisome Proliferator-Activated Receptor-γ Coactivator-1α Signaling Is Redox Sensitive. Free Radic. Biol. Med. 2009, 47, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Mach, J.; Midgley, A.W.; Dank, S.; Grant, R.S.; Bentley, D.J. The Effect of Antioxidant Supplementation on Fatigue during Exercise: Potential Role for NAD+(H). Nutrients 2010, 2, 319. [Google Scholar] [CrossRef]
- Packer, L.; Rimbach, G.; Virgili, F. Antioxidant Activity and Biologic Properties of a Procyanidin-Rich Extract from Pine (Pinus Maritima) Bark, Pycnogenol. Free Radic. Biol. Med. 1999, 27, 704–724. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Beltrán, C.E.; Calderón-Oliver, M.; Pedraza-Chaverri, J.; Chirino, Y.I. Protective Effect of Sulforaphane against Oxidative Stress: Recent Advances. Exp. Toxicol. Pathol. 2012, 64, 503–508. [Google Scholar] [CrossRef]
- Ruhee, R.T.; Ma, S.; Suzuki, K. Protective Effects of Sulforaphane on Exercise-induced Organ Damage via Inducing Antioxidant Defense Responses. Antioxidants 2020, 9, 136. [Google Scholar] [CrossRef]
- Oh, S.; Komine, S.; Warabi, E.; Akiyama, K.; Ishii, A.; Ishige, K.; Mizokami, Y.; Kuga, K.; Horie, M.; Miwa, Y.; et al. Nuclear Factor (Erythroid Derived 2)-like 2 Activation Increases Exercise Endurance Capacity via Redox Modulation in Skeletal Muscles. Sci. Rep. 2017, 7, 12902. [Google Scholar] [CrossRef]
- Komine, S.; Miura, I.; Miyashita, N.; Oh, S.; Tokinoya, K.; Shoda, J.; Ohmori, H. Effect of a Sulforaphane Supplement on Muscle Soreness and Damage Induced by Eccentric Exercise in Young Adults: A Pilot Study. Physiol. Rep. 2021, 9, e15130. [Google Scholar] [CrossRef]
- Aoi, W.; Ogaya, Y.; Takami, M.; Konishi, T.; Sauchi, Y.; Park, Y.Y.; Wada, S.; Sato, K.; Higashi, A. Glutathione Supplementation Suppresses Muscle Fatigue Induced by Prolonged Exercise via Improved Aerobic Metabolism. J. Int. Soc. Sport. Nutr. 2015, 12, 7. [Google Scholar] [CrossRef]
- Dolinsky, V.W.; Jones, K.E.; Sidhu, R.S.; Haykowsky, M.; Czubryt, M.P.; Gordon, T.; Dyck, J.R.B. Improvements in Skeletal Muscle Strength and Cardiac Function Induced by Resveratrol during Exercise Training Contribute to Enhanced Exercise Performance in Rats. J. Physiol. 2012, 590, 2783–2799. [Google Scholar] [CrossRef]
- Alway, S.E.; McCrory, J.L.; Kearcher, K.; Vickers, A.; Frear, B.; Gilleland, D.L.; Bonner, D.E.; Thomas, J.M.; Donley, D.A.; Lively, M.W.; et al. Resveratrol Enhances Exercise-Induced Cellular and Functional Adaptations of Skeletal Muscle in Older Men and Women. J. Gerontol.-Ser. A Biol. Sci. Med. Sci. 2017, 72, 1595–1606. [Google Scholar] [CrossRef]
- Olesen, J.; Gliemann, L.; Biensø, R.; Schmidt, J.; Hellsten, Y.; Pilegaard, H. Exercise Training, but Not Resveratrol, Improves Metabolic and Inflammatory Status in Skeletal Muscle of Aged Men. J. Physiol. 2014, 592, 1873–1886. [Google Scholar] [CrossRef]
- Gliemann, L.; Schmidt, J.F.; Olesen, J.; Biensø, R.S.; Peronard, S.L.; Grandjean, S.U.; Mortensen, S.P.; Nyberg, M.; Bangsbo, J.; Pilegaard, H.; et al. Resveratrol Blunts the Positive Effects of Exercise Training on Cardiovascular Health in Aged Men. J. Physiol. 2013, 591, 5047–5059. [Google Scholar] [CrossRef]
- Jackson, J.R.; Ryan, M.J.; Alway, S.E. Long-Term Supplementation with Resveratrol Alleviates Oxidative Stress but Does Not Attenuate Sarcopenia in Aged Mice. J. Gerontol.-Ser. A Biol. Sci. Med. Sci. 2011, 66, 751–764. [Google Scholar] [CrossRef]
- Hogan, P.S.; Chen, S.X.; Teh, W.W.; Chib, V.S. Neural Mechanisms Underlying the Effects of Physical Fatigue on Effort-Based Choice. Nat. Commun. 2020, 11, 4026. [Google Scholar] [CrossRef] [PubMed]
- Appell, H.J.; Soares, J.M.C.; Duarte, J.A.R. Exercise, Muscle Damage and Fatigue. Sport. Med. Int. J. Appl. Med. Sci. Sport Exerc. 1992, 13, 108–115. [Google Scholar] [CrossRef]
- Supruniuk, E.; Żebrowska, E.; Chabowski, A. Branched Chain Amino Acids—Friend or Foe in the Control of Energy Substrate Turnover and Insulin Sensitivity? Crit. Rev. Food Sci. Nutr. 2021. ahead of print. [Google Scholar] [CrossRef]
- Connell, C.J.W.; Thompson, B.; Turuwhenua, J.; Srzich, A.; Gant, N. Fatigue-Related Impairments in Oculomotor Control Are Prevented by Norepinephrine-Dopamine Reuptake Inhibition. Sci. Rep. 2017, 7, 42726. [Google Scholar] [CrossRef]
- Martin, K.; Meeusen, R.; Thompson, K.G.; Keegan, R.; Rattray, B. Mental Fatigue Impairs Endurance Performance: A Physiological Explanation. Sport. Med. 2018, 48, 2041–2051. [Google Scholar] [CrossRef]
- Thomas, K.; Elmeua, M.; Howatson, G.; Goodall, S. Intensity-Dependent Contribution of Neuromuscular Fatigue after Constant-Load Cycling. Med. Sci. Sport. Exerc. 2016, 48, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.J.; Chan, H.L.; Huang, Y.Z.; Lin, J.H.; Hsu, H.H.; Chang, Y.J. Mechanism of Fatigue Induced by Different Cycling Paradigms with Equivalent Dosage. Front. Physiol. 2020, 11, 545. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, J.; Marcora, S.; De Pauw, K.; Bailey, S.; Meeusen, R.; Roelands, B. The Effects of Mental Fatigue on Physical Performance: A Systematic Review. Sport. Med. 2017, 47, 1569–1588. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.F.S.; Al-Mubarak, B.; Martel, M.A.; McKay, S.; Wheelan, N.; Hasel, P.; Márkus, N.M.; Baxter, P.; Deighton, R.F.; Serio, A.; et al. Neuronal Development Is Promoted by Weakened Intrinsic Antioxidant Defences due to Epigenetic Repression of Nrf2. Nat. Commun. 2015, 6, 7066. [Google Scholar] [CrossRef] [PubMed]
- Somani, S.M.; Ravi, R.; Rybak, L.P. Effect of Exercise Training on Antioxidant System in Brain Regions of Rat. Pharmacol. Biochem. Behav. 1995, 50, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Marosi, K.; Bori, Z.; Hart, N.; Sárga, L.; Koltai, E.; Radák, Z.; Nyakas, C. Long-Term Exercise Treatment Reduces Oxidative Stress in the Hippocampus of Aging Rats. Neuroscience 2012, 226, 21–28. [Google Scholar] [CrossRef]
- Radak, Z.; Sasvari, M.; Nyakas, C.; Kaneko, T.; Tahara, S.; Ohno, H.; Goto, S. Single Bout of Exercise Eliminates the Immobilization-Induced Oxidative Stress in Rat Brain. Neurochem. Int. 2001, 39, 33–38. [Google Scholar] [CrossRef]
- Coşkun, Ş.; Gönül, B.; Güzel, N.A.; Balabanli, B. The Effects of Vitamin C Supplementation on Oxidative Stress and Antioxidant Content in the Brains of Chronically Exercised Rats. Mol. Cell. Biochem. 2005, 280, 135–138. [Google Scholar] [CrossRef]
- Bruna, B.; Lobos, P.; Herrera-Molina, R.; Hidalgo, C.; Paula-Lima, A.; Adasme, T. The Signaling Pathways Underlying BDNF-Induced Nrf2 Hippocampal Nuclear Translocation Involve ROS, RyR-Mediated Ca2+ Signals, ERK and PI3K. Biochem. Biophys. Res. Commun. 2018, 505, 201–207. [Google Scholar] [CrossRef]
- Azevedo, L.V.D.S.; Pereira, J.R.; Silva Santos, R.M.; Rocha, N.P.; Teixeira, A.L.; Christo, P.P.; Santos, V.R.; Scalzo, P.L. Acute Exercise Increases BDNF Serum Levels in Patients with Parkinson’s Disease Regardless of Depression or Fatigue. Eur. J. Sport Sci. 2022, 22, 1296–1303. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Supruniuk, E.; Górski, J.; Chabowski, A. Endogenous and Exogenous Antioxidants in Skeletal Muscle Fatigue Development during Exercise. Antioxidants 2023, 12, 501. https://doi.org/10.3390/antiox12020501
Supruniuk E, Górski J, Chabowski A. Endogenous and Exogenous Antioxidants in Skeletal Muscle Fatigue Development during Exercise. Antioxidants. 2023; 12(2):501. https://doi.org/10.3390/antiox12020501
Chicago/Turabian StyleSupruniuk, Elżbieta, Jan Górski, and Adrian Chabowski. 2023. "Endogenous and Exogenous Antioxidants in Skeletal Muscle Fatigue Development during Exercise" Antioxidants 12, no. 2: 501. https://doi.org/10.3390/antiox12020501
APA StyleSupruniuk, E., Górski, J., & Chabowski, A. (2023). Endogenous and Exogenous Antioxidants in Skeletal Muscle Fatigue Development during Exercise. Antioxidants, 12(2), 501. https://doi.org/10.3390/antiox12020501